The Synergistic Role of Diet and Exercise in the Prevention, Pathogenesis, and Management of Ulcerative Colitis: An Underlying Metabolic Mechanism (original) (raw)
Abstract
Ulcerative colitis (UC) is a biologically complex condition characterized by chronic, relapsing inflammation of the gastrointestinal tract. The relative incidence of this debilitating condition is increasing and sociologically damaging outcomes are a continued reality. Several etiological theories for UC are currently under investigation, spanning between genetic and environmental determinants. From an environmental perspective, previous literature reviews have demonstrated the independent effectiveness of specific diet and exercise patterns in modifying UC immuno-pathophysiology. This article explores the synergistic role of diet and aerobic exercise in the prevention, pathogenesis, and management of UC in the context of recent immunological research. Through a unifying mechanism—that is, microbial influence of colonic inflammation and immuno-pathophysiology—the simultaneous reduction of pro-inflammatory dietary sulfurous amino acid intake (ie methionine, cysteine, homocysteine, and taurine) and the upregulation of aerobic exercise frequency (which spurs the colonization of anti-inflammatory butyrate, acetate, and propionate producing microbial taxa) demonstrate the clinical efficacy of incorporating both diet and exercise modifications for UC prevention and management through pathogenic alterations.
Keywords: ulcerative colitis, diet, exercise, microbiome, immunopathology
Introduction
Inflammatory bowel diseases (IBDs) are a biologically complex set of conditions characterized by chronic, relapsing inflammation of the gastrointestinal (GI) tract.1 Sociological realities attached to those diagnosed with IBDs include altered mental health scores when compared with control groups (other factors being equal) as it relates to the ability to perform daily activities, manage chronic pain, and interact with social circles.2 Also, epidemiologically speaking, there is a general increase in the incidence of IBDs, with current rates of annual cases of ulcerative colitis (UC) and Crohn disease (CD) per 100,000 people at 24.3 and 12.7, 19.2 and 20.2, 6.3 and 5.0, for Europe, North America, and Asia and the Middle East, respectively.3
Symptoms and diagnostic criteria of IBDs
The two main types of IBDs are UC and CD, which are characterized, respectively, by chronic, relapsing inflammation of the colon and rectum (UC) and the mouth, esophagus, stomach, colon, rectum, and anus (CD).1,4-6 Although both diseases can present with rectal bleeding, diarrhea, vomiting, and abdominal pain,7 signs and symptoms specific to CD include porridge-like defecation, pyrexia, fistula, and weight loss8 and those specific to UC include mucus-like bowel movements with blood present, rectal urgency, and tenesmus (perception of non-complete bowel emptying).6 Diagnostic markers indicative of CD typically include terminal ileum, colon, and anal involvement, patchy areas of inflammation, geographically deep serpiginous ulcers, transmural presentation, stenosis, and non-necrotizing, non-peri-intestinal crypt granulomas.5,8 Ulcerative colitis, however, often presents with colon, rectum, and bile duct involvement, continuous areas of inflammation and ulcers, and a shallow, mucosal presentation and without granulomas.7,9
Ulcerative Colitis
Ulcerative colitis is a chronic illness of the colon and rectum ranging in severity from mild to severe, often characterized by intermittent flare-ups of painful abdominal cramps and diarrhea.1 Colon cancers, inflammation of the eyes, liver, or joints, and toxic megacolon contribute potential complications to UC.10
Etiology
Several etiological theories for UC are currently under investigation. Foremost postulations are multifactorial in nature and include areas such as immunogenetics, and, as they relate to this review, environmental aspects. As it were, each etiological category relates to UC in the context of immunological mechanisms (ie cytokine proliferation and autoimmune components)—that is, respectively, mutation of immunologically relevant genes and inflammatory mechanisms as they relate to bioactive components of external food particle intake, in addition to physical exercise and its involvement with energy regulation and colonic microbiota.
Cytokine networks relevant to UC pathophysiology
Various cytokines and immunologically relevant cell lines have been associated with UC. Included in the list are tumor necrosis factor (TNF)-α, TGF-β, interferon (IFN)-γ, interleukin (IL)-4, IL-6, IL-10, IL-13, and IL-17, among several others.11 These cytokines are mechanistically involved in UC pathophysiology in various ways, included among them is apoptosis inhibition, NF-κβ activation, and the manufacture of platelet-activating factor, leukotrienes, nitric oxide, and other inflammatory mediators.11 Th1 and Th2 cells (CD4+ lymphocytes) produce such cytokines. In addition, a T-helper cell subset (Th17) has demonstrated involvement in UC pathophysiology.12 Activation of mast cells and immunoglobulin E (IgE) proliferation as they relate to UC induce an atopic response through IL-4, IL-5, IL-6, IL-10, and IL-13 (produced by Th2 cells).12
As they relate specifically to UC pathophysiology and diagnostic findings, TNF-α, TGF-β, IL-6, IL-10, and IL-13 are, respectively, found in significantly higher levels in inflamed colonic samples, demonstrate increased synthesis by mononuclear cells of the lamina propria in patients with UC, contribute excess inflammation through sIL-6R and STAT3 downstream signaling, act as an anti-inflammatory cytokine and is elevated in UC patients (although is insufficient for counter-immunosuppression), and allows for increased epithelial permeability secondary to Th2 cell activation.13-17
Immunogenetic determinants of UC pathophysiology
Ulcerative colitis demonstrates a 3% concordance rate in dizygotic twins and a 10% concordance rate in monozygotic twins, indicating a partially heritable pattern to the disorder.18 Recent genome-wide association studies (GWAS) have revealed 47 genomic loci linked to UC pathophysiology including genes such as IL1R2, IL8RA/B, IL7R, IL12B, DAP, PRDM1, JAK2, IRF5, GNA12, and LSPI.19
As they relate specifically to UC pathogenesis, altered function/expression of TNFRSF14, TNFRSF9, IL1R2, IL8RA/B, IL7R, DAP, PRDM1, IRF5, GNA12, and LSP1 leads to increased inflammation through, respectively, TNF receptor mutation, increased secretion of IL-2, enhanced ILb1 production (an inflammatory factor), hypermorphic mutation of IL-8 (a neutrophil chemotactic attractor), increased expression of IL-7 (involved in naive and memory T-cell survival), dysregulated autophagy, altered proliferation of B and T cells, induced cytokine production via toll-like receptor (TLR) signaling, modified tight junction assembly, and ineffective neutrophil transmigration.19-28
Environmental determinants of UC pathophysiology
Although several mutated or altered genomic loci account for a statistically significant number of cases of UC (indicating a polygenic inheritance pattern), epidemiological analysis provides evidence to the involvement of environmental determinants, often “pulling the trigger” on a genetically predisposed individual.3 The literature has identified many such environmental triggers for UC pathogenesis and management which act through varied, but underlyingly similar immunological mechanisms.
Several examples include smoking, changes in gut flora, medication use in the form of non-steroidal anti-inflammatory drugs (NSAIDs), oral contraceptive pills (OCPs) and antibiotics, lack of early maternal breastfeeding, and air pollution.4 Respectively, these environmental factors influence UC immuno-pathophysiology by altering T cell and bowel epithelial cells’ nicotinic acetylcholine receptors (currently under investigation), modifying the presence of colonic microbiological populations such as Helicobacter pylori, helminth varieties, among others (hygiene hypothesis), downregulating TNF and stimulating anti-inflammatory cytokines (NSAIDs), causing immunological alterations secondary to estrogen and progesterone stimulation (OCPs), inducing an autoimmune response as a result of early life microbial dysbiosis (antibiotics), reducing access to lactoferrin (an anti-inflammatory compound found in human breast milk), and being exposed to elevated ambient sulfur dioxide levels (an industrial pollutant).29-38
Although the aforementioned triggers contribute to UC immuno-pathophysiology in their own right, the remainder of this review will focus on two such environmental aspects to the disease—diet and exercise—diverse elements of which contribute to UC prevention, pathogenesis, and management in an immuno-microbiological context. Although previous published data and literature reviews have demonstrated the independent effectiveness of specific diet and exercise patterns in modifying UC immuno-pathophysiology, the following exposition will provide empirical and mechanistic evidence for a synergistic role of these lifestyle characteristics.
Immuno-microbiological Underpinnings of Colonic Inflammation
In various ways—both direct and indirect—an individual’s specific dietary intake and quantity of aerobic exercise can influence the initial triggering of UC pathogenesis, quality of the pathophysiological process, and the effective management of disease processes post diagnosis.
From an immuno-microbiological standpoint, there are many mechanisms at play when evaluating colonic inflammation in those patients with UC, which generally involves microbial influence of colonic inflammation and immuno-pathophysiology; ultimately, either pro-inflammatory or anti-inflammatory net effects may ensue as a result of the fermentation of specific compounds.
Butyrate, acetate, and propionate producing microbial taxa
The human colon is home to several different species of bacteria each contributing various bioactive effects on human physiology.39 As a result of the fermentation of dietary fibers, certain species of colonic bacteria produce byproducts such as butyrate, acetate, and propionate—three short-chain fatty acids (SCFAs) which have been shown to reduce colonic inflammation, especially in those with UC pathophysiology.40 Butyrate especially, has attracted attention from the scientific community in recent years.41 Along those lines, several species of bacteria have demonstrated a particular ineptness at producing the anti-inflammatory butyrate compound.
Clostridia are a commensal-type bacterial class of organisms that populate the human microbiome, typically from birth.42 Several subspecies of bacteria under the class Clostridia produce butyrate as a main product of bacterial fermentation, such as the Lachnospiraceae and Erysipelotrichaceae families, as well as Roseburia (a genus classified under the Lachnospiraceae family), which have gained research attention for their anti-inflammatory properties.43-46
Sulfur and bacterial fermentation
As it were, the butyrate-producing nature of the aforementioned bacterial species is conditional; that is, certain bacterial molecular outputs can inhibit others. In recent years, it has been discovered that sulfur-based amino acids lead to competitive inhibition with that of butyrate. Sulfurous amino acids are fermented by such bacterial classes/families as Clostridia, Roseburia, Lachnospiraceae, and Erysipelotrichaceae, which produce hydrogen sulfide as a metabolite.47 (It should be noted that, to date, only in-vitro experimentation has been performed in this area, and therefore more research is required to confirm this aspect). In turn, hydrogen sulfide inhibits the bacteria from producing the anti-inflammatory butyrate compound. Therefore, perhaps, a recipe for low inflammation is as follows: a large proportion of butyrate-producing bacteria in combination with a low proportion of available sulfur-based amino acids. The overall theoretical mechanism can be seen in Figure 1.
Figure 1.
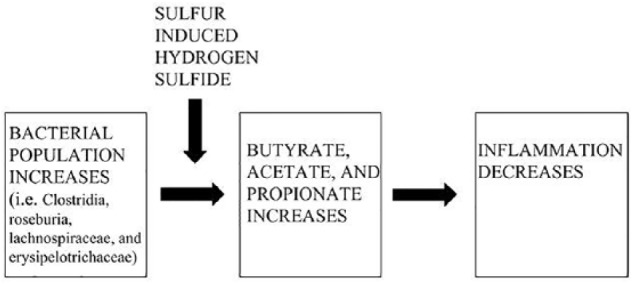
As specific colonic bacterial populations such as Clostridia, Roseburia, Lachnospiraceae, and Erysipelotrichaceae increase (leftmost panel), the production of their respective anti-inflammatory byproducts increases accordingly (middle panel). Ultimately, colonic inflammation decreases in a dose-dependent fashion (rightmost panel). Sulfurous amino acids present in the GI tract undergo anaerobic fermentation resulting in hydrogen sulfide as a metabolite, which inhibits butyrate formation (upper-left insertion), ultimately resulting in an upregulated inflammatory response.
Although the synergistic mechanisms of diet and exercise as they relate to UC immuno-pathophysiology (and butyrate production) will be elaborated in the final section of this literature review, the independent epidemiological evidence for the role of diet and exercise in UC prevention, pathogenesis, and management is provided here.
The Role of Diet in UC Prevention, Pathogenesis, and Management
The composition of an individual’s dietary intake contributes a great deal to their physiological systems, and therefore overall health.
Ultimately, through the sulfur:hydrogen sulfide mechanism described above, high sulfurous amino acid intake (such as methionine, cysteine, homocysteine, and taurine) as a general dietary pattern (ie large mammalian tissue intake) has been significantly associated with UC prevention, pathogenesis, and management.
In a study published in the American Journal of Gastroenterology, researchers followed 67,581 women and their dietary patterns to establish a direct connection between nutrition practices and IBD incidence. High protein intake (specifically mammal-based protein which is proportionally higher in sulfur-based amino acids than plant-based protein) was significantly associated with the onset of UC in the women assessed.48 The data showed a “3.3 fold increased risk for IBD” in women with “moderate to high protein intake, especially of animal origin,” compared with expected rates of UC and CD incidence.48 Notable strengths of the research compared with typical studies involving nutritional science include dietary questionnaires being completed well before IBD onset (as opposed to dietary studies with recall bias), as well as the reproducibility of said questionnaires.49
Of note, the literature is relatively lacking regarding the role of diet for UC prevention, and further studies need to be conducted before conclusions can be drawn. In the aforementioned study, the researchers noted limitations to their study design including timing between dietary data collection and disease onset; however, the researchers explained the weakness of such a potential confound, noting “dietary habits change very little over time in middle-aged adults.”48 Also, to be sure, despite a relative absence of published studies on dietary patterns and UC prevention, the general hypothesis holds promise;50 future epidemiological research will elaborate on the trends.
Several studies have been published of the quality of the UC pathophysiological process as it relates to dietary patterns, especially as it relates to future disease management (ie relapse).51-55
A prospective cohort study published in the journal “Gut” highlights the relationship between sulfur-based amino acid intake (ie mammalian protein) and UC relapse rates. The researchers looked at several different exogenous foodstuff including cereal and cereal products, milk and milk products, eggs, vegetables, fruits, fish and fish products, meat and meat products, red and processed red meat (a separate category), non-alcoholic beverages, alcoholic beverages, sugars, preserves, and snacks. Food groups tested that demonstrated a positive correlation with UC relapse (ie more of the food products resulted in higher incidence of relapse) include milk and milk products, eggs, meat and meat products, red and processed meat, non-alcoholic beverages, and alcoholic beverages. Food groups tested that demonstrated a negative correlation with UC relapse (ie more of the food products resulted in lower incidence of relapse) include cereal and cereal products (negligible difference) vegetables, fruits, fish and fish products (negligible difference), and sugars, preserves, and snacks.56 The researchers concluded that “potentially modifiable dietary factors, such as a high meat or alcoholic beverage intake, have been identified that are associated with an increased likelihood of relapse for UC patients.”56 As it would seem, based on molecular and microbiological processes, a mainstay factor connecting mammalian protein intake with UC immuno-pathophysiology is the level of consumption of sulfurous amino acids (ie methionine, cysteine, homocysteine, and taurine). The overall theoretical mechanism is shown in Figure 2.
Figure 2.
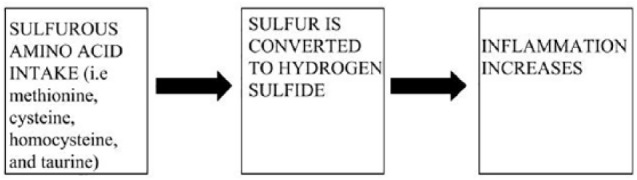
As sulfurous amino acids present in the GI tract such as methionine, cysteine, homocysteine, and taurine increase (leftmost panel), hydrogen sulfide is released as a metabolite (middle panel). Ultimately, colonic inflammation increases in a dose-dependent fashion (rightmost panel).
The Role of Exercise in UC Prevention, Pathogenesis, and Management
As an environmental factor relating to health and disease, few lifestyle characteristics have been evaluated with the same rigor, and with positive results, as has physical exercise.
In addition to various GI disorders including esophagitis peptic ulcers, and constipation, the literature is beginning to uncover strong correlations between exercise and IBD.57 Physical activity has been shown to play a role in several conditions, including that of UC, through exercise-induced myokines (eg irisin, IL-15, leukemia inhibitory factor [LIF], brain-derived neurotrophic factor [BDNF], fibroblast growth factor [FGF]-21, and SPARC [secreted protein acidic and rich in cysteine]).58
For the role of exercise in UC prevention, a retrospective study conducted on German participants highlights the subject well. In the study, 12,014 individuals were evaluated over a 6-year period of time. Men and women with jobs that required high levels of physical activity (ie road construction workers, bricklayers, cleaning and maintenance, and security personnel) displayed lower incidence rates of IBD compared with those individuals with jobs that commanded low levels of physical activity (ie electricians, instrument makers, sales representatives, health care workers, technical assistants, and bakers)59; the effects were observed independent of the subjects’ sex. Also, a study published out of Israel observed a negative correlation between physical exercise and incidence of IBD. A statistically significant number of IBD cases occurred in those with lower levels of physical exercise in the period leading up to disease onset.60
In a more recently published 2013 study evaluating several different environmental factors and IBD onset, increased qualitative and quantitative metrics of physical exercise were associated with a decreased likelihood of UC onset.61 A total of 388 patients (148, UC; 240, CD) were evaluated against 355 control subjects in this case-controlled study out of Slovakia.
Although some studies evaluating physical activity and UC onset did not find such an association,62,63 confounding factors question the validity of their findings; more research will be needed to reach proper conclusions.
In those already experiencing UC pathology, various interventional trials involving aerobic exercise have been completed over the recent years. In one such trial, researchers assigned 15 individuals currently experiencing a quiescent stage of UC to complete an aerobic exercise program among other lifestyle interventions (ie plant-based diet, behavioral techniques, and stress management training). Compared with a control group, those who received intervention demonstrated increased scores on quality-of-life questionnaire, in addition to a reduction in relapse rates.64 To be sure, the other non-exercise lifestyle interventions may have provided confounds to the results.
In addition, a 2018 study discovered that an UC diagnosis reduced patient adherence to exercise programs, resulting ultimately in negative quality-of-life consequences.65 In that regard, the literature has demonstrated an absence of potential adverse effects, or dangers to starting an exercise program with a UC diagnosis; that is to say, only benefits have been shown.66 Current evidence supports microbiological-based immunological alterations to be at the heart of these correlational findings, although more research is needed to determine causality.67
Experimental studies performed on animal models, also, contribute to our current knowledge of UC immuno-pathophysiology from the perspective of aerobic exercise intervention.
A 2013 study published in the journal Brain, Behavior, and Immunity tested mouse models for immunologically related compounds in two groups: forced treadmill running (FTR) and voluntary wheel running (VWR). Contrary to their original hypothesis, the researchers found FTR to increase the gene expression of colonic inflammatory compounds such as IL-6, IL-1β, and IL-17, in addition to higher rates of mouse mortality.68 In those mice allowed to run voluntarily colonic gene expression of such compounds were downregulated.68 Extrapolating to humans, this may indicate voluntary aerobic exercise to be an effective means of downregulating inflammatory cytokines.
Adding to the evidence, a previously held study published in the journal Brain, Behavior, and Immunity set out to determine the mechanistic, immunological pathways involved in long-term aerobic exercise as it relates to colonic inflammation and therefore IBD. The researchers found that, as compared with a control group, those mice who participated in 16 weeks of free wheel training displayed decreased intestinal lymphocyte expression of TNF-α and Caspase 7, and an increased expression of IL-10 and IL-6.69 Despite the variable expression outcomes of the cytokines evaluated, the researchers discovered a net anti-inflammatory result in those mice participating in free wheel training, over the long term.69
Even more, a 2014 study looked at intestinal barrier disruptions in mice due to stress (repeated restraint stress as the researchers described). An attenuation of the leaky gut barrier occurred upon incorporation of 30-minute daily swimming intervals.70 The researchers hypothesized the data to be a result of colonic microbiota-induced anti-inflammatory changes to the gut epithelia.70
Finally, a 2017 study evaluated the effects of exercise on mice with experimentally induced colitis. The researchers found that voluntary exercise reduced the severity of colitis in mice fed a high-fat diet through the release of various biomarkers.71 Along those lines, a 2015 study found similar results: proinflammatory cytokines contributing to UC pathology were suppressed in those mice allowed to participate in voluntary exercise.72
Although not all experimental animal studies demonstrated these effects,73 the totality of the current research supports the hypothesis of a net anti-inflammatory effect of aerobic exercise.
Finally, and of crucial importance to this literature review in particular, a 2018 study published in the Canadian Journal of Gastroenterology discovered an intriguing and vital aspect to the currently proposed theoretical mechanism of exercise and immuno-pathophysiology of UC as it relates to SCFA production (such as butyrate). The researchers attempted to explore the effect of aerobic exercise on the composition of mouse model colonic microbiota; in that regard, a direct connection could be drawn between physical exercise and the anti-inflammatory inducing properties of butyrate through the microbiome. VO2 max values were obtained to determine the level of acute aerobic fitness output in mouse models. Cardiorespiratory fitness levels were varied in test subjects, fecal microbiota profiles were analyzed by means of DNA sequencing and compared with control profiles, and gas chromatography was used to determine the SCFA content in addition to cytokine-related gene expression of the differing mouse subjects. A direct correlation was found between physical activity level and microbial diversity.74 Also, the researchers discovered the primary SCFA in fecal microbiota profiles to be butyrate, which was increased as a result of colonization of Clostridia, Roseburia, Lachnospiraceae, and Erysipelotrichaceae. Regarding cytokine analysis, TGF-β and TNF-α were expressed at an increased rate in those mice that were sedentary compared with those undergoing physical activity.74 The overall theoretical mechanism can be seen in Figure 3.
Figure 3.
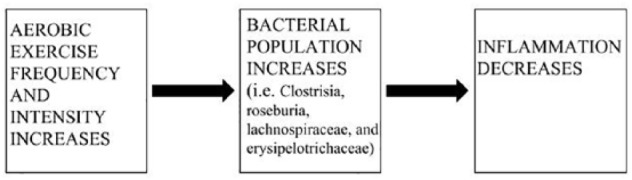
As aerobic exercise frequency and intensity increase (leftmost panel), bacterial populations such as Clostridia, Roseburia, Lachnospiraceae, and Erysipelotrichaceae increase in a dose-dependent manner (middle panel). Ultimately, colonic inflammation decreases accordingly (rightmost panel).
Generally speaking, moderate aerobic exercise in humans has been found to carry both short and long-term immunomodulating effects75 with the precise mechanisms of long-term exercise-induced outcomes currently being researched. For the immunomodulatory effects of diet and exercise, recent research has unveiled several theoretical mechanisms with utter precision;76 the logical, empirical, and mechanistic outline for which we have established provides, in conjunction with the current scholarly literature, strong evidence for its appropriate application to UC prevention, pathogenesis, and management.
Butyrate, IL-17, and T-regulatory Response: The Unifying Mechanism
T-cell subsets (in their functional form) are produced from cytokine-dependent differentiation in addition to antigen-dependent activation. Regulated immunity and inflammation are controlled by CD4+ helper T cells as well as CD25+, FoxP3+, and Tregs.77,78 A unique set of Th cells (in addition to Th1 and Th2 subsets) is the Th17 subset (T helper 17 cells), whose main recognition receptor is IL-17 (along with retinoic acid receptor-related orphan receptor gamma T receptor [RORt]).77 Various original studies have indicated a direct link between Th17 cells (along with IL-17 and IL-23 pathways) and colonic inflammation.79 Moreover, FoxP3+, an intracellular transcription factor, regulates Treg activity through CD4+ and CD25+ on the T-cell receptor.80 Th17 is negatively regulated by Treg activity (especially through IL-10 cytokine production, and in turn responds to TGF-β).81,82 Treg maturation is suppressed by IL-6, which in turn allows for a Th17 response (causing a net pro-inflammatory effect).83 The previously highlighted anti-inflammatory effects of the butyrate molecule have been thought to occur as a result of apoptotic mechanisms, cytokine expression, and immune cell migration which have demonstrated efficacy as it relates to modulating UC immuno-pathophysiology.84,85
A critical study
In their original study titled “Butyrate Inhibits Interleukin-17 and Generates Tregs to Ameliorate Colorectal Colitis in Rats,” Zhang et al provide evidence for several unifying mechanisms for our theory involving the synergistic role of diet and exercise in the prevention, pathogenesis, and management of UC. The authors described an immuno-pathophysiological process connecting the aforementioned notions through in-vitro human monocyte and rat splenocyte analysis in addition to in-vivo butyrate-treated rats.86
In totality, the study uncovered several mechanisms by which butyrate is able to ameliorate colitis in rats (ie by T-cell differentiation and activation).86 In agreement with the previous evidence outlined in this review, lower levels of butyrate were found in those patients with an UC diagnosis. Also, lower levels of inflammation were confirmed through cytokine analysis, thus further supporting the evidence.
Regarding IL-17, previous research has highlighted the effect this cytokine has on UC pathophysiology, with IL-17 blocking therapy currently being evaluated in patients with IBD.87 In the presence of butyrate in typical physiological proportions (around 0.5 mM or 10% of all SCFAs), the authors showed 2,4,6-trinitrobenzene sulfonic acid (TNBS)-induced colitis to be significantly reduced (secondary to IL-17 and RORt) in a supplemental butyrate group, indicating its role in the prevention, pathogenesis, and management of UC. Moreover, TGF-β enforces the differentiation of Th17 cell lineage commitment through IL-6 signaling.77 As plasma IL-23 (sustains Th17 cell stability through bone marrow-derived dendritic cell [BMDC] activation) and IL-6 levels were increased secondary to TNBS treatment (and reduced by butyrate treatment), it can be deduced that, mechanistically speaking, butyrate is targeting the IL-23/Th17/IL-17 pathway, thereby reducing inflammation. The researchers’ evaluation of the BMDC and rat splenocyte analysis confirm these findings: the cytokines released on splenocyte differentiation to Th17 cells (in vitro) were in parallel to those released by colitis immuno-pathophysiology; these cytokines could only be controlled by large doses of supplemental sodium butyrate solution.86 In totality, therefore, the authors suggested that Th17 cell differentiation is inhibited by butyrate.
Finally, TGF-β stimulates Treg differentiation from T helper cell types. Several studies have demonstrated the ability of IL-10 (an anti-inflammatory cytokine) to be protective as it relates to IBD immuno-pathophysiology;81 as such, Tregs (which are maintained by butyrate secondary to a high fiber diet)88,89 produce IL-10 thereby providing negative regulation toward Th17 cells. The authors’ in-vivo studies demonstrating the ability of butyrate to increase the levels of IL-10 and Tregs corroborate these findings. Differentiation of naive T cells to Th17 cells is completed by stimulation of IL-6 and TGF-β. Moreover, confirmed by the human peripheral blood mononuclear cells (PBMC) trail, treatment with butyrate (and Treg proliferation) caused IL-6 levels to decrease and TGF-β levels to increase; ultimately, increased Treg frequency and PBMC-induced TGF-β secretion was caused by high-dose butyrate treatment.
Therefore, as the authors concluded, healthy physiological functioning dictates a tempered balance between Treg and Th17 cell types. Cytokines such as IL-6, TGF-β, and IL-10 determine whether the “Th cell pool” will differentiate into Tregs or Th17. Butyrate helped regulate the balance between Treg and Th17 cell types, thereby demonstrating its importance and clinical relevance for IBD immuno-pathophysiology.
Discussion
Taking together the research of Zhang et al in addition to numerous other studies of similar nature involving butyrate, colitis, inflammation, diet, exercise, epigenetics, and microbiota, several theoretical mechanisms as to how butyrate influences UC immuno-pathophysiology can be drawn.40,45,84,85,90-100
As one theoretical mechanism, butyrate induces Treg cell differentiation leading to a healthy colon, with its absence contributing to colitis through IL-10 and Th17 inhibition as shown in Figure 4.
Figure 4.

Normal immuno-physiological function is a consequence of butyrate and other anti-inflammatory compounds upregulating Treg cell differentiation, which in turn spurs IL-10 release, thereby leading to Th17 cell inhibition, and ultimately, a healthy colon (top box). Altered immuno-physiological function involves the absence of butyrate, which disallows for Treg cell differentiation, subsequent IL-10 release, and Th17 cell inhibition, ultimately resulting in colitis (lower box).
What’s more, butyrate-induced inhibition of IL-6 release may result in a healthy colon, with its absence contributing to colitis through Th17 cell differentiation as shown in Figure 5.
Figure 5.
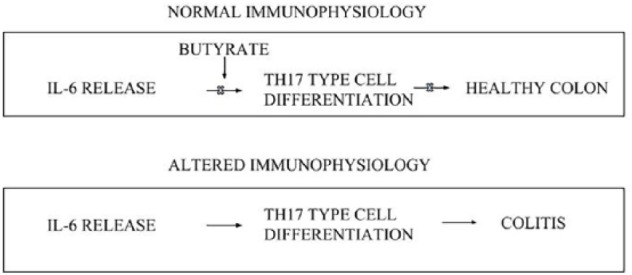
Normal immuno-physiological function is a consequence of butyrate and other anti-inflammatory compounds inhibiting IL-6 release (which contributes to Th17 cell type differentiation and colonic inflammation) resulting in a healthy colon (top box). Altered immuno-physiological function involves the absence of butyrate, which allows for IL-6 release and Th17 cell type differentiation, resulting in colitis (lower box).
Finally, butyrate-induced colitis amelioration may result from the inhibition/interference of IL-23 and IL-17 release as shown in Figure 6.
Figure 6.

Colitis onset spurs splenocyte differentiation and IL-23 and IL-17 release resulting in continued inflammatory pathophysiology. Butyrate suppresses the secretion of IL-23 and IL-17 leading to the amelioration of colitis.
The scholarly literature as it currently stands corroborates greatly and provides well-defined evidence for the independent effectiveness of specific diet and exercise patterns in modifying UC immuno-pathophysiology. In that regard, in consultation with the totality of current literature, we have created a theoretical mechanistic framework as it relates to the synergistic role of diet and aerobic exercise in the prevention, pathogenesis, and management of UC. The theoretical framework, in combining the totality of proposed mechanisms, and as the resultant synergy of diet and exercise patterns, is portrayed in Figure 7.
Figure 7.

From left to right: increased intake of fibrous foods (ie plant-derived sources such as fruits, vegetables, grains, legumes, nuts, and seeds) in addition to the increased frequency of aerobic exercise patterns can increase the colonic colonization of specific bacteria (ie Clostridia, Roseburia, Lachnospiraceae, and Erysipelotrichaceae). In turn, these bacteria produce anti-inflammatory byproducts (ie butyrate, acetate, and propionate) which are blocked due to the presence of hydrogen sulfide secondary to sulfurous amino acid fermentation. Butyrate production leads to upregulated Treg cell differentiation, IL-10 release, and Th17 cell inhibition (top box); suppression of IL-6 release and Th17 cell differentiation (lower box); and inhibition of IL-23 and IL-17 release (middle box). Taken together, these multifactorial elements lead to the statistically greater likelihood of a healthy colon (far right).
Conclusions
Ultimately, the information presented in this literature review portrays a crucial idea: a synergistic role is played by diet and aerobic exercise in the prevention, pathogenesis, and management of UC. To capitalize on butyrate production, and therefore a protective influence on UC, a simultaneous reduction of pro-inflammatory dietary sulfurous amino acid intake and upregulation of aerobic exercise are required. To be sure, although epidemiological research supporting this notion has yet to be conducted, the nature of UC prevalence and incidence rates by geographic area align well with those countries displaying both subpar physical activity and a westernized diet.3 Therefore, these findings should influence clinical advice given to those susceptible, or already experiencing an UC diagnosis.
Acknowledgments
The authors gratefully acknowledge the Yeshiva University Dean’s Office, Biology Department, and the respective faculty for their overall support of our research.
Footnotes
**Funding:**This work was supported by the Yeshiva University Alexander fund #252869 awarded to R.M.
**Declaration of conflicting interests:**The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Author Contributions: JS contributed to conceptualization of theoretical synergistic framework, figure and table creation, literature search, writing up of all drafts of the paper, interpretation of data, and approval of final submission; RM contributed to analysis of theoretical framework, revising it critically for important intellectual content, and final approval of the version to be submitted. Both authors had full access to all the data in the study and had final responsibility for the decision to submit for publication.
References
- 1.Baumgart DC, Carding SR. Inflammatory bowel disease: cause and immunobiology. Lancet. 2007;369:1627–1640. [DOI] [PubMed] [Google Scholar]
- 2.Knowles SR, Graff LA, Wilding H, Hewitt C, Keefer L, Mikocka-Walus A. Quality of life in inflammatory bowel disease: a systematic review and meta-analyses-part I. Inflamm Bowel Dis. 2018;24:742–751. [DOI] [PubMed] [Google Scholar]
- 3.Ye Y, Pang Z, Chen W, Ju S, Zhou C. The epidemiology and risk factors of inflammatory bowel disease. Int J Clin Exp Med. 2015;8:22529–22542. [PMC free article] [PubMed] [Google Scholar]
- 4.Franke A, McGovern DPB, Barrett JC, et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat Genet. 2010;42:1118–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baumgart DC, Sandborn WJ. Inflammatory bowel disease: clinical aspects and established and evolving therapies. Lancet. 2007;369:1641–1657. [DOI] [PubMed] [Google Scholar]
- 6.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature. 2007;448:427–434. [DOI] [PubMed] [Google Scholar]
- 7.Kornbluth A, Sachar DB. Practice Parameters Committee of the American College of Gastroenterology. Ulcerative colitis practice guidelines in adults: American College of Gastroenterology, Practice Parameters Committee. Am J Gastroenterol. 2010;105:501–523, quiz 524. [DOI] [PubMed] [Google Scholar]
- 8.Lichtenstein GR, Hanauer SB, Sandborn WJ; Practice Parameters Committee of American College of Gastroenterology. Management of Crohn’s disease in adults. Am J Gastroenterol. 2009;104:465–483, quiz 464-484. [DOI] [PubMed] [Google Scholar]
- 9.Broome U, Bergquist A. Primary sclerosing cholangitis, inflammatory bowel disease, and colon cancer. Semin Liver Dis. 2006;26:31–41. [DOI] [PubMed] [Google Scholar]
- 10.Wanderas MH, Moum BA, Hoivik ML, Hovde O. Predictive factors for a severe clinical course in ulcerative colitis: results from population-based studies. World J Gastrointest Pharmacol Ther. 2016;7:235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sanchez- Muñoz F. Role of cytokines in inflammatory bowel disease. World J Gastroenterol. 2008;14:4280–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen ML, Sundrud MS. Cytokine networks and T-Cell subsets in inflammatory bowel diseases. Inflamm Bowel Dis. 2016;22:1157–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sands BE, Kaplan GG. The role of TNFalpha in ulcerative colitis. J Clin Pharmacol. 2007;47:930–941. [DOI] [PubMed] [Google Scholar]
- 14.Del Zotto B, Mumolo G, Pronio AM, Montesani C, Tersigni R, Boirivant M. TGF-beta1 production in inflammatory bowel disease: differing production patterns in Crohn’s disease and ulcerative colitis. Clin Exp Immunol. 2003;134:120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reimund JM, Wittersheim C, Dumont S, et al. Mucosal inflammatory cytokine production by intestinal biopsies in patients with ulcerative colitis and Crohn’s disease. J Clin Immunol. 1996;16:144–150. [DOI] [PubMed] [Google Scholar]
- 16.Rogler G, Andus T. Cytokines in inflammatory bowel disease. World J Surg. 1998;22:382–389. [DOI] [PubMed] [Google Scholar]
- 17.Laukoetter MG, Nava P, Nusrat A. Role of the intestinal barrier in inflammatory bowel disease. World J Gastroenterol. 2008;14:401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tysk C, Lindberg E, Jarnerot G, Floderus-Myrhed B. Ulcerative colitis and Crohn’s disease in an unselected population of monozygotic and dizygotic twins. Gut. 1988;29:990–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson CA, Boucher G, Lees CW, et al. Meta-analysis identifies 29 additional ulcerative colitis risk loci, increasing the number of confirmed associations to 47. Nat Genet. 2011;43:246–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steinberg MW, Turovskaya O, Shaikh RB, et al. A crucial role for HVEM and BTLA in preventing intestinal inflammation. J Exp Med. 2008;205:1463–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maerten P, Kwon BS, Shen C, et al. Involvement of 4-1BB (CD137)-4-1BBligand interaction in the modulation of CD4 T cell-mediated inflammatory colitis. Clin Exp Immunol. 2006;143:228–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahida YR, Wu K, Jewell DP. Enhanced production of interleukin 1-beta by mononuclear cells isolated from mucosa with active ulcerative colitis of Crohn’s disease. Gut. 1989;30:835–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams EJ, Haque S, Banks C, Johnson P, Sarsfield P, Sheron N. Distribution of the interleukin-8 receptors, CXCR1 and CXCR2, in inflamed gut tissue. J Pathol. 2000;192:533–539. [DOI] [PubMed] [Google Scholar]
- 24.Schluns KS, Kieper WC, Jameson SC, Lefrançois L. Interleukin-7 mediates the homeostasis of naïve and memory CD8 T cells in vivo. Nat Immunol. 2000;1:426–432. [DOI] [PubMed] [Google Scholar]
- 25.Koren I, Reem E, Kimchi A. DAP1, a novel substrate of mTOR, negatively regulates autophagy. Curr Biol. 2010;20:1093–1098. doi: 10.1016/j.cub.2010.04.041. [DOI] [PubMed] [Google Scholar]
- 26.Pandey AK, Yang Y, Jiang Z, et al. NOD2, RIP2 and IRF5 play a critical role in the type I interferon response to Mycobacterium tuberculosis. PLoS Pathog. 2009;5:e1000500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabath E, Negoro H, Beaudry S, et al. Galpha12 regulates protein interactions within the MDCK cell tight junction and inhibits tight-junction assembly. J Cell Sci. 2008;121:814–824. [DOI] [PubMed] [Google Scholar]
- 28.Liu L, Cara DC, Kaur J, et al. LSP1 is an endothelial gatekeeper of leukocyte transendothelial migration. J Exp Med. 2005;201:409–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frolkis A, Dieleman LA, Barkema HW, et al. Environment and the inflammatory bowel diseases. Can J Gastroenterol. 2013;27:e18–e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Richardson CE, Morgan JM, Jasani B, et al. Effect of smoking and transdermal nicotine on colonic nicotinic acetylcholine receptors in ulcerative colitis. QJM. 2003;96:57–65. [DOI] [PubMed] [Google Scholar]
- 31.Razani-Boroujerdi S, Boyd RT, Davila-Garcia MI, et al. T cells express alpha7-nicotinic acetylcholine receptor subunits that require a functional TCR and leukocyte-specific protein tyrosine kinase for nicotine-induced Ca2+ response. J Immunol. 2007;179:2889–2898. [DOI] [PubMed] [Google Scholar]
- 32.Hunter MM, McKay DM. Review article: helminths as therapeutic agents for inflammatory bowel disease. Aliment Pharmacol Ther. 2004;19:167–177. [DOI] [PubMed] [Google Scholar]
- 33.Luther J, Dave M, Higgins PDR, Kao JY. Association between Helicobacter pylori infection and inflammatory bowel disease: a meta-analysis and systematic review of the literature. Inflamm Bowel Dis. 2010;16:1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Berg DJ, Zhang J, Weinstock JV, et al. Rapid development of colitis in NSAID-treated IL-10-deficient mice. Gastroenterology. 2002;123:1527–1542. [DOI] [PubMed] [Google Scholar]
- 35.Cutolo M, Capellino S, Sulli A, et al. Estrogens and autoimmune diseases. Ann N Y Acad Sci. 2006;1089:538–547. [DOI] [PubMed] [Google Scholar]
- 36.Shaw SY, Blanchard JF, Bernstein CN. Association between the use of antibiotics and new diagnoses of Crohn’s disease and ulcerative colitis. Am J Gastroenterol. 2011;106:2133–2142. [DOI] [PubMed] [Google Scholar]
- 37.Brock JH. The physiology of lactoferrin. Biochem Cell Biol. 2002;80:1–6. [DOI] [PubMed] [Google Scholar]
- 38.Kaplan GG, Hubbard J, Korzenik J, et al. The inflammatory bowel diseases and ambient air pollution: a novel association. Am J Gastroenterol. 2010;105:2412–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449:811–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harig JM, Soergel KH, Komorowski RA, Wood CM. Treatment of diversion colitis with short-chain-fatty acid irrigation. N Engl J Med. 1989;320:23–28. [DOI] [PubMed] [Google Scholar]
- 41.Thibault R, Blachier F, Darcy-Vrillon B, de Coppet P, Bourreille A, Segain JP. Butyrate utilization by the colonic mucosa in inflammatory bowel diseases: a transport deficiency. Inflamm Bowel Dis. 2010;16:684–695. [DOI] [PubMed] [Google Scholar]
- 42.Roberts AK, Chierici R, Sawatzki G, Hill MJ, Volpato S, Vigi V. Supplementation of an adapted formula with bovine lactoferrin: 1. Acta Paediatr. 1992;81:119–124. [DOI] [PubMed] [Google Scholar]
- 43.Hold GL, Pryde SE, Russell VJ, Furrie E, Flint HJ. Assessment of microbial diversity in human colonic samples by 16S rDNA sequence analysis. FEMS Microbiol Ecol. 2002;39:33–39. [DOI] [PubMed] [Google Scholar]
- 44.Pryde SE, Duncan SH, Hold GL, Stewart CS, Flint HJ. The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett. 2002;217:133–139. [DOI] [PubMed] [Google Scholar]
- 45.Machiels K, Joossens M, Sabino J, et al. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut. 2014;63:1275–1283. [DOI] [PubMed] [Google Scholar]
- 46.Pozuelo M, Panda S, Santiago A, et al. Reduction of butyrate- and methane-producing microorganisms in patients with irritable bowel syndrome. Sci Rep. 2015;5:12693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Christl SU, Eisner HD, Dusel G, Kasper H, Scheppach W. Antagonistic effects of sulfide and butyrate on proliferation of colonic mucosa: a potential role for these agents in the pathogenesis of ulcerative colitis. Dig Dis Sci. 1996;41:2477–2481. [DOI] [PubMed] [Google Scholar]
- 48.Jantchou P, Morois S, Clavel-Chapelon F, Boutron-Ruault M-C, Carbonnel F. Animal protein intake and risk of inflammatory bowel disease: the E3N prospective study. Am J Gastroenterol. 2010;105:2195–2201. [DOI] [PubMed] [Google Scholar]
- 49.van Liere MJ, Lucas F, Clavel F, Slimani N, Villeminot S. Relative validity and reproducibility of a French dietary history questionnaire. Int J Epidemiol. 1997;26:S128–S136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Andersen V, Olsen A, Carbonnel F, Tjønneland A, Vogel U. Diet and risk of inflammatory bowel disease. Dig Liver Dis. 2012;44:185–194. [DOI] [PubMed] [Google Scholar]
- 51.Geerling BJ, Dagnelie PC, Badart-Smook A, Russel MG, Stockbrugger RW, Brummer RJ. Diet as a risk factor for the development of ulcerative colitis. Am J Gastroenterol. 2000;95:1008–1013. [DOI] [PubMed] [Google Scholar]
- 52.Tragnone A, Valpiani D, Miglio F, et al. Dietary habits as risk factors for inflammatory bowel disease. Eur J Gastroenterol Hepatol. 1995;7:47–51. [PubMed] [Google Scholar]
- 53.Reif S, Klein I, Lubin F, Farbstein M, Hallak A, Gilat T. Pre-illness dietary factors in inflammatory bowel disease. Gut. 1997;40:754–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Russel MG, Engels LG, Muris JW, et al. Modern life’ in the epidemiology of inflammatory bowel disease: a case-control study with special emphasis on nutritional factors. Eur J Gastroenterol Hepatol. 1998;10:243–249. [DOI] [PubMed] [Google Scholar]
- 55.Persson PG, Ahlbom A, Hellers G. Diet and inflammatory bowel disease: a case-control study. Epidemiology. 1992;3:47–52. [DOI] [PubMed] [Google Scholar]
- 56.Jowett SL, Seal CJ, Pearce MS, et al. Influence of dietary factors on the clinical course of ulcerative colitis: a prospective cohort study. Gut. 2004;53:1479–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bilski J, Mazur-Bialy A, Magierowski M, et al. Exploiting significance of physical exercise in prevention of gastrointestinal disorders. Curr Pharm Des. 2018;24:1916–1925. [DOI] [PubMed] [Google Scholar]
- 58.So B, Kim H-J, Kim J, Song W. Exercise-induced myokines in health and metabolic diseases. Integrat Med Res. 2014;3:172–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sonnenberg A. Occupational distribution of inflammatory bowel disease among German employees. Gut. 1990;31:1037–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klein I, Reif S, Farbstein H, Halak A, Gilat T. Preillness non dietary factors and habits in inflammatory bowel disease. Ital J Gastroenterol Hepatol. 1998;30:247–251. [PubMed] [Google Scholar]
- 61.Hlavaty T, Toth J, Koller T, et al. Smoking, breastfeeding, physical inactivity, contact with animals, and size of the family influence the risk of inflammatory bowel disease: a Slovak case-control study. United European Gastroenterol J. 2013;1:109–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Persson PG, Leijonmarck CE, Bernell O, Hellers G, Ahlbom A. Risk indicators for inflammatory bowel disease. Int J Epidemiol. 1993;22:268–272. [DOI] [PubMed] [Google Scholar]
- 63.Boggild H, Tuchsen F, Orhede E. Occupation, employment status and chronic inflammatory bowel disease in Denmark. Int J Epidemiol. 1996;25:630–637. [DOI] [PubMed] [Google Scholar]
- 64.Elsenbruch S, Langhorst J, Popkirowa K, et al. Effects of mind-body therapy on quality of life and neuroendocrine and cellular immune functions in patients with ulcerative colitis. Psychother Psychosom. 2005;74:277–287. [DOI] [PubMed] [Google Scholar]
- 65.Gatt K, Schembri J, Katsanos KH, et al. Inflammatory bowel disease and physical activity: a study on the impact of diagnosis on the level of exercise amongst patients with IBD [published online ahead of print December 8, 2018]. J Crohns Colitis. doi: 10.1093/ecco-jcc/jjy214. [DOI] [PubMed] [Google Scholar]
- 66.Engels M, Cross RK, Long MD. Exercise in patients with inflammatory bowel diseases: current perspectives. Clin Exp Gastroenterol. 2018;11:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cook MD, Allen JM, Pence BD, et al. Exercise and gut immune function: evidence of alterations in colon immune cell homeostasis and microbiome characteristics with exercise training. Immunol Cell Biol. 2016;94:158–163. [DOI] [PubMed] [Google Scholar]
- 68.Cook MD, Martin SA, Williams C, et al. Forced treadmill exercise training exacerbates inflammation and causes mortality while voluntary wheel training is protective in a mouse model of colitis. Brain Behav Immun. 2013;33:46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hoffman-Goetz L, Pervaiz N, Packer N, Guan J. Freewheel training decreases pro- and increases anti-inflammatory cytokine expression in mouse intestinal lymphocytes. Brain Behav Immun. 2010;24:1105–1115. [DOI] [PubMed] [Google Scholar]
- 70.Luo B, Xiang D, Nieman DC, Chen P. The effects of moderate exercise on chronic stress-induced intestinal barrier dysfunction and antimicrobial defense. Brain Behav Immun. 2014;39:99–106. [DOI] [PubMed] [Google Scholar]
- 71.Mazur-Bialy AI, Bilski J, Wojcik D, et al. Beneficial effect of voluntary exercise on experimental colitis in mice fed a high-fat diet: the role of irisin, adiponectin and proinflammatory biomarkers. Nutrients. 2017;9:E410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu WX, Zhou F, Wang Y, et al. Voluntary exercise protects against ulcerative colitis by up-regulating glucocorticoid-mediated PPAR-γ activity in the colon in mice. Acta Physiol. 2015;215:24–36. [DOI] [PubMed] [Google Scholar]
- 73.Bilski J, Brzozowski B, Mazur-Bialy A, Sliwowski Z, Brzozowski T. The role of physical exercise in inflammatory bowel disease. Biomed Res Int. 2014;2014:429031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Monda V, Villano I, Messina A, et al. Exercise modifies the gut microbiota with positive health effects. Oxid Med Cell Longev. 2017;2017:3831972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gleeson M, Williams C. Intense exercise training and immune function. Nestle Nutr Inst Workshop Ser. 2013;76:39–50. [DOI] [PubMed] [Google Scholar]
- 76.Brolinson PG, Elliott D. Exercise and the immune system. Clin Sports Med. 2007;26:311–319. [DOI] [PubMed] [Google Scholar]
- 77.Mangan PR, Harrington LE, O’Quinn DB, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441:231–234. [DOI] [PubMed] [Google Scholar]
- 78.Gibson DJ, Ryan EJ, Doherty GA. Keeping the bowel regular: the emerging role of Treg as a therapeutic target in inflammatory bowel disease. Inflamm Bowel Dis. 2013;19:2716–2724. [DOI] [PubMed] [Google Scholar]
- 79.Yamagata T, Skepner J, Yang J. Targeting Th17 effector cytokines for the treatment of autoimmune diseases. Arch Immunol Ther Exp. 2015;63:405–414. [DOI] [PubMed] [Google Scholar]
- 80.Zheng Y, Rudensky AY. Foxp3 in control of the regulatory T cell lineage. Nat Immunol. 2007;8:457–462. [DOI] [PubMed] [Google Scholar]
- 81.Glocker E-O, Kotlarz D, Boztug K, et al. Inflammatory bowel disease and mutations affecting the interleukin-10 receptor. N Engl J Med. 2009;361:2033–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fantini MC, Rizzo A, Fina D, et al. Smad7 controls resistance of colitogenic T cells to regulatory T cell-mediated suppression. Gastroenterology. 2009;136:1308–13016, e1-e3. [DOI] [PubMed] [Google Scholar]
- 83.Bettelli E, Carrier Y, Gao W, et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. [DOI] [PubMed] [Google Scholar]
- 84.Berni Canani R, Di Costanzo M, Leone L. The epigenetic effects of butyrate: potential therapeutic implications for clinical practice. Clin Epigenetics. 2012;4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hallert C, Bjorck I, Nyman M, Pousette A, Granno C, Svensson H. Increasing fecal butyrate in ulcerative colitis patients by diet: controlled pilot study. Inflamm Bowel Dis. 2003;9:116–121. [DOI] [PubMed] [Google Scholar]
- 86.Zhang M, Zhou Q, Dorfman RG, et al. Butyrate inhibits interleukin-17 and generates Tregs to ameliorate colorectal colitis in rats. BMC Gastroenterol. 2016;16:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Qiu X, Zhang M, Yang X, Hong N, Yu C. Faecalibacterium prausnitzii upregulates regulatory T cells and anti-inflammatory cytokines in treating TNBS-induced colitis. J Crohns Colitis. 2013;7:e558–e568. [DOI] [PubMed] [Google Scholar]
- 88.Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature. 2013;504:446–450. [DOI] [PubMed] [Google Scholar]
- 89.Arpaia N, Campbell C, Fan X, et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013;504:451–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scheppach W, Sommer H, Kirchner T, et al. Effect of butyrate enemas on the colonic mucosa in distal ulcerative colitis. Gastroenterology. 1992;103:51–56. [DOI] [PubMed] [Google Scholar]
- 91.Vernia P, Cittadini M, Caprilli R, Torsoli A. Topical treatment of refractory distal ulcerative colitis with 5-ASA and sodium butyrate. Dig Dis Sci. 1995;40:305–307. [DOI] [PubMed] [Google Scholar]
- 92.Chapman MA, Grahn MF, Boyle MA, Hutton M, Rogers J, Williams NS. Butyrate oxidation is impaired in the colonic mucosa of sufferers of quiescent ulcerative colitis. Gut. 1994;35:73–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Luhrs H, Gerke T, Muller JG, et al. Butyrate inhibits NF-kappaB activation in lamina propria macrophages of patients with ulcerative colitis. Scand J Gastroenterol. 2002;37:458–466. [DOI] [PubMed] [Google Scholar]
- 94.Butzner JD, Parmar R, Bell CJ, Dalal V. Butyrate enema therapy stimulates mucosal repair in experimental colitis in the rat. Gut. 1996;38:568–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Steinhart AH, Hiruki T, Brzezinski A, Baker JP. Treatment of left-sided ulcerative colitis with butyrate enemas: a controlled trial. Aliment Pharmacol Ther. 1996;10:729–736. [DOI] [PubMed] [Google Scholar]
- 96.Vieira ELM, Leonel AJ, Sad AP, et al. Oral administration of sodium butyrate attenuates inflammation and mucosal lesion in experimental acute ulcerative colitis. J Nutr Biochem. 2012;23:430–436. [DOI] [PubMed] [Google Scholar]
- 97.Vernia P, Monteleone G, Grandinetti G, et al. Combined oral sodium butyrate and mesalazine treatment compared to oral mesalazine alone in ulcerative colitis: randomized, double-blind, placebo-controlled pilot study. Dig Dis Sci. 2000;45:976–981. [DOI] [PubMed] [Google Scholar]
- 98.Allen JM, Mailing LJ, Cohrs J, et al. Exercise training-induced modification of the gut microbiota persists after microbiota colonization and attenuates the response to chemically-induced colitis in gnotobiotic mice. Gut Microbes. 2018;9:115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chiba M, Nakane K, Komatsu M. Westernized diet is the most ubiquitous environmental factor in inflammatory bowel disease. Perm J. 2019;23:18–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Chiba M, Nakane K, Komatsu M. Lifestyle medicine in inflammatory bowel disease. Perm J. 2018;22https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6054412/. [DOI] [PMC free article] [PubMed] [Google Scholar]