Preterm Prelabor Rupture of the Membranes: A Disease of the Fetal Membranes (original) (raw)
. Author manuscript; available in PMC: 2018 Nov 1.
Abstract
Preterm prelabor rupture of the membranes (pPROM) remains a significant obstetric problem that affects 3-4% of all pregnancies and precedes 40% to 50% of all preterm births. pPROM arises from complex, multifaceted pathways. In this review, we summarize some old concepts and introduce some novel theories related to pPROM pathophysiology. Specifically, we introduce the concept that pPROM is a disease of the fetal membranes where inflammation-oxidative stress axis plays a major role in producing pathways that can lead to membrane weakening through a variety of processes. In addition, we report microfractures in fetal membranes that are likely sites of tissue remodeling during gestation; however, increase in number and morphometry (width and depth) of these microfractures in pPROM membranes suggests reduced remodeling capacity of membranes. Microfractures can act as channels for amniotic fluid leak, and inflammatory cell and microbial migration. Further studies on senescence activation and microfracture formation and their role in maintaining membrane homeostasis are needed to fill the knowledge gaps in our understanding of pPROM as well as provide better screening (biomarker and imaging based) tools for predicting women at high risk for pPROM and subsequent preterm birth.
Keywords: Fetal signals, parturition, exosomes, biomarker, aging, fetal membranes, amniochorion
Introduction
Preterm prelabor rupture of the membranes (pPROM) is defined as rupture of the fetal membranes prior to 37 weeks of completed gestation. This significant obstetric problem occurs in about 3-4% of all pregnancies and is directly antecedent to 40% to 50% of all preterm births1,2. Since about 560,000 babies are born prematurely each year in the U.S. (∼12.0% of all births), this correlates to about 150,000 spontaneous preterm births (sPTBs) that are complicated by pPROM3. The number of pPROM cases exceeds that of preeclampsia and gestational diabetes and other iatrogenic preterm births. In addition, neonatal mortality and morbidities are higher in pPROM group than any other subclasses of preterm births. Yet, pPROM is an often-ignored and understudied adverse outcome of pregnancy. Despite remarkable improvements in prenatal care over the past three decades, rates of pPROM and subsequent preterm delivery have worsened4.
While several tests are available to confirm a diagnosis of pPROM post facto (e.g. pooling, fern tests, nitrazine, and Amnisure®), no method to reliably predict pPROM is available. This dilemma is mostly attributable to the fact that precise causes or risk factors are unknown and pathways resulting in pPROM have only recently been delineated4. Empirical treatment approaches that ignore the complexity and heterogeneity of pPROM pathophysiology have to date been futile5. Proper diagnosis and management of pPROM is likely to require thorough investigation of specific exposure-induced pathophysiologic pathways and the development of biomolecular markers that can predict pPROM.
pPROM arises from complex, multifaceted pathways. Several epidemiological and clinical factors are considered precursors to pPROM. These include maternal reproductive tract infections (e.g., bacterial vaginosis [BV], trichomoniasis, gonorrhea, Chlamydia and occult chorioamnionitis), behavioral factors (e.g., cigarette smoking, substance abuse, poor nutritional status, and coitus during pregnancy), obstetric complications (e.g., multiple gestation, polyhydramnios, incompetent cervix, gestational bleeding, prior cervical surgery, and antenatal trauma)6-8. Environmental factors (e.g., stress, toxin exposure) and genetic predisposition also have been proposed. In addition, biochemical signals from the fetus, including endocrine signals that promote fetal membrane apoptosis, have also been implicated in the initiation of pPROM5-9. In this review, we summarize some old concepts and introduce some novel theories related to pPROM pathophysiology.
Fetal membranes: Structure and function
Human fetal membranes, also referred as placental membranes or amniochorionic membranes, is the inner lining of the pregnant intrauterine cavity. These fetal tissues are distinct from placenta and serves as a barrier between the feto-placental and the maternal compartments. Fetal membranes are comprised of the amnion (innermost layer of the intraamniotic cavity) and the chorion (fetal tissue connected to maternal decidua), and are connected by collagen-rich extra cellular matrix (ECM) 9. ECM, which is made up of fibrous proteins along with various types of collagen, provides the architectural and structural framework of the fetal membranes (Figure 1). 9,10 The amnion is constantly bathed in amniotic fluid, signifying its importance as a primary responder to changes in the amniotic cavity. The chorion is in close proximity to maternal decidua and maintains the immune tolerance at the maternal-fetal interface.11-14
Figure 1. Characterization of Fetal Membranes with Non-Linear Optical Microscopy.
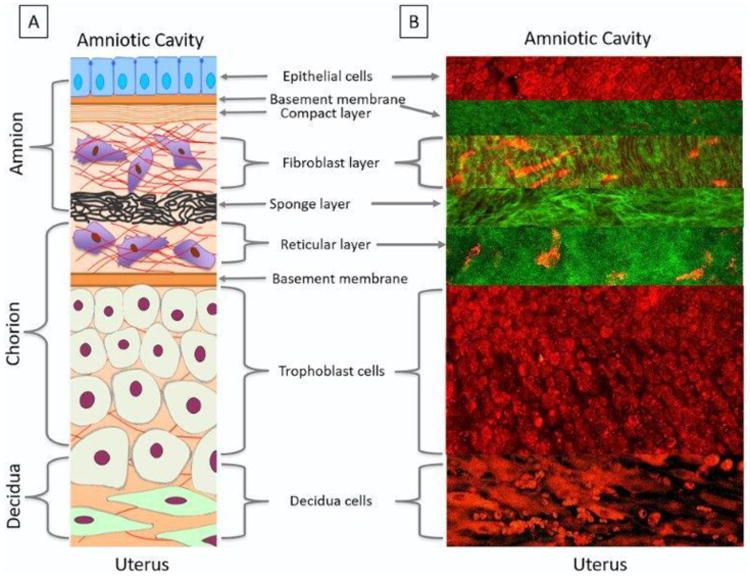
A schematic of the fetal membrane layers (A) compared to the layers imaged by a two-photon laser using multiphoton autofluorescence (MPAM) (red) and second harmonic generation microscopy (SHG) (green) (B).
A: Schematic of the fetal membrane layers: The amnion epithelial layer (blue cells), constantly bathed in amniotic fluid, is a single layer of cuboidal cells held together by neighboring cells gap junctions. Amnion epithelial cells secrete collagen Types III and IV, and glycoproteins to form the basement membrane (orange) on their basal side. This forms a junction and allows communication between the two layers. Stromal cells (purple cells) secreting Type 1 and 3 collagens create the compact layer (orange stripes) and fibroblast layer forming a fibrous skeleton helpful in maintaining amnion integrity. The fibroblast layer is connected to the spongy layer made up of proteoglycans, glycoproteins and Type 3 collagen producing a spongy meshwork (black). The reticular layer, made up of fibrillar bundles of diffused collagen, is attached to the spongy layer on its apical side and the chorion (tan cells) on its basal side. The extra cellular matrix layers, filled with mesenchymal stromal cells, can interact with each other, degrade collagen layers, and communicate with cellular layers to help with the fetal membrane remodeling. The chorion (tan cells) is a typical epithelium layer, with it cell polarity facing the material side connecting to the decidua. The decidua (green cells), a maternally derived tissue, contains enlarged mesenchymal stromal cells and maternal immune cells that play a critical role in regulating inflammation at term. The maternal myometrium, is connected to the basal side of the decidua, and is made up of smooth muscle cells that form contractions around the fetus at the end of gestation.
B: MPAM/SHG: Multiphoton images of the fetal membranes show cellular content in red and collagen in green. Amnion cells are seen in a monolayer with their nuclei appearing darker than the cytoplasm. The basement membrane in green, mimics the outline of the epithelial cells and is connected to the organized/wavy collagen in the fibroblast layer. Elongated stromal cells in red are seen interspersed through the collagen. The spongy layer is made up of tightly packed fibular collagen and is attached to the reticular layer comprised of lose collagen and stromal cells (red). The extra cellular matrix averages around 80 μm in depth. The trophoblast cells are tightly packed close to the basement membrane of the extra cellular matrix and spread out as they approach the decidua. The decidua is made up of enlarged, elongated, mesenchymal cells with intermittent red blood cells.
The amnion and chorion are fetal tissues in origin and play major roles in maintaining pregnancy by providing multi-level protection to the growing fetus. Fetal membranes accommodate constant challenges (immune, structural, mechanical and endocrine) during pregnancy; continue to grow and mechanistically, as well as biochemically, maintains elasticity to the stretch forces experienced during fetal growth. Despite the fact that membranes overlaying the placenta and cervix face distinctly different environments and insults during pregnancy, the membranes still maintain the homeostatic balance necessary to sustain fetal growth without interruption. This companionship between the fetus and the membranes continues until term when the fetus reaches maturity and the membranes reach longevity.
Development of fetal membranes
The development of amnion and chorion begins with embryogenesis, although they do not participate directly in the formation of the embryo or fetus.15 Like the fetus, early growth of the amnion and chorion layers is rapid and independent of each other. The formation of amniochorion as a unit structure is complete by the 12th week of gestation.15 The composition of the membrane and its ability to produce a broad spectrum of biomarkers at different stages of gestation illustrate the possible role of the fetal membrane's influence on the growing fetus, as well as in adverse pregnancy outcomes.10 In addition, fetal membrane cells continue to divide throughout pregnancy and its growth plateaus at term, retaining the capacity for DNA replication.11 Lastly, the presence of stem cells in fetal membranes further depicts the likely important role of fetal membranes during in utero fetal life.9;12-14
pPROM: A disease of the fetal membranes
Unlike the placenta, fetal membranes are not involved in transport of nutrients or other materials. One of the major functions of fetal membranes is to protect the fetus during its growth and development in utero. Specifically, the fetal membrane functions to provide mechanical16,17 and immune protection and acts as a barrier for microbial access18-20. This protective role is supported by the biomarkers that are produced by fetal membranes during gestation and parturition21. Compromise in the immune and mechanical properties of the fetal membranes allows for microbial invasion from genital tract22, activation of host inflammatory response leading to collagenolysis mediated mechanical disruption16,23-25, and membrane weakening predisposing the membranes to pPROM. Abruption associated thrombin, matrix metalloproteinase (MMP) activation and collagenolytic processes have also been reported in fetal membrane weakening and pPROM26. Clearly, the dysfunctional status of fetal membranes is more evident in pPROM than sPTB with no ROM. Thus, pPROM is considered as a disease of the fetal membranes and likely a separate entity from sPTB with no ROM. Current research on this disease of the fetal membranes is focused on addressing three major questions: 1) what are the initiators of proteolytic activity in the fetal membranes resulting in pPROM, 2) what are the effectors (biochemicals) of proteolysis, membrane weakening/rupture and can these biochemicals serve as markers to predict risk of pPROM and 3) how can we reduce the risk of pPROM prior to its occurrence and minimize the impact on maternal-fetal wellbeing27.
Risk factors for pPROM
Approximately 70% of pPROM cases are associated with intraamniotic infection (IAI), as documented by positive amniotic fluid (AF) cultures or by clinical evidence of infection4,28. However, it has been debated whether infection is a cause or consequence of pPROM. Histological and microbiological findings indicate that focal infection and inflammation may play a primary or secondary role in the pathogenesis of pPROM29. Evidence of inflammatory changes is reported to be adjacent to the putative site of membrane rupture, suggesting that bacterial infection may be an initiator of pPROM1. However, bacterial toxins and bacteria-derived matrix degrading enzymes have been discounted as a primary mediators of pPROM for the following reasons: 1) the concentration of bacterial toxins required to produce deleterious effects on isolated membranes are not achieved in AF; 2) pPROM is seen in women with IAI due to organisms that are not known to produce proteases; 3) most bacterial matrix degrading enzymes do not have the catalytic specificity to cleave human ECM collagens; and 4) pPROM has been documented in many cases after effective antibacterial therapy for periodontal disease, BV and IAI30-32. Recent reports have indicated that pPROM may be associated with sterile inflammation in the fetal membranes (an inflammatory condition mimicking infection33 with no evidence of microbial presence either through culture or molecular methods). We hypothesize that certain risk factors of pPROM (ie smoking, bleeding)34, can produce sterile inflammation in the fetal membranes leading to an immunocompromised state that allows for microbial invasion. Infection in pPROM is likely a secondary phenomenon rather than a causal factor. In the remainder of this review, we will provide new perspectives in our understanding of fetal membrane dysfunction and pPROM (that involves factors other than infection-associated inflammation) and provide novel concepts to be tested in future research.
Oxidative stress (OS) and pPROM
One of the pathophysiologic factors that has been overlooked in pPROM is oxidative stress (OS) and generation of reactive oxygen species (ROS) as mediators of OS. Healthy pregnancy, like other physiological states, is characterized by a stable redox balance between reactive oxygen species (ROS) and antioxidants35-39. The two most common sources of ROS are leakage from the electron transport system in the inner mitochondrial membrane during cellular respiration and release by immune cells during phagocytosis. OS increases placental mitochondrial activity and production of ROS when the energy demands of the fetoplacental unit are high, such as high altitude pregnancy, antioxidant nutrient deficiency, or intrinsic microvascular disease. This results in an imbalanced redox state40. Behavioral risk factors associated with pPROM, particularly cigarette smoking, generate superoxide, hydrogen peroxide, hydroxyl ions and nitric oxide that could damage the collagen matrix and consume antioxidant defenses41-48. Collagen has been shown to be a primary target for ROS. Superoxide, stimulates the in vitro cleavage of fibrillar collagen to 4-hydroxyproline49. Chorioamniotic collagenolytic enzymes are vulnerable to ROS activation50, with MMP9 being suppressed by superoxide dismutase (SOD) or the glutathione precursor N-acetylcysteine. Lappas and colleagues have reported inhibition of MMP9 and other inflammatory markers using antioxidant N-Acetyl-cysteine (NAC)51. NAC inhibits an NF-κB-activated pathway and subsequent phospholipid metabolism, proinflammatory cytokine release, and protease activity in human fetal membranes51. The evidence above indicates that OS and inflammation are major phenomena associated with pPROM. These events are initiated in response to exposures with dramatic biochemical consequences that can lower the threshold for membrane rupture.
Collagenolysis, extracellular matrix (ECM) degradation and rupture
Degradation of the collagen-rich ECM, connecting the amnion and chorion layers of the fetal membranes, is one of the key events leading to ROM52-57. This process is mediated by several MMPs, each of which catalyze collagen type-specific substrate turnover. MMP activity is tightly regulated in the fetal membranes by transcription, translation and post-translational modification of MMPs and their endogenous inhibitors, the tissue-specific inhibitors of metalloproteinases (TIMPs)58,59. A balanced MMP/TIMP system in human fetal membranes plays a role both during normal labor and in pPROM. Although studies have yet to clearly define how this balance is disrupted in pPROM, it is clear that proteases associated with infection or inflammation can activate endogenous MMPs in fetal membranes, leading to collagen turnover and membrane weakening23,60-65.
Apoptosis of fetal membranes
Over a decade ago, fetal membrane apoptosis has been reported as a pathological mechanism associated with pPROM66-71. This was further supported by the fact that elements of apoptosis are predominantly seen in membranes from women with pPROM than sPTB67. Infection and other endotoxins are capable of inducing many of these proapoptotic factors during pPROM that are generally absent in membranes collected after sPTB with no ROM69,72,73. Classic DNA fragmentation pattern (used in diagnosing apoptosis) has been reported in fetal membranes obtained from women with pPROM. However, apoptosis is not often associated with the massive inflammatory response that is seen in pPROM. In addition, DNA fragmentation (as shown by TUNEL assay or LmPCR) that is used as evidence of apoptosis can be seen with other forms of tissue/cell death such as necroptosis74,75. OS also causes DNA fragmentation from fetal membrane cells76. These factors raise ambiguity in the role of apoptosis as a pathologic mechanism in predisposing membranes to pPROM. It is likely that pPROM may have elements of all forms of cell deaths as inflammatory mediators can induce various forms of cell deaths depending on their kinetics and dose.
In summary, the roles of infection, inflammation, collagenolytic enzymes and apoptosis in pPROM have been independently characterized with the expectation that pathways responding to specific risk factors should indicate which initiators and effectors of pPROM are operative in any given case. However, to date, findings from such studies have not produced useful biomarkers or improvements in the rates of pPROM and sPTB. We will focus the rest of this review on some novel concepts that can mechanistically lead to pPROM.
Novel Concepts in pPROM
Phenotypic heterogeneity in studies that have evaluated pPROM is one of the major reasons why identifying a biomarker or clinical indicator that predicts pPROM has been elusive. For instance, sPTB and pPROM overlap in several ways; 1) etiologic similarities, 2) association between amniotic fluid and cervico-vaginal fluid genital mycoplasma colonization and infections and sPTB and pPROM, 3) infection-associated inflammation and biochemical markers determining underlying pathophysiology, 4) clinical and histologic evidence of chorioamionitis in cases of sPTB and pPROM. However, approximately 40% of women rupture their membrane prior to labor initiation whereas majority will not experience ROM. This suggests that although similarities may exist in etiologies and clinical presentations, the underlying mechanistic pathways leading to pPROM and sPTB with intact membranes are distinct and therefore they should be considered as independent phenotypes. The following sections will review new evidences form our own laboratories that showed differences in biomarkers between these phenotypes and provide evidence for a novel concept on pPROM pathology. Based on basic and clinical evidences, pPROM is considered a disease of the fetal membranes27. Therefore, we investigated changes to the fetal membranes in pPROM, sPTB with intact membranes and normal term pregnancies (regardless of labor status).
Oxidative stress in pPROM, sPTB and normal term birth
Using fetal membranes as a model, we examine inflammation and OS as underlying pathophysiologies for both pPROM and sPTB77,78. Although inflammation and OS are inseparable components of innate immune mechanism, OS markers (antioxidants, OS induced damages and products of OS induced damages) are not readily investigated compared to pro-inflammatory biomarkers. OS biomarkers are hardly reported in the literature in association with both sPTB and pPROM79. Using clinical specimens (amniotic fluid and fetal membranes) from these two conditions and in vitro model of fetal membrane organ explant system80, we conducted studies to examine OS in these conditions and investigated the source and mechanisms of inflammation. We used normal term membranes as our comparison group and some of the findings from our report are described below.
Generation of ROS as a part of the aerobic energy building process is inevitable and is well balanced (redox status) by an array of enzymatic and non-enzymatic antioxidant systems40,77,78,81-84. Redox balance is maintained through the production and subsequent elimination of ROS. Cells are able to protect themselves against OS by the finely tuned regulation of redox status through endogenous enzymes, antioxidants, and other cellular mechanisms. At low levels, ROS, often generated in biological systems, is essential for cell division and survival, cell signaling, inflammation and immune functions, autophagy, and stress response85,86. However, an overwhelming redox imbalance compromises a biological system's ability to detoxify these highly reactive molecules or to repair any damage caused by them85. Therefore, the former is termed “OS” and the latter “oxidative damage.”38 Oxidative damage due to ROS generation has been linked to the development of adult diseases, including cardiovascular disease, cancer, chronic inflammation, and neurologic disorders.
F2-Isop is a lipid peroxidation product and a biomarker of OS associated damages87-89. To determine OS damage in membranes and release of damaged cellular elements to the tissue environment or biological fluid, we measured F2-isoprostanes (F2-IsoP) in the amniotic fluid of women from pPROM, sPTB and normal term birth90. We found that F2-Isop levels were significantly higher in women with pPROM and normal term birth compared to those who had sPTB with intact membranes90. Similar increases in F2-IsoP were also reported by Longini et al84. We also noted that women who smoked (OS inducer) during pregnancy (regardless of pregnancy outcome) also had elevated F2-IsoP. Cases with infection also had higher F2-IsoP; however, the levels were not so high as seen in women who smoked 90. This suggests that OS damage is a factor associated with pPROM and normal term birth.
Reduction of telomeres in pPROM and normal term birth
Telomeres are cap structures that protect chromosomal ends and their length is gradually reduced as cells divide and age91. Hence, telomere length is considered a measure of biological aging91,92. Telomere length is reduced in response to OS as well93. After documenting OS damages associated with pPROM and normal term birth, we tested the impact of stress on fetal tissue telomere length. Telomere length is not only an indicator of aging it is also a biomarker for OS damage as the guanine repeat sequences (TTAGGG repeats that dominates telomere cap structure) in telomeres are highly vulnerable OS damages. Telomere length analysis in fetal leukocyte DNA (cord blood) and fetal membranes obtained from women with pPROM showed significantly lower telomere length compared to gestational age matched samples obtained from women with sPTB with intact membranes, especially in samples < 34 weeks94. Fetal membrane telomere lengths were also reduced in these conditions and correlated with leukocyte DNA telomere lengths94. These data showing identical telomere lengths in pPROM and normal term births suggest that OS and OS associated telomere length reduction occurs in both conditions. As mentioned above, telomere lengths are indicators of biologic aging and this data led us to hypothesize that pPROM and normal term births are linked to OS and OS damages and these factors can accelerate fetal membrane aging. Aging of fetal membranes is likely a natural and physiologic event in normal term pregnancies; however, this process is likely accelerated in response to various OS inducing risk factors of pPROM.
Oxidative stress at term
The pregnant uterine environment, especially fetal tissues (ie placenta), progress through various states of oxidative status as the fetus matures 95-98. Fetal and placental growth are linked to changing status of oxygen tension and OS in response to various oxidative conditions. ROS generated in response to cellular respiration are involved in cell signaling and growth. Therefore, OS status in the pregnant uterine environment plays a major role in feto-placental growth and development97,99,100. Once the fetal development is complete, OS and inflammation build up at term is a factor responsible for triggering parturition. Oxidative stress and presence of ROS rises at term due to following factors: 1) increased metabolic demands from the fetus who is ready for its independent existence outside the uterus78, 2) depleted fetal antioxidant reserve in the intrauterine compartments101, 3) no change in supply of substrate to meet increased metabolic demands by the fetus102,103 and 4) uterine over-distension and fetal stretching104. In preterm pregnancies, especially those complicated by pPROM, OS buildup can occur in response to risk factors such as infection, nutrient insufficiency (specifically antioxidants), behavioral factors (cigarette smoking, drug and alcohol abuse), and low or high BMI.
OS induces fetal membrane aging at term and in pPROM
Oxidative damage, as described above, affects the functioning of lipids, proteins and nucleic acids along with other cellular structures. In human fetal membranes, OS causes activation of p38 mitogen activated protein kinase (MAPK). p38MAPKs are a family of evolutionarily conserved serine/threonine kinases (mitogen activated protein kinases) that contains four stress response signaling isoforms (p38α, β, γ, δ) whose functions differ significantly105,106. ROS-mediated phosphorylation and activation p38MAPK activates its downstream effectors, p16Ink4 and p19arf, resulting in cell cycle arrest and senescence107-109. Senescence is a mechanism that can predispose to cellular aging. Senescent cells are inflammatory and can persist in cellular environment to cause further damage to the tissues110,111. Examination of fetal membranes obtained from women who underwent labor at term compared to women who did not labor at term showed increased OS39 and induced p38MAPK mediated senescence 112.
Sterile inflammation (also known as senescence associated secretory phenotype – SASP) is a feature of senescence associated aging and this is characterized mostly by the presence inflammatory biomarkers, growth factors and matrix degrading enzymes110. SASP markers are known mediators of uterotonic activity at term and have been widely reported in the OB literature112. In vitro, using amnion cells, we have shown that OS can transition a normal term not in labor membrane into a senescence stage and produce SASP. This transition, histologic and biochemical, is similar to that seen in term labor. Progressive activation of telomere-associated, p38MAPK-mediated development of senescence was also seen in murine pregnancy. Telomere length reduction is maximum in fetal membranes compared to all other tissues (placenta, and uterine decidua) that were tested suggesting that the fetal membranes play a major role in the readiness of parturition. In addition to SASP, senescent cells also release highly inflammatory damage associated molecular pattern markers (DAMPs). These are released in response to senescent associated tissue damage. High mobility group box (HMGB) 1, cell free fetal cell DNA and telomere fragments, uric acid are some of the DAMPs that have been linked to parturition113-115. We postulate that at term, senescence causes increased inflammation due to tissue injury and this enhances the overall inflammatory load in feto-maternal tissues to initiate labor-associated changes.
Although not proven mechanistically or in animal model studies, similar events are expected to happen in membranes prior to rupture. Experimental data has shown that membranes from women with pPROM exhibit the following when compared to membranes obtained from women with sPTB with intact membranes: 1) increased DNA damage and reduced telomere length94,116, 2) reduced antioxidant capacity and increased oxidative stress84,90,117-120 and 3) increased p38MAPK activation and senescence.117 In addition, pPROM is associated with increased matrix metalloproteinase activity, a constituent of SASP that is required to degrade membrane ll23,62,63,65,121
In summary, several molecular and histologic factors are similar at term and pPROM supporting our hypothesis that term labor is associated with a natural and physiologic aging of the fetal membranes rendering them dysfunctional. Inflammation arising from an aging membrane can increase the overall inflammatory load of the uterine cavity. Premature aging of fetal membranes can also cause membrane dysfunction and preterm rupture. Table 1 summarizes the key factors that are common and different in fetal membranes between pPROM, sPTB and term birth. Figure 2 provides pathways in which fetal membranes play a role in normal term birth and pPROM. However, given the data to date, one question still remains, which is if sPTB with intact membranes does devoid of OS? The likely answer is that the extent of OS and OS damage are minimal in sPTB when membranes are intact. This may depend on the type and dose of exposure capable of inducing uterine OS. Lastly, vulnerability of subject to specific risk factors and their innate response to control OS may determine their phenotypic outcome.
Table 1. Oxidative stress, senescence and inflammatory markers in pPROM, PTB and normal term births.
| Comparison between pPROM, spontaneous PTB with intact membranes and normal term birth | ||||
|---|---|---|---|---|
| Measure | Sample | Indication | pPROM vs PTB | pPROM vs Normal term birth |
| F2-IsoP | Amniotic fluid | Oxidative stress | Higher in pPROM | Same as pPROM |
| 3-NT staining | Fetal membranes | Oxidative stress | Higher in pPROM | Higher in pPROM |
| Salivary proteases | Maternal saliva | Proteolysis | Higher in pPROM | Same as pPROM |
| MMPs/TIMPs | Amniotic fluid | Proteolysis | Higher in pPROM | Higher in pPROM |
| Cytokines (IL-1, TNF, IL-6, IL-8) | Amniotic fluid | Inflammation | No Difference | Lower in normal birth |
| Telomere length | Fetal DNA | Senescence | Higher in PTB | Same as pPROM |
| p38MAPK | Fetal membranes | Senescence | Higher in pPROM | Same as pPROM |
| p53 | Fetal membranes | Senescence | Higher in pPROM | Same as pPROM |
| Ras-GTPase | Fetal membranes | Signaling | Higher in PTB | Same as pPROM |
Figure 2.
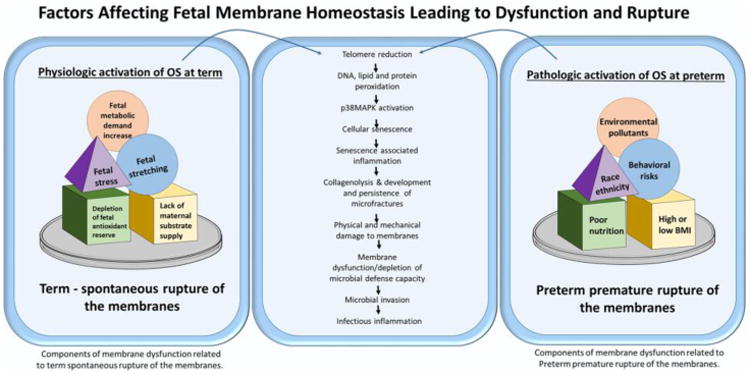
Similarities exist between normal term birth and pPROM. OS increase (factors are depicted in figure) seen towards tem transition a non-labor phenotype into a laboring phenotype. Similarly, increase in intrauterine OS during gestation in response to various risk factors (see figure, list is not complete) can cause similar transition at preterm. Both these processes, culminate in similar pathways leading to telomere attrition, peroxidation of cellular elements and organelles resulting in senescence and senescence associated inflammation. The increased load of inflammation is linked to human parturition122. In pPROM, similar activation of these pathways occur and premature senescence leads to sterile inflammation, membrane dysfunction, loss or reduction in microbial defense capacity. This can lead to microbial invasion and infectious inflammation. Therefore, term parturition is likely linked to sterile inflammation but pPROM may have both sterile and infectious inflammation. This is hard to distinguish as biomarkers of both forms of inflammation are similar.
Microfractures of fetal membranes – sites of membrane architectural damage?
Recently, we examined fetal membrane cellular and collagen structures using nonlinear microscopy from term and preterm labor revealed development of microfractures in fetal membranes123. In this study, the combined methods of multi-photon autofluorescence (MPAM) and second harmonic generation (SHG) were applied to investigate the multilayered 3-dimensional micro-organization of human fetal membranes without the use of exogenous contrast agents. Microfractures are areas of structural changes and are likely initiated by disturbances in the cellular layers. In our study, the following four criteria were used to identify and characterize microfactures: 1) contain altered amnion epithelial layer or site of epithelial shedding, 2) deterioration of basement membrane layer, 3) presence of migrating cells in the ECM portions of the membrane and 4) basement membrane damage and migrating cells develop tunnels in the ECM that extends from basement membrane through the spongy layer. In summary, microfractures are channels or tunnels that appear as extensions of areas often vacated by amnion cells due to shedding or topographically altered due to senescence. Microfractures extend through the spongy layer of the ECM thus connecting its contents with the sparse collagen of the reticular layer. This connection could potently be considered as channels of cross talk between amnion and chorion layers throughout gestation and likely get resealed to facilitate tissue remodeling. The number of microfractures and their dimensions (wider and deeper) are significantly higher in fetal membranes from pPROM than gestational age matched sPTB tissues. We postulate that these microfractures are areas were tissue remodeling was insufficient or ineffective due to underlying pathological reasons or premature senescence. These regions are also associated with the large amounts of collagen degradation. Persistent microfractures can act as channels for amniotic fluid and inflammatory cell infiltration as seen in pPROM. Figure 3 shows an example of microfracture in pPROM membranes that have a 2.5 fold higher prevalence than membranes obtained from women with sPTB or preeclampsia.
Figure 3. Characteristics of microfractures in term and preterm birth.
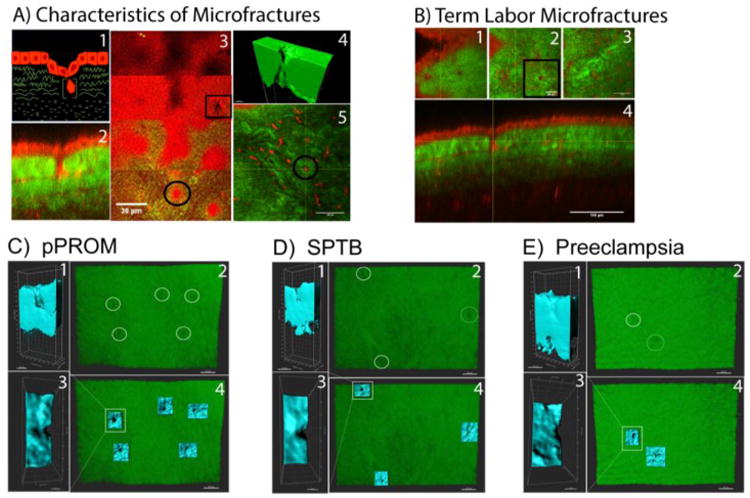
A. Cartoon representation of a microfracture with a cell migrating through a collagen tunnel and a multiphoton image of a microfracture. Cells (red) collagen (green). 1) area of altered amnion morphology. 2) Multiphoton image of a degraded basement membrane (grey arrow) around an area of altered amnion morphology (white astric). 3) Multiphoton image of a cell in a collagen tunnel (grey arrow). Other stromal cells represented in this image are not in collagen tunnels. 4) IMARIS reconstruction of a collagen tunnel. B) Multiphoton image of TL tissue. 1) Amnion morphology above the microfracture. 2) Degraded basement membrane around the migrating cell of interest highlighted with yellow cross hairs. These cross hairs are constant throughout figure 3B. 3) Degraded collagen tunnel with cell migrating through the spongy layer. 4) Orthogonal view of a microfracture with migrating cell in degraded collagen tunnel. Non-Linear Optical Microscopy analysis of collagen degradation by second harmonic generation microscopy (SHG). C) PPROM clinical samples imaged with SHG visualizing areas of collagen degradation (white circles). These areas of collagen degradation, or microfractures, were reconstructed in blue as seen in 1,2, and 4 highlighting the depth, width, and location of microfractures. PPROM samples presented with wider and larger quantify of microfractures compared to SPTB and pre-eclamptic samples. D) SPTB samples only contained one true microfracture that was reconstructed and sliced as seen in images 1,2, and 4. The other blue area in image 4 may show the beginning of microfracture formation due to the indentation but not tunnel formation seen at this location. E) Preeclampsia samples also showed low number and smaller microfractures compared to PPROM samples. Regions of 360um × 260um (9.4×10^4 μm3)were analyzed for each sample.
Summary
pPROM is a disease of the fetal membranes where the inflammation-oxidative stress axis plays a major role in producing pathways that can lead to membrane weakening through a variety of processes. Recent data provide molecular evidences for aging of fetal membranes in response to OS (physiologically at term and pathologically at preterm) can cause a telomere dependent, p38MAPK signaler driven senescence activation of the fetal membranes. Senescence, a mechanism contributing to aging of fetal membranes, produce sterile inflammation that can cause further damage to fetal membranes leading to weakening and or rupture. In addition, we report microfractures in fetal membranes that are likely sites of tissue remodeling during gestation; however, increase in number and morphometry (width and depth) of these microfractures in pPROM membranes suggests reduced remodeling capacity of membranes. In addition, these fractures can act as channels for amniotic fluid leak, and inflammatory cell and microbial migration. Further studies on senescence activation and microfracture formation and their role in maintaining membrane homeostasis are expected to fill more knowledge gaps in our understanding of pPROM pathology as well as provide better screening (biomarker and imaging based) tools for predicting women at high risk for pPROM.
Acknowledgments
This review is supported by 1R01HD084532-01A1 to R Menon
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Naeye RL, Peters EC. Causes and consequences of premature rupture of fetal membranes. Lancet. 1980;1(8161):192–194. doi: 10.1016/s0140-6736(80)90674-1. [DOI] [PubMed] [Google Scholar]
- 2.Mercer BM, Crouse DT, Goldenberg RL, et al. The antibiotic treatment of PPROM study: systemic maternal and fetal markers and perinatal outcomes. American Journal of Obstetrics and Gynecology. 2012;206(2) doi: 10.1016/j.ajog.2011.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beck S, Wojdyla D, Say L, et al. The worldwide incidence of preterm birth: a systematic review of maternal mortality and morbidity. Bull World Health Organ. 2010;88(1):31–38. doi: 10.2471/BLT.08.062554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology and causes of preterm birth. Lancet. 2008;371(9606):75–84. doi: 10.1016/S0140-6736(08)60074-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Menon R. Spontaneous preterm birth, a clinical dilemma: etiologic, pathophysiologic and genetic heterogeneities and racial disparity. Acta Obstet Gynecol Scand. 2008;87(6):590–600. doi: 10.1080/00016340802005126. [DOI] [PubMed] [Google Scholar]
- 6.Macones GA, Parry S, Elkousy M, Clothier B, Ural SH, Strauss JF., III A polymorphism in the promoter region of TNF and bacterial vaginosis: preliminary evidence of gene-environment interaction in the etiology of spontaneous preterm birth. Am J Obstet Gynecol. 2004;190(6):1504–1508. doi: 10.1016/j.ajog.2004.01.001. [DOI] [PubMed] [Google Scholar]
- 7.Burns DN, Landesman S, Muenz LR, et al. Cigarette smoking, premature rupture of membranes, and vertical transmission of HIV-1 among women with low CD4+ levels. J Acquir Immune Defic Syndr. 1994;7(7):718–726. [PubMed] [Google Scholar]
- 8.Silverman RK, Wojtowycz M. Risk factors in premature rupture of membranes. Prim Care Update Ob Gyns. 1998;5(4):181. doi: 10.1016/s1068-607x(98)00092-4. [DOI] [PubMed] [Google Scholar]
- 9.Hay ED. Extracellular matrix. J Cell Biol. 1981;91(3 Pt 2):205s–223s. doi: 10.1083/jcb.91.3.205s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bourne GL, LACY D. Ultra-structure of human amnion and its possible relation to the circulation of amniotic fluid. Nature. 1960;186:952–954. doi: 10.1038/186952a0. [DOI] [PubMed] [Google Scholar]
- 11.Castillo-Castrejon M, Meraz-Cruz N, Gomez-Lopez N, et al. Choriodecidual cells from term human pregnancies show distinctive functional properties related to the induction of labor 2. Am J Reprod Immunol. 2014;71(1):86–93. doi: 10.1111/aji.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.King AE, Kelly RW, Sallenave JM, Bocking AD, Challis JR. Innate immune defences in the human uterus during pregnancy 6. Placenta. 2007;28(11-12):1099–1106. doi: 10.1016/j.placenta.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 13.Vora S, Abbas A, Kim CJ, et al. Nuclear factor-kappa B localization and function within intrauterine tissues from term and preterm labor and cultured fetal membranes 2. Reprod Biol Endocrinol. 2010;8:8. doi: 10.1186/1477-7827-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montenegro D, Romero R, Kim SS, et al. Expression patterns of microRNAs in the chorioamniotic membranes: a role for microRNAs in human pregnancy and parturition 3. J Pathol. 2009;217(1):113–121. doi: 10.1002/path.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mossman HW. Classics revisited: Comparative morphogenesis of the fetal membranes and accessory uterine structures 1. Placenta. 1991;12(1):1–5. doi: 10.1016/0143-4004(91)90504-9. [DOI] [PubMed] [Google Scholar]
- 16.Kumar D, Moore RM, Mercer BM, Mansour JM, Redline RW, Moore JJ. The physiology of fetal membrane weakening and rupture: Insights gained from the determination of physical properties revisited. Placenta. 2016;42:59–73. doi: 10.1016/j.placenta.2016.03.015. [DOI] [PubMed] [Google Scholar]
- 17.Moore RM, Mansour JM, Redline RW, Mercer BM, Moore JJ. The physiology of fetal membrane rupture: insight gained from the determination of physical properties. Placenta. 2006;27(11-12):1037–1051. doi: 10.1016/j.placenta.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 18.Moco NP, Martin LF, Pereira AC, et al. Gene expression and protein localization of TLR-1, -2, -4 and -6 in amniochorion membranes of pregnancies complicated by histologic chorioamnionitis. Eur J Obstet Gynecol Reprod Biol. 2013;171(1):12–17. doi: 10.1016/j.ejogrb.2013.07.036. [DOI] [PubMed] [Google Scholar]
- 19.Keelan JA, Wang K, Chaiworapongsa T, et al. Macrophage inhibitory cytokine 1 in fetal membranes and amniotic fluid from pregnancies with and without preterm labour and premature rupture of membranes. Mol Hum Reprod. 2003;9(9):535–540. doi: 10.1093/molehr/gag068. [DOI] [PubMed] [Google Scholar]
- 20.King AE, Kelly RW, Sallenave JM, Bocking AD, Challis JR. Innate immune defences in the human uterus during pregnancy. Placenta. 2007;28(11-12):1099–1106. doi: 10.1016/j.placenta.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 21.Menon R, NN, Bredeson S, Polettini J. Fetal Membranes: Potential Source of Preterm Birth Biomarkers. In: Preedy Vr PVB, editor. General Methods in Biomarker Research and Their Applications. Springer Science Publisher; 1907. pp. 483–529. [Google Scholar]
- 22.DiGiulio DB, Romero R, Kusanovic JP, et al. Prevalence and diversity of microbes in the amniotic fluid, the fetal inflammatory response, and pregnancy outcome in women with preterm pre-labor rupture of membranes. Am J Reprod Immunol. 2010;64(1):38–57. doi: 10.1111/j.1600-0897.2010.00830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vadillo-Ortega F, Estrada-Gutierrez G. Role of matrix metalloproteinases in preterm labour. BJOG. 2005;112(1):19–22. doi: 10.1111/j.1471-0528.2005.00579.x. [DOI] [PubMed] [Google Scholar]
- 24.Arechavaleta-Velasco F, Ogando D, Parry S, Vadillo-Ortega F. Production of matrix metalloproteinase-9 in lipopolysaccharide-stimulated human amnion occurs through an autocrine and paracrine proinflammatory cytokine-dependent system. Biol Reprod. 2002;67(6):1952–1958. doi: 10.1095/biolreprod.102.004721. [DOI] [PubMed] [Google Scholar]
- 25.Fortunato SJ, Menon R. IL-1 beta is a better inducer of apoptosis in human fetal membranes than IL-6. Placenta. 2003;24(10):922–928. doi: 10.1016/s0143-4004(03)00160-7. [DOI] [PubMed] [Google Scholar]
- 26.Puthiyachirakkal M, Lemerand K, Kumar D, et al. Thrombin weakens the amnion extracellular matrix (ECM) directly rather than through protease activated receptors. Placenta. 2013;34(10):924–931. doi: 10.1016/j.placenta.2013.07.064. [DOI] [PubMed] [Google Scholar]
- 27.Murtha AP, Menon R. Regulation of fetal membrane inflammation: a critical step in reducing adverse pregnancy outcome. Am J Obstet Gynecol. 2015;213(4):447–448. doi: 10.1016/j.ajog.2015.07.008. [DOI] [PubMed] [Google Scholar]
- 28.Mercer BM, Lewis R. Preterm labor and preterm premature rupture of the membranes Diagnosis and management. Infect Dis Clin North Am. 1997;11(1):177–201. doi: 10.1016/s0891-5520(05)70348-2. [DOI] [PubMed] [Google Scholar]
- 29.Savitz DA, Blackmore CA, Thorp JM. Epidemiologic characteristics of preterm delivery: etiologic heterogeneity. Am J Obstet Gynecol. 1991;164(2):467–471. doi: 10.1016/s0002-9378(11)80001-3. [DOI] [PubMed] [Google Scholar]
- 30.McGregor JA, Lawellin D, Franco-Buff A, Todd JK, Makowski EL. Protease production by microorganisms associated with reproductive tract infection. Am J Obstet Gynecol. 1986;154(1):109–114. doi: 10.1016/0002-9378(86)90404-7. [DOI] [PubMed] [Google Scholar]
- 31.McGregor JA, French JI, Lawellin D, Franco-Buff A, Smith C, Todd JK. Bacterial protease-induced reduction of chorioamniotic membrane strength and elasticity. Obstet Gynecol. 1987;69(2):167–174. [PubMed] [Google Scholar]
- 32.French JI, McGregor JA. The pathobiology of premature rupture of membranes. Semin Perinatol. 1996;20(5):344–368. doi: 10.1016/s0146-0005(96)80002-4. [DOI] [PubMed] [Google Scholar]
- 33.Romero R, Miranda J, Chaemsaithong P, et al. Sterile and microbial-associated intra-amniotic inflammation in preterm prelabor rupture of membranes 1. J Matern Fetal Neonatal Med. 2014:1–16. doi: 10.3109/14767058.2014.958463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosen T, Kuczynski E, O'Neill LM, Funai EF, Lockwood CJ. Plasma levels of thrombin-antithrombin complexes predict preterm premature rupture of the fetal membranes. J Matern Fetal Med. 2001;10(5):297–300. doi: 10.1080/714904361. [DOI] [PubMed] [Google Scholar]
- 35.Dennery PA. Effects of oxidative stress on embryonic development. Birth Defects Res C Embryo Today. 2007;81(3):155–162. doi: 10.1002/bdrc.20098. [DOI] [PubMed] [Google Scholar]
- 36.Agarwal A, Gupta S, Sharma RK. Role of oxidative stress in female reproduction. Reprod Biol Endocrinol. 2005;3:28. doi: 10.1186/1477-7827-3-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Myatt L, Cui X. Oxidative stress in the placenta. Histochem Cell Biol. 2004;122(4):369–382. doi: 10.1007/s00418-004-0677-x. [DOI] [PubMed] [Google Scholar]
- 38.Menon R. Oxidative stress damage as a detrimental factor in preterm birth pathology 14. Front Immunol. 2014;5:567. doi: 10.3389/fimmu.2014.00567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chai M, Barker G, Menon R, Lappas M. Increased oxidative stress in human fetal membranes overlying the cervix from term non-labouring and post labour deliveries. Placenta. 2012;33(8):604–610. doi: 10.1016/j.placenta.2012.04.014. [DOI] [PubMed] [Google Scholar]
- 40.Myatt L. Review: Reactive oxygen and nitrogen species and functional adaptation of the placenta. Placenta. 2010;31(Suppl):S66–S69. doi: 10.1016/j.placenta.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wisdom SJ, Wilson R, McKillop JH, Walker JJ. Antioxidant systems in normal pregnancy and in pregnancy-induced hypertension. Am J Obstet Gynecol. 1991;165(6 Pt 1):1701–1704. doi: 10.1016/0002-9378(91)90018-m. [DOI] [PubMed] [Google Scholar]
- 42.Cindrova-Davies T, Yung HW, Johns J, et al. Oxidative stress, gene expression, and protein changes induced in the human placenta during labor. Am J Pathol. 2007;171(4):1168–1179. doi: 10.2353/ajpath.2007.070528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coughlan MT, Permezel M, Georgiou HM, Rice GE. Repression of oxidant-induced nuclear factor-kappaB activity mediates placental cytokine responses in gestational diabetes. J Clin Endocrinol Metab. 2004;89(7):3585–3594. doi: 10.1210/jc.2003-031953. [DOI] [PubMed] [Google Scholar]
- 44.Bennett PR, Rose MP, Myatt L, Elder MG. Preterm labor: stimulation of arachidonic acid metabolism in human amnion cells by bacterial products. Am J Obstet Gynecol. 1987;156(3):649–655. doi: 10.1016/0002-9378(87)90070-6. [DOI] [PubMed] [Google Scholar]
- 45.Ford-Hutchinson AW, Girard Y, Lord A, et al. The pharmacology of L-670, 596, a potent and selective thromboxane/prostaglandin endoperoxide receptor antagonist. Can J Physiol Pharmacol. 1989;67(9):989–993. doi: 10.1139/y89-155. [DOI] [PubMed] [Google Scholar]
- 46.Dietrich M, Block G, Norkus EP, et al. Smoking and exposure to environmental tobacco smoke decrease some plasma antioxidants and increase gamma-tocopherol in vivo after adjustment for dietary antioxidant intakes. Am J Clin Nutr. 2003;77(1):160–166. doi: 10.1093/ajcn/77.1.160. [DOI] [PubMed] [Google Scholar]
- 47.Bauer ME, Jeckel CM, Luz C. The role of stress factors during aging of the immune system. Ann N Y Acad Sci. 2009;1153:139–152. doi: 10.1111/j.1749-6632.2008.03966.x. [DOI] [PubMed] [Google Scholar]
- 48.Hass R, Sohn C. Increased oxidative stress in pre-eclamptic placenta is associated with altered proteasome activity and protein patterns. Placenta. 2003;24(10):979–984. doi: 10.1016/s0143-4004(03)00174-7. [DOI] [PubMed] [Google Scholar]
- 49.Monboisse JC, Braquet P, Randoux A, Borel JP. Non-enzymatic degradation of acid-soluble calf skin collagen by superoxide ion: protective effect of flavonoids. Biochem Pharmacol. 1983;32(1):53–58. doi: 10.1016/0006-2952(83)90651-2. [DOI] [PubMed] [Google Scholar]
- 50.Buhimschi IA, Buhimschi CS, Pupkin M, Weiner CP. Beneficial impact of term labor: nonenzymatic antioxidant reserve in the human fetus 56. Am J Obstet Gynecol. 2003;189(1):181–188. doi: 10.1067/mob.2003.357. [DOI] [PubMed] [Google Scholar]
- 51.Lappas M, Permezel M, Rice GE. N-Acetyl-cysteine inhibits phospholipid metabolism, proinflammatory cytokine release, protease activity, and nuclear factor-kappaB deoxyribonucleic acid-binding activity in human fetal membranes in vitro. J Clin Endocrinol Metab. 2003;88(4):1723–1729. doi: 10.1210/jc.2002-021677. [DOI] [PubMed] [Google Scholar]
- 52.Bryant-Greenwood GD. The extracellular matrix of the human fetal membranes: structure and function. Placenta. 1998;19(1):1–11. doi: 10.1016/s0143-4004(98)90092-3. [DOI] [PubMed] [Google Scholar]
- 53.Bryant-Greenwood GD, Yamamoto SY. Control of peripartal collagenolysis in the human chorion-decidua. Am J Obstet Gynecol. 1995;172(1 Pt 1):63–70. doi: 10.1016/0002-9378(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 54.Fortunato SJ, Menon R, Lombardi SJ. Collagenolytic enzymes (gelatinases) and their inhibitors in human amniochorionic membrane. Am J Obstet Gynecol. 1997;177(4):731–741. doi: 10.1016/s0002-9378(97)70260-6. [DOI] [PubMed] [Google Scholar]
- 55.Vadillo-Ortega F, Gonzalez-Avila G, Furth EE, et al. 92-kd type IV collagenase (matrix metalloproteinase-9) activity in human amniochorion increases with labor. Am J Pathol. 1995;146(1):148–156. [PMC free article] [PubMed] [Google Scholar]
- 56.Draper D, McGregor J, Hall J, et al. Elevated protease activities in human amnion and chorion correlate with preterm premature rupture of membranes. Am J Obstet Gynecol. 1995;173(5):1506–1512. doi: 10.1016/0002-9378(95)90640-1. [DOI] [PubMed] [Google Scholar]
- 57.Lei H, Vadillo-Ortega F, Paavola LG, Strauss JF., III 92-kDa gelatinase (matrix metalloproteinase-9) is induced in rat amnion immediately prior to parturition. Biol Reprod. 1995;53(2):339–344. doi: 10.1095/biolreprod53.2.339. [DOI] [PubMed] [Google Scholar]
- 58.Tsuruda T, Costello-Boerrigter LC, Burnett JC., Jr Matrix metalloproteinases: pathways of induction by bioactive molecules. Heart Fail Rev. 2004;9(1):53–61. doi: 10.1023/B:HREV.0000011394.34355.bb. [DOI] [PubMed] [Google Scholar]
- 59.Vincenti MP, White LA, Schroen DJ, Benbow U, Brinckerhoff CE. Regulating expression of the gene for matrix metalloproteinase-1 (collagenase): mechanisms that control enzyme activity, transcription, and mRNA stability. Crit Rev Eukaryot Gene Expr. 1996;6(4):391–411. doi: 10.1615/critreveukargeneexpr.v6.i4.40. [DOI] [PubMed] [Google Scholar]
- 60.Park KH, Chaiworapongsa T, Kim YM, et al. Matrix metalloproteinase 3 in parturition, premature rupture of the membranes, and microbial invasion of the amniotic cavity. J Perinat Med. 2003;31(1):12–22. doi: 10.1515/JPM.2003.002. [DOI] [PubMed] [Google Scholar]
- 61.Fortunato SJ, LaFleur B, Menon R. Collagenase-3 (MMP-13) in fetal membranes and amniotic fluid during pregnancy. Am J Reprod Immunol. 2003;49(2):120–125. doi: 10.1034/j.1600-0897.2003.00012.x. [DOI] [PubMed] [Google Scholar]
- 62.Fortunato SJ, Menon R, Lombardi SJ. MMP/TIMP imbalance in amniotic fluid during PROM: an indirect support for endogenous pathway to membrane rupture. J Perinat Med. 1999;27(5):362–368. doi: 10.1515/JPM.1999.049. [DOI] [PubMed] [Google Scholar]
- 63.Gomez R, Romero R, Edwin SS, David C. Pathogenesis of preterm labor and preterm premature rupture of membranes associated with intraamniotic infection. Infect Dis Clin North Am. 1997;11(1):135–176. doi: 10.1016/s0891-5520(05)70347-0. [DOI] [PubMed] [Google Scholar]
- 64.Moore RM, Schatz F, Kumar D, et al. Alpha-lipoic acid inhibits thrombin-induced fetal membrane weakening in vitro. Placenta. 2010;31(10):886–892. doi: 10.1016/j.placenta.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Menon R, Fortunato SJ. The role of matrix degrading enzymes and apoptosis in rupture of membranes. J Soc Gynecol Investig. 2004;11(7):427–437. doi: 10.1016/j.jsgi.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 66.Arechavaleta-Velasco F, Mayon-Gonzalez J, Gonzalez-Jimenez M, Hernandez-Guerrero C, Vadillo-Ortega F. Association of type II apoptosis and 92-kDa type IV collagenase expression in human amniochorion in prematurely ruptured membranes with tumor necrosis factor receptor-1 expression. J Soc Gynecol Investig. 2002;9(2):60–67. doi: 10.1016/s1071-5576(01)00159-9. [DOI] [PubMed] [Google Scholar]
- 67.Fortunato SJ, Menon R, Bryant C, Lombardi SJ. Programmed cell death (apoptosis) as a possible pathway to metalloproteinase activation and fetal membrane degradation in premature rupture of membranes. Am J Obstet Gynecol. 2000;182(6):1468–1476. doi: 10.1067/mob.2000.107330. [DOI] [PubMed] [Google Scholar]
- 68.Tanir HM, Sener T, Artan S, Kaytaz B, Sahin-Mutlu F, Ozen ME. Programmed cell death (apoptosis) in placentas from normal pregnancy and pregnancy complicated by term (t) and preterm (p) premature rupture of membranes (PROM) Arch Gynecol Obstet. 2005;273(2):98–103. doi: 10.1007/s00404-005-0028-8. [DOI] [PubMed] [Google Scholar]
- 69.Menon R, Lombardi SJ, Fortunato SJ. TNF-alpha promotes caspase activation and apoptosis in human fetal membranes. J Assist Reprod Genet. 2002;19(4):201–204. doi: 10.1023/A:1014898130008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lonergan M, Aponso D, Marvin KW, et al. Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), TRAIL receptors, and the soluble receptor osteoprotegerin in human gestational membranes and amniotic fluid during pregnancy and labor at term and preterm. J Clin Endocrinol Metab. 2003;88(8):3835–3844. doi: 10.1210/jc.2002-021905. [DOI] [PubMed] [Google Scholar]
- 71.George RB, Kalich J, Yonish B, Murtha AP. Apoptosis in the chorion of fetal membranes in preterm premature rupture of membranes. American Journal of Perinatology. 2008;25(1):29–32. doi: 10.1055/s-2007-1004828. [DOI] [PubMed] [Google Scholar]
- 72.Menon R, Fortunato SJ. Fetal membrane inflammatory cytokines: a switching mechanism between the preterm premature rupture of the membranes and preterm labor pathways. Journal of perinatal medicine. 2004;32(5):391–399. doi: 10.1515/JPM.2004.134. [DOI] [PubMed] [Google Scholar]
- 73.Menon R, Fortunato SJ. Infection and the role of inflammation in preterm premature rupture of the membranes. Best Pract Res Clin Obstet Gynaecol. 2007;21(3):467–478. doi: 10.1016/j.bpobgyn.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 74.Jouan-Lanhouet S, Riquet F, Duprez L, Vanden Berghe T, Takahashi N, Vandenabeele P. Necroptosis, in vivo detection in experimental disease models. Semin Cell Dev Biol. 2014;35:2–13. doi: 10.1016/j.semcdb.2014.08.010. [DOI] [PubMed] [Google Scholar]
- 75.Vanden Berghe T, Grootjans S, Goossens V, et al. Determination of apoptotic and necrotic cell death in vitro and in vivo. Methods. 2013;61(2):117–129. doi: 10.1016/j.ymeth.2013.02.011. [DOI] [PubMed] [Google Scholar]
- 76.Behnia F, Sheller S, Menon R. Mechanistic Differences Leading to Infectious and Sterile Inflammation 3. Am J Reprod Immunol. 2016 doi: 10.1111/aji.12496. [DOI] [PubMed] [Google Scholar]
- 77.Agarwal A, Gupta S, Sekhon L, Shah R. Redox considerations in female reproductive function and assisted reproduction: from molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10(8):1375–1403. doi: 10.1089/ars.2007.1964. [DOI] [PubMed] [Google Scholar]
- 78.Dennery PA. Oxidative stress in development: nature or nurture? Free Radic Biol Med. 2010;49(7):1147–1151. doi: 10.1016/j.freeradbiomed.2010.07.011. [DOI] [PubMed] [Google Scholar]
- 79.Menon R, Torloni MR, Voltolini C, et al. Biomarkers of spontaneous preterm birth: an overview of the literature in the last four decades. Reprod Sci. 2011;18(11):1046–1070. doi: 10.1177/1933719111415548. [DOI] [PubMed] [Google Scholar]
- 80.Fortunato SJ, Menon R, Swan KF, Lyden TW. Organ culture of amniochorionic membrane in vitro. Am J Reprod Immunol. 1994;32(3):184–187. doi: 10.1111/j.1600-0897.1994.tb01112.x. [DOI] [PubMed] [Google Scholar]
- 81.Menon R. Oxidative stress damage as a detrimental factor in preterm birth pathology. Front Immunol. 2014;5:567. doi: 10.3389/fimmu.2014.00567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Buhimschi IA, Kramer WB, Buhimschi CS, Thompson LP, Weiner CP. Reduction-oxidation (redox) state regulation of matrix metalloproteinase activity in human fetal membranes. Am J Obstet Gynecol. 2000;182(2):458–464. doi: 10.1016/s0002-9378(00)70239-0. [DOI] [PubMed] [Google Scholar]
- 83.Dennery PA, Spitz DR, Yang G, et al. Oxygen toxicity and iron accumulation in the lungs of mice lacking heme oxygenase-2. J Clin Invest. 1998;101(5):1001–1011. doi: 10.1172/JCI448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Longini M, Perrone S, Vezzosi P, et al. Association between oxidative stress in pregnancy and preterm premature rupture of membranes. Clin Biochem. 2007;40(11):793–797. doi: 10.1016/j.clinbiochem.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 85.Finkel T. Oxidant signals and oxidative stress. Curr Opin Cell Biol. 2003;15(2):247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 86.Sies H. Role of reactive oxygen species in biological processes. Klin Wochenschr. 1991;69(21-23):965–968. doi: 10.1007/BF01645140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Roberts LJ, Milne GL. Isoprostanes. J Lipid Res. 2009;50(Suppl):S219–S223. doi: 10.1194/jlr.R800037-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Longini M, Perrone S, Vezzosi P, et al. Isoprostane levels in urine of preterm newborns treated with ibuprofen for patent ductus arteriosus closure. Pediatr Nephrol. 2011;26(1):105–109. doi: 10.1007/s00467-010-1651-6. [DOI] [PubMed] [Google Scholar]
- 89.Morrow JD, Roberts LJ. The isoprostanes: unique bioactive products of lipid peroxidation. Prog Lipid Res. 1997;36(1):1–21. doi: 10.1016/s0163-7827(97)00001-5. [DOI] [PubMed] [Google Scholar]
- 90.Menon R, Fortunato SJ, Milne GL, et al. Amniotic fluid eicosanoids in preterm and term births: effects of risk factors for spontaneous preterm labor. Obstet Gynecol. 2011;118(1):121–134. doi: 10.1097/AOG.0b013e3182204eaa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harley CB, Vaziri H, Counter CM, Allsopp RC. The telomere hypothesis of cellular aging. Exp Gerontol. 1992;27(4):375–382. doi: 10.1016/0531-5565(92)90068-b. [DOI] [PubMed] [Google Scholar]
- 92.Hanna CW, Bretherick KL, Gair JL, Fluker MR, Stephenson MD, Robinson WP. Telomere length and reproductive aging. Hum Reprod. 2009;24(5):1206–1211. doi: 10.1093/humrep/dep007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Smith JA, Park S, Krause JS, Banik NL. Oxidative stress, DNA damage, and the telomeric complex as therapeutic targets in acute neurodegeneration. Neurochem Int. 2013;62(5):764–775. doi: 10.1016/j.neuint.2013.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Menon R, Yu J, Basanta-Henry P, et al. Short fetal leukocyte telomere length and preterm prelabor rupture of the membranes. PLoS One. 2012;7(2):e31136. doi: 10.1371/journal.pone.0031136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Tuuli MG, Longtine MS, Nelson DM. Review: Oxygen and trophoblast biology--a source of controversy. Placenta. 2011;32(2):S109–118. doi: 10.1016/j.placenta.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jauniaux E, Poston L, Burton GJ. Placental-related diseases of pregnancy: Involvement of oxidative stress and implications in human evolution. Hum Reprod Update. 2006;12(6):747–755. doi: 10.1093/humupd/dml016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Burton GJ. Oxygen, the Janus gas; its effects on human placental development and function. J Anat. 2009;215(1):27–35. doi: 10.1111/j.1469-7580.2008.00978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ilekis JV, Tsilou E, Fisher S, et al. Placental origins of adverse pregnancy outcomes: potential molecular targets: an Executive Workshop Summary of the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Am J Obstet Gynecol. 2016;215(1 Suppl):S1–S46. doi: 10.1016/j.ajog.2016.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Robins JC, Heizer A, Hardiman A, Hubert M, Handwerger S. Oxygen tension directs the differentiation pathway of human cytotrophoblast cells. Placenta. 2007;28(11-12):1141–1146. doi: 10.1016/j.placenta.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 100.Jauniaux E, Gulbis B, Burton GJ. Physiological implications of the materno-fetal oxygen gradient in human early pregnancy. Reprod Biomed Online. 2003;7(2):250–253. doi: 10.1016/s1472-6483(10)61760-9. [DOI] [PubMed] [Google Scholar]
- 101.Sajjad Y, Leonard M, Doyle M. Antioxidant levels in the cord blood of term fetus. J Obstet Gynaecol. 2000;20(5):468–471. doi: 10.1080/014436100434613. [DOI] [PubMed] [Google Scholar]
- 102.Myatt L. Placental adaptive responses and fetal programming. J Physiol. 2006;572(Pt 1):25–30. doi: 10.1113/jphysiol.2006.104968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Burton GJ, Jaunaiux E. Maternal vascularisation of the human placenta: does the embryo develop in a hypoxic environment? Gynecol Obstet Fertil. 2001;29(7-8):503–508. doi: 10.1016/s1297-9589(01)00179-5. [DOI] [PubMed] [Google Scholar]
- 104.Adams Waldorf KM, Singh N, Mohan AR, et al. Uterine overdistention induces preterm labor mediated by inflammation: observations in pregnant women and nonhuman primates. Am J Obstet Gynecol. 2015;213(6):830 e831–830 e819. doi: 10.1016/j.ajog.2015.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cuenda A, Rousseau S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta. 2007;1773(8):1358–1375. doi: 10.1016/j.bbamcr.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 106.Torres M. Mitogen-activated protein kinase pathways in redox signaling. Front Biosci. 2003;8:d369–d391. doi: 10.2741/999. [DOI] [PubMed] [Google Scholar]
- 107.Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J. 2011;30(8):1536–1548. doi: 10.1038/emboj.2011.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Borodkina A, Shatrova A, Abushik P, Nikolsky N, Burova E. Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells. Aging (Albany NY) 2014;6(6):481–495. doi: 10.18632/aging.100673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wong ES, Le Guezennec X, Demidov ON, et al. p38MAPK controls expression of multiple cell cycle inhibitors and islet proliferation with advancing age. Dev Cell. 2009;17(1):142–149. doi: 10.1016/j.devcel.2009.05.009. [DOI] [PubMed] [Google Scholar]
- 110.Coppe JP, Patil CK, Rodier F, et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 2008;6(12):2853–2868. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Tchkonia T, Zhu Y, van DJ, Campisi J, Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123(3):966–972. doi: 10.1172/JCI64098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Behnia F, Taylor BD, Woodson M, et al. Chorioamniotic Membrane Senescence: A Signal for Parturition? Am J Obstet Gynecol. 2015 doi: 10.1016/j.ajog.2015.05.041. [DOI] [PubMed] [Google Scholar]
- 113.Polettini J, Behnia F, Taylor BD, Saade GR, Taylor RN, Menon R. Telomere Fragment Induced Amnion Cell Senescence: A Contributor to Parturition? PLoS One. 2015;10(9):e0137188. doi: 10.1371/journal.pone.0137188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Menon R, Behnia F, Polettini J, Saade GR, Campisi J, Velarde M. Placental membrane aging and HMGB1 signaling associated with human parturition. Aging (Albany NY) 2016 doi: 10.18632/aging.100891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bredeson S, Papaconstantinou J, Deford JH, et al. HMGB1 promotes a p38MAPK associated non-infectious inflammatory response pathway in human fetal membranes. PLoS One. 2014;9(12):e113799. doi: 10.1371/journal.pone.0113799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ferrari F, Facchinetti F, Saade G, Menon R. Placental telomere shortening in stillbirth: a sign of premature senescence? J Matern Fetal Neonatal Med. 2015:1–6. doi: 10.3109/14767058.2015.1046045. [DOI] [PubMed] [Google Scholar]
- 117.Dutta EH, Behnia F, Boldogh I, et al. Oxidative stress damage-associated molecular signaling pathways differentiate spontaneous preterm birth and preterm premature rupture of the membranes. Mol Hum Reprod. 2016;22(2):143–157. doi: 10.1093/molehr/gav074. [DOI] [PubMed] [Google Scholar]
- 118.Woods JR., Jr Reactive oxygen species and preterm premature rupture of membranes-a review. Placenta. 2001;22(A):S38–S44. doi: 10.1053/plac.2001.0638. [DOI] [PubMed] [Google Scholar]
- 119.Kwiatkowski S, Torbe A, Dolegowska B, et al. Isoprostanes 8-iPF2alpha-III: risk markers of premature rupture of fetal membranes? Biomarkers. 2009;14(6):406–413. doi: 10.1080/13547500903045583. [DOI] [PubMed] [Google Scholar]
- 120.Polettini J, Silva MG, Kacerovsky M, Syed TA, Saade GR, Menon R. Screening of lysyl oxidase (LOX) and lysyl oxidase like (LOXL) enzyme expression and activity in preterm prelabor rupture of fetal membranes. J Perinat Med. 2015 doi: 10.1515/jpm-2014-0337. [DOI] [PubMed] [Google Scholar]
- 121.Maymon E, Romero R, Pacora P, et al. A role for the 72 kDa gelatinase (MMP-2) and its inhibitor (TIMP-2) in human parturition, premature rupture of membranes and intraamniotic infection. J Perinat Med. 2001;29(4):308–316. doi: 10.1515/JPM.2001.044. [DOI] [PubMed] [Google Scholar]
- 122.Menon R, Bonney EA, Condon J, Mesiano S, Taylor RN. Novel concepts on pregnancy clocks and alarms: redundancy and synergy in human parturition. Hum Reprod Update. 2016;22(5):535–560. doi: 10.1093/humupd/dmw022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Richardson L, Vargas G, Brown T, Ochoa L, Sheller-Miller S, Saade GR, Taylor RN, Menon R. Discovery and Characterization of Human Amniochorionic Membrane Microfractures. Obstet Gynecol. 2017 doi: 10.1016/j.ajpath.2017.08.019. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]