Altered Neuronal Mitochondrial Coenzyme A Synthesis in Neurodegeneration with Brain Iron Accumulation Caused by Abnormal Processing, Stability, and Catalytic Activity of Mutant Pantothenate Kinase 2 (original) (raw)
Abstract
Mutations in the pantothenate kinase 2 (PANK2) gene have been identified in patients with neurodegeneration with brain iron accumulation (NBIA; formerly Hallervorden-Spatz disease). However, the mechanisms by which these mutations cause neurodegeneration are unclear, especially given the existence of multiple pantothenate kinase genes in humans and multiple PanK2 transcripts with potentially different subcellular localizations. We demonstrate that PanK2 protein is localized to mitochondria of neurons in human brain, distinguishing it from other pantothenate kinases that do not possess mitochondrial-targeting sequences. PanK2 protein translated from the most 5′ start site is sequentially cleaved at two sites by the mitochondrial processing peptidase, generating a long-lived 48 kDa mature protein identical to that found in human brain extracts. The mature protein catalyzes the initial step in coenzyme A (CoA) synthesis but displays feedback inhibition in response to species of acyl CoA rather than CoA itself. Some, but not all disease-associated point mutations result in significantly reduced catalytic activity. The most common mutation, G521R, results in marked instability of the intermediate PanK2 isoform and reduced production of the mature isoform. These results suggest that NBIA is caused by altered neuronal mitochondrial lipid metabolism caused by mutations disrupting PanK2 protein levels and catalytic activity.
Keywords: pantothenate kinase, neurodegenerative disease, synucleinopathy, lipid metabolism, mitochondria, mitochondrial processing peptidase
Introduction
Neurodegeneration with brain iron accumulation (NBIA), formerly known as Hallervorden-Spatz disease, often begins within the first few years of life and leads to progressive impairment of movement, speech, and cognition (Dooling et al., 1974; Swaiman, 1991). Iron accumulation in the globus pallidus can be visualized by magnetic resonance imaging (MRI), often in a characteristic pattern referred to as the “eye-of-the-tiger” sign (Hayflick et al., 2003). Hypoprebetalipoproteinemia, acanthocytosis, and pigmentary retinopathy are sometimes associated with neurological impairment in NBIA, referred to as HARP syndrome when all three additional features are present (Higgins et al., 1992; Orrell et al., 1995). No therapy currently exists to stop or reverse progression of the disease.
Postmortem examination of NBIA brains reveals a number of pathological features (Halliday, 1995). Histochemical stains demonstrate iron accumulation primarily in the basal ganglia. There is also deposition of proteinaceous material in axonal inclusions termed spheroids. These inclusions have been shown in various case reports to contain α-synuclein (α-syn) and neurofilament proteins (Malandrini et al., 1995; Wakabayashi et al., 1999, 2000; Galvin et al., 2000; Neumann et al., 2000; Saito et al., 2000). Many cases contain α-syn-positive Lewy bodies in addition to axonal spheroids, strengthening the link between NBIA and Parkinson's disease at the pathological level. Neurofibrillary tangles containing the microtubule-associated protein tau have also been observed in the cortex (Wakabayashi et al., 2000). Neuronal loss and gliosis accompany these pathological changes and can be widespread, involving cortical regions as well as basal ganglia and brainstem areas.
The phenotypic features of NBIA suggest that understanding its pathogenesis may yield insight into disease mechanisms underlying inclusion formation, iron accumulation, and oxidative stress that are common to other more prevalent neurodegenerative disorders such as Alzheimer's and Parkinson's disease. The recent identification of pantothenate kinase 2 (PANK2) gene mutations in NBIA as well as HARP syndrome represents a unique opportunity to better understand the pathogenesis of this neurodegenerative disease (Zhou et al., 2001; Ching et al., 2002; Houlden et al., 2003). PANK2 is a member of a family of eukaryotic genes encoding pantothenate kinases, which catalyze the phosphorylation of pantothenate (vitamin B5) to yield phosphopantothenate. This process is the first of five enzymatic steps in the biosynthesis of coenzyme A (CoA), a universal acyl carrier used by perhaps as many as 4% of all enzymes (Begley et al., 2001).
Four putative pantothenate kinase genes have been identified in the human genome. One unique feature of PanK2 may be its mitochondrial localization, predicted by computer sequence analysis and expression of PanK2-GFP fusion proteins in cell lines (Hortnagel et al., 2003). However, alternatively spliced PanK2 transcripts lacking a mitochondrial targeting sequence have also been isolated from human brain (Hortnagel et al., 2003).
To investigate the mechanisms by which PanK2 mutations result in neurodegeneration, we generated specific PanK2 antibodies to characterize the cellular and subcellular localization of PanK2 in human brain. We also used overexpression in 293 cells to characterize the mitochondrial targeting, proteolytic processing, half-life, and catalytic properties of normal PanK2 as well as PanK2 proteins containing NBIA-associated point mutations.
Materials and Methods
Materials. Chemicals, including CoA synthesis intermediates and CoA esters, were obtained from Sigma (St. Louis, MO) unless otherwise indicated. Secondary antibodies were obtained from Jackson ImmunoResearch (West Grove, PA).
Plasmids. The full-length human PanK2 coding sequence was amplified by PCR from human brain Marothon-Ready cDNA (BD Biosciences, Palo Alto, CA). The forward primer AAGAATTCACCATGAGGAGGCTCGGGCCCTTC contained an _Eco_RI restriction site 5′ to the ATG start site, and the reverse primer TATCTAGATGATCACGGGATCTTCAACAG contained an _Xba_I site downstream of the stop codon. The coding sequence was also amplified using the same forward primer in combination with a reverse primer TATCTAGACGGGATCTTCAACAGCTCAAG that lacked a stop codon to enable fusion of a myc epitope tag and poly-histidine tag to the C terminus of PanK2. PCR products were subcloned into the pGEM-T vector and sequenced completely. The _Eco_RI and _Xba_I digested inserts were then ligated into either pcDNA 3.1 or pcDNA 3.1 myc-his. Point mutations were generated in the pcDNA 3.1 PanK2-myc-his plasmid according to the Quickchange Site-Directed Mutagenesis protocol (Stratagene, La Jolla, CA), using the following oligonucleotides (sequences shown for sense strand): CGCGCCCATGGGGCGCCACGGCA for E134G, GGTTTGGACTGGATATCGTTGGAACTCTGGTCAAG for G219V, TGAACCCAAAGACATCGCTGCTGAAGAAGAAGAGG for T234A, GGGCTGTGGCTTCAAACTTTGGAAACATGATGAGC for S471N, CCAGGTGGTATTTGTTAGAAATTTCTTGAGAATTAATACGATCGC for G521R, and TGGAAATTTCTTGAGAATTAATATGATCGCCATGCGGCTTTTG for T528M. For production of recombinant human PanK2 in Escherichia coli, PanK2 coding sequence beginning at a predicted alternative CTG start site (Zhou et al., 2001) was inserted into pET-30a vector (Novagen, Madison, WI) by cloning a _Bam_HI-_Eco_RI fragment from pMJ2 (a gift from Susan Hayflick, Oregon Health and Science University, Portland, OR) into _Bam_HI-_Eco_RI digested pET30a.
Production of bacterial recombinant PanK2 protein. Host BL21(DE3) E. coli was transformed with pET30a-PANK2 expression plasmid and incubated overnight after plating. The next morning, transformed colonies were resuspended from the plate in two washes of 5-10 ml of Luria broth (LB) and inoculated into a flask with 200 ml of LB with kanamycin. When the culture reached an OD600 of 0.6 (∼2 h), isopropyl-β-d-thiogalactopyranoside was added to 0.4 mm, and the culture was incubated for an additional 2 h. Inclusion bodies containing insoluble PanK2 protein were purified as described previously (Nagai and Thogersen, 1987), and protein was solubilized in 10 mm Tris buffer, pH 8.0, containing 6 m urea, 5 mm EDTA, and 1% β-mercaptoethanol.
PanK2 polyclonal antibody production. Rabbit polyclonal antibodies to PanK2 were produced using two PanK2 peptides lacking significant homology to PanK1, PanK3, and PanK4. PanK2 549 was generated from the peptide CEQAAGDPEGRRQEPLRRRAS, corresponding to amino acids 9-28 of mature human PanK2. PanK2 550 was generated from the peptide CENPADSEKCQKLPFDLKNP, corresponding to amino acids 224-243 of mature human PanK2. Peptides were conjugated to maleimide-activated KLH and BSA protein (Pierce, Rockford, IL) according to the manufacturer's instructions, and equal quantities of the peptide conjugates were injected subcutaneously and intramuscularly every 3 weeks for a total of four injections. Injection of antigen and collection of antisera were performed by Covance Research Products (Denver, PA). Peptide antisera were affinity purified using bacterial recombinant PanK2 protein coupled to Affi-Gel 10 (Bio-Rad, Hercules, CA) according to the manufacturer's instructions.
PanK2 monoclonal antibody production. Monoclonal antibodies were generated against bacterial recombinant human PanK2. Mice received initial subcutaneous injections of 100 μg of protein emulsified in Freund's complete adjuvant, followed by additional subcutaneous injections of 25 μg of protein emulsified in Freund's incomplete adjuvant on days 21 and 42, and an intraperitoneal injection of 25 μg of protein in PBS on day 109. On day 112, the spleen was removed, and the lymphocytes were fused to myeloma cells (line Sp2/0-Ag14) using polyethylene glycol 1500 (Tu et al., 1998). Ascites fluid was produced from monoclonal cell line 25.1, and IgG was purified using Hitrap Protein G Sepharose column (Amersham Biosciences, Piscataway, NJ).
Human brain extraction and Western blot analysis. Protein was extracted from various brain regions of histopathologically normal control brains obtained from the Center for Neurodegenerative Disease Research (CNDR) brain bank at the University of Pennsylvania. Brain tissue samples of 0.15 g were homogenized in 0.3 ml of radioimmunoprecipitation assay (RIPA) buffer (50 mm Tris, pH 8.0, 150 mm NaCl, 5 mm EDTA, 1% NP-40, 0.5% sodium deoxycholate, and 0.1% SDS) that contained a mixture of protease inhibitors (PI) (1 μg/ml each of pepstatin-A, leupeptin, _N_-α-tosyl-l-phenylalanine chloromethyl ketone, _N_-tosyl-l-lysine chloromethyl ketone, soybean trypsin inhibitor, and EDTA), 1 mm phenylmethylsulfonyl fluoride (PMSF), and _O_-phenanthroline. Homogenates were centrifuged at 100,000 × g to remove insoluble material. For Western blot analysis, samples were separated on SDS-PAGE and transferred to nitrocellulose as described previously (Kotzbauer et al., 2004). SDS-PAGE was performed in Criterion precast 10% polyacrylamide gels. Antibody concentrations used for Western blot analysis were 1:5000 for affinity-purified PanK2 549, 1:2500 for affinity-purified PanK2 550, and 1:5000 for protein G-purified PanK2 25.1. Blocking buffer was 5% milk in TTBS (50 mm Tris, pH 7.5, 150 mm NaCl, and 0.01% Tween 20) for PanK2 549 and 550. For PanK2 25.1, blocking buffer was 4% bovine serum albumin in TTBS.
Immunohistochemistry. Immunohistochemistry with antibodies to human PanK2 was performed on histopathologically normal control brain tissue from the CNDR brain bank, which was formalin-fixed, paraffin-embedded, and processed for immunohistochemistry as described previously (Duda et al., 2000). Antibodies used for immunohistochemistry were diluted in blocking buffer [2% fetal bovine serum (FBS) in 0.1 m Tris buffer] (1:250 for PanK2 549 and 25.1 and 1:125 for PanK2 550).
Immunoelectron microscopy. For immunoelectron microscopy, selected brain sections were labeled with antibody PanK2 549 (1:100) and sequentially incubated with a biotinylated goat anti-rabbit antibody and HRP-conjugated streptavidin. After chromogen reaction with diaminobenzidine (DAB)-containing medium, reaction products were enhanced by the silver methenamine method as described previously (Rodriguez et al., 1984). Sections were postfixed with 1.5% glutaraldehyde and 1% osmium tetraoxide, dehydrated in graded ethanols, and embedded in Epon. Ultrathin sections were prepared and examined by electron microscopy (EM).
Cell culture and generation of PanK2-expressing cell lines. QBI 293 cells were cultured in DMEM containing 10% FBS, penicillin, and streptomycin. Cells were transfected in six-well tissue culture plates (200,000 cells per well) using Geneporter II reagent (Gene Therapy Systems, San Diego, CA) according to the manufacturer's instructions, combining 17.5 μl of Geneporter reagent with 5 μg of plasmid DNA in a final volume of 1 ml per well of DMEM without serum or antibiotics. Three hours after addition of transfection mixture, an additional 1 ml of DMEM containing 20% FBS was added to each well. The DNA liposome-containing medium was replaced with growth medium after an additional 16 h. Stably transfected cells were selected in growth medium containing 500 μg/ml (active) G418, and individual colonies were expanded and screened for expression. Clonal lines expressing wild-type (WT) or mutant PanK2 were used in experiments examining localization and processing of PanK2, with replication of results in at least one additional clonal line.
Subcellular fractionation of QBI 293 cells. QBI 293 cells were grown to confluence in 15 cm tissue culture plates. Medium was removed, and cells were washed with 10 ml of PBS and gently scraped into 2 ml of PBS per plate. Cells were centrifuged at 1500 × g for 5 min, and the cell pellet was resuspended in TS buffer (0.1 m Tris, pH 8.0, 0.25 m sucrose, 1.5 mm DTT, 10 mm NaF, and PI mixture). Cells were passed through a 25 gauge needle five times and homogenized using a Dounce B homogenizer. Nuclei were pelleted at 1000 × g. The supernatant was removed and centrifuged at 12,000 × g. A Dounce A homogenizer was used to resuspend the mitochondrial pellet in TS buffer.
Purification of recombinant PanK2 protein. Mitochondrial fractions from 293 cells were diluted in four volumes of NiNTA binding buffer (NBB) (50 mm Tris, pH 8.0, 500 mm NaCl, 0.1% Triton X-100, 10 mm NaF, and PI) containing 25 mm imidazole. The mitochondrial suspension was sonicated and centrifuged at 12,000 × g for 10 min. The supernatant was added to NiNTA agarose (Qiagen, Valencia, CA) equilibrated in NBB with 20 mm imidazole and incubated for 30 min with rocking in a disposable column. After allowing extract containing unbound protein to flow through the resin bed, the column was washed with 10 volumes of NBB containing 20 mm imidazole and then eluted with two volumes of NBB containing 250 mm imidazole.
PanK2 enzymatic assay. Catalytic activity of recombinant PanK2 protein was determined based on an assay described for PanK1 (Rock et al., 2000, 2002). Protein preparations were assayed in a mixture containing 45.5 μm d-[1-14C]pantothenate (55 mCi/mmol; American Radiolabeled Chemicals, St. Louis, MO), 2.5 mm ATP, 2.5 mm MgCl, 0.5 mg/ml BSA, 1.5 mm DTT, and 0.1 m Tris, pH 7.5, in a total volume of 40 μl. The mixture was incubated for 10 min at 37°C and then applied to a Whatman (Maidstone, UK) DE81 ion-exchange filter disc. The filter discs were washed three times in 95% ethanol with 1% acetic acid to remove pantothenate substrate. Filter bound 4′ phosphopantothenate was quantitated by scintillation counting. Background counts were determined by parallel assays performed in the absence of added protein and subtracted from counts obtained for assays of PanK2 protein preparations. Measured enzymatic activity was linear with respect to time and Pank2 protein concentration.
N-terminal sequence analysis of PanK2 intermediate and PanK2 mature. Recombinant PanK2-myc-his protein purified from 293 cells (4 μg) was concentrated in a Microcon centrifuge concentrator (Millipore, Billerica, MA) and resolved by SDS-PAGE using a 10% acrylamide gel with the addition of 0.1 mm thioglycolate to the cathode buffer. Protein was transferred to Sequi-Blot polyvinylidene difluoride (PVDF) membrane (BioRad). The membrane was stained with amido black, and bands corresponding to PanK2 intermediate (PanK2i) and PanK2 mature (PanK2m) were excised. N-terminal amino acid sequencing was performed by the W. M. Keck Facility at Yale University (New Haven, CT).
Ion exchange separation of PanK2i and PanK2m. Mitochondrial fractions of QBI 293 cells expressing human PanK2 were diluted in four volumes of SP buffer (50 mm HEPES, pH 7.4, 0.1% Triton X-100, 10 mm NaF, 1.5 mm DTT, and PI mixture) containing 100 mm NaCl and applied to a 1 ml HiTrap SP Sepharose column (Amersham Biosciences) at a flow rate of 0.5 ml/min. The column was washed with SP buffer containing 0.1 m NaCl, and bound protein was eluted with a 20 ml linear gradient of 0.1-1.0 m NaCl in SP buffer. Fractions of 0.5 ml were collected and analyzed by Western blot analysis. Recombinant PanK2 isoforms were concentrated from 0.2 ml of each fraction by immunoprecipitation with PanK2 549 and protein A-Sepharose beads as described above. After washing the beads once with enzymatic assay buffer minus 14Cpantothenate, 40 μl of enzymatic assay mixture was added to protein ASepharose-bound protein from each fraction and incubated for 10 min at 37°C with shaking. The assay mixture was then separated from the beads and analyzed for production of 14C-phosphopantothenate as described above.
Gel filtration analysis of PanK2. NiNTA-purified human recombinant PanK2-myc-his was applied to a Superose 6 column (Amersham Biosciences) equilibrated in running buffer (50 mm Tris, pH 7.5, 150 mm NaCl, 0.1% Triton X-100, 10 mm NaF, 1.5 mm DTT, and PI mixture). Proteins were resolved at a flow rate of 0.4 ml/min. Fractions were collected, and levels of PanK2 were analyzed quantitatively by SDS-PAGE and Western blot using PanK2 549 and 125I-labeled protein A detection of rabbit primary antibody.
Pulse-chase experiments. QBI 293 cell lines were plated at 3 × 105 cells per well in six-well plates and grown to confluence over 3-4 d. Cells were starved for 30 min in 1 ml/well DMEM minus methionine and cysteine containing 10% dialyzed FBS. Cells were then pulse-labeled for 30 min in 1 ml/well of the above medium containing 250 μCi/ml 35S-labeled methionine and cysteine (35S Express Tag, 14 mCi/ml; Amersham Biosciences). At the end of the 30 min labeling period, the labeling medium was replaced with DMEM and 10% FBS growth medium, and cultures were incubated for additional time periods as indicated. At each time point, cells were washed once in cold PBS, lysed in 0.4 ml/well RIPA buffer containing _O_-phenanthroline, PI mixture, and PMSF, and immediately frozen on dry ice. Extracts were sonicated and centrifuged at 16,000 × g for 10 min, and the supernatants (200 μl) were analyzed by immunoprecipitation with PanK2 549.
Immunoprecipitation. Extracts were preincubated with protein A-Sepharose beads (Amersham Biosciences), and the beads were removed by centrifugation. Extracts were then incubated for 1.5 h at 4°C with PanK2 549 (0.5 μl of antibody per 200 μl of extract). Protein A beads (10 μl) were added, and samples were incubated with rotation for 1.5 h at 4°C. Beads were pelleted by centrifugation at 2000 × g and washed three times by resuspension in 400 μl of RIPA buffer, rotation at 4°C for 10 min, and repeat centrifugation. Bound PanK2 protein was released from the beads by boiling in 15 μl of 1× SDS sample buffer and resolved in a 10% SDS polyacrylamide gel. Gels were dried and exposed to a PhosphorImager plate (Molecular Dynamics, Sunnyvale, CA). Quantitation of individual PanK2 isoforms at each time point was performed using Imagequant software (Amersham Biosciences). Half-lives of PanK2 isoforms were estimated by fitting a one-phase exponential decay curve to the data by nonlinear regression using Prism 4 software (GraphPad, San Diego, CA).
Results
PanK2 is localized to neuronal mitochondria in human brain
We developed a panel of both monoclonal and polyclonal antibodies specific for human PanK2 protein. As shown in Figure 1, polyclonal antibodies raised to two distinct PanK2 peptides and a monoclonal antibody raised against recombinant human PanK2 protein all recognize a 48 kDa protein in Western blots of human postmortem brain extracts (Fig. 1_A_). We used these antibodies to characterize cellular and subcellular patterns of PanK2 expression in human brain. Immunohistochemistry performed with each of the three antibodies generated similar staining patterns in postmortem brain sections. We observed punctate staining of neuronal cell bodies and proximal dendrites throughout the brain. Intensity of staining varied among different neuronal populations but was most prominent in neurons of the cortex, globus pallidus, nucleus basalis of Meynert, and pontine nuclei (Fig. 1_B-I_). Immunoelectron microscopy demonstrated localization of PanK2 to mitochondria of cortical and globus pallidus neurons in postmortem human brain (Fig. 1_J-M_).
Figure 1.
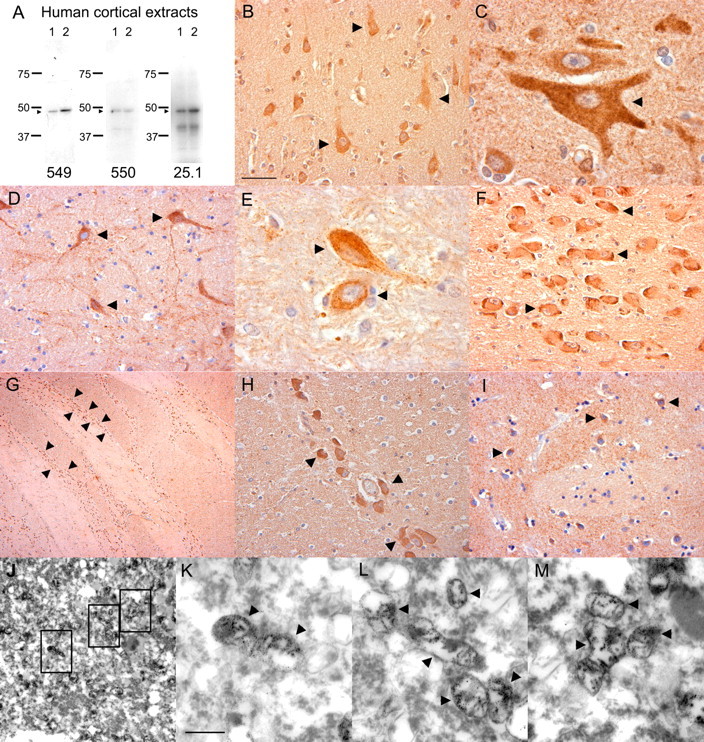
Localization of a 48 kDa PanK2 protein to neuronal mitochondria in human brain.A, Multiple PanK2 antibodies specifically recognize a 48 kDa protein in human brain. Samples of frozen cortex from two different pathologically normal postmortem cases (lanes 1 and 2) were extracted in RIPA buffer and analyzed by Western blot with each of the indicated PanK2 antibodies, revealing a commonly recognized 48 kDa band. B-I, Immunohistochemical analysis demonstrates prominent neuronal expression of PanK2 protein in human brain. Antibodies PanK2 549, 550, and 25.1 were used with brown DAB chromagen detection of bound antibody and produced consistent staining patterns throughout the brain. Prominent staining of neuronal cytoplasm and proximal dendrites (arrowheads) was found in multiple brain areas, including the cortex (B, C), globus pallidus (D, E), nucleus basalis of Meynert (F), pontine nuclei in the basis pontis (G, H), and putamen (I). J-M, Immunoelectron microscopy demonstrates localization of PanK2 protein to neuronal mitochondria in human brain. Sections of human motor cortex were stained with PanK2 549 as described above, developed with silver enhancement of DAB chromagen, and processed for EM as described. Illustrative photographs show numerous mitochondria within a large cortical neuron that were strongly labeled with silver grains specific for PanK2 staining. Similar mitochondrial staining was observed in globus pallidus neurons (data not shown). Boxes in J indicate fields shown at higher magnification in K-M. Scale bars: (in B) B, D, F, H, I, 50 μm; C, E, 20 μm; G, 500 μm; (in K) J, 2.5 μm; K-M, 0.5 μm.
Multiple PanK2 isoforms observed in brain and transfected cells
The 48 kDa band observed in Western blots was significantly smaller than predicted based on a transcript previously identified in human brain in which initiation of translation from the most 5′ ATG codon would generate a 63 kDa protein (Hortnagel et al., 2003). Computer analysis by the MITOPROT algorithm (Claros and Vincens, 1996) predicts proteolytic cleavage of the 4 kDa mitochondrial targeting sequence to yield a 59 kDa protein after targeting to the mitochondria. Alternatively, an ∼48 kDa protein can be predicted after initiation from a CTG codon 300 bp downstream from the ATG codon (Zhou et al., 2001). Using reverse-transcribed human brain RNA, we were able to PCR amplify the same fulllength PanK2 transcript identified by Hortnagel et al. (2003), encoding the predicted 63 kDa PanK2 protein when translation is initiated at the first ATG. To characterize the localization and proteolytic processing of protein encoded by this transcript, we generated stably transfected human 293 cells with an expression vector containing the PanK2 coding sequence beginning with the ATG start site. Immunofluorescence staining demonstrated efficient targeting of the expressed protein to the mitochondria based on precise colocalization with mitotracker red fluorescent dye (Fig. 2_A-C_).
Figure 2.

Transfection of 293 cells with PanK2 cDNA produces multiple PanK2 isoforms that are targeted to the mitochondria and catalyze pantothenate phosphorylation. A-C, Immunofluorescence staining demonstrates colocalization of PanK2 and mitotrackerred staining. Stably transfected 293 cells were stained with PanK2 (A) and mitotracker red CMXros (B). Merged red and green images (C) demonstrate colocalization of staining. D, Multiple PanK2 isoforms are present in extracts of transfected 293 cells. Extracts of transiently transfected and stably transfected 293 cells were analyzed by Western blot using PanK2 549 antibody. Although transiently transfected cells displayed variable subcellular localization of PanK2 (data not shown) and contained predominantly 63 and 48 kDa isoforms, stably transfected cells contained the 48 kDa isoform in combination with a 59 kDa isoform. E, The 48 kDa PanK2 isoform is present in human brain and stably transfected 293 cells. Extracts from PanK2-transfected 293 cells and from human cortex (cases 1 and 2) were analyzed by Western blot analysis with PanK2 25.1. ECL detection with a longer exposure time reveals 63, 59, and 48 kDa isoforms in 293 cells. The 48 kDa isoform is present in human brain as well as 293 cells. F, Mitochondrial extracts of PanK2-transfected cells catalyze the phosphorylation of pantothenate in vitro. Subcellular fractions of 293 cells were assayed for pantothenate kinase activity in vitro. Enzymatic activity corresponded to the predominantly mitochondrial distribution of PanK2 protein observed by Western blot analysis of subcellular fractions (inset). A similar distribution profile was observed by Western blot analysis for the mitochondrial protein pyruvate dehydrogenase (data not shown).
Western blot analysis of stably transfected cell extracts revealed an unexpected combination of 59 and 48 kDa protein species (Fig. 2_D_). The 48 kDa protein was ∼10-fold more abundant than the 59 kDa species. On longer exposures of ECL Western blots, a less abundant 63 kDa band was detected in stably transfected cells (data not shown) (Fig. 2_E_). The 48 kDa isoform in 293 cells was identical in size to the 48 kDa isoform present in brain extracts (Fig. 2_E_). Transient transfection of PanK2 expression vectors, in which the level of protein production per cell was significantly higher, generated a combination of 63 and 48 kDa proteins (Fig. 2_D_).
Mitochondrial PanK2 catalyzes the phosphorylation of pantothenate
After subcellular fractionation of transfected 293 cells by differential centrifugation, PanK2-immunoreactive bands were found predominantly in the mitochondrial fraction (Fig. 2_F_). We used an in vitro enzymatic assay to determine whether PanK2 protein expressed in 293 cells contained pantothenate kinase enzymatic activity. Incubation of extracts in the presence of 14C-labeled pantothenate and ATP was followed by ion exchange separation of phosphopantothenate product from pantothenate substrate. As shown in Figure 2_F_, although extracts of vector-transfected cells contained a detectable amount of catalytic activity, extracts of PanK2-transfected 293 cells possessed significantly higher pantothenate kinase enzymatic activity. Furthermore, the pantothenate kinase enzymatic activity was found predominantly in mitochondrial extracts of PanK2-transfected cells with a distribution nearly identical to that of PanK2 protein in Western blots.
Three PanK2 isoforms produced by sequential proteolytic cleavage
We hypothesized that the three different PanK2 isoforms might be generated by proteolytic processing of a 63 kDa protein translated from the 5′ ATG initiation site, rather than by initiation of translation at the predicted alternate CTG site in addition to the 5′ATG. To distinguish between these two possibilities, a pulsechase experiment was performed, in which newly synthesized PanK2 was pulse-labeled with 35S-methionine and monitored by immunoprecipitation and SDS-PAGE at multiple time points over 24 h during the chase period.
Consistent with the proteolytic processing hypothesis, this experiment revealed a clear temporal relationship between the three isoforms, which we have labeled PanK2 precursor (PanK2p), PanK2i, and PanK2m (Fig. 3). The 63 kDa PanK2p is the first species to be detected during the pulse-labeling period, and its levels rapidly decline during the chase period. Initial synthesis of a 52 kDa protein predicted to originate from the CTG start site was not observed. PanK2i appears next during the pulse, and its levels continue to increase during the chase, as PanK2p declines. PanK2m levels rise well into the chase period as the levels of Pank2i decline. Furthermore, PanK2m appears to be very long-lived. Once its peak is reached, it declines by only 50% over the following 15 h period, suggesting a half-life of 30 h. A band of lower molecular weight than PanK2m was also detected with a temporal profile parallel to PanK2m; however, this band appeared to be less abundant in Western blots of rapidly prepared cell extracts, suggesting that it may be a degradative product generated during cell extraction and immunoprecipitation.
Figure 3.

Pulse-chase 35Slabeling of PanK2-myc-his-transfected 293 cells demonstrates sequential proteolytic processing of newly synthesized PanK2 protein. The 293 cells were labeled with 35S-methionine for 30 min and then chased with unlabeled amino acids. A, Cells were harvested at the indicated times after the initiation of labeling and analyzed by immunoprecipitation with PanK2 549 followed by SDS-PAGE. Migration of molecular weight markers is indicated on the left. The positions of the three major PanK2 isoforms are indicated on the right (note the 3 kDa upward shift in molecular weight caused by myc-his tag). A 40 kDa band was inconsistently observed and may arise from proteolysis of the mature isoform during sample processing. B, Quantitation of relative amounts of labeled isoforms at each time point demonstrates the temporal relationship of PanK2 isoforms. PanK2p is the first isoform to appear during the labeling period, and its levels rapidly decline during the chase period as levels of PanK2i rise. The PanK2m isoform rises in a delayed manner corresponding to the decline of the PanK2 intermediate isoform. Similar results were obtained using multiple clonal lines in more than three separate experiments with both native and myc-his-tagged human PanK2.
Identification of PanK2 cleavage sites implicates the mitochondrial processing peptidase
N-terminal amino acid sequence was obtained from purified PanK2i and PanK2m to define the cleavage sites for PanK2 proteolytic processing. Sequencing results revealed cleavage between residues 31 and 32 to generate PanK2i and cleavage between 140 and 141 to generate PanK2m (Fig. 4). Both cleavage sites fit consensus sequences for cleavage by the mitochondrial processing peptidase (MPP) (Gakh et al., 2002). The first cleavage occurs two amino acids downstream of an arginine residue (R-2 motif), and the second cleavage occurs three amino acids downstream of an arginine residue (R-3 motif). PanK2m contains 82 amino acids preceding the 350 amino acid core catalytic domain that is highly conserved among PanK1, PanK2, and PanK3 (Fig. 4 D). By analogy with PanK1, this unique N-terminal domain of PanK2 may function to regulate the catalytic activity of the protein (Rock et al., 2002).
Figure 4.

N-terminal sequence analysis of PanK2i and PanK2m identifies cleavage sites, implicating the mitochondrial processing peptidase in both cleavage steps. PanK2 protein purified from stably transfected 293 cells was resolved on SDS-PAGE and transferred to a PVDF membrane. Bands corresponding to those indicated on a Coomassie-stained gel (A) were excised and subjected to amino acid sequencing. B, Predicted cleavage site in PanK2p based on N-terminal amino acid sequence obtained for PanK2i. An arginine residue at position -2 relative to the cleavage site is predicted to direct cleavage by MPP. C, Predicted cleavage site in PanK2i based on N-terminal sequence of PanK2m. An arginine residue at position -3 relative to the cleavage site is predicted to direct cleavage by MPP. A less abundant peptide with sequence beginning at residue 143 was also detected during analysis of mature PanK2. D, Schematic model of PanK2 protein indicating the position of cleavage sites in relation to predicted catalytic and regulatory domains of the mature enzyme. The position of NBIA-associated point mutations is indicated with amino acid numbers corresponding to the ATG start site rather than the CTG start site (Hayflick et al., 2003).
PanK2i and PanK2m were chromatographically separated on a cation exchange column to evaluate the catalytic activity of each isoform in an in vitro enzymatic assay. As shown in Figure 5, PanK2i and PanK2m possessed comparable catalytic activity, suggesting that the second proteolytic processing step is not necessary to generate a catalytically active protein. Steadystate levels of PanK2p were not high enough to allow evaluation of its catalytic activity. Bacterial pantothenate kinase protein, which possesses <3% sequence homology to PanK2 is a dimer, and displays cooperative binding of 2 ATP molecules per dimer (Song and Jackowski, 1994). Fractionation of mychis-tagged PanK2m by size-exclusion chromatography revealed a relative molecular weight of 109 kDa, double the calculated monomeric size of 51 kDa, indicating that PanK2 exists as a dimer under native conditions (Fig. 5_C,D_).
Figure 5.

Biochemical characterization of PanK2 protein. A, B, Chromatographic separation of PanK2i and PanK2m demonstrates catalytic activity for both isoforms. Mitochondrial fractions from stably transfected 293 cells were fractionated using a SP Sepharose cation exchange column, and fractions were analyzed by Western blot analysis (A) as well as by immunoprecipitation and enzymatic assay (B). Two peaks in catalytic activity correspond to peaks in PanK2m and PanK2i. C, D, Gel filtration analysis of PanK2 indicates that mature PanK2 is a homodimer. Purified PanK2-myc-his produced in 293 cells was fractionated using a Superose 6 gel filtration column. C, Levels of PanK2 protein in individual fractions were determined by quantitative Western blot using 125I detection. D, Analysis of PanK2 elution time in relation to molecular weight standards results in a predicted molecular weight of 109 kDa, approximately twice the molecular weight of the myc-his-tagged PanK2 protein. E, CoA esters regulate the enzymatic activity of PanK2. Pantothenate kinase enzymatic activity was measured in the presence of various concentrations of dephospho-CoA, CoA, and palmitoyl CoA. As shown on the graph, PanK2 enzymatic activity was negatively regulated by palmitoyl CoA much more than CoA, and displayed little sensitivity to dephospho-CoA. Error bars indicate SD (n = 3). Similar results were obtained in three independent analyses for each compound.
Acyl CoAs inhibit PanK2 catalytic activity
In both prokaryotic and eukaryotic systems, pantothenate kinase activity has been shown to be rate limiting in the synthesis of CoA (Jackowski and Rock, 1981; Rock et al., 2000). Because initial enzymatic reactions are often subject to feedback regulation in biosynthetic pathways, we examined the regulation of PanK2 catalytic activity by CoA and CoA esters using purified recombinant PanK2 (Fig. 5_E_). PanK2 catalytic activity was significantly decreased in the presence of CoA with an IC50 value of ∼50 μm. This effect of CoA appeared to be specific, because there was no significant effect of dephospho-CoA, the penultimate intermediate in CoA biosynthesis. However, the effect of CoA on PanK2 catalytic activity was modest compared with CoA esters such as palmitoyl CoA (Fig. 5_E_), acetyl CoA, and malonyl CoA (data not shown). Each of these species of acyl CoA inhibited PanK2 catalytic activity at a much lower concentration with an IC50 value of ∼1 μm. Furthermore, the maximum inhibitory effect was dramatic. PanK2 catalytic activity is reduced to 0.5% of basal activity in the presence of 20 μm palmitoyl CoA.
Disease-associated mutations alter processing, stability, and catalytic activity of PanK2
More than 50 different mutations in the PanK2 gene have been identified in patients with NBIA (Hayflick et al., 2003). Although several mutations result in a frameshift or premature stop codon early in the PanK2 coding sequence, >30 mutations resulting in single amino acid substitutions have been reported. We evaluated the effect of several disease-associated point mutations on the mitochondrial localization, proteolytic processing, and catalytic activity of PanK2 [amino acid numbers used to designate point mutations are increased by the 110 amino acids preceding the predicted CTG start site used for numbering of amino acids in Hayflick et al. (2003)]. Proteins containing the E134G, G219V, T234A, S471N, G521R, and T528M mutations were all efficiently targeted to mitochondria, as assessed by colocalization of immunofluorescent staining with mitotracker red (shown for G521R in Fig. 6_A-C_).
Figure 6.

Altered stability and processing for G521R mutant PanK2. A-C, G521R mutant PanK2 is targeted to the mitochondria in stably transfected 293 cells. Staining with PanK2 549 is shown in green (A), and staining with mitotracker red CMXros is shown in red (B). The merged image (C) demonstrates colocalization similar to that seen for wild-type PanK2. D, The G521R mutation results in a reduced steady-state ratio of mature to intermediate isoforms in 293 cells. Western blot analysis of 293 cells stably transfected with wild-type or G521R mutant PanK2-myc-his demonstrates marked reduction in the mature isoform relative to the intermediate isoform with the G521R mutation, in contrast to wild-type PanK2, in which the mature isoform is ∼10-fold more abundant than the intermediate isoform. E, Pulse-chase labeling of G521R PanK2-myc-his-transfected 293 cells reveals altered stability and processing of G521R compared with WT. 293 cells were labeled and analyzed by immunoprecipitation and SDS-PAGE as in Figure 3. F, Levels of each isoform for mutant and WT PanK2 were normalized to the total amount of labeled PanK2 (all three isoforms) present at the end of the labeling period (30 min time point) and graphed over time. Similar results were obtained in two independent experiments.
We observed altered steady-state levels of the mature and intermediate isoforms for G521R mutant protein (Fig. 6_D_). In both transient and stable transfection of 293 cells, levels of G521R PanK2 were significantly lower than WT PanK2. Furthermore, the ratio of intermediate to mature G521R PanK2 was ∼1:1 rather than the 1:10 ratio typically observed for WT PanK2. We compared the proteolytic processing and stability for G521R and WT protein in pulse-chase experiments (Fig. 6_E,F_). For G521R PanK2, we observed marked instability of PanK2i, reduced processing to PanK2m, and instability of PanK2m. The half-life of G521R PanK2i was estimated to be 27 min, compared with 156 min for WT PanK2i (Fig. 6_F_). In contrast to the delayed rise in WT PanK2m during the chase period, levels of G521R PanK2m declined, suggesting that cleavage of the intermediate isoform is impaired. Quantitative analysis revealed that, as a percentage of total PanK2 protein (all three isoforms) present at the end of the 30 min pulse-labeling period, the amount of mature PanK2 produced after 360 min was <2% of that produced in parallel pulse-chase experiments with WT PanK2 (Fig. 6F) (compare Fig. 3_B_, 6 E).
We compared specific enzymatic activity of several mutant PanK2 proteins to WT PanK2 protein in transient transfection experiments. Expression levels of WT and mutant PanK2 proteins in extracts of transiently transfected cells were determined by quantitative Western blots with detection by 125I-labeled sec-ondary antibodies. Analysis by in vitro enzymatic assay of normalized concentrations of WT and mutant PanK2 proteins revealed that three point mutations (G219V, S471N, and G521R) resulted in a ≥90% reduction in enzymatic activity (Fig. 7). However, the specific activities of PanK2 containing the E134G, T234A, and T528M mutations did not differ significantly from WT in these experiments, and additional in vitro experiments using purified recombinant protein confirm no loss in activities of these mutants. We were also unable to detect significant differences in the sensitivity of these three mutant proteins to inhibition by acetyl CoA (data not shown). Quantitative analyses of pulse-chase experiments also revealed no alterations in processing or stability of these three mutant proteins (data not shown).
Figure 7.

Multiple disease-associated point mutations disrupt PanK2 catalytic activity. QBI 293 cells were transiently transfected with expression plasmids containing wild-type PanK2 or PanK2 containing various disease-associated point mutations. Extracts of transfected cells were prepared, and the concentration of PanK2 protein in each extract was determined by quantitative Western blot using PanK2 549 antibody and 125I protein A detection of bound primary antibody. After normalizing the volumes of extract to equalize total PanK2 protein, catalytic activity was determined by pantothenate kinase enzymatic assay. Because QBI 293 cells produce endogenous panthothenate kinases, catalytic activity was measured for corresponding volumes of vector-transfected QBI 293 cells and subtracted from values obtained for each of the PanK2-transfected cell extracts. Error bars indicate 95% confidence intervals for each mean (n = 3). Similar results were obtained in two independent experiments.
Discussion
The identification of mutations in the PanK2 gene offers a unique opportunity to investigate mechanisms of NBIA pathogenesis. Because NBIA shares several pathological features with other more common neurodegenerative diseases, such as inclusion formation and iron accumulation, it may also share mechanisms of pathogenesis. The consequences of the many NBIA-associated point mutations on the function of PanK2 protein has not been investigated. Furthermore, although mitochondrial targeting of Pank2 fusion proteins has been demonstrated in cell culture experiments (Hortnagel et al., 2003), the cellular and subcellular localization of PanK2 in human brain has not been examined.
Our results demonstrate that PanK2 protein is expressed in the mitochondria of neurons in those brain regions that are impaired in NBIA. They also demonstrate that some, but not all, disease-associated mutations alter PanK2 function by disrupting maturation and stability of the mitochondrially targeted protein and by disrupting catalytic activity. Interestingly, we observed that several point mutations do not alter PanK2 catalytic activity as measured in vitro. This suggests that there may be additional mechanisms by which mutations can alter PanK2 function in the mitochondria. Because the T234A and T528M mutations have been most frequently associated with later onset and more slowly progressive disease (Zhou et al., 2001; Hayflick et al., 2003), these mutations might alter PanK2 function by a mechanism that becomes significant only in the context of environmental factors or increasing age.
The sequential proteolytic processing observed after targeting of PanK2 to the mitochondria is unusual, particularly if the MPP is responsible for both cleavage steps as predicted by the identification of the cleavage sites. Both the mitochondrial intermediate peptidase (MIP) and the mitochondrial inner membrane peptidase (IMP) can process specific subsets of proteins after initial cleavage by MPP. IMP cleavage can occur for nucleus-encoded proteins targeted to the inner membrane. MIP is responsible for cleavage of an N-terminal octapeptide after initial cleavage of a targeting sequence by MPP. However, neither of these peptidases appears to be involved in PanK2 cleavage based on the position and sequence of the cleavage site for PanK2i.
Sequential cleavage by MPP has been observed for one other mammalian protein, human frataxin, which is mutated in the neurodegenerative disease Friedreich's ataxia (Branda et al., 1999). As we have observed for PanK2, the second cleavage of frataxin occurs more slowly than the initial cleavage and limits the rate of mature frataxin production (Cavadini et al., 2000). Furthermore, an interaction between the β-MPP subunit and the precursor form of frataxin has been observed in a yeast two-hybrid screen (Koutnikova et al., 1998). Frataxin is involved in the maturation of iron-sulfur cluster proteins and mitochondrial metabolism (Pandolfo, 2002), an interesting link to the iron accumulation that occurs with PanK2 mutations.
Although PanK1, PanK3, and PanK4 also appear to be expressed in brain by Northern blot analysis (Zhou et al., 2001), they do not possess mitochondrial-targeting sequences. By virtue of its mitochondrial localization, PanK2 may function to regulate the biosynthesis of CoA and ultimately lipid metabolism according to specific mitochondrial metabolic needs. PanK1 enzymatic activity is tightly regulated by levels of CoA and CoA esters in vitro, and the response to CoA differs between two PanK1 protein isoforms generated by alternate mRNA splicing (Rock et al., 2000, 2002). Furthermore, as for bacterial PanK, mammalian PanK1 activity limits the rate of CoA synthesis in cell lines, suggesting that this first in a series of five enzymatic steps is a point of regulation at which CoA synthesis is titrated to the metabolic needs of the cell. The ability to assess PanK2 enzymatic activity in vitro may allow investigation of regulatory mechanisms unique to PanK2, perhaps providing further insight into mechanisms by which this enzyme might regulate CoA synthesis within the mitochondrial environment.
The mitochondrial localization of PanK2 and the regulation of PanK2 activity by species of acyl CoA may have potential relevance to a separate pathway for fatty acid synthesis within the mitochondria. The fatty acid synthase complex uses acetyl and malonyl CoA in addition to acyl carrier protein (ACP), which requires a CoA-derived phosphopantetheine group covalently linked to a serine residue for its function as an acyl carrier. Although the cytosolic ACP and fatty acid synthetase complex have been characterized as the major proteins involved in fatty acid synthesis, a mitochondrial ACP protein distinct from cytosolic ACP has also been identified and shown to contain a phosphopantetheine prosthetic group (Sackmann et al., 1991; Zhang et al., 2003). The precise role of a separate mitochondrial pathway for fatty acid synthesis is not yet clear, but there is evidence in fungal systems that it may be essential for maintenance of phospholipids of the mitochondrial membranes (Schneider et al., 1995, 1997). Alternatively, other fatty acyl CoA derivatives may be important products of this pathway.
Our results suggest that alterations in mitochondrial metabolism and perhaps specific pathways of mitochondrial lipid metabolism may underlie the neurodegenerative process in NBIA. The link between PanK2 mutations and iron accumulation in specific brain regions remains unclear. Mitochondria are the predominant consumers of imported cellular iron, where it is incorporated into heme groups and ironsulfur clusters, which are intrinsic to the activity of numerous mitochondrial enzymes, including the respiratory chain complexes. It is possible that products of mitochondrial lipid synthesis are required for iron metabolism or that altered levels of specific lipid species may directly mediate regulation of iron transport proteins. Generation of cellular and animal models of PanK2 loss of function may allow further investigation of altered iron transport in NBIA.
Mitochondrial DNA mutations are responsible for a number of degenerative neurological conditions, but neuropathological studies of these diseases have not revealed iron accumulation or inclusion formation similar to NBIA (Betts et al., 2004). Alterations in iron metabolism do occur in other hereditary neurodegenerative diseases, such as aceruloplaminemia and Friedreich's ataxia, but with anatomic patterns of iron accumulation and neurodegeneration that are distinct from NBIA (Roy and Andrews, 2001; Kaneko et al., 2002). Thus, distinct neuronal populations may be selectively vulnerable to disruption of specific metabolic processes.
Deficits in mitochondrial complex I have been detected in postmortem brain and other tissues in Parkinson's disease (Bindoff et al., 1989; Parker et al., 1989; Schapira et al., 1990; Shoffner et al., 1991; Mann et al., 1992; Cardellach et al., 1993). Furthermore, partial inhibition of complex I with rotenone causes neurodegeneration in rats that includes the substantia nigra and other regions (Betarbet et al., 2000; Hoglinger et al., 2003). Recently, mutations in PTEN-induced kinase 1, a mitochondrially localized serinethreonine protein kinase, were identified in a rare familial parkinsonian syndrome (Valente et al., 2004).
Specific links between the mitochondrial defects of these various diseases remain undefined. However, further investigation of the neurodegenerative mechanisms in NBIA and other disorders may reveal common metabolic alterations that lead to common pathological features such as protein fibrillization, oxidative stress, and iron accumulation.
Footnotes
This work was supported by grants from the Parkinson's Disease Foundation (P.T.K.), National Institute of Neurological Disorders and Stroke (P.T.K.), and National Institute on Aging (J.Q.T. and V.M.-Y.L.). P.T.K. was a Howard Hughes Medical Institute Physician Postdoctoral Fellow. J.Q.T. is the Measey-Schnabel professor of Aging research, and V.M.-Y.L. is the John H. Ware III professor of Alzheimer's research. We thank Alexxai Kravitz and Chi Li for technical assistance.
Correspondence should be addressed to Paul T. Kotzbauer, Center for Neurodegenerative Disease Research, University of Pennsylvania School of Medicine, Maloney Building, Third Floor, 3600 Spruce Street, Philadelphia, PA 19104. E-mail: kotzbaue@mail.med.upenn.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250689-10$15.00/0
References
- Begley TP, Kinsland C, Strauss E (2001) The biosynthesis of coenzyme A in bacteria. Vitam Horm 61: 157-171. [DOI] [PubMed] [Google Scholar]
- Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT (2000) Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci 3: 1301-1306. [DOI] [PubMed] [Google Scholar]
- Betts J, Lightowlers RN, Turnbull DM (2004) Neuropathological aspects of mitochondrial DNA disease. Neurochem Res 29: 505-511. [DOI] [PubMed] [Google Scholar]
- Bindoff LA, Birch-Machin M, Cartlidge NE, Parker Jr WD, Turnbull DM (1989) Mitochondrial function in Parkinson's disease. Lancet 2: 49. [DOI] [PubMed] [Google Scholar]
- Branda SS, Cavadini P, Adamec J, Kalousek F, Taroni F, Isaya G (1999) Yeast and human frataxin are processed to mature form in two sequential steps by the mitochondrial processing peptidase. J Biol Chem 274: 22763-22769. [DOI] [PubMed] [Google Scholar]
- Cardellach F, Marti MJ, Fernandez-Sola J, Marin C, Hoek JB, Tolosa E, Urbano-Marquez A (1993) Mitochondrial respiratory chain activity in skeletal muscle from patients with Parkinson's disease. Neurology 43: 2258-2262. [DOI] [PubMed] [Google Scholar]
- Cavadini P, Adamec J, Taroni F, Gakh O, Isaya G (2000) Two-step processing of human frataxin by mitochondrial processing peptidase. Precursor and intermediate forms are cleaved at different rates. J Biol Chem 275: 41469-41475. [DOI] [PubMed] [Google Scholar]
- Ching KH, Westaway SK, Gitschier J, Higgins JJ, Hayflick SJ (2002) HARP syndrome is allelic with pantothenate kinase-associated neurodegeneration. Neurology 58: 1673-1674. [DOI] [PubMed] [Google Scholar]
- Claros MG, Vincens P (1996) Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem 241: 779-786. [DOI] [PubMed] [Google Scholar]
- Dooling EC, Schoene WC, Richardson Jr EP (1974) Hallervorden-Spatz syndrome. Arch Neurol 30: 70-83. [DOI] [PubMed] [Google Scholar]
- Duda JE, Giasson BI, Gur TL, Montine TJ, Robertson D, Biaggioni I, Hurtig HI, Stern MB, Gollomp SM, Grossman M, Lee VM, Trojanowski JQ (2000) Immunohistochemical and biochemical studies demonstrate a distinct profile of alpha-synuclein permutations in multiple system atrophy. J Neuropathol Exp Neurol 59: 830-841. [DOI] [PubMed] [Google Scholar]
- Gakh O, Cavadini P, Isaya G (2002) Mitochondrial processing peptidases. Biochim Biophys Acta 1592: 63-77. [DOI] [PubMed] [Google Scholar]
- Galvin JE, Giasson B, Hurtig HI, Lee VM, Trojanowski JQ (2000) Neurodegeneration with brain iron accumulation, type 1 is characterized by alpha-, beta-, and gamma-synuclein neuropathology. Am J Pathol 157: 361-368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday W (1995) The nosology of Hallervorden-Spatz disease. J Neurol Sci [Suppl] 134: 84-91. [DOI] [PubMed] [Google Scholar]
- Hayflick SJ, Westaway SK, Levinson B, Zhou B, Johnson MA, Ching KH, Gitschier J (2003) Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med 348: 33-40. [DOI] [PubMed] [Google Scholar]
- Higgins JJ, Patterson MC, Papadopoulos NM, Brady RO, Pentchev PG, Barton NW (1992) Hypoprebetalipoproteinemia, acanthocytosis, retinitis pigmentosa, and pallidal degeneration (HARP syndrome). Neurology 42: 194-198. [DOI] [PubMed] [Google Scholar]
- Hoglinger GU, Feger J, Prigent A, Michel PP, Parain K, Champy P, Ruberg M, Oertel WH, Hirsch EC (2003) Chronic systemic complex I inhibition induces a hypokinetic multisystem degeneration in rats. J Neurochem 84: 491-502. [DOI] [PubMed] [Google Scholar]
- Hortnagel K, Prokisch H, Meitinger T (2003) An isoform of hPANK2, deficient in pantothenate kinase-associated neurodegeneration, localizes to mitochondria. Hum Mol Genet 12: 321-327. [DOI] [PubMed] [Google Scholar]
- Houlden H, Lincoln S, Farrer M, Cleland PG, Hardy J, Orrell RW (2003) Compound heterozygous PANK2 mutations confirm HARP and Hallervorden-Spatz syndromes are allelic. Neurology 61: 1423-1426. [DOI] [PubMed] [Google Scholar]
- Jackowski S, Rock CO (1981) Regulation of coenzyme A biosynthesis. J Bacteriol 148: 926-932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko K, Yoshida K, Arima K, Ohara S, Miyajima H, Kato T, Ohta M, Ikeda SI (2002) Astrocytic deformity and globular structures are characteristic of the brains of patients with aceruloplasminemia. J Neuropathol Exp Neurol 61: 1069-1077. [DOI] [PubMed] [Google Scholar]
- Kotzbauer PT, Giasson BI, Kravitz AV, Golbe LI, Mark MH, Trojanowski JQ, Lee VM (2004) Fibrillization of alpha-synuclein and tau in familial Parkinson's disease caused by the A53T alpha-synuclein mutation. Exp Neurol 187: 279-288. [DOI] [PubMed] [Google Scholar]
- Koutnikova H, Campuzano V, Koenig M (1998) Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase. Hum Mol Genet 7: 1485-1489. [DOI] [PubMed] [Google Scholar]
- Malandrini A, Cavallaro T, Fabrizi GM, Berti G, Salvestroni R, Salvadori C, Guazzi GC (1995) Ultrastructure and immunoreactivity of dystrophic axons indicate a different pathogenesis of Hallervorden-Spatz disease and infantile neuroaxonal dystrophy. Virchows Arch 427: 415-421. [DOI] [PubMed] [Google Scholar]
- Mann VM, Cooper JM, Krige D, Daniel SE, Schapira AH, Marsden CD (1992) Brain, skeletal muscle and platelet homogenate mitochondrial function in Parkinson's disease. Brain 115 (Pt 2): 333-342. [DOI] [PubMed] [Google Scholar]
- Nagai K, Thogersen HC (1987) Synthesis and sequence-specific proteolysis of hybrid proteins produced in _Escherichia coli_Methods Enzymol 153: 461-481. [DOI] [PubMed] [Google Scholar]
- Neumann M, Adler S, Schluter O, Kremmer E, Benecke R, Kretzschmar HA (2000) Alpha-synuclein accumulation in a case of neurodegeneration with brain iron accumulation type 1 (NBIA-1, formerly Hallervorden-Spatz syndrome) with widespread cortical and brainstem-type Lewy bodies. Acta Neuropathol (Berl) 100: 568-574. [DOI] [PubMed] [Google Scholar]
- Orrell RW, Amrolia PJ, Heald A, Cleland PG, Owen JS, Morgan-Hughes JA, Harding AE, Marsden CD (1995) Acanthocytosis, retinitis pigmentosa, and pallidal degeneration: a report of three patients, including the second reported case with hypoprebetalipoproteinemia (HARP syndrome). Neurology 45: 487-492. [DOI] [PubMed] [Google Scholar]
- Pandolfo M (2002) Iron metabolism and mitochondrial abnormalities in friedreich ataxia. Blood Cells Mol Dis 29: 536-547. [DOI] [PubMed] [Google Scholar]
- Parker Jr WD, Boyson SJ, Parks JK (1989) Abnormalities of the electron transport chain in idiopathic Parkinson's disease. Ann Neurol 26: 719-723. [DOI] [PubMed] [Google Scholar]
- Rock CO, Calder RB, Karim MA, Jackowski S (2000) Pantothenate kinase regulation of the intracellular concentration of coenzyme A. J Biol Chem 275: 1377-1383. [DOI] [PubMed] [Google Scholar]
- Rock CO, Karim MA, Zhang YM, Jackowski S (2002) The murine pantothenate kinase (Pank1) gene encodes two differentially regulated pantothenate kinase isozymes. Gene 291: 35-43. [DOI] [PubMed] [Google Scholar]
- Rodriguez EM, Yulis R, Peruzzo B, Alvial G, Andrade R (1984) Standardization of various applications of methacrylate embedding and silver methenamine for light and electron microscopy immunocytochemistry. Histochemistry 81: 253-263. [DOI] [PubMed] [Google Scholar]
- Roy CN, Andrews NC (2001) Recent advances in disorders of iron metabolism: mutations, mechanisms and modifiers. Hum Mol Genet 10: 2181-2186. [DOI] [PubMed] [Google Scholar]
- Sackmann U, Zensen R, Rohlen D, Jahnke U, Weiss H (1991) The acylcarrier protein in Neurospora crassa mitochondria is a subunit of NADH: ubiquinone reductase (complex I). Eur J Biochem 200: 463-469. [DOI] [PubMed] [Google Scholar]
- Saito Y, Kawai M, Inoue K, Sasaki R, Arai H, Nanba E, Kuzuhara S, Ihara Y, Kanazawa I, Murayama S (2000) Widespread expression of alphasynuclein and tau immunoreactivity in Hallervorden-Spatz syndrome with protracted clinical course. J Neurol Sci 177: 48-59. [DOI] [PubMed] [Google Scholar]
- Schapira AH, Cooper JM, Dexter D, Clark JB, Jenner P, Marsden CD (1990) Mitochondrial complex I deficiency in Parkinson's disease. J Neurochem 54: 823-827. [DOI] [PubMed] [Google Scholar]
- Schneider R, Massow M, Lisowsky T, Weiss H (1995) Different respiratory-defective phenotypes of Neurospora crassa and Saccharomyces cerevisiae after inactivation of the gene encoding the mitochondrial acyl carrier protein. Curr Genet 29: 10-17. [DOI] [PubMed] [Google Scholar]
- Schneider R, Brors B, Massow M, Weiss H (1997) Mitochondrial fatty acid synthesis: a relic of endosymbiontic origin and a specialized means for respiration. FEBS Lett 407: 249-252. [DOI] [PubMed] [Google Scholar]
- Shoffner JM, Watts RL, Juncos JL, Torroni A, Wallace DC (1991) Mitochondrial oxidative phosphorylation defects in Parkinson's disease. Ann Neurol 30: 332-339. [DOI] [PubMed] [Google Scholar]
- Song WJ, Jackowski S (1994) Kinetics and regulation of pantothenate kinase from _Escherichia coli_J Biol Chem 269: 27051-27058. [PubMed] [Google Scholar]
- Swaiman KF (1991) Hallervorden-Spatz syndrome and brain iron metabolism. Arch Neurol 48: 1285-1293. [DOI] [PubMed] [Google Scholar]
- Tu PH, Galvin JE, Baba M, Giasson B, Tomita T, Leight S, Nakajo S, IwatsuboT, Trojanowski JQ, Lee VM (1998) Glial cytoplasmic inclusions in whitematter oligodendrocytes of multiple system atrophy brains contain insoluble alpha-synuclein. Ann Neurol 44: 415-422. [DOI] [PubMed] [Google Scholar]
- Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, Gonzalez-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, et al. (2004) Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science 304: 1158-1160. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Yoshimoto M, Fukushima T, Koide R, Horikawa Y, Morita T, Takahashi H (1999) Widespread occurrence of alpha-synuclein/NACP-immunoreactive neuronal inclusions in juvenile and adult-onset Hallervorden-Spatz disease with Lewy bodies. Neuropathol Appl Neurobiol 25: 363-368. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, Fukushima T, Koide R, Horikawa Y, Hasegawa M, Watanabe Y, Noda T, Eguchi I, Morita T, Yoshimoto M, Iwatsubo T, Takahashi H (2000) Juvenile-onset generalized neuroaxonal dystrophy (Hallervorden-Spatz disease) with diffuse neurofibrillary and Lewy body pathology. Acta Neuropathol (Berl) 99: 331-336. [DOI] [PubMed] [Google Scholar]
- Zhang L, Joshi AK, Smith S (2003) Cloning, expression, characterization, and interaction of two components of a human mitochondrial fatty acid synthase. Malonyltransferase and acyl carrier protein. J Biol Chem 278: 40067-40074. [DOI] [PubMed] [Google Scholar]
- Zhou B, Westaway SK, Levinson B, Johnson MA, Gitschier J, Hayflick SJ (2001) A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet 28: 345-349. [DOI] [PubMed] [Google Scholar]