Cyclic Polyhydroxy Ketones. I. Oxidation Products of Hexahydroxybenzene (Benzenehexol) (original) (raw)
Abstract
Reliable procedures are given for the preparation and purification of hexahydroxybenzene (benzenehexol), tetrahydroxy-_p_-benzoquinone, rhodizonic acid, triquinoyl (cyclohexanehexone), croconic acid, and leuconic acid (cyclopentanepentone). Certain derivatives and color tests, as well as infrared and ultraviolet spectra, are reported for their identification.
1. Introduction
On oxidation, cyclohexanehexols (inositols) yield keto derivatives which, by successive enolization and elimination reactions, can be transformed to polyhydroxybenzenes [1-5].1 In progressing from cyclohexane derivatives through cyclic olefinic intermediates to aromatic compounds, reactions and materials are encountered which are eminently suited for the study of reaction mechanisms and basic concepts of theoretical organic chemistry [6].
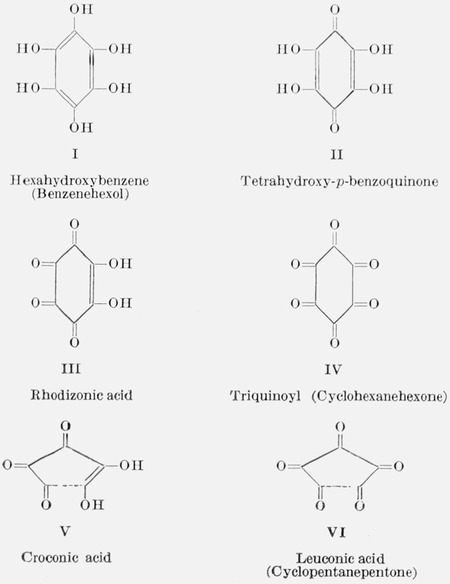
Hexahydroxybenzene (benzeneliexol, I) can be prepared from inositols (cyclohexanehexols) and from tetrahydroxy-_p_-benzoquinone (II, obtained by self-condensation of glyoxal in alkaline solution). Oxidation of I yields II, which is further oxidized to rhodizonic acid (III), and this is oxidized to triquinoyl (cyclohexanehexone, IV). Tetrahydroxy-_p_-benzoquinone and rhodizonic acid are not only interesting in their own right, but also serve as intermediates in the preparation of croconic acid (V), a dihydroxy triketone having a five-carbon ring. Oxidation of croconic acid yields leuconic acid (VI). Because these compounds, needed as model substrates in a study of antioxidant and oxygen-carrier properties, were not available in a suitable state of purity, the methods reported in the experimental part were developed for their synthesis and purification.
2. Hexahydroxybenzene (Benzenehexol, I) [7]
2.1. Discussion
Hexahydroxybenzene (I), the enolic form of the trihydroxytriketocyclohexanes, can be prepared by the self-condensation of potassium carbonyl followed by hydrolysis [8, 9], by nitration of di-_O_-acetylhydroquinone followed by reduction and hydrolysis [10], by oxidation of _myo_-inositol followed by enolization [11], and by reduction of tetrahydroxy-_p_-benzoquinone (II) by means of stannous chloride [12].
2.2. Preparation of Hexahydroxybenzene [13]2
Stannous chloride dihydrate (100 g, 0.44 mole) is added in one portion to a boiling, mechanically stirred solution of 10 g (0.058 mole) of tetrahydroxy-p_-benzoquinone (II) in 200 ml of 2.5_-N hydro-chloric acid. In a few minutes, the red color of II disappears and long, needle-like crystals of hexahydroxybenzene (I) begin to form. Concentrated hydrochloric acid (250 ml; specific gravity 1.08) is added, and the stirred suspension is heated to boiling, mixed with 600 ml of concentrated hydrochloric acid, cooled in ice, and filtered with suction on a fritted-glass filter. As much solvent as possible is removed by suction, without exposure to air, by covering the filter with an inverted funnel and introducing nitrogen, and by using a polyethylene dam to remove the final traces of the solvent.
For purification, the crude product is dissolved in 450 ml of hot 2.5-N hydrochloric acid containing 3 g of stannous chloride dihydrate. Decolorizing carbon is added, the hot suspension is filtered, the insoluble matter is washed with 75 ml of hot water, and the combined filtrate and washings are mixed with 1 liter of concentrated hydrochloric acid and cooled in ice. The resulting crystals are collected on a fritted-glass filter, in an atmosphere of nitrogen, washed with 100 ml of a cold, 1:1 mixture of ethanol and concentrated hydrochloric acid, and dried over sodium hydroxide in a vacuum desiccator to give 7 to 8 g (about 75%) of colorless crystals which do not melt3 when placed on a hot plate at 310° C. Infrared and ultraviolet absorption spectra are recorded on pages 158 and 160.
Hexahydroxybenzene can also be recrystallized from 2-methoxyethanol by the addition of benzene; the crystals are separated, washed with acetone, and dried in a vacuum desiccator. The yield is about the same as in the previous method.
Characteristic derivatives of hexahydroxybenzene are the hexaacetate, mp 203 to 205 °C [10, 15], the hexabenzoate, mp 323 to 325 °C [9], and the hexarnethyl ether, mp 81 °C [16].
The following convenient color tests may be used for identification of hexahydroxybenzene:
- (Developed in this laboratory.) A solution of ferric chloride in 1:1 aqueous methanol is slowly added dropwise to a stirred, aqueous solution of hexahydroxybenzene under a stream of nitrogen. As addition of the ferric chloride proceeds, the color of the solution passes successively through the following characteristic series, representing oxidation steps: magenta or cherry red (II); green (III); and, finally, a stable, deep blue (presumably a mixture of III and IV). When the solution is cooled in ice for several hours, colorless crystals of the octahydrate of IV gradually separate.
- [9] One ml of a concentrated, aqueous solution of barium chloride is mixed with 0.5 ml of a dilute, aqueous solution of ferric chloride containing one or two drops of acetic acid (or of dilute hydrochloric acid). To the mixed solution are added one or two drops of an aqueous solution of hexahydroxybenzene. A bright-red precipitate of barium rhodizonate results.
- [9] One ml of a dilute, aqueous solution of hexahydroxybenzene is mixed with 1 ml of a saturated, aqueous solution of magnesium nitrate, and a few drops of dilute ammonia are added. A carminered precipitate results.
3. Tetrahydroxy-_p_-benzoquinone (II) [17]
3.1. Discussion
The preparation of tetrahydroxy-_p_-benzoquinone is based on the self-condensation of glyoxal in the presence of sodium sulfite, air, and a base. The procedure was originated by Homolka [15], and has been studied and modified by subsequent workers [18, 19, 20]. Presumably, hexahydroxybenzene is formed initially; this is oxidized to tetrahydroxy-_p_-benzoquinone, and this, in turn, to rhodizonic acid. As shown by Fatiadi and Sager [13], when sodium bicarbonate is used as a buffer, the disodium salt of tetrahydroxy-_p_-benzoquinone is formed in a yield of 8 to 11 percent. When the reaction mixture is highly alkaline, substantial quantities of disodium rhodizonate and disodium croconate are formed, in addition to the disodium salt of tetrahydroxy-_p_-benzoquinone.
3.2. Preparation of the Disodium Salt of Tetrahydroxy-_p_-benzoquinone [13]
A solution of 400 g (3.17 moles) of anhydrous sodium sulfite and 150 g (1.79 moles) of sodium bicarbonate in 3 liters of water is placed in a 5-liter, three-necked flask fitted with a thermometer, an air inlet tube (1-cm diam), and a tube connected to a water aspirator. The solution is heated to 40 to 45 °C, and 600 g of a 30-percent, aqueous solution of glyoxal (3.1 moles) is added. A brisk stream of air is drawn through the solution for 1 hr without heating, after which the mixture is gradually heated to 90 °C. Aeration is then stopped, and the mixture is heated to incipient boiling and cooled to room temperature. The resulting, crystalline disodium salt of II is removed by filtration, washed successively with 50 ml of cold, 50-percent, aqueous methanol and 50 ml of methanol, and dried in a vacuum desiccator. The yield (22 to 24 g) corresponds to about 10 percent of the theoretical, based on the glyoxal used. The product can be used without purification for the preparation of tetrahydroxybenzene, rhodizonic acid, or croconic acid. It is sensitive to oxidation by air and, if stored in a loosely stoppered bottle, is completely oxidized to disodium rhodizonate in several months. Tetrahydroxy-_p_-benzoquinone is, however, relatively stable in air.
3.3. Preparation of Tetrahydroxy-_p_-benzoquinone
The crude disodium salt of II (20 g) is dissolved in 220 ml of 2.5-N hydrochloric acid by heating. On cooling, the solution yields glistening, black crystals of tetrahydroxy-p_-benzoquinone which are collected by filtration, washed with ice water, and dried in a vacuum desiccator; wt, 12 to 15 g. The product is recrystallized from warm, 2_-N hydrochloric acid to give lustrous, dark crystals [8]; alternatively, it may be recrystallized from acetone by the addition of petroleum ether to give small, dark crystals. Spectrograms are given on pages 158 and 160.
4. Rhodizonic Acid (III) [21]
4.1. Discussion
Heretofore, rhodizonic acid (III), the oxidation product of II, has been prepared by oxidation of _myo-_inositol with nitric acid [11], by reduction of IV with sulfurous acid [10, 16, 20], and by synthesis from glyoxal [15]. Described below are the preparation of ammonium rhodizonate [21] from glyoxal, and of rhodizonic acid from the disodium salt of II by oxidation in air.
4.2. Preparation of Tetrahydroxy-_p_-benzoquinone and Ammonium Rhodizonate from Glyoxal
Aqueous glyoxal (600 g of a 30-percent solution) is self-condensed in the presence of sodium sulfite, oxygen, and sodium bicarbonate as described in section 3.2. After 1 hr of aeration, 100 g of sodium hydroxide is added. The alkaline solution (p_H 10) is heated, with aeration, to incipient boiling, and cooled to room temperature. The mixture of crystalline sodium salts of II and III is filtered off and washed successively with 50 ml of cold, 50-percent aqueous methanol and 50 ml of methanol. The air-dried salts (24 to 26 g) are dissolved in 200 ml of 2.5_-N hydrochloric acid by boiling, and the solution is immediately cooled in ice.4 The resulting crystalline II (8 to 9 g) is separated by filtration, washed with 20 ml of ice water, and dried.
The ice-cold, dark-red filtrate is brought to _p_H 5.5 to 6.5 with ammonia. The resulting, dark-blue, crystalline ammonium rhodizonate is separated by filtration, washed with 25 ml of ice water, and air-dried; wt, 7 to 8 g. It is recrystallized from 750 ml of hot water.
4.3. Preparation of Rhodizonic Acid from the Disodium Salt of II
The disodium salt of II (40 g), prepared as described in section 3.2, is placed in a large, flat-bottomed dish and heated in an oven at 170 to 180 °C for 24 hr. The residue is suspended, with mechanical stirring, in 200 ml of warm, 2.5-N hydrochloric acid, and after several minutes, 1 g of decolorizing carbon is added; the solution is cooled in ice and filtered through a filter paper precoated with paper pulp. The brownish-yellow solution is concentrated under diminished pressure at 35 °C to a semicrystalline mass, and the aqueous hydrochloric acid is removed by successive addition and evaporation of three 50-ml portions of a 1:1 mixture of benzene and acetone. The residue is triturated with 50 ml of acetone, the suspension is filtered, and the insoluble product is washed with sufficient cold acetone to remove all colored impurities. The crystalline residue, consisting of sodium chloride and crude rhodizonic acid dihydrate, is air-dried and pulverized, and the powder is added to 200 ml of dioxane pre-heated to 80 °C. The suspension is stirred for 5 min and then filtered through a filter paper precoated with decolorizing carbon. The insoluble matter is washed with 50 ml of warm dioxane, and the slightly colored filtrate is cooled to about 20 °C. Pentane (about 150 ml) is added to incipient turbidity, and crystallization of rhodizonic acid dihydrate is induced by scratching the inside walls of the container with a glass rod. The mixture is cooled in ice for about 30 min, the suspension is filtered, and the crystals are washed with 50 ml of a cold, 1:1 mixture of dioxane and pentane, and air-dried; wt, 25 to 35 g.
The crude rhodizonic acid dihydrate is recrystallized from warm dioxane by the addition of pentane and ether; by concentration of the filtrate, an additional crop is obtained. The air-dried product, which still contains dioxane, is dried for 48 hr in a vacuum desiccator over sulfuric acid; total yield, about 21 g (60%) of colorless crystals. Measurements of infrared and ultraviolet absorption are reported on pages 158 and 160.
The purity of the product may be determined by titration with iodine [20]; 155 mg of rhodizonic acid dihydrate consume 14.97 ml of 0.1-N I3− solution; found, 15.07 ml.
If subjected to a high vacuum (0.1 mm) at room temperature the compound is converted into the scarlet-red crystals of anhydrous rhodizonic acid in as little as 5 min. The same transformation can be accomplished at higher temperatures by vacuum sublimation [20]. Rhodizonic acid gives a colorless solution in 2.5-N hydrochloric acid, whereas tetrahydroxy-_p_-benzoquinone gives a cherry-red solution.
5. Triquinoyl (Cyclohexαnehexone, IV) [23]
5.1. Discussion
In 1862, Lerch [24] obtained a product, now known as triquinoyl, by oxidation of either hexahydroxybenzene (I) or tetrahydroxy-_p_-benzoquinone (II) with nitric acid or chlorine. The substance has been characterized by Nietzki and coworkers [8, 10, 25], and studied by Henle [26], Bergel [27], and Eistert and coworkers [20, 22]. The procedure described here is based on the original work of Lerch and of Nietzki. Because some workers have experienced difficulty in the preparation and purification of IV, the procedures are given in detail.
5.2. Preparation of Triquinoyl Octahydrate
Recrystallized tetrahydroxy-_p_-benzoquinone (10 g) is added in small increments during 5 min to a mechanically stirred solution of 100 ml of concentraten nitric acid and 25 ml of water kept at 10 °C. The reaction is continued for an additional 5 min, 25 ml of water is added, and the mixture is cooled in ice for 15 min. The crystalline triquinoyl octahydrate that separates is filtered off, washed successively with 20 ml of ice water and 30 ml of an ice-cold, 1:1 mixture of acetone and ether, and air-dried; wt, 12 to 12.5 g; mp 96 to 97 °C.
The substance is recrystallized in the following manner: The octahydrate (5 g) is suspended in 50 ml of methanol containing 3 ml of concentrated nitric acid, and the suspension is heated to 55 to 60 °C, with stirring. When dissolution is complete, about 200 mg of decolorizing carbon is added, the suspension is filtered, and the insoluble matter is washed with 25 ml of water. The combined filtrate and washings are evaporated under diminished pressure to about 25 ml, and the solution is cooled in ice until crystallization begins, and then gradually diluted with 20 ml of acetone. The crystals are collected on a filter, washed successively with 10 ml of cold 1:1 acetone-water mixture and 20 ml of cold acetone, and air-dried; wt, 2.1 g. By concentration of the mother liquor, additional material (0.6 g) is obtained. The octahydrate consists of colorless, lustrous plates or prisms, mp 99 to 100 °C (decomposition). Infrared and ultraviolet absorption spectra are recorded on pages 159 and 161.
A characteristic color test [26] for IV involves heating the compound with a saturated, aqueous solution of barium chloride; bright-red barium rhodizonate is precipitated, following disproportionation [8, 22].
6. Croconic Acid (V) [28]
6.1. Discussion
Croconic acid (V) is one of the first enediolic acids to have been reported. Since its discovery by Gmelin [29] in 1825, it has engaged the attention of many workers [10, 30–39]. As pointed out by Hirata and coworkers [36], the anion has an unusual resonance structure. Croconic acid is formed from a variety of polyhydroxybenzene derivatives by oxidation followed by a rearrangement of the benzylic acid type. It can be efficiently prepared by oxidation of tetrahydroxy-_p_-benzoquinone or rhodizonic acid with manganese dioxide. In the process, the benzene ring is degraded, with elimination of carbon dioxide. The reaction was discovered by Nietzki [32] and has since been used by many workers. The procedure given here resembles one described by Yamada and Hirata [40], and differs from prior methods in that it starts with the crude mixture of sodium salts obtained from the condensation of glyoxal (instead of with pure sodium rhodizonate) and uses a special type [41] of active manganese dioxide as the oxidant.
6.2. Preparation of Active Manganese Dioxide
Commercial manganese carbonate (500 g) in a flat-bottomed tray or dish is heated in an oven at 295 to 310°C for 12 to 18 hr. After being cooled, the product is treated with sufficient aqueous nitric acid (15 to 20 ml of concentrated acid per 100 ml of solution) to decompose any residual carbonate and to give a strongly acid solution. After 30 to 45 min, the manganese dioxide is filtered off, thoroughly washed with water, and dried at 150 to 160 °C for 18 to 24 hr. After its recovery from reaction mixtures, active manganese dioxide can be used repeatedly if it is redried at 150 to 160 °C for 18 to 24 hr. Manganese dioxide prepared by other methods proved to be less satisfactory.
6.3. Barium Croconate Hydrate
To a solution of 40 g of sodium hydroxide in 1.2 liters of water are added, with stirring, 21 g (0.1 mole) of crude disodium tetrahydroxy-_p_-benzoquinone (see section 3.2) and 55 g of active manganese dioxide. After 5 min, the mixture is heated to boiling, refluxed for 45 min, and filtered, and the manganese dioxide is washed with 800 ml of hot water. The combined filtrate and washings are treated with about 210 ml of concentrated hydrochloric acid in portions, giving a bright-yellow solution. To this is added a hot solution of 50 g of barium chloride dihydrate in 150 ml of water, with stirring; the mixture is heated to 85 to 90 °C, cooled, and kept at room temperature. The resulting, lustrous, yellow plates of barium croconate monohydrate are collected on a filter, washed successively with water and ethanol, and air-dried; wt, 22 to 23 g.
6.4. Conversion of Barium Croconate to Sodium Croconate
Barium croconate (17.5 g; 0.06 mole) is added in small portions, with stirring, to 150 ml of a 10-percent aqueous solution of anhydrous sodium carbonate. The mixture is boiled for 5 min and filtered; the barium carbonate is washed with 30 ml of hot water, and the combined yellow filtrate and washings are diluted with 20 ml of glacial acetic acid, heated to boiling, and cooled in ice. Disodium croconate trihydrate separates in long, yellow crystals, which are collected on a filter, washed with 40 ml of ice water, and air-dried; wt, about 10 g. A second crop (2.5 to 3.5 g) is obtained by adding about 200 ml of 95-percent ethanol to the mother liquor and cooling the solution.
Dipotassium croconate may be prepared similarly.
6.5. Croconic Acid Trihydrate
Barium croconate (55 g) is added in small portions to 200 ml of mechanically stirred, aqueous sulfuric acid (containing 20 ml of concentrated acid) at 55 to 60 °C; stirring is continued for 30 to 45 min. The barium sulfate is separated by filtration, washed with 25 ml of hot water, and discarded. The combined filtrate and washings are evaporated to dryness under diminished pressure, the residue is dissolved in a hot solution of 25 ml of absolute ethanol and 150 ml of dioxane, and the solution is treated with a small quantity of decolorizing carbon and filtered. The filtrate is concentrated under diminished pressure at 40 °C until crystallization begins, and the mixture is diluted with benzene to incipient turbidityand cooled. After several hours, the resulting crystals are separated by filtration, washed with benzene, and dried in a vacuum desiccator. By concentration of the mother liquor, additional crystals are obtained; the combined yield of croconic acid trihydrate is about 20 g. Infrared and ultraviolet absorption spectra are recorded on pages 159 and 161.
Anliydrous croconic acid is prepared by heating the trihydrate at 120 °C for 2 to 4 hrs; it slowly decomposes above 150 °C. Croconic acid may be characterized as the dimethyl ether, mp 113 °C [35].
Small quantities of croconic acid may be prepared by passing a solution of 5 g of disodium croconate in 300 ml of water through 80 ml of a cation-exchange resin (Amberlite 120–H+), concentrating the effluent under diminished pressure, and crystallizing the product from dioxane with the addition of ether, benzene, or pentane.
6.6. Preparation of Manganese Croconate Trihydrate
A solution of 5 g of croconic acid trihydrate in 50 ml of hot water is treated, with stirring, with a hot, saturated, aqueous solution of manganous acetate until a precipitate forms. The mixture is cooled to room temperature, and the resulting greenish-yellow crystals are separated by filtration. The product is recrystallized from water by the addition of acetone; the yield is almost quantitative.
Analysis:
Calculated for C5MnO5.3H2O: C, 24.1; H, 2.41. Found: C, 24.0; H, 2.5.
6.7. Analysis of Croconic Acid
The purity of samples of croconic acid may be determined by titration with a standard solution of potassiuin permanganate (see eq (1)).
| 12 KMnO4+23 H2SO4+5 Na2C5O5→6 K2SO4+12 MnSO4+25 CO2+5 Na2SO4+23 H2O | (1) |
|---|
A 50-mg sample of disodium croconate, previously dried at 120 °C for 1 hr, is transferred to a 250-ml Erlenmeyer flask with 25 to 50 ml of water. Sulfuric acid (50 ml of 3.6 N) is added, and the solution is heated to 80 °C and rapidly titrated with 0.1-N potassium permanganate to the complete disappearance of the original yellow color. Then 5 ml of a 5-percent solution of zinc chloride is added, and the addition of permanganate is continued dropwise with swirling, until a faint pink color, stable for about 1 min, is produced. A 50-mg sample of disodium croconate monohydrate consumed 29.20 ml of w 0.1-N potassium permanganate; calculated for Na2C5O5 H2O: 29.40 ml.
7. Leuconic Acid (Cyclopentαnepentone, VI) [42]
7.1. Discussion
Leuconic acid was first prepared by Will [43] in 1861. Particularly noteworthy investigations were made by Nietzki [32], Homolka [44], and Eistert and coworkers [22]. The procedure given in the present paper is a modification of that of Nietzki.
7.2. Preparation of Leuconic Acid Pentahydrate
Purified anhydrous croconic acid (2 g), or an equivalent amount of the trihydrate, is added in portions, with stirring, to 20 ml of ice-cold, concentrated nitric acid during 3 min, and the mixture is stirred until evolution of nitrogen dioxide ceases (about 10 min). Then 20 ml of ice-cold methanol is added, and the mixture is stirred while crystallization of leuconic acid pentahydrate occurs; after 10 min, the crystals are separated by filtration, washed with methanol, and air-dried; wt, about 2.7 g
For recrystallization, the compound is dissolved in the minimal quantity of water; the solution is treated with decolorizing carbon and filtered, and the filtrate is concentrated under diminished pressure at 40 °C to about 5 ml. After the addition of several drops of concentrated nitric acid, the solution, 011 cooling, yields colorless crystals of leuconic acid pentahydrate. Infrared and ultraviolet absorption spectra are given on pages 159 and 161.
Crystalline leuconic acid pentahydrate may be isolated from concentrated, aqueous solution by adding a 1:1 mixture of 2-propanol and ethanol; this product is devoid of nitric acid.
The pentahydrate melts with dehydration at 115 to 118 °C, followed by slow decomposition at 158 to 162 °C. Leuconic acid may be characterized as the pentaoxime [32], mp 172 °C. In the present study, it has been found that the oxime may conveniently be recrystallized from N,_N_-dimethylformamide.
When a saturated solution of sodium carbonate is added to an aqueous solution of leuconic acid, a pink color is produced, followed by deposition of a difficultly soluble, white precipitate of the sodium salt of mesoxalic acid [44].
8. Spectrophotometric Measurements
Figure 1 reports infrared spectrograms for the freshly prepared, crystalline compounds in Nujol mulls. The measurements were made with a Perkin-Elmer Infracord Model 137 (double beam) spectrophotometer equipped with a prism of sodium chloride for the 2- to 15-μ range.
Figure 1. Infrared spectrograms of materials in Nujol mulls.
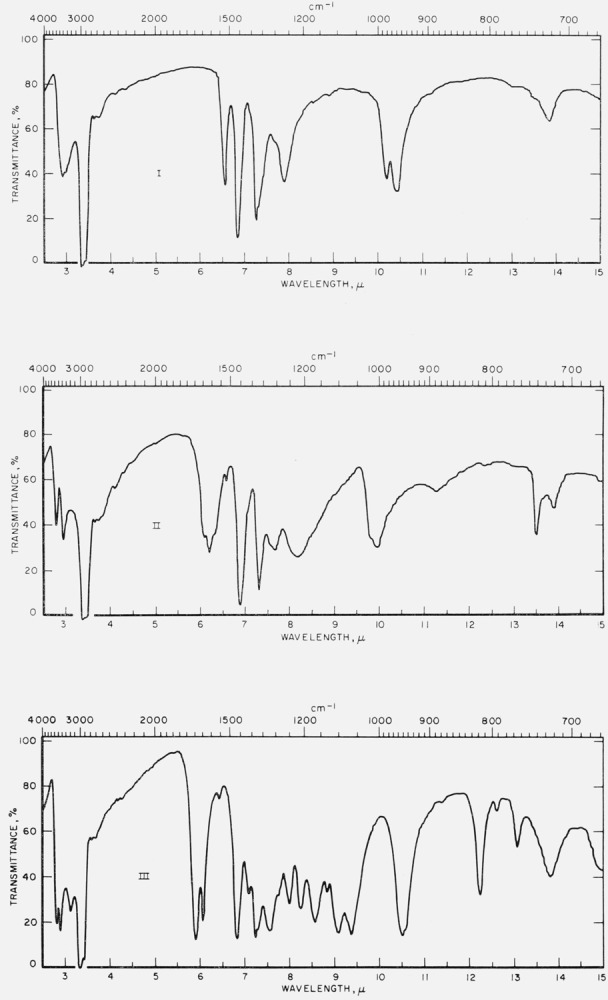
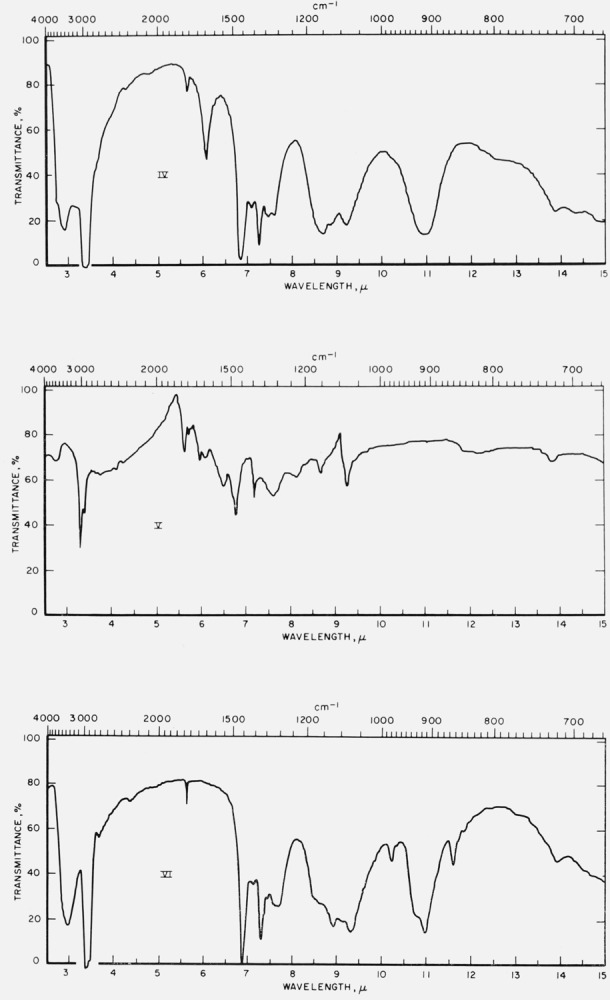
I, Hexahydroxybenzene; II, tetrahydroxyquinone; III, rhodizonic acid dihydrate.
IV, Triquinoyl octahydrate; V, croconic acid trihydratc; VI, leuconic acid pentahydrate.
The ultraviolet absorption measurements given in figure 2 were made with a Beckman DK-2 spectrophotometer with matched, 1-cm quartz cells and the appropriate solvent as the reference standard. In the course of time, the spectra for some of the compounds change, presumably from oxidation reactions. To minimize oxidation, freshly boiled solvents were used, but complete exclusion of oxygen was not attempted.
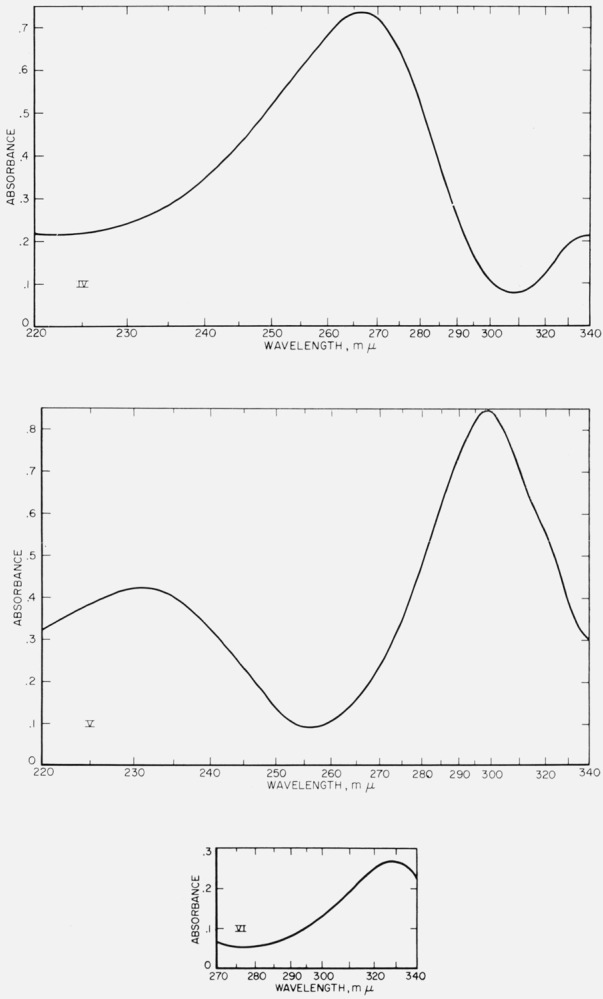
Figure 2. Ultraviolet spectrograms of materials.
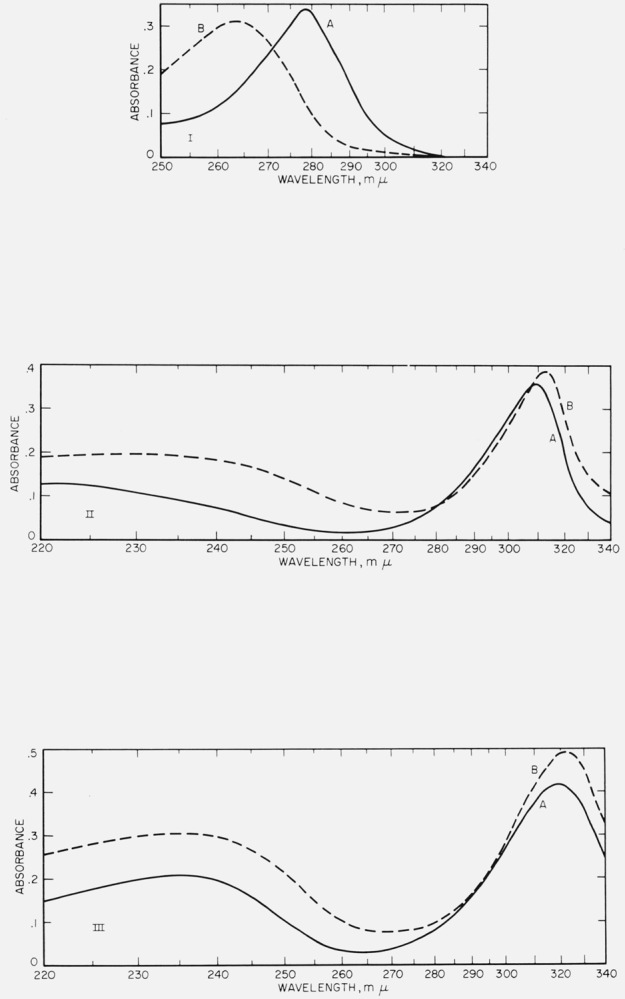
I. Hexahydroxybenzene. Curve A, 7.39 mg of I in 100 ml of 6_N_ HCl in methanol, 3 min after dissolution; λmax at 278 m_μ_. Curve B, 67 mg of the hexaacetate of I in 200 ml of glacial acetic acid, 10 min after dissolution; λmax at 263 m_μ_.
II. Tetrahydroxyquinone. Curve A, 3.82 mg of II in 1000 ml of 2_N_ aqueous HCl, 24 hr after dissolution; λmax at 308 m_μ_. Curve B, 7 mg of II in 1000 ml of methanol, 30 min after dissolution; λmax at 312 m_μ_.
III. Rhodizonic acid dihydrate. Curve A, 6.87 mg of III in 1000 ml of 2_N_ aqueous HCl 24 hr after dissolution; λmax at 235 m_μ_ and at 319 m_μ_. Curve B, 8.58 mg of III in 1000 ml of methanol, 5 min after dissolution; λmax at 235 m_μ_ and at 323 m_μ_. In the visible region, III shows absorption at 443 m_μ_ and at 487 m_μ_ (sh).
IV. Triquinoyl octahydrate. 3.24 mg of IV in 100 ml of 2_N_ aqueous NaCl 1.5 hr after dissolution; λmax at 267 mμ. In the visible region, IV shows marked absorption at 363 m_μ_, 440 m_μ_ (sh) and 480 m_μ_.
V. Croconic acid trihydrate. 5.87 mg of V in 100 ml of 2_N_ aqueous HCl, 10 min after dissolution; λmax at 231 m_μ_ and at 298 m_μ_.
VI. Leuconic acid pentahydrate. 156.45 mg of VI in 50 ml of water, 5 min after dissolution; λmax at 328 m_μ_.
Footnotes
*
The George Washington University, Washington, D. C.
1
Figures in brackets indicate the literature references at the end of this paper.
2
This procedure was developed in 1959 by one of us (A.J.F.), under the direction of Dr. William F. Sager at George Washington University [13], and was briefly described by Fieser and Fieser [14].
3
Unless the product is thoroughly washed, it may contain traces of tin salts. The presence of tin can be determined by dissolving a sample in nitric acid, evaporating the solution, and igniting the residue. When the recrystallization is conducted properly, this test does not provide any appreciable final residue. Use of stannous chloride in the recrystallization prevents the formation of undesired oxidation products.
4
Prolonged heating causes disproportionation [22].
9. References
- [1].Posternak T., Helv. Chim. Acta 19, 1333 (1936); 24, 1045 (1941). [Google Scholar]
- [2].Posternak T. and Deshusses J., Helv. Chim. Acta 44, 2089 (1961). [Google Scholar]
- [3].Stanacev N. Z. and Kates M., J. Org. Chem. 26, 912 (1961). [Google Scholar]
- [4].Isbell H. S., Ann. Rev. Biochem. 12, 213 (1943). [Google Scholar]
- [5].Isbell H. S., J. Research NBS 32, 45 (1944). [Google Scholar]
- [6].West R. and Hsien-Ying Niu J. Am. Chem. Soc. 84, 1324 (1962); [Google Scholar]; Chem. Eng. News 40, 40 (1962). [Google Scholar]
- [7].Handb Beilsteins. Org. Chemie 6, 1198, Springer, Berlin: (1923). [Google Scholar]
- [8].Nietzki R. and Benckiser T., Ber. 18, 1833 (1885). [Google Scholar]
- [9].Fatiadi A. J. (with Sager W. F.), M. S. Thesis, 1959, The George Washington University, Washington, D.C. [Google Scholar]
- [10].Nietzki R. and Benckiser T., Ber. 18, 499 (1885). [Google Scholar]
- [11].Preisler P. W. and Berger L., J. Am. Chem. Soc. 64, 67 (1942). [Google Scholar]
- [12].Anderson R. C. and Wallis E. S., J. Am. Chem. Soc. 70, 2931 (1948). [DOI] [PubMed] [Google Scholar]
- [13].Fatiadi A. J. and Sager W. F., Org. Syntheses 42, (a)66, (b)90 (1962). [Google Scholar]
- [14].Fieser Louis F. and Fieser Mary, Advanced Organic Chemistry, p. 757 (Reinhold Publ. Corp, New York, N.Y., 1961). [Google Scholar]
- [15].Homolka B., Ber. 54, 1393 (1921). [Google Scholar]
- [16].Robinson R. and Vasey C., J. Chem. Soc. 1941, 660. [Google Scholar]
- [17].Handb Beilsteins. Org. Chemie 8, 534, Springer, Berlin: (1925). [Google Scholar]
- [18].Backer H. J. and Baan S.v.d., Rec. Trav. Chim. 56, 1161 (1937). [Google Scholar]
- [19].Kuhn R., Quadbeck G., and Rohm E., Ann. 565, 1 (1949). [Google Scholar]
- [20].Eistert B. and Bock G., Angew. Chem. 70, 595 (1958). [Google Scholar]
- [21].Handb Beilsteins. Org. Chemie 8, 535, Springer, Berlin: (1925). [Google Scholar]
- [22].Eistert B., Bock G., Kosch E., and SpaIink F., Chem. Ber. 93, 1451 (1960). [Google Scholar]
- [23].Handb Beilsteins. Org. Chemie 7, 907, Springer, Berlin: (1925). [Google Scholar]
- [24].Lerch J., Ann. 124, 34 (1862). [Google Scholar]
- [25].Nietzki R. and Kehrman F., Ber. 20, 322 (1887). [Google Scholar]
- [26].Henle F., Ann. 350, 330 (1906). [Google Scholar]
- [27].Berge1 F., Ber. 62, 490 (1929). [Google Scholar]
- [28].Handb Beilsteins. Org. Chemie 8, 489, Springer, Berlin: (1925). [Google Scholar]
- [29].Gmelin L., Ann. Phys. 4, 31 (1825). [Google Scholar]
- [30].Liebig J., Ann. 11, 182 (1834). [Google Scholar]
- [31].Nietzki R. and Benckiser T., Ber. 19, 293 (1886). [Google Scholar]
- [32].Nietzki R., Ber. 20, 1617 (1887). [Google Scholar]
- [33].Nietzki R., Ber. 23, 3136 (1890). [Google Scholar]
- [34].Gelormini O. and Artz E., J. Am. Chem. Soc. 52, 2483 (1930). [Google Scholar]
- [35].Malacbowski R. and Prebendowski S., Ber. 71, 2241 (1938). [Google Scholar]
- [36].Hirata Y., Inukai K., and Tsujiuchi T., J. Chem. Soc. Japan 69, 63 (1948); C. A. 47, 5902 (1953). [Google Scholar]
- [37].Arcamone F., Prevost C., and Souchay P., Bull. Soc. Chim. France 1953, 891. [Google Scholar]
- [38].Ruthkowski S., Rocz. Chem. 36, 169 (1962). [Google Scholar]
- [39].Carlquist B. and Dyrssen D., Acta Chem. Scancl. 16, 94 (1962). [Google Scholar]
- [40].Yamada K. and Hirata Y., Bull. Chem. Soc. Japan 31, 550 (1958). [Google Scholar]
- [41].Harfenist M., Barley A., and Lazier W. A., J. Org. Chem. 19, 1608 (1954). [Google Scholar]
- [42].Handb Beilsteins. Org. Chemie 7, 905, Springer, Berlin: (1925). [Google Scholar]
- [43].Will H., Ann. 118, 177 (1861). [Google Scholar]
- [44].Homolka B., Ber. 55, 1310 (1922). [Google Scholar]