Recent highlights in the development of new antiviral drugs (original) (raw)
Abstract
Twenty antiviral drugs, that is about half of those that are currently approved, are formally licensed for clinical use in the treatment of human immunodeficiency virus infections (acquired immune deficiency syndrome). The others are used in the treatment of herpesvirus (e.g. herpes simplex virus, varicella zoster virus and cytomegalo virus), hepatitis B virus, hepatitis C virus or influenza virus infections. Recent endeavours have focussed on the development of improved antiviral therapies for virus infections that have already proved amenable to antiviral drug treatment, as well as for virus infections for which, at present, no antiviral drugs have been formally approved (i.e. human papilloma viruses, adenoviruses, human herpesvirus type 6, poxviruses, severe acute respiratory syndrome coronavirus and hemorrhagic fever viruses).
Introduction
Of the ∼40 antiviral drugs that have been formally licensed for clinical use [1], 20 are used in the treatment of human immunodeficiency virus (HIV) infections (acquired immune deficiency syndrome; AIDS). The anti-HIV compounds fall into five categories: first, the nucleoside reverse transcriptase inhibitors (NRTIs), zidovudine, didanosine, zalcitabine, stavudine, lamivudine, abacavir and emtricitabine; second, the nucleotide reverse transcriptase inhibitor, tenofovir disoproxil fumarate; third, the non-nucleoside reverse transcriptase inhibitors (NNRTIs), nevirapine, delavirdine and efavirenz; fourth, the protease inhibitors (PIs), saquinavir, ritonavir, indinavir, nelfinavir, amprenavir, lopinavir (combined at a 4 to 1 ratio with ritonavir), atazanavir and fosamprenavir; and fifth, the fusion inhibitor, enfuvirtide. Lamivudine and adefovir dipivoxil have been approved for the treatment of hepatitis B virus infections. Of the anti-herpesvirus agents, acyclovir, valaciclovir, famciclovir, brivudin, penciclovir, idoxuridine and trifluridine are used (the latter three only topically) in the treatment of herpes simplex virus (HSV) and/or varicella-zoster virus (VZV) infections; ganciclovir, valganciclovir, foscarnet, cidofovir and fomivirsen (the latter only by intravitreal injection) have proven useful in the treatment of cytomegalovirus (CMV) infections in immunocompromised patients (i.e. AIDS patients that have CMV retinitis). Following the progression of amantadine and rimantadine (matrix 2 protein blockers) onto the market, the neuraminidase inhibitors zanamivir and oseltamivir have become available for the therapy (and prophylaxis) of influenza virus infections. Ribavirin has been used (topically as an aerosol) in the treatment of respiratory syncytial virus infections, and the combination of ribavirin with (pegylated) interferon-α has received increased acceptance for the treatment of hepacivirus (hepatitis C virus; HCV) infections.
There are, however, several other important virus infections for which no antiviral drugs have been developed; even for those that are amenable to antiviral drug therapy there is still considerable room for improvement in terms of higher potency and/or increased selectivity or safety. In this review, I will highlight some of the most recent approaches towards the treatment of human papilloma virus (HPV) infections, adenovirus infections, HSV and VZV infections, human herpesvirus type 6 (HHV-6) infections, poxvirus infections, hepacivirus infections, corona virus infections (i.e. severe acute respiratory syndrome [SARS]), influenza virus infections, hemorrhagic fever virus infections and HIV infections (AIDS).
Human papilloma virus infections
Although cidofovir, which is also known as (S)-1-(3-hydroxy-2-phosphonylmethoxypropyl)cytosine (HPMPC; Figure 1), is formally licensed only for the intravenous treatment of CMV retinitis in AIDS patients, ‘off label’ it has been used successfully in the systemic and topical treatment of several other DNA virus infections, such as polyoma-, papilloma-, adeno-, herpes- and pox-virus [2]. Particularly striking are the effects that have been obtained with cidofovir (topical gel application or direct intralesional injection) in the treatment of HPV-associated papillomatous lesions, such as hypopharyngeal and esophageal papilloma, laryngeal papilloma, recurrent respiratory papillomatosis (in children), anogenital HPV infections (i.e. condylomata acuminata), bowenoid papulosis (perianal intraepithelial neoplasia), cervical and vulvar intraepithelial neoplasia grade III, recalcitrant warts (including plantar warts), and other mucocutaneous HPV lesions that did not respond to conventional therapies [3•]. In most of these cases, a virtually complete and durable resolution of the lesions was achieved following the application of cidofovir (as a 1% gel or cream).
Figure 1.
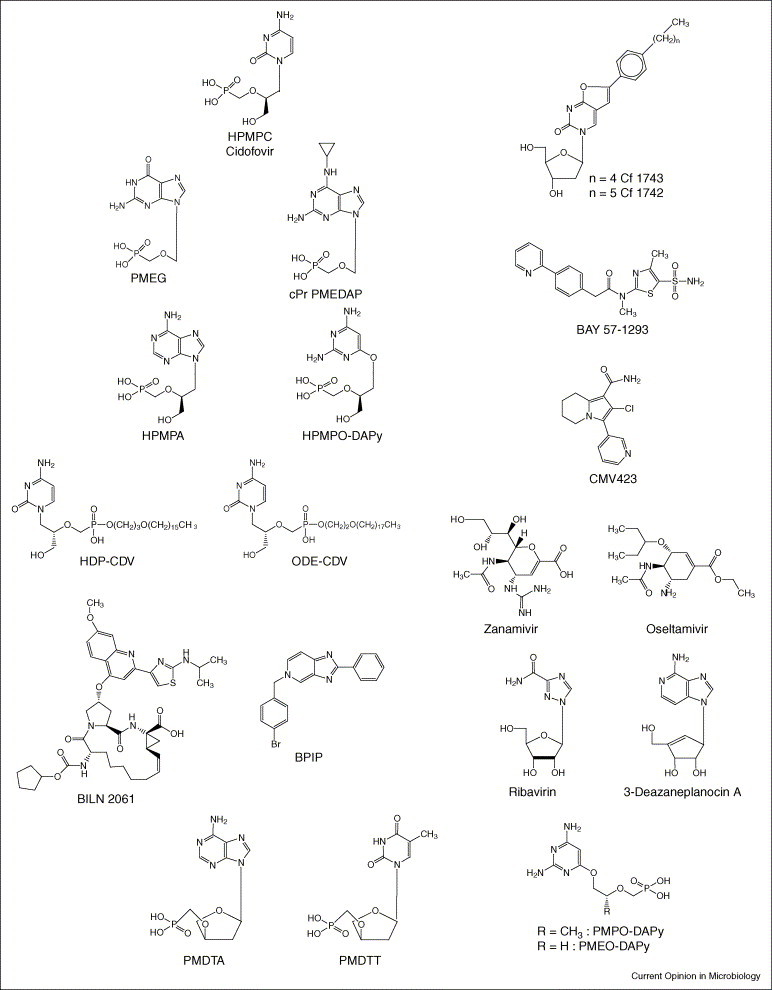
Structures of the compounds indicated in text.
Although the (compelling) evidence for the effectiveness of cidofovir against HPV infections stems from in vivo observations in an increasing number of patients, in vitro experiments with HPV-infected keratinocytes have ascertained that cidofovir specifically ‘kills’ HPV-positive cells by the induction of apoptosis. This effect might be related to the ability of cidofovir to restore the function of the tumor suppressor proteins p53 and pRb (which are neutralized by the oncoproteins E6 and E7, respectively) in HPV-infected cells [3•]. In addition to cidofovir, there are several other acyclic nucleoside phosphonates, for example 9-(2-phosphonylmethoxyethyl)guanine (PMEG; Figure 1) and 9-(2-phosphonylmethoxyethyl)-N6-cyclopropyl-2,6-diaminopurine (cPr PMEDAP; Figure 1), which, like cidofovir, are able to specifically induce apoptosis in HPV-infected keratinocytes. PMEG and cPr PMEDAP are currently being investigated for their potential in the treatment of HPV-associated papillomatous lesions.
Adenovirus infections
Adenovirus infections in immunocompetent individuals are generally self-limiting, but in immunocompromised patients and, in particular, allogeneic hematopoietic stem cell transplant recipients, adenovirus infections can be severe and life-threatening. At present, cidofovir (HPMPC) appears to be the only licensed antiviral drug that can be successfully used to treat adenovirus infections [3•].
In addition to cidofovir, a few other acyclic nucleoside phosphonates such as (S)-9-(3-hydroxy-2-phosphonylmethoxypropyl)adenine (HPMPA; Figure 1) and (S)-2,4-diamino-6-[3-hydroxy-2-phosphonomethoxy)propoxy]pyrimidine (HPMPO-DAPy; Figure 1) have proved to be potent and selective inhibitors of the in vitro replication of adenoviruses [4]. Also, the N7-substituted acyclic nucleoside 2-amino-7-(1,3-dihydroxy-2-propoxymethyl)purine S-2242 and the 2′,3′-dideoxynucleoside analogues zalcitabine (2′,3′-dideoxycytidine; ddC) and alovudine (3′-fluoro-2′,3′-dideoxythymidine; also known as FddT and FLT) were found to inhibit in vitro adenovirus replication, whereas no anti-adenovirus activity was observed for several other antiviral compounds, such as ribavirin, foscarnet, acyclovir, penciclovir and brivudin [4]. It might be worthwhile to further examine HPMPA, HPMPO-DAPy, S-2242, ddC and FLT in both experimentally and clinically oriented studies.
Also, the ether lipid ester (i.e. hexadecyloxypropyl [HDP] and octadecyloxyethyl [ODE]) prodrugs of cidofovir (HPMPC) and HPMPA should be evaluated in the future for their potential to treat adenovirus infections in humans; these compounds were found to inhibit adenovirus replication in vitro at significantly lower concentrations and with significantly higher selectivity indices than those noted for the parent compounds (HPMPC and HPMPA) [5].
Herpes simplex virus and varicella-zoster virus infections
Standard antiviral drugs currently used in the treatment of HSV and/or VZV infections include acyclovir, valaciclovir (the oral prodrug of acyclovir), famciclovir (the oral prodrug of penciclovir) and brivudin (BVDU). The latter is only available in some European countries, and, although exquisitely active against VZV, its use is restricted to patients that have herpes zoster or HSV-1 infections. BVDU cannot be administered concomitantly with 5-fluorouracil (which is a cytostatic agent used in the treatment of certain cancers) because through the release of its degradation product, BVU, BVDU can potentiate the toxicity of 5-fluorouracil (through inhibition of dihydropyrimidine dehydrogenase, a key enzyme in the degradation of 5-fluorouracil). The newly discovered bicyclic furo(2,3-d)pyrimidine nucleoside analogues (BCNAs; i.e. Cf 1368, Cf 1369, Cf 1742 [Figure 1] and Cf 1743 [Figure 1]) do not have this drawback. Although the BCNAs have proven to be as active, and Cf 1742 and Cf 1743 are 10-fold more active against VZV than BVDU [6, 7], they are unlikely to enhance the toxicity of 5-fluorouracil for two reasons: first, they are not degraded to the free pyrimidine base, and second, the latter was shown not to interfere with the dihydropyrimidine dehydrogenase that initiates the catabolism of 5-fluorouracil [6]. BCNAs are extremely selective in their anti-VZV activity, in that they inhibit only VZV replication and not the replication of any other viruses such as HSV. With a selectivity index (ratio of 50% cytotoxic concentration to 50% inhibitory concentration against viral replication) in excess of 100 000, the BCNAs offer an unprecedented potential for the treatment of VZV infections.
New anti-HSV agents that target the viral helicase–primase complex, such as the thiazolylphenyl derivatives BILS 179BS and, especially, BAY 57-1293 (Figure 1), were recently reported to have in vivo efficacy in animal models of HSV-1 and HSV-2 infections [8, 9]. These compounds appear to function by enhancing the affinity of the helicase–primase complex for the HSV DNA. This complex comprises three viral proteins — the HSV UL5, UL-8 and UL52 gene products — which together unwind the double-stranded viral DNA and generate the primer/templates for DNA synthesis by the viral DNA polymerase. It has been claimed that the antiviral potency of BAY-1293 is superior to all other compounds currently used for the treatment of HSV infections [10]. This by itself validates the further pursuit of helicase–primase inhibitors for the treatment of HSV infections.
A further extension of this approach involves the molecules that interact with the formation of the HSV DNA polymerase complex (i.e. BP5, which inhibits the interaction between UL-30 and UL42) [11]. Protein–protein interactions constitute a new target for the design of antiviral agents to be further explored for their potential as antiviral drugs.
Human herpesvirus type 6 infections
HHV-6, like CMV, has been increasingly recognized as an important pathogen in immunocompromised patients (in which it might cause life-threatening complications); however, unlike for CMV infections, no compounds have been formally approved for the treatment of HHV-6 infections [12•]. The drugs clinically used against HHV-6 are the same as those used in CMV therapy (or prophylaxis) and consist of ganciclovir, valganciclovir, and, to a lesser extent, acyclovir, valaciclovir, cidofovir and foscarnet. All these compounds are targeted at the viral DNA polymerase and can be considered (following phosphorylation) as substrate analogues (except for foscarnet, which acts as a pyrophosphate analogue).
Another class of compounds, the 4-oxo-dihydroxyquinolines, act as non-nucleoside inhibitors of herpesvirus DNA polymerases. They exhibit broad-spectrum antiviral activity against most human herpesviruses (including HCMV), with the exception of HHV-6 and HHV-7. Their lack of activity against HHV-6 is owing to a single amino acid change in the conserved domain III of the HHV-6 DNA polymerase [13].
We have recently discovered a new antiviral agent, CMV423 (2-chloro-3-pyridin-3-yl-5,6,7,8-tetrahydroindolizine-1-carboxamide; Figure 1), which demonstrated potent and selective in vitro activity against all three human β-herpesviruses — CMV, HHV-6 and HHV-7 [14]. Compared to ganciclovir and foscarnet, CMV423 showed a higher potency and lower cytotoxicity. However, its antiviral action appeared to be cell-dependent. CMV423 must be targeted at an event that follows viral entry but that precedes viral DNA replication. CMV423 was also found to inhibit total cellular protein tyrosine kinase activity. It was concluded that CMV423 exerts its activity against HHV-6 through inhibition of a cellular process that is crucial in the early stage of viral replication and that might involve protein tyrosine kinase activity [14].
Poxvirus infections
The family of poxviridae encompasses orthopoxviruses (e.g. variola, vaccinia, cowpox, monkeypox and camelpox), parapoxviruses (e.g. orf) and mollusciviruses (e.g. molluscum contagiosum virus). Several nucleoside analogues (i.e. S2242, 8-methyladenosine and idoxuridine) and nucleotide analogues (including HPMPC [cidofovir] and HPMPO-DAPy) are effective in various animal models of poxvirus infections [15]. In particular, cidofovir has shown high efficacy in protection of mice from a lethal respiratory infection of either vaccinia or cowpox, even after administration of a single systemic (intraperitoneal) or intranasal (aerosolized) dose. Cidofovir has demonstrated high effectiveness in the treatment of disseminated progressive vaccinia in athymic-nude mice [16]. In humans, cidofovir has been used successfully by both the topical and intravenous route in the treatment of orf and recalcitrant molluscum contagiosum in immunocompromised patients (reviewed by De Clercq [17]). These data indicate that cidofovir will be effective in the therapy and prophylaxis of smallpox (variola) and related poxviruses in humans, as well as in the treatment of the complications of vaccinia that can arise in immunocompromised patients inadvertently inoculated with the smallpox vaccine (vaccinia) [17].
However, as cidofovir is a phosphonate analogue it has limited oral bioavailability. In an outbreak of smallpox, it would be useful to have an orally active drug at hand that could be self-administered [18•]. Therefore, hexadecyloxypropyl-cidofovir (HDP-CDV) and octadecyloxyethyl-cidofovir (ODE-CDV) were designed and developed as potential (oral) drugs for the prophylaxis and treatment of variola virus infection (Figure 1) [18•]. These alkyloxyalkyl esters of cidofovir were found to significantly enhance inhibition of orthopoxvirus replication (i.e. vaccinia and cowpox) in vitro [19]. When given orally, HDP-CDV and ODE-CDV were as effective as cidofovir is when administered parenterally for the treatment of vaccinia and cowpox infections [20]; HDP-CDV has also proven effective in the treatment of lethal vaccinia virus respiratory infections in mice [21]. Furthermore, when administered orally, HDP-CDV and ODE-CDV proved highly efficacious in a lethal mousepox (aerosol ectromelia virus) model, further attesting the potential usefulness of other lipid prodrugs of cidofovir in the oral therapy and prophylaxis of poxvirus infections.
Hepacivirus infections (hepatitis C)
The present therapy for chronic hepatitis C consists of pegylated interferon (IFN)-α2a (180 μg, once weekly) combined with ribavirin (1000 or 1200 mg, daily) [22, 23]. This treatment regimen produces a sustained virologic response in at least 50% of the patients infected with HCV genotype 1 and 2 (and 80% of the patients infected with another genotype of HCV). Patients infected with HCV genotype 1 should be treated for at least 48 weeks [24], but this length of time might be reduced to 24 weeks for patients infected with HCV genotypes 2 or 3. In this combination, ribavirin is assumed to act as an immunomodulator, whereas the IFN would act as an antiviral agent targeted at a phosphoprotein encoded by the non-structural NS5A gene of the HCV genome [25].
In addition to the NS5A gene product, there are a few other non-structural proteins such as the NS3-encoded NTPase–helicase and serine protease and the NS5B-encoded RNA-dependent RNA polymerase (RdRp, which is also known as RNA replicase) that could be envisaged as targets for inhibitors of HCV replication. Proof-of-principle that compounds targeted at the NS3 protease could reduce plasma concentrations of HCV RNA has been provided for the protease inhibitor BILN 2061 (Figure 1) [26•].
Equally attractive as a target for the development of HCV inhibitors is the HCV RNA replicase. Highly selective antiviral agents targeted at the viral RNA replicase have been described to inhibit the replication of bovine viral diarrhoea virus (BVDV), a pestivirus that could be considered as a surrogate virus for HCV [27, 28]. We have recently described a novel series of compounds (e.g. prototype 5-[(4-bromophenyl)methyl]-2-phenyl-5_H_-imidazo[4, 5, 6, 7, 8, 9, 10, 11, 12•, 13, 14, 15, 16, 17, 18•, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40•, 41, 42, 43, 44•, 45, 46, 47, 48, 49, 50, 51, 52, 53•, 54, 55, 56•, 57, 58, 59, 60••]pyridine [also known as BPIP; Figure 1]) that act as ‘non-nucleoside’ RdRp inhibitors; these inhibit the replication of BVDV with high efficiency [29]. From this class of compounds, based on a different structure–activity relationship, new congeners have been derived that act equally efficiently against HCV replication. In the future treatment of HCV infections, these non-nucleoside RdRp inhibitors are likely to be combined with the ‘nucleoside’ RdRp inhibitors. An example of the latter, albeit a relatively weak inhibitor of BVDV and HCV, is N4-hydroxycytidine [30].
Coronavirus infections
There are several HCV proteins encoded by the SARS coronavirus (SARS-CoV) that could be considered as targets for chemotherapeutic intervention: the spike (S) protein, the SARS-CoV main protease (also known as the 3C-like protease), the NTPase/helicase, the RNA-dependent RNA polymerase (RdRp) and, possibly, other viral (or cellular) protein-mediated processes [31]. The SARS-CoV S protein mediates infection of permissive cells by controlling the interaction of its S1 domain with cells that express the SARS-CoV receptor angiotensin-converting enzyme 2 (ACE2) [32]. A 193-amino acid fragment of the S protein (that corresponds to amino acid residues 318–510) was found to block S protein-mediated infection [33], and a human monoclonal antibody to the S1 domain was found to neutralize SARS-CoV by blocking its association with the SARS-CoV receptor ACE2 [34]. In addition to ACE2, the C-type lectin CD209L (also called L-SIGN) was identified recently as a receptor for SARS-CoV (as well as for other enveloped viruses, including Ebola); thus, CD209L, like ACE2, might be a target for blocking SARS-CoV infection. Also, to the extent that the fusogenic mechanism of HIV is similar to that of SARS-CoV (e.g. with regard to heptad repeat interactions and six-helix bundle formation), it should be feasible to develop SARS-CoV fusion inhibitors analogous to enfuvirtide, which is used for treatment of HIV [35].
Knowledge of the crystal structure of the SARS-CoV main protease offers a solid basis for rational drug design of SARS-CoV protease inhibitors [36]. For other potential targets, for example the NTPase/helicase and RNA replicase, such a structural basis still has to be elaborated. Meanwhile, a variety of ‘old’ and ‘new’ compounds have been reported to inhibit the in vitro replication of SARS-CoV when present in relatively high concentrations (≥1 μM) [37]. There is no shortage of small molecules that are active in the 1–10 μM concentration range [38], but whether any of these molecules will prevent or suppress the infection in vivo remains to be established.
Should SARS re-emerge, now two years after its first apparition (and subsequent disappearance), perhaps the pegylated form of IFN-α would be a logical choice for the prophylaxis and early post-exposure management of SARS: IFN-α inhibits SARS-CoV replication in vitro [39], and pegylated IFN-α was recently shown to reduce viral replication and excretion, to decrease viral antigen expression by type-1 pneumocytes, and to reduce the attendant pulmonary damage in cynomolgus macaques infected experimentally with SARS-CoV [40•].
Influenza virus infections
Given the persistent epidemics of influenza A strains H3N2 and H1N1 and the threat of a pandemic with the avian influenza A strain H5N1, combined with the fact that no vaccination is available for the latter, much emphasis has been recently put on the chemotherapy (and prophylaxis) of influenza virus infections. There are, in principle, three classes of compounds that could be considered for this purpose: ribavirin, amantadine and rimantadine, and the neuraminidase inhibitors zanamivir and oseltamivir (Figure 1).
Although ribavirin is active in vitro against the replication of influenza virus, it has not been actively pursued for the management of influenza virus infections in vivo. Instead, it has been developed for inhalation as a small-particle aerosol formulation in patients (primarily young infants) that are at (high) risk of bronchopneumonitis caused by respiratory syncytial virus infection.
The matrix (M2) hydrogen ion channel blockers amantadine and rimantadine have been used for many years for the prophylaxis and therapy of influenza A virus infections. However, in the past they did not gain wide acceptance, mainly for the following reasons: they rapidly lead to the emergence of drug-resistant virus mutants; they are not active against influenza B; and it has been demonstrated, particularly for amantadine, that they exert side effects on the central nervous system.
At present, only the neuraminidase inhibitors zanamivir and oseltamivir are practically available for the therapy and/or prophylaxis of influenza A and B virus infections. Influenza virus has adopted a unique replication strategy: it uses one of its surface glycoproteins, hemagglutinin (H), to bind to the target cell receptor, which contains a terminal sialic acid (_N_-acetylneuraminic acid), and another surface glycoprotein, neuraminidase, to cleave off the terminal sialic acid of the host cell receptor, allowing the virus particles to leave the cells after the viral replicative cycle has been completed [37, 41]. Neuraminidase inhibitors block the release of newly formed virus particles, thus preventing their further spread to other host cells.
The benefits offered by the neuraminidase inhibitors are substantial. When used therapeutically, they lead to a reduction in illness in 1–2 days, a reduction in virus transmission to household or healthcare contacts, and a reduction in the frequency and severity of complications (such as sinusitis and bronchitis); they therefore decrease the requirement for antibiotics. When used prophylactically, they significantly reduce the number of new cases that have influenza. Although resistance to neuraminidase inhibitors can develop [42], there is no evidence of naturally occurring resistance to either zanamivir or oseltamivir [43]. Zanamivir and oseltamivir are both able to block influenza A and B viruses, and would, in theory, also be effective against the 1918 pandemic influenza A (H1N1) virus [44•]. Zanamivir, which must be taken by (oral) inhalation, and in particular oseltamivir, which can be more conveniently administered as oral capsules, should be stockpiled to affront a potential influenza A pandemic in the future.
Hemorrhagic fever virus infections
The most important hemorrhagic fever viruses fall within the families of the flaviviridae (i.e. yellow fever and dengue), arenaviridae (i.e. Lassa, Junin, Machupo, Guanarito and Sabia), bunyaviridae (i.e. Rift Valley, Crimean-Congo and Hantaan) and filoviridae (i.e. Ebola, Marburg).
Prospects for the therapy of flavivirus infections are not overwhelming [37]. Ribavirin has only weak activity against flaviviruses. At present, IFN-α, whether pegylated or not, and IFN inducers, such as poly(I)·poly(C) and ampligen, offer an appreciable potential for the prophylaxis and/or therapy of flavivirus infections as they decrease virus-induced morbidity (paralysis) and mortality (caused by progressive encephalitis) in an experimental flavivirus encephalitis model in severe combined immunodeficient mice [45]. Another approach, based on the conjugation of phosphorodiamidate morpholino oligomers with arginine-rich peptides to increase their cellular uptake, has been described to inhibit the replication of several RNA viruses in vitro, including dengue virus types 1–4 [46], but it remains to be demonstrated whether or not this antisense approach also offers in vivo efficacy.
For arenaviruses and bunyaviruses, ribavirin (Figure 1) can be accredited with some efficacy in experimental animal models as well as in humans (in the latter case, against Lassa fever [37]). Various mechanisms of action have been proposed to explain the antiviral activity of ribavirin. The most fascinating of these is the theory of ‘error catastrophe’ [47, 48]. According to this hypothesis, ribavirin would act as an RNA virus mutagen, forcing the RNA virus into gaining a lethal accumulation of errors. This error catastrophe has been shown only with poliovirus; it has not been demonstrated with other RNA viruses such as HIV or HCV. For flaviviruses (such as yellow fever) and paramyxoviruses (such as parainfluenza), the predominant mechanism by which ribavirin exerts its antiviral activity in vitro appears to be based on inhibition of inosine 5’-monophosphate dehydrogenase [41, 49].
For the treatment of Ebola (or Marburg) virus infections, _S_-adenosylhomocysteine hydrolase inhibitors (such as carbocyclic 3-deazaadenosine and 3-deazaneplanocin A; Figure 1) might be worth pursuing: even when administered as a single dose on the first or second day after infection, 3-deazaneplanocin A was found to protect almost all the mice against a lethal Ebola virus infection, and this protective effect was accompanied, and probably mediated, by an enhanced production of IFN in the Ebola virus-infected animals [50, 51]. A totally different approach towards the treatment of Ebola virus infection is based on the use of a recombinant nematode anticoagulant protein C2, a potent inhibitor of tissue factor-initiated blood coagulation. This strategy targets the disease process rather than viral replication [52].
Human immunodeficiency virus infections
HIV has generated more efforts towards antiviral drug development than any other virus. In recent years, we have witnessed the development of novel approaches as well as newly emerging (candidate) drugs for anti-HIV therapy [53•, 54]. The compounds that are currently under (pre)clinical investigation target the same specific viral proteins as the licensed compounds, target specific novel viral proteins, or target cellular proteins [54]. Also, new acyclic nucleoside phosphonates (i.e. the 6-[2-(phosphonomethoxy)alkoxy]-2,4-diamino pyrimidines PMPO-DAPy and PMEO-DAPy [55]) and deoxythreosyl nucleoside phosphonates (i.e. phosphonomethyldeoxythreosyl-adenine [PMDTA] and -thymine [PMDTT]; Figure 1) [56•] have been described as potent and selective anti-HIV agents.
Some compounds, such as cyanovirin-N and thiocarboxanilide UC-781 are being actively pursued as topical (i.e. vaginal) microbicides. Also, the plant lectins (i.e. Galanthus nivalis agglutinin and Hippeastrum hybrid agglutinin) represent potential candidate anti-HIV microbicides: they show marked stability at relatively low pH and high temperatures for prolonged time periods, they directly interact with the viral envelope, and they prevent entry of HIV into its target cell [57]. Upon prolonged exposure (in cell culture, in vitro) of HIV to Hippeastrum hybrid agglutinin or to Galanthus nivalis agglutinin, the virus acquires resistance mutations in the gp120 glycoprotein that are predominantly located at the _N_-glycosylation (asparagine) sites [58]. In addition to their role as topical microbicides, plant lectins might, due to their propensity to deplete the viral envelope of its glycan shield [59], force the virus to convert to a gp120 phenotype that can no longer escape the immune surveillance of the host.
As a final highlight in the search for new anti-HIV agents, I would like to cite the multidisciplinary approach that, over a period of 17 years, led to the identification of what can now be claimed to be (one of) the most potent anti-HIV agents ever discovered: the NNRTI R-278474, which is also known as rilpivirine [60••]. This chemical evolution starting from TIBO R-86183 in 1987 has yielded compound R-278474, which shows activity against both wild-type and NNRTI mutant (K103N + Y181C) HIV-1 strains at sub-nanomolar concentrations. The discovery of R-278474 emanated from a coordinated multidisciplinary effort that involved medicinal chemists, virologists, crystallographers, molecular modellers, toxicologists, analytical chemists and pharmacists, among others [60••]. R-278474 highlights the important requirements of novel anti-HIV drugs: high antiviral potency against wild-type and mutant viruses, high oral bioavailability (allowing once-daily administration), minimal side effects, and ease of synthesis and formulation.
Conclusions
In recent years, considerable progress has been made towards the treatment of a wide variety of virus infections, irrespective of whether or not they are currently amenable to antiviral therapy. Highlighted in this review are: the potential of cidofovir and other acyclic nucleoside phosphonates for the treatment of HPV-associated papillomatous lesions, future prospects for the treatment of adenovirus, HSV, VZV and HHV-6 infections, and the potential of oral cidofovir prodrugs for the prophylaxis and therapy of poxvirus infections. I have also discussed therapeutic approaches, both present and future, for HCV infections, antiviral strategies for the management of SARS, and the importance of having neuraminidase inhibitors at hand to affront future influenza virus epidemics. Possible strategies for the prevention and treatment of hemorrhagic fever virus infections are identified, and the many new (candidate) antiviral drugs that have been developed for the treatment of HIV infections (AIDS) are also reviewed.
References and recommended reading
Papers of particular interest, published within the annual period of review, have been highlighted as:
- • of special interest
- •• of outstanding interest
Acknowledgements
I am grateful to Christiane Callebaut for her invaluable editorial assistance.
References
- 1.De Clercq E. Antiviral drugs in current clinical use. J Clin Virol. 2004;30:115–133. doi: 10.1016/j.jcv.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 2.De Clercq E. Potential of acyclic nucleoside phosphonates in the treatment of DNA virus and retrovirus infections. Expert Rev Anti Infect Ther. 2003;1:21–43. doi: 10.1586/14787210.1.1.21. [DOI] [PubMed] [Google Scholar]
- 3.De Clercq E. Clinical potential of the acyclic nucleoside phosphonates cidofovir, adefovir, and tenofovir in treatment of DNA virus and retrovirus infections. Clin Microbiol Rev. 2003;16:569–596. doi: 10.1128/CMR.16.4.569-596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naesens L., Lenaerts L., Andrei G., Snoeck R., Van Beers D., Holý A., Balzarini J., De Clercq E. Antiadenovirus activities of several classes of nucleoside and nucleotide analogues. Antimicrob Agents Chemother. 2005;49:1010–1016. doi: 10.1128/AAC.49.3.1010-1016.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartline C.B., Gustin K.M., Wan W.B., Ciesla S.L., Beadle J.R., Hostetler K.Y., Kern E.R. Ether lipid-ester prodrugs of acyclic nucleoside phosphonates: activity against adenovirus replication in vitro. J Infect Dis. 2005;191:396–399. doi: 10.1086/426831. [DOI] [PubMed] [Google Scholar]
- 6.De Clercq E. Highly potent and selective inhibition of varicella-zoster virus replication by bicyclic furo[2,3-d]pyrimidine nucleoside analogues. Med Res Rev. 2003;23:253–274. doi: 10.1002/med.10035. [DOI] [PubMed] [Google Scholar]
- 7.Andrei G., Sienaert R., McGuigan C., De Clercq E., Balzarini J., Snoeck R. Susceptibilities of several clinical varicella-zoster virus (VZV) isolates and drug-resistant VZV strains to bicyclic furano pyrimidine nucleosides. Antimicrob Agents Chemother. 2005;49:1081–1086. doi: 10.1128/AAC.49.3.1081-1086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crute J.J., Grygon C.A., Hargrave K.D., Simoneau B., Faucher A.M., Bolger G., Kibler P., Liuzzi M., Cordingley M.G. Herpes simplex virus helicase–primase inhibitors are active in animal models of human disease. Nat Med. 2002;8:386–391. doi: 10.1038/nm0402-386. [DOI] [PubMed] [Google Scholar]
- 9.Kleymann G., Fischer R., Betz U.A., Hendrix M., Bender W., Schneider U., Handke G., Eckenberg P., Hewlett G., Pevzner V. New helicase–primase inhibitors as drug candidates for the treatment of herpes simplex disease. Nat Med. 2002;8:392–398. doi: 10.1038/nm0402-392. [DOI] [PubMed] [Google Scholar]
- 10.Betz U.A., Fischer R., Kleymann G., Hendrix M., Rübsamen-Waigmann H. Potent in vivo antiviral activity of the herpes simplex virus primase–helicase inhibitor BAY 57-1293. Antimicrob Agents Chemother. 2002;46:1766–1772. doi: 10.1128/AAC.46.6.1766-1772.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pilger B.D., Cui C., Coen D.M. Identification of a small molecule that inhibits herpes simplex virus DNA polymerase subunit interactions and viral replication. Chem Biol. 2004;11:647–654. doi: 10.1016/j.chembiol.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 12•.De Bolle L., Naesens L., De Clercq E. Update on human herpesvirus 6 biology, clinical features, and therapy. Clin Microbiol Rev. 2005;18:217–245. doi: 10.1128/CMR.18.1.217-245.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]; Comprehensive review on all aspects, including therapy, of HHV-6 infections.
- 13.Thomsen D.R., Oien N.L., Hopkins T.A., Knechtel M.L., Brideau R.J., Wathen M.W., Homa F.L. Amino acid changes within conserved region III of the herpes simplex virus and human cytomegalovirus DNA polymerases confer resistance to 4-oxo-dihydroquinolines, a novel class of herpesvirus antiviral agents. J Virol. 2003;77:1868–1876. doi: 10.1128/JVI.77.3.1868-1876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Bolle L., Andrei G., Snoeck R., Zhang Y., Van Lommel A., Otto M., Bousseau A., Roy C., De Clercq E., Naesens L. Potent, selective and cell-mediated inhibition of human herpesvirus 6 at an early stage of viral replication by the non-nucleoside compound CMV423. Biochem Pharmacol. 2004;67:325–336. doi: 10.1016/j.bcp.2003.08.042. [DOI] [PubMed] [Google Scholar]
- 15.De Clercq E., Neyts J. Therapeutic potential of nucleoside/nucleoide analogues against poxvirus infections. Rev Med Virol. 2004;14:289–300. doi: 10.1002/rmv.439. [DOI] [PubMed] [Google Scholar]
- 16.Neyts J., Leyssen P., Verbeken E., De Clercq E. Efficacy of cidofovir in a murine model of disseminated progressive vaccinia. Antimicrob Agents Chemother. 2004;48:2267–2273. doi: 10.1128/AAC.48.6.2267-2273.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Clercq E. Cidofovir in the treatment of poxvirus infections. Antiviral Res. 2002;55:1–13. doi: 10.1016/S0166-3542(02)00008-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18•.Painter G.R., Hostetler K.Y. Design and development of oral drugs for the prophylaxis and treatment of smallpox infection. Trends Biotechnol. 2004;22:423–427. doi: 10.1016/j.tibtech.2004.06.008. [DOI] [PubMed] [Google Scholar]; Rationale for the use of cidofovir derivatives that could be administered perorally to control (prevent and treat) poxvirus infections.
- 19.Kern E.R., Hartline C., Harden E., Keith K., Rodriguez N., Beadle J.R., Hostetler K.Y. Enhanced inhibition of orthopoxvirus replication in vitro by alkoxyalkyl esters of cidofovir and cyclic cidofovir. Antimicrob Agents Chemother. 2002;46:991–995. doi: 10.1128/AAC.46.4.991-995.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Quenelle D.C., Collins D.J., Wan W.B., Beadle J.R., Hostetler K.Y., Kern E.R. Oral treatment of cowpox and vaccinia virus infections in mice with ether lipid esters of cidofovir. Antimicrob Agents Chemother. 2004;48:404–412. doi: 10.1128/AAC.48.2.404-412.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smee D.F., Wong M.-H., Bailey K.W., Beadle J.R., Hostetler K.Y., Sidwell R.W. Effects of four antiviral substances on lethal vaccinia virus (IHD strain) respiratory infections in mice. Int J Antimicrob Agents. 2004;23:430–437. doi: 10.1016/j.ijantimicag.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 22.Fried M.W., Shiffman M.L., Reddy K.R., Smith C., Marinos G., Gonçales F.L., Häussinger D., Diago M., Carosi G., Dhumeaux D. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347:975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 23.McHutchison J.G., Manns M., Patel K., Poynard T., Lindsay K.L., Trepo C., Dienstag J., Lee W.M., Mak C., Garaud J.-J. Adherence to combination therapy enhances sustained response in genotype-1-infected patients with chronic hepatitis C. Gastroenterology. 2002;123:1061–1069. doi: 10.1053/gast.2002.35950. [DOI] [PubMed] [Google Scholar]
- 24.Hadziyannis S.J., Sette H., Jr., Morgan T.R., Balan V., Diago M., Marcellin P., Ramadori G., Bodenheimer H., Jr., Bernstein D., Rizzetto M. Peginterferon-α2a and ribavirin combination therapy in chronic hepatitis C: a randomized study of treatment duration and ribavirin dose. Ann Intern Med. 2004;140:346–355. doi: 10.7326/0003-4819-140-5-200403020-00010. [DOI] [PubMed] [Google Scholar]
- 25.Tan S.L., Pause A., Shi Y., Sonenberg N. Hepatitis C therapeutics: current status and emerging strategies. Nat Rev Drug Discov. 2002;1:867–881. doi: 10.1038/nrd937. [DOI] [PubMed] [Google Scholar]
- 26.Lamarre D., Anderson P.C., Bailey M., Beaulieu P., Bolger G., Bonneau P., Bos M., Cameron D.R., Cartier M., Cordingley M.G. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature. 2003;426:186–189. doi: 10.1038/nature02099. [DOI] [PubMed] [Google Scholar]
- 27.Baginski S.G., Pevear D.C., Seipel M., Sun S.C., Benetatos C.A., Chunduru S.K., Rice C.M., Collett M.S. Mechanism of action of a pestivirus antiviral compound. Proc Natl Acad Sci USA. 2000;97:7981–7986. doi: 10.1073/pnas.140220397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun J.H., Lemm J.A., O’Boyle D.R., II, Racela J., Colonno R., Gao M. Specific inhibition of bovine viral diarrhea virus replicase. J Virol. 2003;77:6753–6760. doi: 10.1128/JVI.77.12.6753-6760.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paeshuyse J., Leyssen P., Mabery E., Boddeker N., Donis R., Ansari I.H., Koenen F., Kerkhofs P., Gil L., Letellier C. A novel highly selective inhibitor of pestivirus replication that targets the viral RNA-dependent RNA polymerase. Antiviral Res. 2004;62:A84–A85. doi: 10.1128/JVI.80.1.149-160.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stuyver L.J., Whitaker T., McBrayer T.R., Hernandez-Santiago B.I., Lostia S., Tharnish P.M., Ramesh M., Chu C.K., Jordan R., Shi J. Ribonucleoside analogue that blocks replication of bovine viral diarrhea and hepatitis C viruses in culture. Antimicrob Agents Chemother. 2003;47:244–254. doi: 10.1128/AAC.47.1.244-254.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stadler K., Masignani V., Eickmann M., Becker S., Abrignani S., Klenk H.D., Rappuoli R. SARS — beginning to understand a new virus. Nat Rev Microbiol. 2003;1:209–218. doi: 10.1038/nrmicro775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li W., Moore M.J., Vasilieva N., Sui J., Wong S.K., Berne M.A., Somasundaran M., Sullivan J.L., Luzuriaga K., Greenough T.C. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454. doi: 10.1038/nature02145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wong S.K., Li W., Moore M.J., Choe H., Farzan M. A 193-amino acid fragment of the SARS coronavirus S protein efficiently binds angiotensin-converting enzyme 2. J Biol Chem. 2004;279:3197–3201. doi: 10.1074/jbc.C300520200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Siu J., Li W., Murakami A., Tamin A., Matthews L.J., Wong S.K., Moore M.J., St Clair Tallarico A., Olurinde M., Choe H. Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association. Proc Natl Acad Sci USA. 2004;101:2536–2541. doi: 10.1073/pnas.0307140101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu S., Xiao G., Chen Y., He Y., Niu J., Escalante C.R., Xiong H., Farmar J., Debnath A.K., Tien P. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. 2004;363:938–947. doi: 10.1016/S0140-6736(04)15788-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yang H., Yang M., Ding Y., Liu Y., Lou Z., Zhou Z., Sun L., Mo L., Ye S., Pang H. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc Natl Acad Sci USA. 2003;100:13190–13195. doi: 10.1073/pnas.1835675100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Clercq E. Antivirals and antiviral strategies. Nat Rev Microbiol. 2004;2:704–720. doi: 10.1038/nrmicro975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu C.-Y., Jan J.-T., Ma S.-H., Kuo C.-J., Juan H.-F., Cheng Y.-S.E., Hsu H.-H., Huang H.-C., Wu D., Brik A. Small molecules targeting severe acute respiratory syndrome human coronavirus. Proc Natl Acad Sci USA. 2004;101:10012–10017. doi: 10.1073/pnas.0403596101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ströher U., DiCaro A., Li Y., Strong J.E., Aoki F., Plummer F., Jones S.M., Feldmann H. Severe acute respiratory syndrome-related coronavirus is inhibited by interferon-α. J Infect Dis. 2004;189:1164–1167. doi: 10.1086/382597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40•.Haagmans B.L., Kuiken T., Martina B.E., Fouchier R.A., Rimmelzwaan G.F., van Amerongen G., van Riel D., de Jong T., Itamura S., Chan K.H. Pegylated interferon-α protects type 1 pneumocytes against SARS coronavirus infection in macaques. Nat Med. 2004;10:290–293. doi: 10.1038/nm1001. [DOI] [PMC free article] [PubMed] [Google Scholar]; If SARS re-emerges, in the wake of any approved vaccine or treatment, pegylated IFN-α, now available for the treatment of chronic hepatitis C, should be the drug of choice to prevent and/or control a new SARS epidemic.
- 41.De Clercq E. Strategies in the design of antiviral drugs. Nat Rev Drug Discov. 2002;1:13–25. doi: 10.1038/nrd703. [DOI] [PubMed] [Google Scholar]
- 42.Kiso M., Mitamura K., Sakai-Tagawa Y., Shiraishi K., Kawakami C., Kimura K., Hayden F.G., Sugaya N., Kawaoka Y. Resistant influenza A viruses in children treated with oseltamivir: descriptive study. Lancet. 2004;364:759–765. doi: 10.1016/S0140-6736(04)16934-1. [DOI] [PubMed] [Google Scholar]
- 43.McKimm-Breschkin J., Trivedi T., Hampson A., Hay A., Klimov A., Tashiro M., Hayden F., Zambon M. Neuraminidase sequence analysis and susceptibilities of influenza virus clinical isolates to zanamivir and oseltamivir. Antimicrob Agents Chemother. 2003;47:2264–2272. doi: 10.1128/AAC.47.7.2264-2272.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44•.Oxford J., Balasingam S., Lambkin R. A new millenium conundrum: how to use a powerful class of influenza anti-neuraminidase drugs (NAIs) in the community. J Antimicrob Chemother. 2004;53:133–136. doi: 10.1093/jac/dkh037. [DOI] [PMC free article] [PubMed] [Google Scholar]; Neuraminidase inhibitors (zanamivir and/or oseltamivir) should be made available (stockpiled) for prophylactic and therapeutic use in case of an influenza epidemic.
- 45.Leyssen P., Drosten C., Paning M., Charlier N., Paeshuyse J., De Clercq E., Neyts J. Interferons, interferon inducers, and interferon-ribavirin in treatment of flavivirus-induced encephalitis in mice. Antimicrob Agents Chemother. 2003;47:777–782. doi: 10.1128/AAC.47.2.777-782.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kinney R.M., Huang CY.-H., Rose B.C., Kroeker A.D., Dreher T.W., Iversen P.L., Stein D.A. Inhibition of dengue virus serotypes 1 to 4 in Vero cell cultures with morpholino oligomers. J Virol. 2005;79:5116–5128. doi: 10.1128/JVI.79.8.5116-5128.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crotty S., Maag D., Arnold J.J., Zhong W., Lau J.Y.N., Hong Z., Andino R., Cameron C.E. The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat Med. 2000;6:1375–1379. doi: 10.1038/82191. [DOI] [PubMed] [Google Scholar]
- 48.Crotty S., Cameron C.E., Andino R. RNA virus error catastrophe: direct molecular test by using ribavirin. Proc Natl Acad Sci USA. 2001;98:6895–6900. doi: 10.1073/pnas.111085598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leyssen P., Balzarini J., De Clercq E., Neyts J. The predominant mechanism by which ribavirin exerts its antiviral activity in vitro against flaviviruses and paramyxoviruses is mediated by inhibition of IMP dehydrogenase. J Virol. 2005;79:1943–1947. doi: 10.1128/JVI.79.3.1943-1947.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bray M., Driscoll J., Huggins J.W. Treatment of lethal Ebola virus infection in mice with a single dose of an S-adenosyl-l-homocysteine hydrolase inhibitor. Antiviral Res. 2000;45:135–147. doi: 10.1016/s0166-3542(00)00066-8. [DOI] [PubMed] [Google Scholar]
- 51.Bray M., Raymond J.L., Geisbert T., Baker R.O. 3-Deazaneplanocin A induces massively increased interferon-α production in Ebola virus-infected mice. Antiviral Res. 2002;55:151–159. doi: 10.1016/s0166-3542(02)00018-9. [DOI] [PubMed] [Google Scholar]
- 52.Geisbert T.W., Hensley L.E., Jahrling P.B., Larsen T., Geisbert J.B., Paragas J., Young H.A., Fredeking T.M., Rote W.E., Vlasuk G.P. Treatment of Ebola virus infection with a recombinant inhibitor of factor VIIa/tissue factor: a study in rhesus monkeys. Lancet. 2003;362:1953–1958. doi: 10.1016/S0140-6736(03)15012-X. [DOI] [PubMed] [Google Scholar]
- 53•.De Clercq E. New approaches toward anti-HIV chemotherapy. J Med Chem. 2005;48:1297–1313. doi: 10.1021/jm040158k. [DOI] [PubMed] [Google Scholar]; What's new in the anti-HIV chemotherapy field? An updated review on current anti-HIV drugs, as a tribute to the late Dr. Paul Janssen.
- 54.De Clercq E. Emerging anti-HIV drugs. Expert Opin Emerg Drugs. 2005;10:241–274. doi: 10.1517/14728214.10.2.241. [DOI] [PubMed] [Google Scholar]
- 55.De Clercq E, Andrei G, Balzarini J, Leyssen P, Naesens L, Neyts J, Pannecouque C, Snoeck R, Ying Y, Hocková D et al.: Antiviral potential of a new generation of acyclic nucleoside phosphonates, the 6-[2-(phosphonomethoxy)alkoxy]-2,4-diaminopyrimidines. Nucleosides Nucleotides Nucleic Acids 2005, in press. [DOI] [PubMed]
- 56•.Wu T., Froeyen M., Kempeneers V., Pannecouque C., Wang J., Busson R., De Clercq E., Herdewijn P. Deoxythreosyl phosphonate nucleosides as selective anti-HIV agents. J Am Chem Soc. 2005;127:5056–5065. doi: 10.1021/ja043045z. [DOI] [PubMed] [Google Scholar]; Deoxyribonucleoside (as well as ribonucleoside) phosphonates might represent a new approach for the therapy of HIV and hepatitis B virus (as well as HCV) infections.
- 57.Balzarini J., Hatse S., Vermeire K., Princen K., Aquaro S., Perno C.-F., De Clercq E., Egberink H., Vanden Mooter G., Peumans W. Mannose-specific plant lectins from the Amaryllidaceae family qualify as efficient microbicides for prevention of human immunodeficiency virus infection. Antimicrob Agents Chemother. 2004;48:3858–3870. doi: 10.1128/AAC.48.10.3858-3870.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Balzarini J., Van Laethem K., Hatse S., Vermeire K., De Clercq E., Peumans W., Van Damme E., Vandamme A.-M., Bolmstedt A., Schols D. Profile of resistance of human immunodeficiency virus to mannose-specific plant lectins. J Virol. 2004;78:10617–10627. doi: 10.1128/JVI.78.19.10617-10627.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Balzarini J., Van Laethem K., Hatse S., Froeyen M., Van Damme E., Bolmstedt A., Peumans W., De Clercq E., Schols D. Marked depletion of glycosylation sites in HIV-1 gp120 under selection pressure by the mannose-specific plant lectins of Hippeastrum hybrid and Galanthus nivalis. Mol Pharmacol. 2005;67:1556–1565. doi: 10.1124/mol.104.005082. [DOI] [PubMed] [Google Scholar]
- 60••.Janssen P.A., Lewi P.J., Arnold E., Daeyaert F., de Jonge M., Heeres J., Koymans L., Vinkers M., Guillemont J., Pasquier E. In search of a novel anti-HIV drug: multidisciplinary coordination in the discovery of 4-[[4-[[4-[(1E)-2-cyanoethenyl]-2,6-dimethylphenyl]amino]-2-pyrimidinyl]amino]benzonitrile (R278474, rilpivirine) J Med Chem. 2005;48:1901–1909. doi: 10.1021/jm040840e. [DOI] [PubMed] [Google Scholar]; Representative example of the unabated medicinal chemistry approach towards the development of the ‘cure’ for AIDS: the legacy of Dr. Paul Janssen.