Rapid chemical decontamination of infectious CJD and scrapie particles parallels treatments known to disrupt microbes and biofilms (original) (raw)
Abstract
Neurodegenerative human CJD and sheep scrapie are diseases caused by several different transmissible encephalopathy (TSE) agents. These infectious agents provoke innate immune responses in the brain, including late-onset abnormal prion protein (PrP-res) amyloid. Agent particles that lack detectable PrP sequences by deep proteomic analysis are highly infectious. Yet these agents, and their unusual resistance to denaturation, are often evaluated by PrP amyloid disruption. To reexamine the intrinsic resistance of TSE agents to denaturation, a paradigm for less resistant viruses and microbes, we developed a rapid and reproducible high yield agent isolation procedure from cultured cells that minimized PrP amyloid and other cellular proteins. Monotypic neuronal GT1 cells infected with the FU-CJD or 22L scrapie agents do not have complex brain changes that can camouflage infectious particles and prevent their disruption, and there are only 2 reports on infectious titers of any human CJD strain treated with chemical denaturants. Infectious titers of both CJD and scrapie were reduced by >4 logs with Thiourea-urea, a treatment not previously tested. A mere 5 min exposure to 4M GdnHCl at 22°C reduced infectivity by >5 logs. Infectious 22L particles were significantly more sensitive to denaturation than FU-CJD particles. A protocol using sonication with these chemical treatments may effectively decontaminate complicated instruments, such as duodenoscopes that harbor additional virulent microbes and biofilms associated with recent iatrogenic infections.
Keywords: CJD, decontamination, Guanidine HCl, surgical instruments, Thiourea, Urea, viruses
Abbreviations
TSEs
Transmissible encephalopathies
CJD
Creutzfeldt-Jakob Disease
BSE
Bovine Spongiform Encephalopathy
ThU
Thiourea
GdnSCN
Guanidine thiocyanate
PK
Proteinase K
PrP
Prion protein
Introduction
Prusiner introduced and expanded the hypothesis that a host membrane protein of 34 kd (known as prion protein or PrP) spontaneously misfolds into an amyloid or “prion” form that is the causal infectious agent of transmissible encephalopathies (TSE), including human Creutzfeldt-Jakob Disease (CJD), sheep scrapie, and “mad cow” disease (BSE).1,2 There are at least 4 distinct infectious human CJD agents that show major differences in incubation time and neuropathology when compared in normal wt mice.3 These 4 agents are the worldwide sporadic CJD (sCJD) agent, the more virulent Asiatic CJD strain (FU-CJD) that has not been identified in the Western hemisphere, the kuru agent of New Guinea, and variant CJD (vCJD) caused by the epidemic UK BSE agent. Misfolded prion amyloid resists limited proteinase K (PK) digestion (PrP-res), and presumably encodes all the different TSE agent-strains that infect mammals.2 This “protein-only” hypothesis has encouraged numerous detailed studies of PrP structure, with the presumption that it will reflect both intrinsic strain-specific agent properties as well as quantitative infectious titer changes. Our view is that a separate foreign viral particle of ~22 nm binds to its essential PrP membrane receptor to induce abnormal PrP-res amyloid.4
While PrP-res is an excellent surrogate marker for agent-induced disease in established animal and culture models, e.g.,3,5-8 strain-specific PrP folding patterns have not been apparent (reviewed in ref 9). Moreover, PrP itself is insufficient for infection. Misfolded recombinant PrP is not reproducibly infectious in animal or culture assays, even though massive amounts of PrP-res amyloid are readily generated in a test tube.4,10-13 Huge increases in PrP-res can also coincide with a >5 log loss of FU-CJD agent from chronically infected living cell cultures.12 This PrP-res is indistinguishable from the “infectious” PrP-res form, yet it is unable to produce any detectable agent in sensitive infectivity culture assays. PrP-res amyloid is also ultrastructurally separate from virus-like 20-25 nm dense particles.9,14-17 Additionally, destruction of PrP further underscores a biochemical separation between PrP-res and the infectious agent; protease or PK digestion of all detectable PrP did not significantly reduce either BSE or FU-CJD brain titers.18-20 A variety of other proteases also has shown little detrimental effect on 263K or RML scrapie agent brain titers in different material contexts.21,22 Indeed, PK + Pronase yielded negligible destruction of RML infectivity without first pre-heating samples at 100°C for 15 min in 4% SDS.22 In a physiological context, TSE agents, like many viruses, can survive proteolytic digestive juices whereas PrP does not.23,24
The above data emphasizes the critical importance of infectivity measurements for assessing agent denaturation and effective decontamination. Because there are many known discrepancies between PrP gel band patterns and infectious titers in different tissues, animal models, and material contexts,4,25-27 we sought to further define chemical treatments that can prevent iatrogenic infection by evaluating the titer of isolated agent particles with reduced PrP and PrP-res amyloid that can trap and/or impede chemical denaturation of infectious particles.
Decontamination has become a critical public health problem with the increasing use of new mucosal and gastrointestinal (GI) instrument probes. A GI transmitted epidemic of BSE was well underway in the UK by 1996 when the first human cases of BSE were recognized (in humans it is called vCJD). Although the BSE epidemic dramatically subsided after culling ~1 million cows, endemic sheep scrapie remains prevalent, and decontamination of farms with chlorine has failed to prevent de novo scrapie infections within 18 months.28 A long asymptomatic phase after exposure is typical for TSE infections, and even after direct brain surgery, a human transmission from sCJD contaminated electrodes took 15 months to become clinically evident.29,30 Because of the enormous waste from BSE infected cows in the UK, and the need to effectively decontaminate such materials, we previously tested chemical treatments that disrupt viruses for their ability to destroy hamster brain homogenates with high sCJD infectivity titers. Even without heat, 4M Guanidine thiocyanate (GdnSCN) reduced the infectious titer by >5 logs in animal assays,31 and longer hamster observation showed ≥7 logs of infectivity were destroyed. This GdnSCN treatment was considerably more effective than an SDS-acetic acid-65°C treatment of sCJD in transgenic mice overexpressing PrP,32 the only other titered CJD decontamination study we can find. GdnSCN did not corrode small fine surgical instruments, unlike other recommended sterilizing procedures, such as immersion in 1N NaOH, 5-10% bleach, or extended autoclaving. GdnSCN can be toxic, and is not easy to safely dispose of. Hence we sought improved decontamination methods, including those that might be effective for new complex 3-D instruments bearing cameras and non-disposable parts.
Recent problematic outbreaks of deadly infections have been traced to incompletely sterilized instruments such as duodenoscopes that are exposed to large numbers of microorganisms and biofilms, and in the USA these are used ~500,000 times per year.33 Such expensive instruments are destroyed by heat, bleach, 1M NaOH and other recommended TSE decontamination procedures, and standard decontamination with aldehydes is not sufficient to inactivate either conventional microorganisms or TSE agents.34 Because BSE as well as other TSEs, including FU-CJD, also transmit via mucus membranes and the GI tract,23,35,36 rapid non-destructive liquid treatments that can simultaneously prevent a variety of infectious agents are desirable.
TSE agents are believed to be extraordinarily resistant to inactivation as compared to other infectious agents such as bacteria and viruses, and hence their destruction may be extended to other viruses that are difficult to sterilize in a hospital setting, such as Hepatitis B.31,37 There is only one report since 1993 outside our laboratory on quantitative titer reductions of any TSE agent by guanidine salts, and there are none on urea or thiourea. Moreover, in tests of proteases and/or detergents, it is almost impossible to compare or confirm results because the preparations tested are so different from each other. This includes brain homogenates, PrP-enriched fibrils,38 the presence of diverse detergents, and material dried on wires8 in viruses in addition to the variety of agent strains and species used. Even in a single laboratory very discordant data have been reported. For example, boiling in SDS did not decrease titer39 and also produced a >2 log (>99%) titer loss40 in comparable PrP-res preparations from 263K scrapie infected hamster brain. By comparison, we found an ∼3 log titer loss in sCJD brain particles by animal assay after 5 min at 95°C in 1% SDS, and this treatment released uninfected intact PrP amyloid multimers as well as nucleic acids and other proteins.41 In sum, the agent strain and the agent’s material context can have a large impact on effective decontamination, and this underscores the importance of first testing isolated infectious particles for their intrinsic resistance.
We found no reports on chemical treatments of highly infectious particles with substantially reduced host components, including PrP, that confirm the extraordinary resistance ascribed to TSE agents. To further investigate agent particle structure and improve universal decontamination methods, we developed neuronal GT1 mouse cell cultures that stably support a variety of different TSE agent-strains.3,42 We also devised a rapid and reproducible quantitative end-point tissue culture assay for representative CJD and scrapie agents that is only slightly less sensitive (by 0.5 logs or 3-fold) as compared to very expensive end-point animal assays that take ~600 days.7,19 This small difference in end-point sensitivity is statistically and biologically insignificant, especially for the 4 to >5 log (10,000 to >100,000 fold) titer losses reported here. Moreover, despite cell passaging for >1 year, GT1 cells infected with different agents have validated the maintenance of strain-specific characteristics that are completely faithful to the original agent phenotype, i.e., when infected GT1 cell homogenates are re-inoculated in mice they produce the highly specific incubation time and neuropathology for that agent including animal assayed titers.43 Even the in vivo TSE strain-specific prevention of superinfection by a second challenge agent strain is recapitulated in GT1 cell cultures.42
Because GT1 cells are monotypic and show no infection-induced cytotoxic changes, infectious particles can be isolated free of complex brain components and extensive secondary neurodegenerative sequelae. GT1 cells are also ideal for fair comparison of different agent-strains because the species and cell type are kept constant. We here report extensive repeated assays showing reproducible chemical effects on 2 distinct TSE agent-strains that replicate exponentially in GT1 cells: the human FU-CJD and sheep scrapie 22L strains whose high infectious titers were also verified in wt mice.42,43 Although kuru and vCJD (the BSE agent) have also been serially passaged in GT1 cells, and in wt mice, their lower titers are insufficient for evaluating a >5 log loss, even with the new method described here for concentrating infectious particles. Others have also acknowledged this problem in their own BSE studies, and therefore used the RML scrapie agent as a substitute for calculating BSE titer losses.22
To best evaluate intrinsic infectious particle resistance, we first developed a rapid particle isolation procedure for infected cells that removes most PrP and other host proteins while retaining essentially all of the starting cell infectivity. This is documented in multiple experiments below. Several chemical treatments of these particles were effective decontaminants in a short time frame (5 min to 1 hr), and these chemicals should be useful on complex non-disposable instruments. Remarkably, some of these chemicals, without any detergent or heating, are known to disrupt amyloid, biofilms and other virulent bacteria that are difficult to completely remove by mechanical and other currently recommended methods.
Results
Because brain has a high molecular and cellular complexity, and also develops multiple degenerative changes, we isolated infectious particles from monotypic GT1 cells infected with mouse adapted FU-CJD and 22L scrapie agents. Both agents consistently produce high titers. In fact, FU-CJD infected cells produce 10 fold more tissue culture infectious doses (TCID) per cell than FU-CJD infected brain,19 allowing assays over a greater range of agent destruction. After rapidly concentrating infectious particles we tested their resistance to different chemicals. Analyses included multiple repeated quantitative infectivity assays with parallel partitioning/sedimentation changes of PrP/PrP-res and other proteins. Although PrP itself is not infectious, we thought residual particle-linked PrP-res or PrP amyloid solubility might still be a useful indicator of agent disruption, and therefore each was evaluated
Rapid quantitative isolation of infectious particles
Figure 1A outlines the new procedure that reproducibly concentrates infectious particles from cultured cells without requiring sucrose gradient fractionation.44 The procedure outlined can generate large amounts of infectious particles in one working day with minimal equipment, and is not limited to purification of a single agent strain. Briefly: 1) lysis of washed whole cells with 1% Triton X-100 in 8% sucrose followed by a 500 g spin removes most nuclei; 2) a 10,000 g spin of the 500 g supernatant sediments many cellular contaminants, such as lysosomes and membranes, leaving 90% of the starting infectivity in the s10 supernatant; 3) RNase treatment of the s10 supernatant facilitates concentration of all infectious particles by pelleting at 18,000 g x 30 min (p18). An additional sucrose step gradient can further purify >80% of the starting infectious particles from brains, N2a cells, and GT1 cells.20,44
Figure 1.
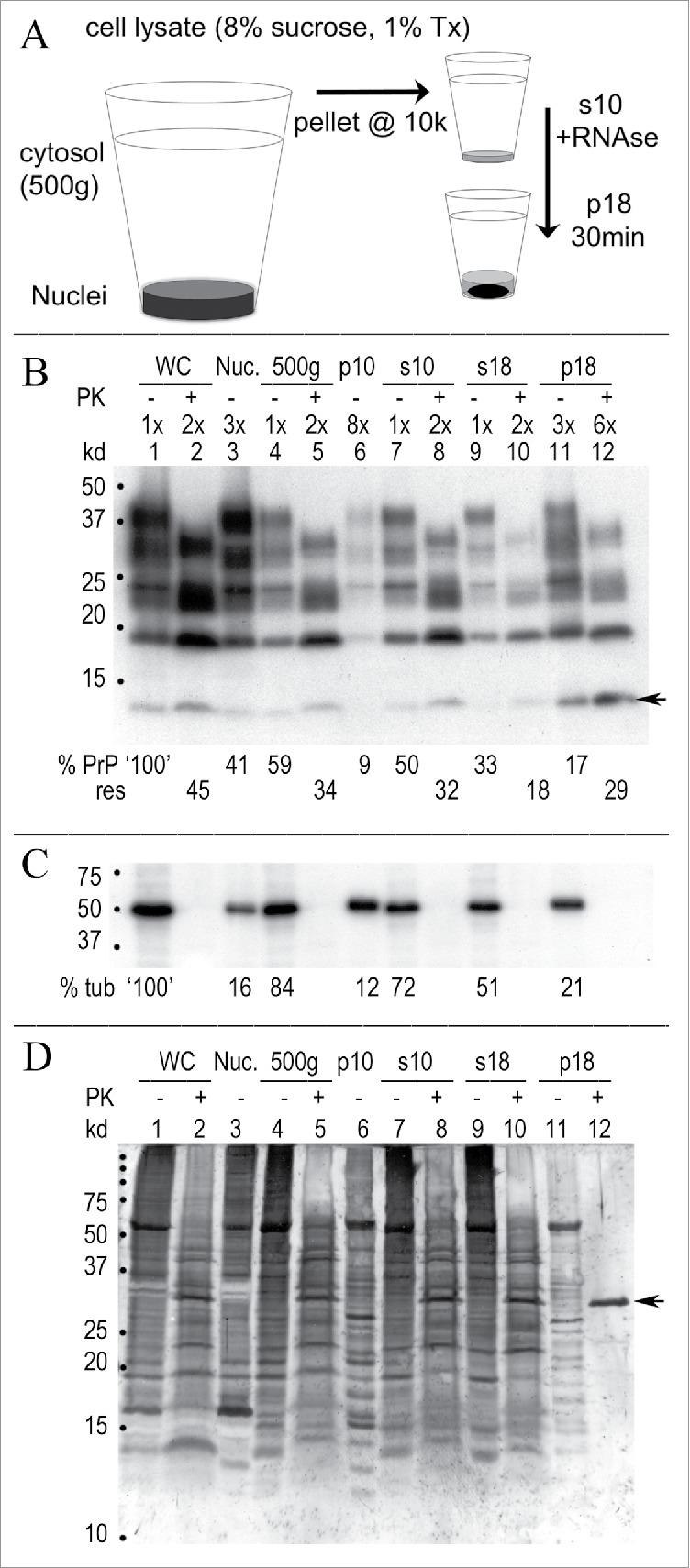
Rapid concentration of FU-CJD infectious particles from GT1 neuronal cells. (A) Simplified diagram of subcellular fractionation that yields highly infectious p18 particles with reduced protein, PrP/PrP-res and nucleic acids. (B) Representative Western blot of sequential purification steps where the cell equivalent loads are indicated at the top of each lane. The % of starting whole cell PrP is calculated at the bottom of each lane and the % of that fraction that is PrP-res (adjacent +PK lanes) is indicated below this. For example, PrP-res is 45% (or 0.45x) of the starting whole cell PrP (lane 1, 100%), and the s18 retains 33% of the whole cell PrP (lane 9) with 18% of this in the PrP-res form (lane 10). Note the increased 3x-6x lane loads needed to detect PrP and PrP-res in the concentrated p18 particles. The FU-CJD agent specific 13 kd PrP band in GT1 cells 3,42 is at arrow. (C) % tubulin in above blot shows the partitioning in each sequential fraction (PK – lanes), and tubulin is completely digested by PK (+ lanes). (D) Gold stain of the same blot demonstrates the markedly decreased protein in concentrated p18 particles. The p18 pattern of bands is clearly different than the non-infectious s18 (compare lanes 9 and 11). After digestion, the intense PK band at 29 kd (arrow) is dominant, with few other remaining bands. Starting whole cells (WC), nuclei (Nuc) and markers (kd) indicated.
Table I summarizes >20 independent FU-CJD cell preparations titered by serial dilution for average infectivity (line A) in each of the sequential fractions. Pertinent molecular components are shown in lines B, C and D. The first 500 g nuclear pellet contains only 5-10% of the total infectivity, some of which is likely due to incompletely lysed cells. Large PrP amyloid aggregates, identified ultrastructurally in the cytoplasm of GT1 infected cells,14 can also sediment at these low g forces. More than 85% of the starting whole cell infectivity is routinely recovered in the p18, with negligible infectivity in the s18 supernatant. Most cellular proteins (line B) were also removed from the p18 particles, with only 5% remaining in the p18 pellet. Line C shows that a large percent (35%) of starting PrP sedimented with low infectivity nuclei, and 30% of this crude nuclear fraction PrP, i.e., 0.3x the “nuclear” PrP, was in the PK resistant PrP-res form. In contrast, the PrP/PrP-res of the highly infectious concentrated p18 particles was only 15% of the starting whole cell PrP, again with 30% (0.3x) of this fraction in a PrP-res form. Uninfected cells yielded a small amount of starting PrP in the p18 (~2%), but no PrP-res was detectable in this or any other control fraction. Thus PrP-res was a reliable marker of agent-induced disease in these monotypic cells. Although the vast majority of host nucleic acids were removed in the p18 (line D), residual mitochondrial and nuclear DNA could still be detected by gel analysis of >5e6 cell equivalents (CE) on a Sybr Gold stained agarose gel lane, as well as by PCR of long mitochondrial DNA as previously detailed.45 Since there are 3 TCID/cell of the FU-CJD agent prior to nucleic acid extraction, this represents a load of ≥e7 TCID on a lane, far more than others have loaded on a gel to conclude that there are no nucleic acids of >50nt in infectious preparations.
A representative Western blot analysis for PrP and other host proteins, all analyzed on a single blot, is shown in Fig. 1B, 1C and 1D. These results showed little variability in quantitative Western blot analyses of >20 preparations made by different individuals in our lab with an SEM of <2% for the p18 PrP. Figure 1B shows PrP and its PrP-res component (PK+ lanes) in each sequential particle isolation step. As previously documented, the M20 PrP antibody used here faithfully targets the amyloid core, and allows detection of both full length glycosylated and PK truncated PrP amyloid, unlike antibodies directed to the NH2 terminus of PrP.43 The cytosol (500 g) in this individual preparation contains 59% of the total starting whole cell PrP (WC: lane 1), where 45% of this is PrP-res (lane 2). The low infectivity s18 supernatant (lane 9) contains 33% of the starting whole cell PrP, while the highly infectious p18 pellet, with a 3 fold greater CE lane load, contains 17% of the starting PrP (lane 11). It also has less PK+ PrP-res than the starting cells (lane 12, 29% versus 45% starting PrP-res). TrisCl pH 8.9 washes of the p18, done before particle disruption treatments, removed residual Triton X-100 along with an additional 3% of total starting cell PrP, or 20% of the p18 before washes. Figure 1C shows tubulin, with 21% of the starting whole cell tubulin retained in the p18, a higher level than for PrP. No tubulin is detected after PK+ digestion. Figure 1D shows the colloidal gold stain. Whereas silver staining is often used to assess other proteins in PrP-res preparations, colloidal gold reveals many proteins that silver does not stain. Such non-PrP proteins have also been confirmed by deep proteomic sequencing of 100 µ filtered FU-CJD brain particles that are free of collagen and lipid, but retain 80% of the starting brain titer.20,46 Colloidal gold (Figure 1D) shows additional host proteins are markedly reduced in the p18 (compare s18 to p18 in lanes 9 and 11). In sum, relatively low PrP/PrP-res and other cell proteins are retained by concentrated infectious p18 particles.
The above strategy for rapidly isolating infectious particles is not limited to a single TSE strain. Comparable results were found with 22L scrapie infected GT1 cells that previously required sucrose gradient searation of large volumes of s10 particles for agent purification.44 We also successfully modified this rapid procedure to isolate highly infectious p18 particles from brain.20 These particles had >5 fold higher agent recoveries than PrP-res enriched brain fractions.20 Because PrP-res amyloid fibrils and other cellular components can impede or complicate chemical denaturation effects, the low PrP and protein of particles isolated here were advantageous for assessing the intrinsic resistance of the FU-CJD and 22L scrapie agent particles.
Effect of Urea, Thiourea and β-ME on p18 proteins
The overall denaturation strategy was to first assess intrinsic agent structure by exposing p18 to chemicals, and then to re-sediment particle components at 18,000 g for analysis of solubilized (sol) and insoluble (insol) molecules. PrP/PrP-res, tubulin, and total gold stained proteins were quantitated in these 2 partitions on the same Western blot. Urea is a non-charged protein denaturant that disrupts noncovalent protein bonds and unfolds them to reveal their hydrophobic cores. It can progressively separate specific sets of membrane proteins,47,48 and because normal PrP is predominantly a membrane protein, as demonstrated by ultrastructural PrP antibody labeling,14 it was relevant to evaluate the effects of different urea concentrations on both protein structure and particle infectivity. Thiourea (ThU), an alternative and more powerful denaturant,49 was also tested, along with 5% β-ME that reduces S-S bonds, including those of PrP that contribute to PrP-res misfolding and amyloid aggregation. ThU alone, or in combination, has never been tested on any TSE agent preparation for either changes in titer or PrP solubilization.
Figure 2 shows an individual Western blot of urea and ThU treatments of a single p18 preparation for fair comparison with control buffer-incubated p18 particles. Resedimentation after treatment was used to first assess the % disruption (solubilization) and band pattern of particle components as compared to the starting treated p18. In each condition, the same blot is analyzed for A) PrP/PrP-res, B) tubulin and C) gold stained proteins. For simplicity, the washed p18 particles are taken as 100%, even though the PrP and other cell proteins in the p18 constitute only a small fraction of the starting whole cell PrP, i.e., an average of 15% for PrP, and 5% for total proteins. The FU-CJD control buffer incubation (Fig. 2A, untreated lanes 1-6) shows that the majority of PrP and other cell proteins are recovered in the re-sedimented p18 (insol) fraction, typically 78-81%, with 78% in this preparation. PrP partitioning in the p18 sol plus insol fractions also shows 100% recovery of the PrP in the starting particles (PK- lanes 3 and 5; 22%+78% PrP), and also confirms the precision and consistency of the Western blot quantitation. Unlike total PrP, the aggregated PrP-res amount increases by 1.5x after sedimentation (compare Fig. 2A, PK+ lanes 2 and 6, 19% to 30%). Pellet induced increases in PrP-res have been observed in multiple preparations, and thus the new protocol minimized the easily sedimented PrP-res by avoiding infectious particle sedimentation until the final p18 concentration step.
Figure 2.
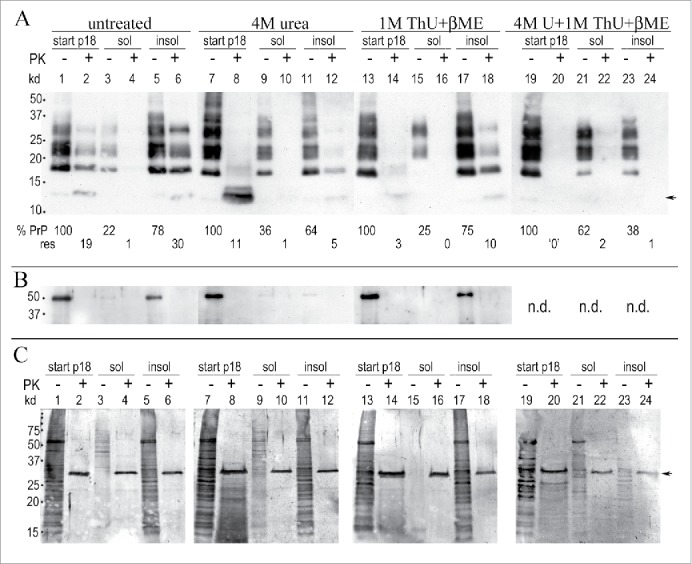
Urea and thiourea (ThU) effects after 1 hr at 22°C on p18 proteins. (A): Representative Western blot shows residual PrP/PrP-res in the p18 (here taken as “100%” for simplicity) with the % of the p18 solubilized (sol) and the % of the p18 that pellets (insol) after chemical treatment. Buffer treated control particles (labeled untreated) are shown in lanes 1-6; 4M urea in lanes 7-12, 1M thiourea (ThU) + 5% β-ME in lanes 13-18, with 4M urea, 1M ThU + 5% β-ME in lanes 19-24. The %PrP and %PrP-res amounts in each p18 partition are shown at the bottom of each lane (- and +PK). Faint standard PrP-res bands of 17-27 kd are present but reproduce poorly in these less exposed films (lanes 4, 10 and 12). (B): Tubulin shows equal loads for each treatment (p18 start lanes); missing tubulin band in lanes 11-12 due to inadequate antibody exposure as evident by its white background vs. the rest of blot. (C): Gold stained proteins. Strong PK band at 29 kd (arrow) is seen in the +PK lanes. Note in Fig. 2A and C the low kd smears with 4M urea treatment + PK (lanes 8) that contrasts to the standard banding pattern seen in urea before PK (lanes 7); the typical PrP pattern and the 17 kd PrP-res reappear when urea is removed from the pellet (lanes 11 and 12). Even more robust PrP-res band restoration is seen when 1M ThU + β-ME is removed from the pellet (lane 18). Multiple preparations showed highly reproducible %PrP in treatments (see Fig. 3A, SEMs).
Treatment with 4M urea (Fig. 2A, lanes 7-12) shows a very different picture from the buffer control, even though 100% of total PrP was again recovered in the sol + insol fractions (PK- lanes 9 and 11; 36%+64%). First, the total PrP before resedimentation shows a standard banding pattern despite the presence of 4M urea in the starting p18 sample (lane 7). Second, the total PrP in the 4M urea treated particles after PK digestion migrates as a prominent smear at only ~13 kd (lane 8) instead of the typical PrP-res pattern of multiple bands from 17-27 kd. Because the PK digestion is done before SDS and heat denaturation of samples for gel electrophoresis, the low Mr of PrP in the urea treated PK digested samples shows that 4M urea exposes previously buried sites in the amyloid resistant core that are now unfolded and more susceptible to PK. This core is targeted by the M20 PrP antibody here. The low Mr PrP smear after PK cannot be caused by the presence of urea itself because the non-digested PrP has a standard banding pattern in 4M urea (lane 7). Although the 4M urea insoluble pellet contains the majority of PrP (64%, lane 11), with only 5% PrP-res (lane 12), this insoluble PrP-res now shows a typical 19 kd PrP-res band, along with faint higher kd PrP bands that were obvious on longer film exposures and confirmed by densitometry. This is evidence that after removal of urea from the insoluble pellet, a portion of PrP refolded sufficiently to generate typical higher Mr PK-resistant fragments. Gold stained p18 proteins in 4M urea show behavior that parallels PrP; the starting p18 gold stained proteins display an intense smear of markedly low kd species with PK (Fig. 2C, lane 8).
In contrast to urea, 1M thiourea (ThU) + β–ME allowed PK to digest even more PrP in the starting p18 (Fig. 2A, lane 14) yielding only 3% PrP-res vs. 19% in the buffer control (lane 2) and 11% in 4M Urea (lane 8). Unambiguous standard PrP bands of 17-27 kd reappeared after ThU was removed. This allowed reburial of the PrP amyloid core sequences. Refolding accounted for a 3.3 fold increase in PrP-res (compare lanes 14 and 18 with 3% and 10% PrP-res respectively). The 3 chemical combination of 1M ThU, 4M Urea + β-ME was even more irreversibly disruptive of PrP-res folding. No detectable PrP-res bands were refolded or reconstituted in the corresponding insoluble pellet (lane 24). Again, this reagent combination, as other treatments (lanes 7 & 13) failed to modify the starting PrP band pattern (lane 19).
Finally, Figure 2B shows the tubulin in these samples, and demonstrates equal p18 aliquots were used for each of the chemical treatments. In both 4M Urea and 1M ThU + β–ME, an average of 80% of the tubulin pelleted after treatment. Tubulin in 4M urea (lanes 11 and 12) was compromised by uneven antibody exposure but multiple experiments showed a range of 75-85% insoluble tubulin in 4M urea. In sum, treatment of the p18 infectious particles with urea and thiourea facilitates PrP susceptibility to PK, and removal of 1M ThU + β–ME restores more PK resistant higher kd PrP bands indicative of rapid PrP refolding.
Urea and thiourea reduce FU-CJD infectivity
Because the above data did not lead us to any clear predictions for large quantitative infectivity changes in the total treated p18 particles, or in the derivative sol and insol fractions, we evaluated the treated total p18 for changes in infectious titer. At the same time we analyzed the solubilization and resistance characteristics of PrP systematically and quantitatively for reproducibility. The top panel of Figure 3 graphs the quantitative Western blot analyses after indicated treatments with urea and/or thiourea (>5 independent p18 preparations). Note the excellent reproducibility of quantitative PrP Western blot data with small SEMs for each fraction from many different preparations. The bottom panel shows the corresponding tissue culture infectious doses (TCID) using ≥3 of these preparations for each infectivity determination. For comparison with standard measurements of infectivity in the literature, the TCID are graphed per gram of brain, equivalent to e9 cells. Significant changes induced by different treatments are indicated (*). The high titer of FU-CJD cells (3e9 TCID/gm) and the high concentration of infectivity in the p18 particles enabled definitive titer losses of >4 logs. These high titers also allowed dilution of denaturants to very low non-toxic levels for TCID assays.
Figure 3.
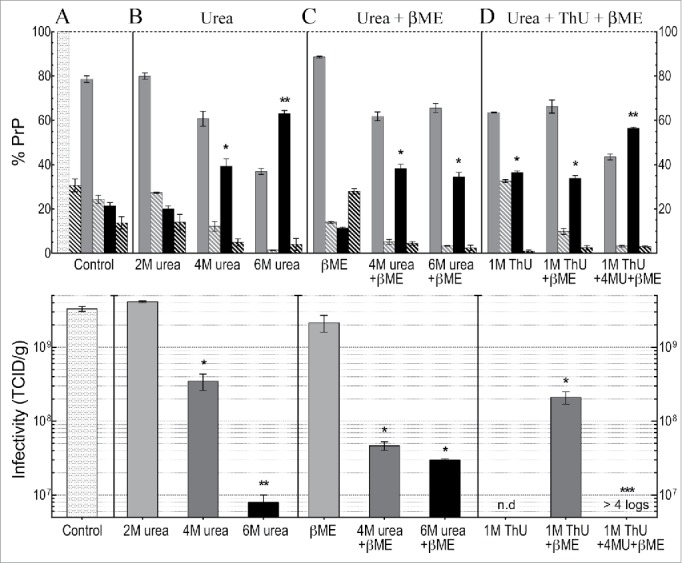
Western blot PrP quantitation and infectivity (TCID) determinations for FU-CJD based on 3-6 independent p18 preparations for each treatment, including SEMs. Top panel shows analysis of % PrP partitioning in the insol pellet and in the solubilized supernatant after treatments (A): Untreated PrP reference buffer control where the starting p18 PrP is plotted as 100% with the insol component (medium gray bars), and solubilized PrP (black bars); (B): increasing urea concentrations as indicated; (C): β-ME alone, and β-ME + urea; (D): ThU and/or in combination with urea + β-ME. The %PrP-res component of each partition is shown in adjacent hatched bars for each fraction. Bottom panel shows infectivity per gram (e9 CE) of the total p18 controls and the total p18 titer after chemical treatments. The TCID in the control p18 before and after washing was the same (Table 1) and thus is shown only once as a control (lightest gray bar) in panel A. In the treatment panels B to D, increasing shades of gray to black are used to show increasing chemical concentrations and/or additional components. 2M urea produces no significant decrease in titer, whereas 4M urea significantly decreases titer (*), with further significant decreases** in 6M urea. Addition of β-ME to 6M urea shows no significant titer difference (p=0.5 logs). However, a dramatic loss of infectivity (>4 logs) is induced by the combination of 4M urea, ThU + β-ME, even though 43% of PrP remains aggregated and insoluble (top panel D).
The starting PrP in the washed p18 particles (lightest gray bar with 100% PrP, Fig. 3A, top panel) and its PrP-res component (adjacent striped bar at 30%) is shown as a reference for all the subsequent control buffer (panel A) and other chemical treatments (panels B, C and D in the top graph). Note the small SEMs. The resedimented insoluble particle PrP (medium gray bar in top panel) and the solubilized PrP (black bar) is shown for each chemical set and treatment variation, e.g., 2M, 4M and 6M urea. The PrP-res component of the sol and insol fractions is again shown by striped bars. Although 22% of total starting p18 PrP is removed or solubilized by simple buffer washes (Fig. 3A, top panel) the washed control p18 titer showed no loss (lightest gray control bar, bottom panel A). Buffer washed particles, as plotted, contained the same 3e9 TCID/gm as unwashed p18 particles (composite data in Table 1). Because the current experiments showed no loss of infectivity with buffer washing in any sample this TCID control bar is not repeated in each of the treatment panels. In each of the TCID panels with chemical treatments, progressive shades from gray to black indicate the increasingly disruptive conditions or additives on p18 titers.
Table 1.
Sequential subcellular fractionation and purification of FU-CJD infectious neuronal cell (GT1) particles from total starting whole cell lysate (100%). Recoveries of different molecular components in each fraction is shown with (A) % infectivity, (B) % protein, (C) % PrP and its PrP-res component (PrP/PrP-res) and (D) nucleic acids. The PrP-res component is calculated as follows: 0.3x denotes that 30% of PrP in that fraction is in the PK resistant PrP-res form. Note the marked reductions in all the different molecular components in the highly infectious p18 particles. Multiple independent cell preparations and infectious titers (TCID) varied by < 3-fold in >30 preparations assayed by duplicate serial dilutions and both the p18 and the p18 buffer washed particles consistently contained an average of 3e9 TCID/gm/e9 cells. Although PrP was reduced, no infectivity was lost by washing (n=27). Protein and PrP/PrP-res amounts are an average of >30 preparations.
| Starting total (100%) | Nuclei | cytosol | s10 | s18 | p18 | p18 washed |
|---|---|---|---|---|---|---|
| A. Infectivity | 5–10% | 90–95% | 90% | <5% | >85% | >85% |
| B. Protein | 18–15% | 82–85% | 75–80% | 70–75% | 5% | nd |
| C. %PrP /PrP-res | 35% / 0.3x | 65% / 0.25x | 57% / 0.25x | 42% / 0.25x | 15% / 0.25x | 12%/ 0.3x |
| D. Nucleic acids | ≥98% | nd | nd | nd | <0.5% | <0.5% |
After 2M urea, p18 particles showed the same ~20% solubilization of PrP as the buffer control, and 2M urea also did not reduce the infectious titer (p > 0.99). In contrast, PrP solubility was significantly increased by 4M urea (to 40%, black bars), and 6M urea increased PrP solubility even more in a semi-linear fashion to 63% (**p < 0.0001). However, infectivity in the total p18 was reduced by only 20 fold in 4M urea, whereas it was reduced by 300 fold (2.5 log) in 6M urea (*p < 0.0001). Panel C shows that β-ME added to 4M urea did not further enhance solubility (compare 4M urea in panels B and C) although titer was slightly lower (by <1 log). Finally, panel D shows the effects of 1M ThU alone, in combination with β-ME, and after exposure to a combination of all 3 chemicals (ThU, urea + β-ME). The 3 chemical combination solubilized 57% of the p18 PrP (** black bar, p <0.0001), i.e., less than solubilized by 6M urea alone, and both had comparable amounts of insoluble PrP-res (striped bars). Yet the 3 chemical combination produced a profound >4 log (>10,000 fold) loss of infectivity with no apparent infectivity detected even after 13 cell passages. In this TCID assay, a 4 log reduction of the FU-CJD agent shows a positive signal at culture passage 13.19,50 The insoluble PrP and its clearly present PrP-res (>1%) also did not predict the FU-CJD titer losses which were >10,000 fold, or <0.01% of the control infectivity. In sum, neither the PrP-res amount or solubility can be used to assess titer.
The above infectivity data also show that FU-CJD particles have an intrinsic and significant sensitivity to ThU disruption. The addition of 1M ThU to 4M urea caused extreme agent destruction (>4 logs), and ThU is known to have a co-operative enhanced unfolding effect on many proteins and structures when combined with urea.51,52 The above data further showed that 6M urea itself can be part of a decontamination strategy for CJD agents. Moreover, the new combination of urea with ThU may be even more effective for processing delicate instruments. In this context we tested small fine surgical instruments, including ultrafine superthin style 5 tweezers, in this 3 chemical combination for 24 hrs. Even with intermittent bath sonication, no degradation of their delicate tips was apparent when inspected at 10x magnification. Furthermore, bath sonication in this solution enhanced the release of small adherent particles, and thus the combination of 1M ThU, 4M urea + β-ME should be useful as a first step to reduce infectivity by >4 logs (>99.99%) prior to a second agent-destroying treatment (vide infra).
22L scrapie agent is more sensitive to urea and thiourea denaturation than the FU-CJD agent
Because denaturation and destruction of TSE and other resistant microorganisms has become critical for the reuse of expensive instruments in gastrointestinal, brain and other tissue invasive procedures, it was important to evaluate another TSE strain. For fair agent comparisons, only the TSE agent strain was varied by using identical GT1 cells infected with 22L scrapie; p18 particle preparation and solubilization protocols were also identical. Figure 4A shows residual PrP in the control incubated p18 particles (again taken as 100%), and in the soluble supernatant (black bars) and insoluble pellet (gray bars) after treatments. In contrast to FU-CJD particles, 2M urea significantly increased the solubility of PrP to 45% (p <0.0001). Yet this higher PrP solubilization did not significantly decrease the 22L scrapie agent titer (0.7 logs, or 5 fold), as shown in corresponding Figure 4B. Thus intrinsic resistance of the 22L scrapie agent was comparable to that of FU-CJD in 2M urea despite its greater loss of PrP. With 4M urea, PrP solubility of 22L particles increased only slightly, to 55%, yet there was now a significant 2.9 log (800 fold) loss of the control infectivity (Fig. 4B). Additionally, 6M urea + β-ME solubilized less PrP than 4M urea (33% versus 55% respectively), yet infectivity was reduced to undetectable levels (>4 log loss) in 6M urea (Fig. 4B). These 22L scrapie results again give clear evidence that 22L infectious particles are easier to inactivate than are PrP aggregates.
Figure 4.
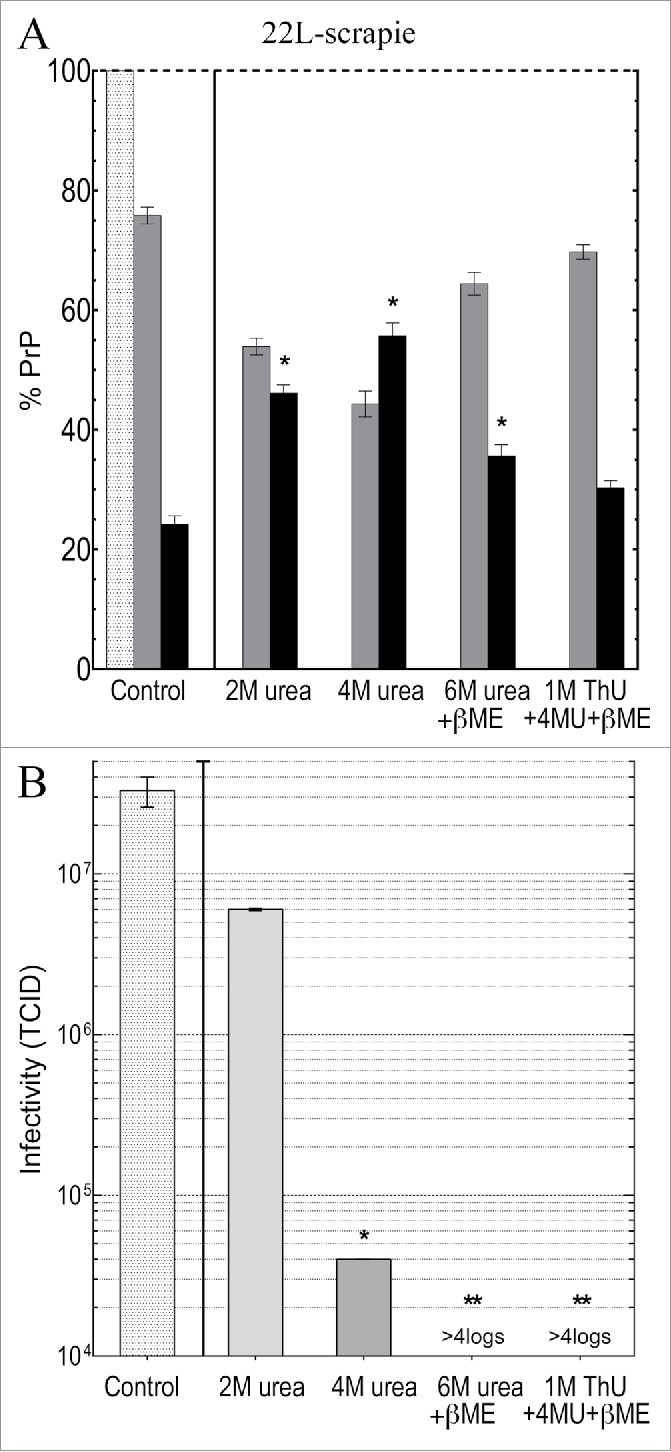
(A) shows PrP solubilization in 22L scrapie infected p18 particles, and (B) shows the corresponding TCID/gm after treatment with the denaturants indicated. The light gray bar is the residual p18 particle PrP (taken as 100%) and the black bars show the PrP solubilized by buffer control, and after parallel chemical denaturants. As much as 65% of PrP remains insoluble (medium gray bars) with treatments that induce the greatest >4 log loss of titer, e.g., with 6M urea + β-ME and with the 3 chemical combination of ThU, Urea + β-ME. The 22L scrapie agent is clearly more sensitive than the FU-CJD agent to destruction by 4M urea, and the 22L agent titer is also undetectable after 6M urea + βME, unlike FU-CJD. All p-values <0.05 (including * compared with **).
Additional data demonstrates that the 22L scrapie agent is more sensitive to denaturation than FU-CJD agent. In the 22L experiments, >10 fold more particles/ml were treated to ensure that a >4 logs loss of titer would be detectable. This higher particle concentration, if anything, should decrease a chemical’s effective disruption on a molecule to particle ratio. Yet 4M urea caused a much more dramatic titer loss in 22L scrapie than in FU-CJD (2.9 log or 800 fold compared to a 1.1 log or only 20 fold loss in FU-CJD). A more dramatic loss of 22L scrapie infectivity was also seen with 6M urea + β-ME (>4 logs). Finally, the 3 chemical combination of ThU, urea + β-ME, as with FU-CJD, reduced the 22L scrapie titer by >4 logs, even though this treatment yielded comparable levels of aggregated insoluble PrP as the highly infectious control (69% and 75%). In sum, 22L scrapie particles are inherently more sensitive to denaturation than FU-CJD particles, and this suggests subtle structural or molecular difference between 22L and FU-CJD particles, and/or their cellular ligands other than PrP. The combination of 4M urea, ThU + β-ME that denatured >4logs of infectivity in both FU-CJD and 22L scrapie also shows that these 2 very different TSE agents have common structural features that are likely to be conserved in other TSE strains.
Sarkosyl, SDS and Guanidine HCl effects on infectious particles
It was also relevant to examine the effects of detergents and guanidine salts on infectious particles from GT1 cells. Sarkosyl was tested here as a solubilization standard because it has been used in many TSE disaggregation and infectivity purification schemes with a variety of agent-strains from different tissue sources and species. Sarkosyl did not reduce infectivity in any of these studies. On the other hand, although SDS at 22°C did not significantly reduce titer of sporadic CJD (sCJD) hamster brain particles, it did induce an ~3 log titer loss after 95°C for 5 min even though abnormal PrP multimers were preserved.41 In PrP-res enriched 263k scrapie hamster brain fractions, SDS without heat also caused both no loss in titer, and a >4 log loss.39,53 A much greater titer loss (~7 logs) in sCJD infected brain homogenates treated with >3M GdnSCN,31 and a >99.6% loss using the less chaotropic 2.5M GdnHCl on less well-purified sCJD brain particles,41 led us to evaluate GdnHCl disruption more systematically on the isolated FU-CJD particles here. FU-CJD is a good TSE agent to test for high intrinsic resistance because it is more stable than 22L scrapie, and is also more difficult to sterilize than the high titer sCJD agent in hamster brain. In >25 years of experience working with both the sCJD and FU-CJD agents in animals, only FU-CJD has given us an instrument cross-contamination. Additionally, its resistance to denaturation by heat is similar to BSE (unpublished data). Thus we used ascending concentrations of GdnHCl to assess FU-CJD agent disruption. GdnHCl is less toxic, more stable, and safer to dispose of than GdnSCN, and even heating fine surgical instruments at 75°C in the more chaotropic GdnSCN does not corrode them.31 Similarly, 4M GdnHCl did not corrode instruments. All treatments of FU-CJD particles by GdnHCl were conveniently and rapidly accomplished at 22°C for only 5 min prior to dilution of the treated particles for infectivity assays.
Figure 5A plots the effects of increasing concentrations of sarkosyl, SDS and GdnHCl on PrP solubility in FU-CJD particles at 22°C. Solubility of PrP increases in an almost linear manner with increasing sarkosyl. With only 0.2% sarkosyl, >90% of the p18 PrP becomes solubilized (p < 0.0001). SDS shows a much steeper and sudden increase in PrP solubility at lower concentrations, and as little as 0.03% SDS solubilizes >80% of the residual PrP in p18 particles. Essentially all residual p18 PrP is solubilized by 0.1% SDS, and although PrP-res is still detected after PK, it is reduced by 93%. Infectivity titrations of FU-CJD particles in these 2 detergents is shown in Figure 5B. Sarkosyl solubilization of PrP at 0.2% caused no reduction in infectious titer (p > 0.999). Moreover, despite solubilization, as well as the enormous loss of PrP-res in 0.2% SDS, the FU-CJD titers fell by only <0.7 logs (p < 0.004). This minor <5-fold decrease is not significantly different from the very slight titer loss of sCJD brain particles treated with 1% SDS at room temperature.41 Thus, as in urea-ThU studies, solubilization of residual p18 particle-linked PrP/PrP-res molecules was misleading for assessing agent destruction. Additionally, these studies clearly show that detergents alone, without heating, are not useful decontaminants. They can also leave an undesirable film in instrument crevices.
Figure 5.
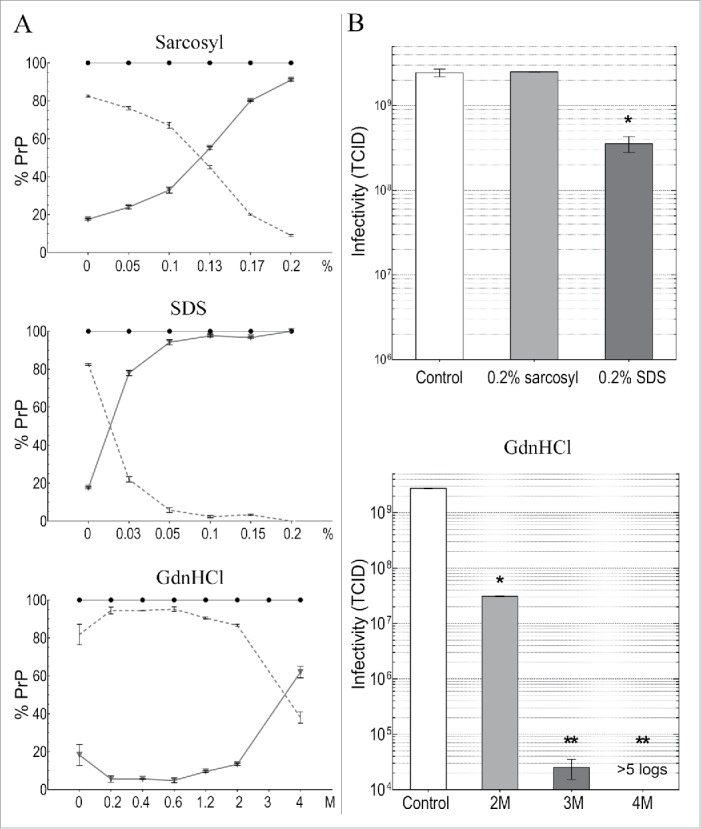
Detergent and GdnHCl treatments for 5 min at 22°C of FU-CJD p18 particles. Column A graphs the % of PrP solubilized at indicated detergent concentrations where the sol partition is shown by triangles with solid line and the insol % on the dotted line. Both components added to 100% PrP (black circles). Column B plots TCID of untreated control (white bar) and after indicated treatments. As little as 0.2% SDS without heat reduced p18 TCID by ~1 log, whereas 0.2% sarkosyl treatment, which also solubilizes most PrP, retains all infectivity. At 2M GdnHCl there is a 2 log decrease in titer, while 3M and 4M GdnHCl treatments caused >5 log reductions. SEM shown with all p-values <0.05 (*).
Figure 5 shows that GdnHCl also induced a sudden sharp rise in solubility of PrP between 2M and 4M, but in contrast to the detergents, only ~60% of PrP was solubilized by 4M GdnHCl, and PrP-res was still detected. We therefore compared infectivity at 2M, 3M and 4M GdnHCl where PrP solubilization was induced by a strong chaotropic chemical. Even though 2M GdnHCl solubilized only 15% of the residual p18 particle PrP, titer fell by 2 logs to 1% of the control (p < 0.0001). Treatment with 3M GdnHCl reduced FU-CJD titer by >4 logs, and with 4M GdnHCl p18 infectivity was reduced by >5 logs (undetectable at serial passage 15).
Representative TCID determinations
Validated quantitative TCID assays for infected brain and cells were used to determine titer losses in chemically treated p18 particles. The TCID assay has been verified in parallel animal inoculations, and indicator cells infected by different agents also recapitulate agent-specific titer differences by animal assay.3,7,12,19,20,42-44,50 Figure 6 shows 2 representative assays at early passages after infecting indicator cells. Figure 6A shows the urea and Figure 6B the GdnHCl effects on titer. As in animal incubation time assays, and in others’ scrapie cell assays,8,54 a low titer requires more time to induce PrP-res pathology. The TCID is calculated from published standard dilution curves that give a progressively increasing linear PrP-res response in indicator cells at sequential passages, as determined for the FU-CJD and 22L agents, e.g..7,50 Even though an infectivity assay is less precise than a direct analysis of cellular molecules, note the comparable %PrP-res elicited in duplicate wells exposed to the same sample. In Figure 6A, the non-infectious mock control sample induces no PrP-res (+ PK lane 2, 10x load) in indicator cells. In contrast, the FU-CJD buffer controls (chemically untreated p18 samples) induce a strong PrP-res response even at a lower 4x lane load (lanes 4 and 6). The 2M urea sample also induces a similarly high PrP-res response (lanes 8 and 10), demonstrating retention of 94% of the TCID of the high infectivity control. In contrast, 6M urea treated particles don’t induce appreciable PrP-res (lanes 12 and 14), signifying major reductions in titer of this sample. These 6M urea treated samples required a 10x load to induce a <1.5% de novo PrP-res response in indicator cells at the same assay passage. Later passages showed the 6M urea sample induced a PrP-res signal that was in a linear range adequate for accurate titer quantitation.50 Figure 6B shows GdnHCl effects on FU-CJD infectivity. The mock control lane again induces no detectable PrP-res, while the FU-CJD in untreated lanes induces high de novo production of PrP-res by passage 4. In contrast, the 2M GdnHCl lanes show only a low amount of detectable PrP-res (lanes 8 and 10), and the 3M GdnHCl (lane 12) and the 4M GdnHCl treated particles (lane 14, with a 12x load) have undetectable amounts of agent-induced PrP-res at this passage. This represents huge decreases in titer. At passage 15, indicator PrP-res remained undetectable in the 4M GdnHCl treated particles, consistent with an ~6 log titer loss.
Figure 6.
Representative TCID Western blot assays for FU-CJD where only a TSE infectious agent, but not PrP-res amyloid of cured cells, can provoke progressively increasing PrP-res. (A): Duplicates of control untreated FU-CJD and 2M and 6M urea treated samples inoculated on indicator cells and then passaged. Assay cell passage 5 shows the mock uninfected sample (PK+ lane 2), elicits no PrP-res response. In contrast, the infected FU-CJD p18 control (PK+ lanes 4 and 6) shows a strong response, with 100% of the TCID. The 2M urea treated p18 samples (PK+ lanes 8 and 10) also shows a strong PrP-res response, equivalent to 94% of the TCID of the buffer treated FU-CJD control. In comparison, 6M urea treated samples (PK+ lanes 12 and 14) show only a small PrP-res response that was insufficient for quantitating TCID at this passage (nd). (B): FU-CJD control and 2M, 3M and 4M GdnHCl treated p18 particles at assay passage 4. GdnHCl at 2M was effective at markedly reducing infectivity (PK+ lanes 8 and10) while 3M and 4M GdnHCl reductions were even more pronounced. Even by passage 15 no PrP-res was detectable after 4M GdnHCl treatment of particles, demonstrating a >5 log titer loss. Mock uninfected cells again induced no PrP-res signal (PK+ lane 2). Markers in kd.
In sum, urea, a non-charged chaotropic molecule, in combination with ThU + β-ME causes >4 log TSE agent reductions, and this treatment can be a useful first step for fine instrument decontamination, with or without sarkosyl. Alternatively, only 5 min exposure to 4M GdnHCl, without any heat or detergents, is even more efficient at reducing infectivity by >5 logs, and it may be sufficient by itself for decontamination in some settings. The comparative lack of PrP solubilization by agent destructive 4M GdnHCl, in addition to other data here, indicates that structural disruption of TSE agent particles is independent of PrP denaturation, a conclusion consistent with previous studies demonstrating that digestion of all detectable PrP induces no significant loss of infectivity.18-20
Discussion
The current paper presents a new method for rapidly concentrating TSE infectious particles from monotypic cell cultures. Previously, only complex brain material has been tested for chemical denaturation (see Introduction). The infectious particles used here had markedly reduced cell components, and this facilitated comparison of the intrinsic chemical resistance of 2 very different mouse-adapted agent-strains, the human derived FU-CJD strain and the sheep derived 22L scrapie strain. No previous reports of others have evaluated either of these agents for titer losses, nor has there been any study of the effects of Thiourea alone or in combination with urea. We identified reproducible titer reductions of >10,000 fold for both the FU-CJD and 22L scrapie agents using short room temperature exposures to inexpensive chemicals that do not degrade standard fine surgical instruments. These treatments can be easily tested by manufacturers on their duodenoscopes of various design, and other expensive instruments, for non-corrosiveness. Unlike standard and more expensive proteolytic treatments for TSE agent decontamination, detergents and heat were not required. These TSE-decontamination procedures can also be further validated using infected tissue in susceptible animal models, and in different material contexts and cellular matrixes, including brain homogenates baked on brain electrodes. Previous experiments have already shown that the lower concentration of 2.5M GdnHCl reduces infectivity of 120S CJD brain particles by >99.5%, and this reduction coincided with the solubilization of particle-associated nucleic acids and the disruption of endogenous retroviral particles.41 We also identified ThU and urea conditions here that induced large titer losses. These similarly disrupted and solubilized particle nucleic acids (unpublished data).
The progressive decreases in TSE agent titers with increasing concentrations of urea and GdnHCl was clear with both TSE agents, but 22L scrapie was consistently more sensitive than FU-CJD. This is an unambiguous strain determined property because the identical cells and particle isolation procedures were used to propagate both agents. Both strains induced large PrP-res fibril accumulations within the GT1 cell cytoplasm ultrastructurally, and by Western blot.3,14 The amount of residual PrP and PrP-res in p18 particles of both strains was also the same. Thus total PrP-res levels did not account for this strain-specific sensitivity. We are aware of only one study of increasing concentrations of urea on a TSE agent that was titered for infectivity, and this study tested aggregated PrP-res fibrils made from hamster 263K scrapie infected brain.39 Despite the difference between that material and the cell-derived TSE particles here, 2M urea treatment of 263K brain scrapie also produced no significant titer loss after a 2 hr exposure; this paralleled the lack of disruption by 2M urea for both FU-CJD and 22L isolated particles here. Interestingly, in that 263K fibril study, 4M urea without detergent caused an ~1log (or 10-20 fold) titer loss at 2 hr, and similarly, FU-CJD particle titers, like 263K scrapie, decreased by only 1.5 logs (30 fold) in 4M urea. In contrast, concentrated 22L scrapie particles exposed to 4M urea for only 1hr showed a significantly enhanced ~3 log (1,000 fold) titer loss. These results further underscore differences among these agents, and again demonstrate that the 263K scrapie agent like the FU-CJD strain, is more resistant to disruption than 22L scrapie. Hence FU-CJD particles represent a fair agent choice to determine the more effective semi-universal TSE disruption treatments. Other strains with high resistance to heat, such as the BSE agent, should be tested with urea and other chemicals here as soon as a high titer brain or culture model is developed that is economically feasible.
Because the p18 PrP is more tightly linked to infectious particles, we thought p18 PrP disruption might reflect agent titer losses. This was not the case. Both insoluble aggregated PrP and PrP-res were clearly detected when titers were reduced >10,000 fold. Interesting, PrP was unfolded sufficiently to become highly susceptible to PK digestion with urea and ThU treatments, and standard PK resistant bands, an unambiguous sign of refolding and/or masking of amyloid core sites, was seen after removal of denaturants from the insoluble sediment. This was most clear in studies with 1M ThU + β-ME exposure, and was not due to a chemically induced change in PrP band migration on gels. This refolding parallels the relative ease of producing PrP-res forms, but not infectivity, in a test-tube.4,10-12 Dilution of the chemical denaturants here did not restore infectivity. While restoration of TSE agent infectivity has been tried for many years in various preparations,39,55 a small amount of infectivity was recently restored by combining phenol purified 263K brain nucleic acids with recombinant PrP.13 Non-PrP proteins that are particle-associated20,46 may also be needed to effectively transmit an agent genome from among the many nucleic acids present in all highly infectious preparations.41,45
Thiourea (ThU) alone, or combined with urea, is very effective for destroying >4 logs of TSE infectivity within 1 hr at 22°C, and ThU has not been used previously for TSE agent disruption. Notably, this chemical combination disrupts keratins,56 and a keratin-like protein structure(s) may protect TSE agents. In brain homogenates without detergent, a keratinase from Nocardiopsis (NAD), but not PK, also destroyed >3 logs of FU-CJD infectivity in both brain and GT1 cell homogenates within 2 hr at 50°C even though PrP and PrP-res were still visible.19 Other keratinases also significantly reduced the mouse adapted ME7 and the sheep SSBP1 agent titers in brain homogenates, unlike several proteases (see Introduction), but these required incubations at 65°C with detergents or surfactants for more complete destruction of infectivity.54,57
By comparison with the ThU-urea combination or NAD keratinase, only 5 min in 4M GdnHCl at 22°C was required to irreversibly denature >5 logs of the FU-CJD titer. Hence GdnHCl was the most effective single treatment for decontamination here. GdnHCl is charged, while urea and ThU are neutral, and charge affects protein stability beyond its competition for H-bonds. If the protein is highly charged with salt bridges, then it will be more destabilized by GdnHCl than by the same amount of urea.58 Many viral capsid and coat proteins are highly charged, contain salt bridges, and thus are disrupted by GdnHCl to release their nucleic acid genomes. Our results with 4M GdnHCl are completely in accord with its very high and widespread disruptive effects. GdnHCl at 4-6M has also been used for many years to effectively disrupt bacteria, fungi and lysosomes, and also to inactivate RNase. Unlike the RNase protein, whose activity is restored by removal of Gdn salts, 263K scrapie infectivity could not be restored after GdnSCN was removed.39 Similarly, 2.5M GdnHCl denaturation of less purified sCJD brain particles showed no restoration of infectivity when GdnHCl was removed after pelleting.41 Moreover, brain PrP aggregates after pelleting in 2.5M GdnHCl contained abundant PrP-res multimers but lost ~3 logs of infectivity by animal bioassays. Only one other study tested GdnHCl concentrations on infectivity, and it used 263K scrapie PrP-res enriched brain fractions. In marked contrast to our results, infectivity increased after exposure to 2.5M-3M GdnHCl, and all infectivity was recovered in the pellet.59 Paradoxically, 4M GdnHCl reduced 263k titer by <3 logs, but left no detectable PrP-res. In contrast, we found a rapid >5 log (100-fold greater) decrease in FU-CJD titer with 4M GdnHCl and the progressive loss of titer in p18 cell particles with increasing GdnHCl concentrations here was consistent and highly reproducible in assays of multiple independent particle preparations. It also was in complete accord with bioassayed disruption of brain sCJD agent in 2.5M GdnHCl. The above results further suggest that GdnHCl, as GdnSCN, may be effective in a variety of different matrices, including brain.
In sum, we have identified 2 inexpensive and simple chemical treatments which when used sequentially should theoretically destroy >9 logs of infectivity in both FU-CJD and 22L scrapie, a vast excess over the typical contaminations of delicate instruments. Unfortunately, there is no way to test such a large additive effect because there is currently no known TSE agent particle or other preparation with e9 infectious particles in a 20-50μl volume with low detergents or toxic chemicals. Nevertheless, both treatments here are likely to denature additional TSE agent strains although these remain to be tested.
A protocol to inactivate many different microbes
With the exception of this paper, TSE agent decontamination studies have been limited to crude brain homogenates or PrP-res fibrils, and there are no systematic studies of TSE agent destruction in the context of either GI or mucous membrane materials or other relevant matrixes for non-brain medical interventions. However, the known sensitivities of many microbes, viruses and biofilms (vide infra) indicate that the following protocol, without the use of heat or detergents, is likely to be effective not only for TSE agents, but for many other microorganisms. Relevant common microorganisms, including MRSAs and viruses, can rapidly be tested using various medical instruments with the following protocol that is aimed at preventing iatrogenic infection, and therefore avoids drying or baking materials on instruments. On the other hand, the following protocol can also be tested on electrodes with dried brain implanted into the CNS with and without immersions and sonications as described below.
First, instruments should be immediately immersed in water in a sealable pouch, and then bath sonicated for a few minutes to reduce tissue debris. Bath sonication is a validated dental instrument procedure for this purpose,60 and bath sonication was previously proposed to further enhance chemical decontamination of TSE agents.31 Indeed, the chemical solutions above can be quite effective in solubilizing and/or dislodging particles, and sonication reduces the need for laborious manual cleaning. Second, instruments are transferred/immersed in the 3 chemical combination of 4M urea, 1M ThU, + β-ME, incubated at room temperature for 1 hr, and bath sonicated for 5 min before immersing in excess water. Conveniently, instruments also can be left overnight in the 3 chemical solution if it doesn’t affect optics and cements on endoscopes. Third, after H2O washing to minimize interference from the first chemical, instruments are then immersed in 4M GdnHCl for 5-10 min, and again sonicated, and finally immersed several times in large volumes of water to remove any residual salt. The whole procedure should take less than 2 hr, and fluid changes and sonication times probably can be automated.
The above treatments, as reported by others, have been effective on many microbes and viruses, including hepatitis B that can be unexpectedly resistant for an enveloped virus, and that has caused outbreaks especially in hospital settings.37 Hepatitis B resistance is due to the observed hysteresis of hepatitis B capsid assemblies, and these require 4M urea or >2.5M GdnHCl for denaturation.61 Both conditions are fulfilled by the above protocol for decontamination. Ultrastructural studies also similarly show that hepatitis B particles are irreversibly disrupted only by >3.5M GdnHCl or 4M urea,62 i.e., a strong parallel to the empirical conditions demonstrated here for TSE agent disruption. Non-enveloped viruses can be more difficult to denature than enveloped viruses. Nevertheless, even an infectious coxsackie enterovirus, a structure most relevant for GI non-enveloped viruses, was degraded and inactivated by 3M urea in only 15 min at pH 9.0,63 comparable to the pH 8.9 conditions used here with more concentrated urea. Although we have not ourselves tested virulent MRSA bacteria, 6M GdnHCl which is commonly used in laboratories, is known to lyse all tested microbes and even plant cell walls.
Finally, GdnHCl and Urea-ThU can effectively disrupt biofilms that coat the walls of instruments and water conducting systems. Biofilms are resistant to recommended disinfectants, and also require mechanical cleaning that can be difficult and time consuming.64 Remarkably, urea strongly disturbs biofilm structure, and GdnHCl is even better at disrupting model biofilms.65 Thus again, either of the 2 chemical treatment, or the double treatment protocol should be effective. Although there are probably a few microbes and viruses that can escape denaturation by this “universal” procedure, it is important to communicate the findings here for public health. TSEs are not the only viral-sized infectious agents that can be highly resistant, and newly discovered viral structures survive even more extremes of heat than TSE agents.66 These viruses can also be rapidly tested with this protocol. Finally, the evaluation of more purified TSE agent particles here, without confounding brain and other complex cellular components, demonstrates these TSE agents intrinsically are not some extraordinary and indestructible Andromeda strain.
Materials and Methods
Cell culture, Western blot and infectivity assays
Uninfected GT1 cells, or those infected with the FU-CJD agent (GT1-FU) or the 22L scrapie agent (GT1-22L) were split at 1:4 every 4 days as previously described.14,19 Samples were analyzed on conventional Western blots and probed with goat anti-PrP antibody (M20, Santa Cruz Biotechnology) that binds to a region near the PrP C-terminus within amyloid core fibrils, as verified by immunocytochemistry and ultrastructure.14,67 Primary mouse anti-tubulin antibodies were used to confirm equal loads on undigested sample lanes. As previously, limited PK digestion was used to obtain maximal PrP-res signals (25 μg/ml for 30 min in 0.5%NP40-0.5%DOC lysis buffer) and reactions were stopped with PMSF before conventional denaturation in SDS for gel electrophoresis and blotting.43 After blocking blots, primary antibody, washes and HRP secondary antibodies (e.g., HRP Bovine anti- goat IgG for PrP) were applied sequentially, followed by chemiluminescence detection. As in many reproducible experiments over the last 10 years here, and in the present studies (see PrP/PrP-res SEMs, Fig. 3), Chemiglow developer (Alpha Innotech) was used for quantitative photon signal detection and analysis on a flat field lens equipped Alpha Innotech imager calibrated to discriminate chemiluminescent signals over 3 orders of magnitude. A BioRad analyzer has also been used with the same results. Colloidal gold staining of blots was done after antibody detections, and standard BCA protein assays also confirmed gel loads and subcellular recoveries.
Tissue culture infectious dose (TCID) assays were performed using standard reference end-point dilution curves previously determined for dilutions of the FU-CJD and 22L scrapie agent in mouse brain and in GT1 cells.7,44,50 Briefly, control and treated cell equivalent (CE) subcellular fractions were serially diluted, then applied to target GT1 cells, and passaged. Replicate wells of at least 3 different passages in the linear range (at least 6 samples for Western blot) were used to determine TCID, and replicate wells all showed comparable results (<10% TCID differences). Stably infected FU-CJD GT1 cells typically contain a titer of 3 TCID per cell, while the 22L scrapie infected GT1 cells reproducibly contain 0.03 TCID per cell (100 fold less than FU-CJD) by both animal and culture titrations.7,19 To avoid cell toxicity, final concentrations were <0.01% for sarkosyl, <0.002% SDS, <2 mM GdnHCl, <2.5 mM urea, and <0.5 mM thiourea.
Simplified Sequestration of Infectious Particles from Cultured GT1 Cells
Infectious particles from GT1 cells were made by simplifying a previous protocol for scrapie infected N2a cells 44. Briefly, (see Fig. 1A) mock GT1, FU-CJD infected GT1 and 22L infected GT1 cells were washed in PBS, collected by scraping and pelleted at 1500 rpm (450 g) × 10 min at 22°C. The cell pellet (~5-10×107 cells) was gently suspended by tapping, and 6 mL of lysis buffer [1% Triton-X 100, 0.1 mM PMSF, 8% sucrose in PBS] was added. The lysate was then spun at 1600 rpm (500 g) × 5 min at 22°C; the cytoplasmic supernatant (500 g) was carefully removed, and 1 mL of TrisCl pH 8.9 was added to resuspend the loose nuclear pellet. After a 2800 rpm (1500 g) × 5 min spin at 22°C, both supernatants were pooled (~7 mL total per ~0.5-1×108 cells). The 500 g supernatant was further clarified at 10,000 g ×10 min at 22°C to yield a 10k supernatant (s10) that was further processed immediately, or frozen at −80°C for later processing with comparable results. The s10 was digested with 120μg/ml RNase A (10 U/mL; Calbiochem) for 1 hr at 37°C which abolished almost all host RNAs and also facilitated rapid quantitative concentration of infectious particles by centrifugation at 18,000 g × 30 min at 22°C into a 20 µl 30% sucrose cushion. The 18k pellet (p18) was resuspended thoroughly by bath sonication in TrisCl 8.9 and washed twice with TrisCl 8.9 and resedimented to remove any residual Triton X 100 and sucrose. At each sequential purification step an aliquot was taken for protein, infectivity and nucleic acid analyses. A pH of 8.9 was chosen because previous experiments have shown it facilitates disaggregation of PrP-res aggregates that are extremely susceptible to PK.68
Incubation with Denaturants and/or Detergents
TrisCl 8.9 washed p18 particles were suspended in denaturants (urea, thiourea, β-ME, GdnHCl) and detergents (sarkosyl, SDS), re-pelleted at 18,000 g × 30 min and each resulting soluble supernatant and insoluble pellet further analyzed. Briefly, ~e8 CE of FU-CJD particles were suspended in 50-100μl of chemicals at final concentrations indicated, e.g., 2M, 4M, 6M urea with or without β-ME (5%) and/or thiourea (1M). All urea and thiourea (ThU) treated particles and untreated control particles were incubated for 1 hr at 22°C. Incubations for sarkosyl, SDS and GdnHCl treated particles were only 5 min at 22°C, and treated aliquots diluted and assayed for infectivity and protein analysis. Control and treated samples were then pelleted at 18,000 g × 30 min at 22°C, the supernatant (sol) was separated, and the insoluble pellet further washed with a small amount of Tris 8.9 and respun at 18,000 g × 5 min. The combined wash and soluble supernatants were pooled, the insoluble pellet re-suspended in TrisCl 8.9 and aliquots taken for analysis. Urea and ThU were in 50 mM Tris 8.9, while sarkosyl, SDS and GdnHCl were diluted in MilliQ water. Identical procedures were applied for 22L scrapie particles. Significance (p values) were determined by both Anova and t-test with comparable results.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
Portions of this work were supported by NIH NS RO1 012674-34, the William Prusoff Foundation, and a Yale OHSE grant.
References
- 1.Prusiner SB. Molecular biology of prion diseases. Science 1991; 252:1515-22; PMID:1675487; http://dx.doi.org/ 10.1126/science.1675487 [DOI] [PubMed] [Google Scholar]
- 2.Prusiner S. Prions. Proc Natl Acad Sci USA 1998; 95:13363-83; PMID:9811807; http://dx.doi.org/ 10.1073/pnas.95.23.13363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Manuelidis L, Chakrabarty T, Miyazawa K, Nduom NA, Emmerling K. The kuru infectious agent is a unique geographic isolate distinct from Creutzfeldt-Jakob disease and scrapie agents. Proc Natl Acad Sci U S A 2009; 106:13529-34; PMID:19633190; http://dx.doi.org/ 10.1073/pnas.0905825106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manuelidis L. Infectious particles, stress, and induced prion amyloids: a unifying perspective. Virulence 2013; 4:373-83; PMID:23633671; http://dx.doi.org/ 10.4161/viru.24838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kimberlin RH, Walker CA, Fraser H. The genomic identity of different strains of mouse scrapie is expressed in hamsters and preserved on reisolation in mice. J Gen Virol 1989; 70:2017-25; PMID:2504883; http://dx.doi.org/ 10.1099/0022-1317-70-8-2017 [DOI] [PubMed] [Google Scholar]
- 6.Manuelidis L, Lu ZY. Virus-like interference in the latency and prevention of Creutzfeldt-Jakob disease. Proc Natl Acad Sci USA 2003; 100:5360-5; PMID:12692308; http://dx.doi.org/ 10.1073/pnas.0931192100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, Sun R, Chakrabarty T, Manuelidis L. A rapid accurate culture assay for infectivity in Transmissible Encephalopathies. J Neurovirol 2008; 14:352-61; PMID:18989813; http://dx.doi.org/ 10.1080/13550280802105283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edgeworth JA, Jackson GS, Clarke AR, Weissmann C, Collinge J. Highly sensitive, quantitative cell-based assay for prions adsorbed to solid surfaces. Proc Natl Acad Sci U S A 2009; 106:3479-83; PMID:19204279; http://dx.doi.org/ 10.1073/pnas.0813342106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Manuelidis L. A 25 nm virion is the likely cause of Transmissible Spongiform Encephalopathies. J Cell Biochem 2007; 100:897-915; PMID:17044041; http://dx.doi.org/ 10.1002/jcb.21090 [DOI] [PubMed] [Google Scholar]
- 10.Timmes AG, Moore RA, Fischer ER, Priola SA. Recombinant prion protein refolded with lipid and RNA has the biochemical hallmarks of a prion but lacks in vivo infectivity. PLoS One 2013; 8:e71081; PMID:23936256; http://dx.doi.org/ 10.1371/journal.pone.0071081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ladner-Keay CL, Griffith BJ, Wishart DS. Shaking alone induces de novo conversion of recombinant prion proteins to β-sheet rich oligomers and fibrils. PLoS One 2014; 9:e98753; PMID:24892647; http://dx.doi.org/ 10.1371/journal.pone.0098753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyazawa K, Kipkorir T, Tittman S, Manuelidis L. Continuous production of prions after infectious particles are eliminated: implications for Alzheimer's disease. PLoS One 2012; 7:e35471; PMID:22509412; http://dx.doi.org/ 10.1371/journal.pone.0035471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Simoneau S, Thomzig A, Ruchoux MM, Vignier N, Daus ML, Poleggi A, Lebon P, Freire S, Durand V, Graziano S, et al. . Synthetic Scrapie Infectivity: Interaction between Recombinant PrP and Scrapie Brain-Derived RNA. Virulence 2015; 6:132-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manuelidis L, Yu ZX, Barquero N, Mullins B. Cells infected with scrapie and Creutzfeldt-Jakob disease agents produce intracellular 25-nm virus-like particles. Proc Natl Acad Sci USA 2007; 104:1965-70; PMID:17267596; http://dx.doi.org/ 10.1073/pnas.0610999104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bignami A, Parry H. Aggregations of 35-nanometer particles associated with neuronal cytopathic changes in natural scrapie. Science 1971; 171:389-90; PMID:5099604; http://dx.doi.org/ 10.1126/science.171.3969.389 [DOI] [PubMed] [Google Scholar]
- 16.David-Ferreira J, David-Ferreira K, Gibbs C, Morris J. Scrapie in mice: ultrastructural observations in the cerebral cortex. Proc Soc Exp Biol Med 1968; 127:313-20; PMID:5689462; http://dx.doi.org/ 10.3181/00379727-127-32680 [DOI] [PubMed] [Google Scholar]
- 17.Liberski P, Jeffrey M. Tubulovesicular structures—the ultrastructural hallmark for transmissible spongiform encephalopathies (prion diseases). Folia Neuropathologica 2004; 42 Supplement B:96-108; PMID:16903145 [PubMed] [Google Scholar]
- 18.McLeod AH, Murdoch H, Dickinson J, Dennis MJ, Hall GA, Buswell CM, Carr J, Taylor DM, Sutton JM, Raven ND. Proteolytic inactivation of the bovine spongiform encephalopathy agent. Biochem Biophys Res Commun 2004; 317:1165-70; PMID:15094392; http://dx.doi.org/ 10.1016/j.bbrc.2004.03.168 [DOI] [PubMed] [Google Scholar]
- 19.Miyazawa K, Emmerling K, Manuelidis L. High CJD infectivity remains after prion protein is destroyed. J Cell Biochem 2011; 112:3630-7; PMID:21793041; http://dx.doi.org/ 10.1002/jcb.23286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kipkorir T, Tittman S, Botsios S, Manuelidis L. Highly Infectious CJD Particles Lack Prion Protein but Contain Many Viral-Linked Peptides by LC-MS/MS. J Cell Biochem 2014; 115:2012-21; PMID:24933657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.McDonnell G, Dehen C, Perrin A, Thomas V, Igel-Egalon A, Burke PA, Deslys JP, Comoy E. Cleaning, disinfection and sterilization of surface prion contamination. J Hospital Infect 2013; 85:268-73; PMID:24074640; http://dx.doi.org/ 10.1016/j.jhin.2013.08.003 [DOI] [PubMed] [Google Scholar]
- 22.Jackson GS, McKintosh E, Flechsig E, Prodromidou K, Hirsch P, Linehan J, Brandner S, Clarke AR, Weissmann C, Collinge J. An enzyme-detergent method for effective prion decontamination of surgical steel. J General Virol 2005; 86:869-78; PMID:15722550; http://dx.doi.org/ 10.1099/vir.0.80484-0 [DOI] [PubMed] [Google Scholar]
- 23.Jeffrey M, Gonzalez L, Espenes A, Press C, Martin S, Chaplin M, Davis L, Landsverk T, MacAldowie C, Eaton S, et al. . Transportation of prion protein across the intestinal mucosa of scrapie-susceptible and scrapie-resistant sheep. J Pathol 2006; 209:4-14; PMID:16575799; http://dx.doi.org/ 10.1002/path.1962 [DOI] [PubMed] [Google Scholar]
- 24.Scherbel C, Pichner R, Groschup MH, Mueller-Hellwig S, Scherer S, Dietrich R, Maertlbauer E, Gareis M. Infectivity of Scrapie Prion Protein (PrPSc) Following In vitro Digestion with Bovine Gastrointestinal Microbiota. Zoonoses Public Health 2007; 54:185-90; PMID:17542960; http://dx.doi.org/ 10.1111/j.1863-2378.2007.01040.x [DOI] [PubMed] [Google Scholar]
- 25.Dobie K, Barron R. Dissociation between transmissible spongiform encephalopathy (TSE) infectivity and proteinase K-resistant PrP(Sc) levels in peripheral tissue from a murine transgenic model of TSE disease. J Virol 2013; 87:5895-903; PMID:23487470; http://dx.doi.org/ 10.1128/JVI.03469-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barron RM, Campbell SL, King D, Bellon A, Chapman KE, Williamson RA, Manson JC. High titers of transmissible spongiform encephalopathy infectivity associated with extremely low levels of PrPSc in vivo. J Biol Chem 2007; 282:35878-86; PMID:17923484; http://dx.doi.org/ 10.1074/jbc.M704329200 [DOI] [PubMed] [Google Scholar]
- 27.Baker CA, Martin D, Manuelidis L. Microglia from CJD brain are infectious and show specific mRNA activation profiles. J Virol 2002; 76:10905-13; PMID:12368333; http://dx.doi.org/ 10.1128/JVI.76.21.10905-10913.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hawkins SA, Simmons HA, Gough KC, Maddison BC. Persistence of ovine scrapie infectivity in a farm environment following cleaning and decontamination. Vet Rec 2015; 176:99; PMID:25362003; http://dx.doi.org/ 10.1136/vr.102743 [DOI] [PubMed] [Google Scholar]
- 29.Bernoulli C, Siegfried J, Baumgartner G, Regli F, Rabinowicz T, Gajdusek DC, CJ G. Danger of accidental person-to-person transmission of Creutzfeldt-Jakob disease by surgery. Lancet 1977; 8009:478-9; http://dx.doi.org/ 10.1016/S0140-6736(77)91958-4 [DOI] [PubMed] [Google Scholar]
- 30.Will RG. Acquired prion disease: iatrogenic CJD, variant CJD, kuru. Br Med Bull 2003; 66:255-65; PMID:14522863; http://dx.doi.org/ 10.1093/bmb/66.1.255 [DOI] [PubMed] [Google Scholar]
- 31.Manuelidis L. Decontamination of Creutzfeldt-Jakob Disease and other transmissible agents. J NeuroVirol 1997; 3:62-5; PMID:9147822; http://dx.doi.org/ 10.3109/13550289709015793 [DOI] [PubMed] [Google Scholar]
- 32.Peretz D, Supattapone S, Giles K, Vergara J, Freyman Y, Lessard P, Safar JG, Glidden DV, McCulloch C, Nguyen HO, et al. . Inactivation of prions by acidic sodium dodecyl sulfate. J Virol 2006; 80:322-31; PMID:16352557; http://dx.doi.org/ 10.1128/JVI.80.1.322-331.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisler P. Deadly bacteria outbreak inflames disinfection concerns. USA Today; 2015 [Google Scholar]
- 34.CDC Healthcare Infection Control Practices Advisory Committee (HICPAC);Concerns. Centers for Disease Control and Infection; 2009 [Google Scholar]
- 35.Radebold K, Chernyak M, Martin D, Manuelidis L. Blood borne transit of CJD from brain to gut at early stages of infection. BMC Infect Dis 2001; 1:20-5; PMID:11716790; http://dx.doi.org/ 10.1186/1471-2334-1-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shlomchik MJ, Radebold K, Duclos N, Manuelidis L. Neuroinvasion by a Creutzfeldt-Jakob disease agent in the absence of B cells and follicular dendritic cells. Proc Natl Acad Sci U S A 2001; 98:9289-94; PMID:11470899; http://dx.doi.org/ 10.1073/pnas.161055198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McKinnon D. Thirty new cases of Hepatitis B found. All had EEGs at six clinics, officials say. Toronto Star 1996:SC4 [Google Scholar]
- 38.Gabizon R, McKinley MP, Prusiner SB. Purified prion proteins and scrapie infectivity copartition into liposomes. Proc Natl Acad Sci U S A 1987; 84:4017-21; PMID:3108886; http://dx.doi.org/ 10.1073/pnas.84.12.4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Prusiner SB, Groth D, Serban A, Stahl N, Gabizon R. Attempts to restore scrapie prion infectivity after exposure to protein denaturants. Proc Natl Acad Sci 1993; 90:2793-7; PMID:8464892; http://dx.doi.org/ 10.1073/pnas.90.7.2793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Prusiner SB, Groth DF, Bolton DC, Kent SB, Hood LE. Purification and structural studies of a major scrapie prion protein. Cell 1984; 38:127-34; PMID:6432339; http://dx.doi.org/ 10.1016/0092-8674(84)90533-6 [DOI] [PubMed] [Google Scholar]
- 41.Manuelidis L, Sklaviadis T, Akowitz A, Fritch W. Viral particles are required for infection in neurodegenerative Creutzfeldt-Jakob disease. Proc Natl Acad Sci (USA) 1995; 92:5124-8; http://dx.doi.org/ 10.1073/pnas.92.11.5124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishida N, Katamine S, Manuelidis L. Reciprocal interference between specific CJD and scrapie agents in neural cell cultures. Science 2005; 310:493-6; PMID:16239476; http://dx.doi.org/ 10.1126/science.1118155 [DOI] [PubMed] [Google Scholar]
- 43.Arjona A, Simarro L, Islinger F, Nishida N, Manuelidis L. Two Creutzfeldt-Jakob disease agents reproduce prion protein-independent identities in cell cultures. Proc Natl Acad Sci USA 2004; 101:8768-73; PMID:15161970; http://dx.doi.org/ 10.1073/pnas.0400158101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun R, Liu Y, Zhang H, Manuelidis L. Quantitative recovery of scrapie agent with minimal protein from highly infectious cultures. Viral Immunol 2008; 21:293-302; PMID:18788938; http://dx.doi.org/ 10.1089/vim.2008.0039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Manuelidis L. Nuclease resistant circular DNAs copurify with infectivity in scrapie and CJD. J Neurovirol 2011; 17:131-45; PMID:21165784; http://dx.doi.org/ 10.1007/s13365-010-0007-0 [DOI] [PubMed] [Google Scholar]
- 46.Kipkorir T, Colangelo CM, Manuelidis L. Proteomic analysis of host brain components that bind to infectious particles in Creutzfeldt-Jakob disease. Proteomics 2015; 15:2983-98; PMID:25930988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Klug CS, Su W, Liu J, Klebba PE, Feix JB. Denaturant unfolding of the ferric enterobactin receptor and ligand-induced stabilization studied by site-directed spin labeling. Biochemistry 1995; 34:14230-6; PMID:7578022; http://dx.doi.org/ 10.1021/bi00043a030 [DOI] [PubMed] [Google Scholar]
- 48.Hong H, Tamm LK. Elastic coupling of integral membrane protein stability to lipid bilayer forces. Proc Natl Acad Sci U S A 2004; 101:4065-70; PMID:14990786; http://dx.doi.org/ 10.1073/pnas.0400358101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gordon JA, Jencks WP. The relationship of structure to the effectiveness of denaturing agents for proteins. Biochemistry 1963; 2:47-57; PMID:13949214; http://dx.doi.org/ 10.1021/bi00901a011 [DOI] [PubMed] [Google Scholar]
- 50.Miyazawa K, Emmerling K, Manuelidis L. Replication and spread of CJD, kuru and scrapie agents in vivo and in cell culture. Virulence 2011; 2:188-99; PMID:21527829; http://dx.doi.org/ 10.4161/viru.2.3.15880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rabilloud T. Use of thiourea to increase the solubility of membrane proteins in two-dimensional electrophoresis. Electrophoresis 1998; 19:758-60; PMID:9511858 [DOI] [PubMed] [Google Scholar]
- 52.Natarajan S, Xu C, Caperna TJ, Garrett WM. Comparison of protein solubilization methods suitable for proteomic analysis of soybean seed proteins. Anal Biochem 2005; 342:214-20; PMID:15953580; http://dx.doi.org/ 10.1016/j.ab.2005.04.046 [DOI] [PubMed] [Google Scholar]
- 53.Riesner D, Kellings K, Post K, Wille H, Serban H, Groth D, Baldwin MA, Prusiner SB. Disruption of prion rods generates 10-nm spherical particles having high a-helical content and lacking scrapie infectivity. J Virol 1996; 70:1714-22; PMID:8627692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Okoroma EA, Purchase D, Garelick H, Morris R, Neale MH, Windl O, Abiola OO. Enzymatic formulation capable of degrading scrapie prion under mild digestion conditions. PLoS One 2013; 8:e68099; PMID:23874511; http://dx.doi.org/ 10.1371/journal.pone.0068099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Millson G, Hunter G, Kimberlin R. The physico-chemical nature of the scrapie agent. In: Kimberlin R, ed. Slow virus diseases of animals and man. Amsterdam: North-Holland, 1976:243-66 [PubMed] [Google Scholar]
- 56.Nakamura A, Mrimoto M, Takeuchi K, Fujii T. A Rapid Extraction Procedure of Human Hair Proteins and Identification of Phosphorylated Species. Biol Pharm Bull 2002; 25:569-72; PMID:12033494; http://dx.doi.org/ 10.1248/bpb.25.569 [DOI] [PubMed] [Google Scholar]
- 57.Koga Y, Tanaka S, Sakudo A, Tobiume M, Aranishi M, Hirata A, Takano K, Ikuta K, Kanaya S. Proteolysis of abnormal prion protein with a thermostable protease from Thermococcus kodakarensis KOD1. Appl Microbiol Biotechnol 2014; 98:2113-20; PMID:23880875; http://dx.doi.org/ 10.1007/s00253-013-5091-7 [DOI] [PubMed] [Google Scholar]
- 58.Emadi S, Behzadi M. A comparative study on the aggregating effects of guanidine thiocyanate, guanidine hydrochloride and urea on lysozyme aggregation. Biochem Biophys Res Commun 2014; 450:1339-44; PMID: 25003319; http://dx.doi.org/ 10.1016/j.bbrc.2014.06.133 [DOI] [PubMed] [Google Scholar]
- 59.Caughey B, Raymond GJ, Kocisko DA, Lansbury PT Jr. Scrapie infectivity correlates with converting activity, protease resistance, and aggregation of scrapie-associated prion protein in guanidine denaturation studies. J Virol 1997; 71:4107-10; PMID:9094691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Walker JT, Dickinson J, Sutton JM, Raven ND, Marsh PD. Cleanability of dental instruments–implications of residual protein and risks from Creutzfeldt-Jakob disease. British Dental J 2007; 203:395-401; PMID:17934424; http://dx.doi.org/ 10.1038/bdj.2007.893 [DOI] [PubMed] [Google Scholar]
- 61.Singh S, Zlotnick A. Observed hysteresis of virus capsid disassembly is implicit in kinetic models of assembly. J Biol Chem 2003; 278:18249-55; PMID:12639968; http://dx.doi.org/ 10.1074/jbc.M211408200 [DOI] [PubMed] [Google Scholar]
- 62.Lee KW, Tan WS. Recombinant hepatitis B virus core particles: association, dissociation and encapsidation of green fluorescent protein. J Virol Methods 2008; 151:172-80; PMID:18584885; http://dx.doi.org/ 10.1016/j.jviromet.2008.05.025 [DOI] [PubMed] [Google Scholar]
- 63.Philipson L, Beatrice ST, Crowell RL. A structural model for picornaviruses as suggested from an analysis of urea-degraded virions and procapsids of coxsackievirus B3. Virol 1973; 54:69-79; PMID:4351615; http://dx.doi.org/ 10.1016/0042-6822(73)90115-3 [DOI] [PubMed] [Google Scholar]
- 64.Exner M, Tuschewitzki GJ, Scharnagel J. Influence of biofilms by chemical disinfectants and mechanical cleaning. Zentralbl Bakteriol Mikrobiol Hyg B 1987; 183:549-63; PMID:3109156 [PubMed] [Google Scholar]
- 65.Mayer C, Moritz R, Kirschner C, Borchard W, Maibaum R, Wingender J, Flemming HC. The role of intermolecular interactions: studies on model systems for bacterial biofilms. Int J Biol Macromol 1999; 26:3-16; PMID:10520951; http://dx.doi.org/ 10.1016/S0141-8130(99)00057-4 [DOI] [PubMed] [Google Scholar]
- 66.Di Maio F, Yu X, Resen E, Krupovic M, Prangishvili D, Egelman E. Virology. A virus that infects a hyperthermophile encapsidates A-form DNA. Science 2015; 348:914-7; PMID:25999507; http://dx.doi.org/ 10.1126/science.aaa4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Manuelidis L, Fritch W, Xi YG. Evolution of a strain of CJD that induces BSE-like plaques. Science 1997; 277:94-8; PMID:9204907; http://dx.doi.org/ 10.1126/science.277.5322.94 [DOI] [PubMed] [Google Scholar]
- 68.Sklaviadis TL, Manuelidis EE, Manuelidis L. Physical properties of the Creutzfeldt-Jakob disease agent. J Virol 1989; 63:1212-22; PMID:2492609 [DOI] [PMC free article] [PubMed] [Google Scholar]