Cancer Risks for PMS2-Associated Lynch Syndrome (original) (raw)
Abstract
Purpose
Lynch syndrome due to pathogenic variants in the DNA mismatch repair genes MLH1, MSH2, and MSH6 is predominantly associated with colorectal and endometrial cancer, although extracolonic cancers have been described within the Lynch tumor spectrum. However, the age-specific cumulative risk (penetrance) of these cancers is still poorly defined for _PMS2_-associated Lynch syndrome. Using a large data set from a worldwide collaboration, our aim was to determine accurate penetrance measures of cancers for carriers of heterozygous pathogenic PMS2 variants.
Methods
A modified segregation analysis was conducted that incorporated both genotyped and nongenotyped relatives, with conditioning for ascertainment to estimates corrected for bias. Hazard ratios (HRs) and corresponding 95% CIs were estimated for each cancer site for mutation carriers compared with the general population, followed by estimation of penetrance.
Results
In total, 284 families consisting of 4,878 first- and second-degree family members were included in the analysis. PMS2 mutation carriers were at increased risk for colorectal cancer (cumulative risk to age 80 years of 13% [95% CI, 7.9% to 22%] for males and 12% [95% CI, 6.7% to 21%] for females) and endometrial cancer (13% [95% CI, 7.0%–24%]), compared with the general population (6.6%, 4.7%, and 2.4%, respectively). There was no clear evidence of an increased risk of ovarian, gastric, hepatobiliary, bladder, renal, brain, breast, prostate, or small bowel cancer.
Conclusion
Heterozygous PMS2 mutation carriers were at small increased risk for colorectal and endometrial cancer but not for any other Lynch syndrome–associated cancer. This finding justifies that _PMS2_-specific screening protocols could be restricted to colonoscopies. The role of risk-reducing hysterectomy and bilateral salpingo-oophorectomy for PMS2 mutation carriers needs further discussion.
INTRODUCTION
Lynch syndrome is most commonly associated with colorectal cancer and endometrial cancer. However, when first described in 1913, the observation of the co-occurrence of gastric cancer and endometrial cancer led to the initial identification of these families, underlining the apparently diverse phenotype.1 The genetic background of Lynch syndrome is now known, and it is caused by heterozygous germline mutations in one of the four mismatch repair (MMR) genes, MLH1, MSH2, MSH6, and PMS2, or EPCAM deletions. The broad Lynch syndrome–associated tumor spectrum includes not only colorectal cancer and endometrial cancer but also gastric, ovarian, small bowel, brain, urothelial cell, skin, pancreas, prostate, and biliary tract cancers.2,3 The involvement of germline MMR mutations in the development of breast cancer is still a subject of debate.4-7 Although the reported cumulative risk (penetrance) to age 70 years for these noncolorectal, nonendometrial cancers in MMR gene mutation carriers is generally < 10%, mutation carriers still have a higher risk relative to the general population.3 The Lynch syndrome–associated tumor phenotypes and their penetrance could depend on the type of MMR gene mutated or the specific variant.8,9
For heterozygous PMS2 mutation carriers, accurate estimation of penetrance, especially for extracolonic cancers, has been hampered both by difficulties in variant analysis related to the existence of multiple pseudogenes and, perhaps more importantly, by problems in identifying PMS2 mutation carriers because of a markedly lower penetrance.10-12 Our previous study of penetrance for PMS2 mutation carriers, using 98 PMS2 families ascertained through family cancer clinics in several European countries, reported standardized incidence ratios for extracolonic cancers and found an increased _PMS2_-related risk of cancer of the small bowel, ovaries, renal pelvis and—most notably—of the breast.11 Although that study presented the largest data set then available, we were unable to generate reliable estimates of penetrance for these cancers because of their infrequency. In addition, there was an ascertainment bias in this cohort because of the recruitment via family cancer clinics. Another study from Iceland reported significant increases in the risk of colorectal, endometrial, and ovarian cancer for two pathogenic PMS2 founder variants.13 This study and others have reported relatively high prevalence of PMS2 variants in the population,13-15 thus underlining the need for _PMS2_-specific cancer risks. In the current study, we have expanded the previous study database to 284 families, including several that were identified through a population-based ascertainment, with the aim of generating accurate penetrance estimates of colorectal, endometrial, and other cancers for patients with _PMS2_-associated Lynch syndrome.
METHODS
Data Collection
European data set.
Pedigree data on families with a segregating pathogenic variant were originally collected between 2009 and 2012, as previously described.11 These data were supplemented with PMS2 families identified between 2012 and 2017. Briefly, data were collected in collaboration with the Netherlands Foundation for the Detection of Hereditary Tumors and with clinical genetic departments in the Netherlands, Norway, Germany, Sweden, Denmark, and Spain. Data collection from patient records included demographic data, family pedigrees, age and location of cancer diagnosis, polypectomy, and hysterectomy if applicable. When available, clinical and pathologic diagnoses were confirmed using patient records. Data collection and subsequent analysis protocols were approved by the local ethical review board (Leiden University Medical Center Ethics Review Board, protocol ID: P01.019).
Ohio State data sets.
For the Ohio State data sets, the first set of patients included both population-based patients with colorectal and endometrial cancer from Columbus, Ohio, as described elsewhere,10,16-19 and patients with cancer identified at family cancer clinics with absence of PMS2 only on immunohistochemistry. The second set of patients from Ohio included only population-based patients with colorectal and endometrial cancer from 50 hospitals throughout the state of Ohio, as described previously.20 All patients provided informed consent (Ohio State University Biomedical Sciences Institutional Review Board protocol IDs: 1999C0051, 1999C0245, and 2012C0123).
Colon Cancer Family Registry data set.
The study cohort from the Colon Cancer Family Registry has been described in detail elsewhere.21-23 Between 1998 and 2012, the Colon Cancer Family Registry recruited families via population-based probands, recently diagnosed with colorectal cancer, in state or regional population cancer registries in the United States (Washington, California, Arizona, Minnesota, Colorado, New Hampshire, North Carolina, and Hawaii), Australia (Victoria), and Canada (Ontario). In addition, clinic-based probands were enrolled from multiple-case families referred to family cancer clinics in the United States (Mayo Clinic, Rochester, Minnesota and Cleveland Clinic, Cleveland, Ohio), Canada (Ontario), Australia (Melbourne, Adelaide, Perth, Brisbane, Sydney, and Newcastle), and New Zealand (Auckland). Probands were asked for permission to contact their relatives to seek their enrollment in the Cancer Family Registry (detailed in Newcomb et al21). Informed consent was obtained from all study participants, and the study protocol was approved by the institutional research ethics review board at each registry. Information on demographics, personal characteristics, personal and detailed family history of cancer in first- and second-degree relatives, cancer-screening history, history of polyps, polypectomy, and other surgeries was obtained by questionnaires from all probands and participating relatives. Participants were followed approximately every 5 years after baseline to update this information. For the current study, each individual’s lifetime cancer history was based on the most recent data (baseline or most recent follow-up). Reported cancer diagnoses and age at diagnosis were confirmed using pathology reports, medical records, cancer registry reports, and death certificates, where possible.
Mutation Analysis and Clinical Variant Classification
Probands included in the cohorts were screened for point mutations as well as large genomic rearrangements in the PMS2 gene (Data Supplement). Relatives of probands were tested for the specific family mutation. A detailed description of specific variants detected and their classification can be found in the Data Supplement.
Statistical Analysis
For estimation of the hazard ratios (HRs) and age-specific cumulative risks (penetrance), we used a modified segregation analysis.24 This analytical method is not subject to population stratification, can rigorously adjust for ascertainment, and uses data on all study participants, whether genotyped or not, thereby maximizing statistical power. Models were fitted by the method of maximum likelihood with the statistical package MENDEL 3.2.25 Estimates were appropriately adjusted for the ascertainment of families using a combination of retrospective likelihood and ascertainment-corrected joint likelihood. A conditional likelihood was maximized, in which each pedigree’s data were conditioned on the proband’s PMS2 mutation status, cancer history, and ages of cancer diagnoses (for population-based families) or on the proband’s PMS2 mutation status and the cancer history and ages of cancer diagnoses of all family members (for clinic-based families).
For the purposes of analysis, we restricted included individuals to the first- and second-degree relatives of the probands. Observation time started at birth and stopped at age at diagnosis of cancer for affected, and last known age or age at death for unaffected, family members. Because age information for each family member was required for the pedigree analysis, missing values were estimated using a defined protocol as follows. If an exact age was unknown but an age range was provided, age was estimated as the midpoint of that range. If age at diagnosis was unknown, it was assumed to be the same as age at death (if the relative was deceased) or the mean age at diagnosis for the specific cancer (if the relative was alive and older than the mean age at diagnosis). For relatives for whom last known age was unknown, ages were censored at the time they were last known to be alive (eg, at the age at a cancer diagnosis). In the absence of any age information, it was assumed that both parents of the proband were born in the same year, that years of birth differed by 25 years in each generation (eg, at birth of proband, parents were age 25 years and grandparents were age 50 years), and the ages of the siblings were the same. As a sensitivity analysis, we conducted analyses with and without imputing missing age; the results did not differ materially, and therefore results from the nonimputed analysis were not shown in detail.
To calculate HRs, we used a likelihood-based approach in which age-specific incidence for PMS2 mutation carriers was divided by that for noncarriers. Incidence rates for noncarriers were assumed to be the same as age-, sex-, and country-specific population incidence rates (Australia, Canada, United States, The Netherlands, Germany) for the period 1998 to 2002, as obtained from Cancer Incidence in Five Continents.26 The period of 1998 to 2002 was selected for analysis because it was the closest available data set to the mean calendar year of cancer diagnoses in the sample. For each cancer, the age at cancer diagnosis was modeled as a random variable whose hazard was the relevant population incidence multiplied by a cancer-specific HR. For colorectal cancer, HRs for carriers were assumed to be continuous, piece-wise linear functions of age that are constant before age 40 years; linear in the intervals 40 to 50, 50 to 60, and 60 to 70 years; and constant after age 70 years. For all other cancer sites, HRs were assumed to be independent of age. HRs for colorectal cancer, endometrial cancer, and other cancers were estimated simultaneously to allow proper adjustment for colorectal cancer–based ascertainment schemes when estimating the risks of noncolorectal cancers and to increase power (by helping the model identify likely carriers from the placement of Lynch syndrome–associated cancers within each family). HRs were assumed to be independent of country of recruitment.
Age-specific cumulative risks (penetrance) of each cancer site for PMS2 mutation carriers were calculated separately for males and females, using the formula:
where λ(t) is the HR multiplied by the US population incidence.27 Corresponding CIs were calculated using a parametric bootstrap. More specifically, 5,000 draws were taken from the multivariate normal distribution that the maximum likelihood estimates would be expected to follow under asymptotic likelihood theory. For each age, corresponding values of the cumulative risk were calculated, and the 95% CI for the cumulative risks to that age were taken to be the 2.5th and 97.5th percentile of this sample.
RESULTS
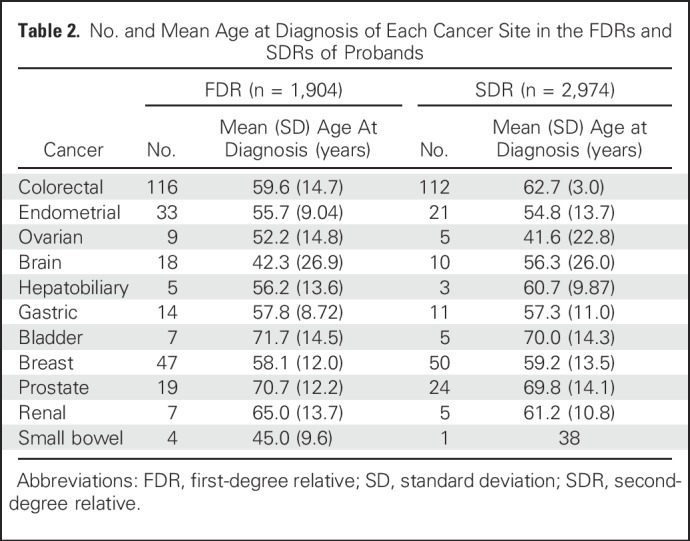
The final analysis included 284 families (211 from the European, 19 from the Ohio State, and 54 from the CCFR data set), with 1,904 first- and 2,974 second-degree family members, in whom 513 were confirmed carriers (Table 1). The numbers and mean ages at diagnosis of each cancer site in first- and second-degree relatives are depicted in Table 2.
Table 1.
Study Data Set Description
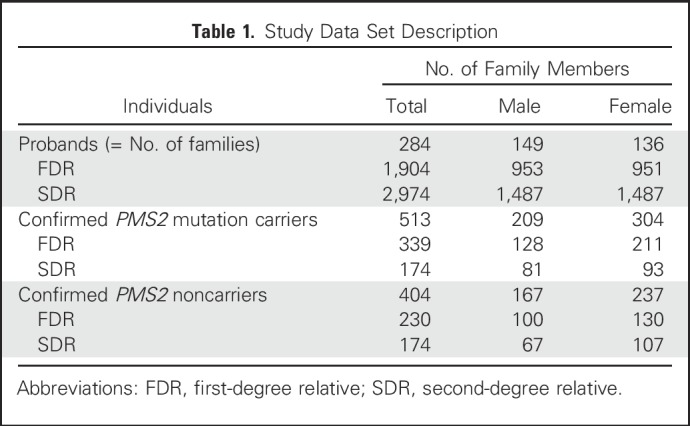
Table 2.
No. and Mean Age at Diagnosis of Each Cancer Site in the FDRs and SDRs of Probands
Colorectal Cancer
PMS2 mutation carriers were at increased risk of developing colorectal cancer, with an HR depending on age and sex of the mutation carrier: 6.51 (95% CI, 2.03 to 20.9) for males younger than 40 years; 1.70 (95% CI, 0.89 to 3.24) for males older than 70 years; 6.48 (95% CI, 2.24 to 18.8) for females younger than 40 years; and 2.23 (95% CI, 1.21 to 4.12) for females older than 70 years. Estimated cumulative risks of colorectal cancer to age 80 years for PMS2 mutation carriers were approximately 13% (95% CI, 7.9% to 22%) for male carriers and 12% (95% CI, 6.7% to 21%) for female carriers (general population 6.6% and 4.7%, respectively; Fig 1A).
Fig 1.

Cumulative risks (solid lines) and corresponding 95% CIs (dotted lines) of (A) colorectal cancer, and (B) endometrial cancer for heterozygous PMS2 mutation carriers, and for the US general population (dashed lines). Blue and gold represent males and females, respectively.
Gynecologic Cancers
PMS2 mutation carriers were also at small increased risk of endometrial cancer, with an HR of 5.73 (95% CI, 2.98 to 11.0) and estimated cumulative risk to age 80 years of approximately 13% (95% CI, 7.0% to 24%), compared with females from the general population (2.4%; Fig 1B). There was no clear evidence of increase in the risk of ovarian cancer (HR, 1.52; 95% CI, 0.45 to 5.05; Fig 2).
Fig 2.

Hazard ratios (HRs) and corresponding 95% CIs of extracolonic cancers for PMS2 mutation carriers.
Other Cancers
There was no clear increase in risk of gastric, hepatobiliary, bladder, renal, brain, breast, or prostate cancer for PMS2 mutation carriers (HR for each cancer shown in Fig 2). There were too few occurrences of small bowel cancer (n = 5) to generate an HR.
DISCUSSION
On the basis of the results from this large, international study of heterozygous PMS2 mutation carriers, the _PMS2_-associated Lynch syndrome spectrum seems to be restricted to colorectal and endometrial cancer only, underlining the distinct phenotype for PMS2 mutation carriers. We have also shown that PMS2 mutation carriers have much lower cancer risks compared with other MMR gene mutation carriers.
The previous two studies of PMS2 mutation carriers have estimated cumulative risks to age 70 years of 11% to 20% for colorectal cancer and 12% to 15% for endometrial cancer.10,11 Our current analysis has confirmed that PMS2 carriers are at small increased risk of colorectal and endometrial cancer. These penetrance estimates are considerably lower than those for other MMR gene mutation carriers, which have been estimated at 35% to 55% for colorectal cancer and 10% to 45% for endometrial cancer.3 A recent report from the Prospective Lynch Syndrome Database described cancer risk and survival for all patients with Lynch syndrome.7 This report included 124 PMS2 mutation carriers, with 524 observation years. The findings support our study data, in that endometrial cancer was the sole cancer type observed. Notably, colorectal cancer did not occur in any of the PMS2 mutation carriers undergoing regular colonoscopic screening. This, together with our penetrance estimates, could justify consideration of less-frequent colonoscopy screening for PMS2 mutation carriers. This, together with our low penetrance estimates (Fig 1) could justify modification of the colonoscopy surveillance protocol, for example starting at age 35 to 40 years, every 2 to 3 years, similar to what has been proposed in the National Comprehensive Cancer Network guidelines.28
The Prospective Lynch Syndrome Database showed that endometrial cancer survival for all MMR pathogenic variant carriers was excellent, with a 10-year survival of 93% (95% CI, 85% to 97%). The reported survival for ovarian cancer in patients with Lynch syndrome was lower, at 74% (95% CI, 44% to 90%), but still better than that for sporadic ovarian cancer cases. Current surveillance guidelines advise that risk-reducing hysterectomy and bilateral salpingo-oophorectomy should be considered in women with Lynch syndrome, because transvaginal ultrasound with or without biopsy is ineffective in reducing risk of endometrial cancer and ovarian cancer and might not have a strong influence on survival.29 The good survival rates for endometrial cancer, combined with the data presented in the current study showing no evidence of a clinically relevant increase in ovarian cancer risk for PMS2 mutation carriers, raise questions about the justification of risk-reducing hysterectomy and bilateral salpingo-oophorectomy, which may be too rigorous in carriers of heterozygous pathogenic PMS2 mutations.
In our previous study of patients with _PMS2_-associated Lynch syndrome, we found increased standardized incidence ratios (SIRs) for cancer of the small bowel, ovary, renal pelvis, and breast.11 However, that study was limited by inclusion of confirmed mutation carriers identified through family cancer clinics and a limited number of cancer events. The first factor in particular could have been a potential source of ascertainment bias, because a strong family history of cancer and/or early-onset disease increases the likelihood of inclusion and PMS2 testing. In that report, we did not adjust for this potential ascertainment bias when estimating SIRs for extracolonic cancers. Traditionally, a strong family history of colorectal and endometrial cancers prompted suspicion of Lynch syndrome, and consequently patients were tested for tumor MMR deficiency followed by MMR gene mutation testing. Currently, family histories of other cancers are increasingly being ascertained by clinical genetic centers for additional evaluation as possible Lynch syndrome. Therefore, it is important to take into account and adjust for ascertainment bias when estimating risks of cancers other than colorectal or endometrial cancer. Furthermore, pathogenic PMS2 variants are relatively frequently observed using extensive gene panel testing for women with hereditary breast and ovarian cancer.30-33 Nevertheless, we could not confirm an increased SIR for breast cancer in the current study (HR, 1.30; 95% CI, 0.79 to 2.16), and the discrepancy with earlier reports can probably be attributed to a high prevalence of PMS2 (and MSH6) mutations in the general population. Conversely, the relative infrequency of PMS2 variants among patients with Lynch syndrome can be explained by the milder phenotype, which makes ascertainment by family cancer clinics less likely.
The current study is the largest to date, to our knowledge, in estimation of cancer risks for heterozygous PMS2 mutation carriers. Previous studies have shown that analyses of retrospective data from clinic-based families (ie, ascertained because of family history of cancer) without (statistical) adjustment can lead to overestimation of cancer risks for mutation carriers.24,34,35 In the current study, we used a high-level statistical approach to properly adjust for such ascertainment bias. The modified segregation method used data on all family members, regardless of whether they were genotyped, thereby maximizing statistical power while avoiding survival bias.
A potential limitation of the current study was the use of unverified cancer diagnoses that were self- or proband-reported, thus potentially affecting the accuracy of estimates. However, previous studies showed a high probability of agreement between proband-reported cancer status in first-degree relatives and the validated report (for example, 95.4% [95% CI, 92.6 to 98.3] for breast cancer, 83.3% [95% CI, 72.8 to 93.8] for ovarian cancer, and 79.3% [95% CI, 70.0 to 88.6] for prostate cancer).36 Another possible limitation is that our analysis did not take into account a potential role for genetic or environmental modifiers of risk. The existence of such modifiers is plausible, because a high degree of variability in penetrance and phenotype has been observed,24 and modifiers of cancer risk such as lifestyle, genetic modifiers, and phenotype-genotype correlations have been identified previously.37-40 Our study estimated cancer risks of all variants combined; however, it is plausible that not all PMS2 variants confer the same risk. A previous study in a selection of the currently analyzed cohort investigated genotype-phenotype correlations and found no difference in risk between the group of variants with retained versus loss of RNA expression.40 However, this study did report that those carrying a variant with loss of RNA expression were diagnosed with colorectal cancer on average 9 years younger than those with retained expression. The influence of these modifiers is still not well understood, especially for PMS2 mutation carriers, although efforts are currently ongoing to better define such factors and their potential role in modifying disease risk. Our study results highlight that studies of penetrance modifiers should take the specific MMR gene mutated into account.
In the current study, we analyzed the first data set, to our knowledge, large enough to generate the unbiased estimates for the risk of each extracolonic cancer for PMS2 mutation carriers. Our results show that PMS2 carriers are only at small increased risk of colorectal and endometrial cancer. This underlines the importance of gene-specific genetic counseling of patients with Lynch syndrome and the development of appropriate clinical guidelines.
ACKNOWLEDGMENT
We thank all study participants of the Colon Cancer Family Registry and staff for their contributions to this project.
Footnotes
Supported by Grant No. UL-2012-5515 from the Dutch Cancer Society and Grant No. UM1 CA167551 from the National Cancer Institute and through cooperative agreements with the following Colon Cancer Family Registry centers: Australasian Colorectal Cancer Family Registry Grants No. U01 CA074778 and U01/U24 CA097735; Mayo Clinic Cooperative Family Registry for Colon Cancer Studies Grant No. U01/U24 CA074800; Ontario Familial Colorectal Cancer Registry Grant No. U01/U24 CA074783; Seattle Colorectal Cancer Family Registry Grant No. U01/U24 CA074794; University of Hawaii Colorectal Cancer Family Registry Grant No. U01/U24 CA074806; and University of Southern California Consortium Colorectal Cancer Family Registry Grant No. U01/U24 CA074799. Seattle Colon Cancer Family Registry research was also supported by the Cancer Surveillance System of the Fred Hutchinson Cancer Research Center, which was funded by Control Nos. N01-CN-67009 (1996 to 2003) and N01-PC-35142 (2003 to 2010) and Contract No. HHSN2612013000121 (2010 to 2017) from the SEER Program of the National Cancer Institute, with additional support from the Fred Hutchinson Cancer Research Center. The Columbus-area Hereditary Non-Polyposis Colorectal Cancer study cohort in Ohio was supported by Grants No. NCI R01-CA67941 and CA16058. The Ohio Colorectal Cancer Prevention Initiative was supported by Pelotonia. The collection of cancer incidence data for the State of Hawaii used in this study was supported by the Hawaii Department of Health as part of the statewide cancer reporting program mandated by Hawaii Revised Statutes; the National Cancer Institute’s SEER under Control Nos. N01-PC-67001 (1996 to 2003) and N01-PC-35137 (2003 to 2010) and Contract Nos. HHSN26120100037C (2010 to 2013) and HHSN261201300009I (2010 to current) awarded to the University of Hawaii. The collection of cancer incidence data used in this study was supported by the California Department of Public Health as part of the statewide cancer reporting program mandated by California Health and Safety Code Section 103885; the National Cancer Institute’s SEER Program under Contract No. HHSN261201000140C awarded to the Cancer Prevention Institute of California, Contract No. HHSN261201000035C awarded to the University of Southern California, and Contract No. HHSN261201000034C awarded to the Public Health Institute; and the Centers for Disease Control and Prevention’s National Program of Cancer Registries, under agreement No. U58DP003862-01 awarded to the California Department of Public Health. Data from Sweden were collected with support of the Swedish Cancer Society and the Stockholm County Council (ALF project). This work was also supported by the Spanish Ministry of Economy and Competitiveness, cofunded by European Regional Development Fund (FEDER) funds–a way to build Europe– Grant No. SAF2015-68016-R (G.C.); Carlos III National Health Institute (CIBERONC); the Government of Catalonia (Pla estratègic de recerca i innovació en salut and 2014SGR338); and the Scientific Foundation Asociación Española Contra el Cáncer. A.K.W. is a National Health and Medical Research Council (NHMRC) Career Development Fellow. M.A.J. is an NHMRC Senior Research Fellow. J.L.H. is an NHMRC Senior Principal Research Fellow. D.D.B. is an NHMRC Career Development Fellow and a University of Melbourne Research at Melbourne Accelerator Program (R@MAP) Senior Research Fellow. C.R. is a Jeremy Jass Pathology Fellow. M.N. is a Dutch Cancer Society Fellow.
Presented at the Biennial meeting of International Society for Gastrointestinal Hereditary Tumours (InSiGHT), Florence, Italy, July 8, 2017.
The ideas and opinions expressed herein are those of the author(s), and endorsement by the State of Hawaii Department of Health, the National Cancer Institute, SEER Program, the State of California Department of Public Health, the Centers for Disease Control and Prevention, or their Contractors and Subcontractors is not intended nor should it be inferred. The content of this manuscript does not necessarily reflect the views or policies of the National Cancer Institute or any of the collaborating centers in the Colon Cancer Family Registry, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government or the Colon Cancer Family Registry. Authors had full responsibility for the design of the study, the collection of the data, the analysis and interpretation of the data, the decision to submit the manuscript for publication, and the writing of the manuscript.
M.N. and A.K.W. are joint last authors.
AUTHOR CONTRIBUTIONS
Conception and design: Sanne W. ten Broeke, Frederik J. Hes, Juul T. Wijnen, Maartje Nielsen, Aung Ko Win
Provision of study materials or patients: Sanne W. ten Broeke, Carli M.J. Tops, Stefan Aretz, Inge Bernstein, Daniel D. Buchanan, Albert de la Chapelle, Gabriel Capella, Mark Clendenning, Christoph Engel, Steven Gallinger, Encarna Gomez Garcia, Jane C. Figueiredo, Robert Haile, Heather L. Hampel, Liselotte van Hest, John L. Hopper, Nicoline Hoogerbrugge, Magnus von Knebel Doeberitz, Loic Le Marchand, Tom G.W. Letteboer, Mark A. Jenkins, Annika Lindblom, Noralane M. Lindor, Arjen R. Mensenkamp, Pål Møller, Polly A. Newcomb, Theo A.M. van Os, Rachel Pearlman, Marta Pineda, Nils Rahner, Egbert J.W. Redeker, Maran J.W. Olderode-Berends, Christophe Rosty, Hans K. Schackert, Rodney Scott, Leigha Senter, Liesbeth Spruijt, Verena Steinke-Lange, Manon Suerink, Stephen Thibodeau, Yvonne J. Vos, Anja Wagner, Ingrid Winship, Frederik J. Hes, Hans F.A. Vasen, Aung Ko Win
Collection and assembly of data: Sanne W. ten Broeke, Carli M.J. Tops, Stefan Aretz, Inge Bernstein, Daniel D. Buchanan, Albert de la Chapelle, Gabriel Capella, Mark Clendenning, Christoph Engel, Steven Gallinger, Encarna Gomez Garcia, Jane C. Figueiredo, Robert Haile, Heather L. Hampel, John L. Hopper, Nicoline Hoogerbrugge, Magnus von Knebel Doeberitz, Loic Le Marchand, Tom G.W. Letteboer, Mark A. Jenkins, Annika Lindblom, Noralane M. Lindor, Arjen R. Mensenkamp, Pål Møller, Polly A. Newcomb, Theo A.M. van Os, Rachel Pearlman, Marta Pineda, Nils Rahner, Egbert J.W. Redeker, Maran J.W. Olderode-Berends, Christophe Rosty, Hans K. Schackert, Rodney Scott, Leigha Senter, Liesbeth Spruijt, Verena Steinke-Lange, Manon Suerink, Stephen Thibodeau, Yvonne J. Vos, Anja Wagner, Ingrid Winship, Frederik J. Hes, Hans F.A. Vasen, Juul T. Wijnen, Maartje Nielsen, Aung Ko Win
Data analysis and interpretation: Sanne W. ten Broeke, Heleen M. van der Klift, Maartje Nielsen, Aung Ko Win
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Cancer Risks for _PMS2_-Associated Lynch Syndrome
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Sanne W. ten Broeke
No relationship to disclose
Heleen M. van der Klift
No relationship to disclose
Carli M.J. Tops
No relationship to disclose
Stefan Aretz
Consulting or Advisory Role: AstraZeneca
Inge Bernstein
No relationship to disclose
Daniel D. Buchanan
Consulting or Advisory Role: Merck Sharp & Dohme
Albert de la Chapelle
No relationship to disclose
Gabriel Capella
Leadership: VCN Biosciences
Stock or Other Ownership: VCN Biosciences
Consulting or Advisory Role: VCN Biosciences
Research Funding: VCN Biosciences (Inst)
Mark Clendenning
No relationship to disclose
Christoph Engel
No relationship to disclose
Steven Gallinger
No relationship to disclose
Encarna Gomez Garcia
No relationship to disclose
Jane C. Figueiredo
No relationship to disclose
Robert Haile
No relationship to disclose
Heather L. Hampel
Stock or Other Ownership: Genome Medical
Consulting or Advisory Role: BeaconLBS, InVitae, Genome Medical
Research Funding: Myriad Genetics (Inst)
Liselotte van Hest
No relationship to disclose
John L. Hopper
No relationship to disclose
Nicoline Hoogerbrugge
No relationship to disclose
Magnus von Knebel Doeberitz
Honoraria: Merck Sharp & Dohme
Research Funding: NEC Europe, Merck Sharp & Dohme (Inst)
Loic Le Marchand
No relationship to disclose
Tom G.W. Letteboer
No relationship to disclose
Mark A. Jenkins
No relationship to disclose
Annika Lindblom
No relationship to disclose
Noralane M. Lindor
Honoraria: UpToDate (I)
Patents, Royalties, Other Intellectual Property: Royalties as editor for section in UpToDate (I)
Travel, Accommodations, Expenses: Intercept Pharmaceuticals (I)
Arjen R. Mensenkamp
No relationship to disclose
Pål Møller
No relationship to disclose
Polly A. Newcomb
No relationship to disclose
Theo A.M. van Os
No relationship to disclose
Rachel Pearlman
Research Funding: Myriad Genetics, InVitae
Marta Pineda
No relationship to disclose
Nils Rahner
No relationship to disclose
Egbert J.W. Redeker
No relationship to disclose
Maran J.W. Olderode-Berends
No relationship to disclose
Christophe Rosty
No relationship to disclose
Hans K. Schackert
No relationship to disclose
Rodney Scott
No relationship to disclose
Leigha Senter
Honoraria: Ambry Genetics
Consulting or Advisory Role: AstraZeneca
Liesbeth Spruijt
No relationship to disclose
Verena Steinke-Lange
Consulting or Advisory Role: AstraZeneca
Manon Suerink
No relationship to disclose
Stephen Thibodeau
No relationship to disclose
Yvonne J. Vos
No relationship to disclose
Anja Wagner
No relationship to disclose
Ingrid Winship
No relationship to disclose
Frederik J. Hes
No relationship to disclose
Hans F.A. Vasen
No relationship to disclose
Juul T. Wijnen
No relationship to disclose
Maartje Nielsen
No relationship to disclose
Aung Ko Win
No relationship to disclose
REFERENCES
- 1.Lynch HT, Snyder CL, Shaw TG, et al. : Milestones of Lynch syndrome: 1895-2015. Nat Rev Cancer 15:181-194, 2015 [DOI] [PubMed] [Google Scholar]
- 2.Umar A, Boland CR, Terdiman JP, et al. : Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst 96:261-268, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barrow E, Hill J, Evans DG: Cancer risk in Lynch syndrome. Fam Cancer 12:229-240, 2013 [DOI] [PubMed] [Google Scholar]
- 4.Harkness EF, Barrow E, Newton K, et al. : Lynch syndrome caused by MLH1 mutations is associated with an increased risk of breast cancer: A cohort study. J Med Genet 52:553-556, 2015 [DOI] [PubMed] [Google Scholar]
- 5.Win AK, Lindor NM, Young JP, et al. : Risks of primary extracolonic cancers following colorectal cancer in Lynch syndrome. J Natl Cancer Inst 104:1363-1372, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Scott RJ, McPhillips M, Meldrum CJ, et al. : Hereditary nonpolyposis colorectal cancer in 95 families: Differences and similarities between mutation-positive and mutation-negative kindreds. Am J Hum Genet 68:118-127, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Møller P, Seppälä TT, Bernstein I, et al. Mallorca Group : Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut 67:1306-1316, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lindor NM, Petersen GM, Spurdle AB, et al. : Pancreatic cancer and a novel MSH2 germline alteration. Pancreas 40:1138-1140, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stuckless S, Parfrey PS, Woods MO, et al. : The phenotypic expression of three MSH2 mutations in large Newfoundland families with Lynch syndrome. Fam Cancer 6:1-12, 2007 [DOI] [PubMed] [Google Scholar]
- 10.Senter L, Clendenning M, Sotamaa K, et al. : The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 135:419-428, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.ten Broeke SW, Brohet RM, Tops CM, et al. : Lynch syndrome caused by germline PMS2 mutations: Delineating the cancer risk. J Clin Oncol 33:319-325, 2015 [DOI] [PubMed] [Google Scholar]
- 12.Møller P, Seppälä T, Bernstein I, et al. : Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: First report from the prospective Lynch syndrome database. Gut 66:464-472, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Haraldsdottir S, Rafnar T, Frankel WL, et al. : Comprehensive population-wide analysis of Lynch syndrome in Iceland reveals founder mutations in MSH6 and PMS2. Nat Commun 8:14755, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Espenschied CR, LaDuca H, Li S, et al. : Multigene panel testing provides a new perspective on Lynch syndrome. J Clin Oncol 35:2568-2575, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Win AK, Jenkins MA, Dowty JG, et al. : Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol Biomarkers Prev 26:404-412, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hampel H, Frankel WL, Martin E, et al. : Feasibility of screening for Lynch syndrome among patients with colorectal cancer. J Clin Oncol 26:5783-5788, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hampel H, Frankel WL, Martin E, et al. : Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). N Engl J Med 352:1851-1860, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Hampel H, Panescu J, Lockman J, et al. : Comment on: Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res 67:9603, 2007 [DOI] [PubMed] [Google Scholar]
- 19.Hampel H, Stephens JA, Pukkala E, et al. : Cancer risk in hereditary nonpolyposis colorectal cancer syndrome: Later age of onset. Gastroenterology 129:415-421, 2005 [DOI] [PubMed] [Google Scholar]
- 20.Pearlman R, Frankel WL, Swanson B, et al. : Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early-onset colorectal cancer. JAMA Oncol 3:464-471, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Newcomb PA, Baron J, Cotterchio M, et al. : Colon Cancer Family Registry: An international resource for studies of the genetic epidemiology of colon cancer. Cancer Epidemiol Biomarkers Prev 16:2331-2343, 2007 [DOI] [PubMed] [Google Scholar]
- 22.Jenkins MA, Win AK, Templeton AS, et al. : Cohort profile: The Colon Cancer Family Registry Cohort (CCFRC). Int J Epidemiol 47:387-388i, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colon CFR: Colon Cancer Family Registry Informatics Center. www.coloncfr.org.
- 24.Dowty JG, Win AK, Buchanan DD, et al. : Cancer risks for MLH1 and MSH2 mutation carriers. Hum Mutat 34:490-497, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lange K, Weeks D, Boehnke M: Programs for pedigree analysis: MENDEL, FISHER, and dGENE. Genet Epidemiol 5:471-472, 1988 [DOI] [PubMed] [Google Scholar]
- Ferlay J, Bray F, Steliarova-Foucher E, et al (eds): Cancer Incidence in Five Continents: CI5plus. IARC CancerBase No. 9. Lyon, International Agency for Research on Cancer, 2014. http://ci5.iarc.fr/Default.aspx.
- doi: 10.1053/j.gastro.2014.01.022. Win AK, Dowty JG, Cleary SP, et al: Risk of colorectal cancer for carriers of mutations in MUTYH, with and without a family history of cancer. Gastroenterology 146:1208-1211.e1-5, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Provenzale D, Gupta S, Ahnen DJ, et al. : Genetic/familial high-risk assessment: Colorectal version 1.2016, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 14:1010-1030, 2016 [DOI] [PubMed] [Google Scholar]
- 29.Renkonen-Sinisalo L, Bützow R, Leminen A, et al. : Surveillance for endometrial cancer in hereditary nonpolyposis colorectal cancer syndrome. Int J Cancer 120:821-824, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Caminsky NG, Mucaki EJ, Perri AM, et al. : Prioritizing variants in complete hereditary breast and ovarian cancer genes in patients lacking known BRCA mutations. Hum Mutat 37:640-652, 2016 [DOI] [PubMed] [Google Scholar]
- 31.Desmond A, Kurian AW, Gabree M, et al. : Clinical actionability of multigene panel testing for hereditary breast and ovarian cancer risk assessment. JAMA Oncol 1:943-951, 2015 [DOI] [PubMed] [Google Scholar]
- 32.Castéra L, Krieger S, Rousselin A, et al. : Next-generation sequencing for the diagnosis of hereditary breast and ovarian cancer using genomic capture targeting multiple candidate genes. Eur J Hum Genet 22:1305-1313, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tung N, Battelli C, Allen B, et al. : Frequency of mutations in individuals with breast cancer referred for BRCA1 and BRCA2 testing using next-generation sequencing with a 25-gene panel. Cancer 121:25-33, 2015 [DOI] [PubMed] [Google Scholar]
- 34.Antoniou AC, Goldgar DE, Andrieu N, et al. : A weighted cohort approach for analysing factors modifying disease risks in carriers of high-risk susceptibility genes. Genet Epidemiol 29:1-11, 2005 [DOI] [PubMed] [Google Scholar]
- 35.Vos JR, Oosterwijk JC, Aalfs CM, et al. : Bias explains most of the parent-of-origin effect on breast cancer risk in BRCA1/2 mutation carriers. Cancer Epidemiol Biomarkers Prev 25:1251-1258, 2016 [DOI] [PubMed] [Google Scholar]
- 36.Ziogas A, Anton-Culver H: Validation of family history data in cancer family registries. Am J Prev Med 24:190-198, 2003 [DOI] [PubMed] [Google Scholar]
- 37.Talseth-Palmer BA, Wijnen JT, Brenne IS, et al. : Combined analysis of three Lynch syndrome cohorts confirms the modifying effects of 8q23.3 and 11q23.1 in MLH1 mutation carriers. Int J Cancer 132:1556-1564, 2013 [DOI] [PubMed] [Google Scholar]
- 38.Win AK, Hopper JL, Buchanan DD, et al. : Are the common genetic variants associated with colorectal cancer risk for DNA mismatch repair gene mutation carriers? Eur J Cancer 49:1578-1587, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.van Duijnhoven FJ, Botma A, Winkels R, et al. : Do lifestyle factors influence colorectal cancer risk in Lynch syndrome? Fam Cancer 12:285-293, 2013 [DOI] [PubMed] [Google Scholar]
- doi: 10.1038/gim.2015.83. Suerink M, van der Klift HM, Ten Broeke SW, et al: The effect of genotypes and parent of origin on cancer risk and age of cancer development in PMS2 mutation carriers. Genet Med 18:405-409, 2016 [Erratum: Genet Med 18:108, 2016] [DOI] [PubMed] [Google Scholar]