Chemokine profiling of Japanese encephalitis virus-infected mouse neuroblastoma cells by microarray and real-time RT-PCR: Implication in neuropathogenesis (original) (raw)
Abstract
Japanese encephalitis (JE) is one of the leading causes of acute encephalopathy affecting children and adolescents in the tropics. JE virus (JEV) infection causes prominent neurological sequelae in approximately one-third of the survivors. In humans, the inflammatory response of CNS consequent to JEV induced viral encephalitis is mediated through chemokines released by various cells of CNS. In the present study, the chemokine profiles of mouse neuroblastoma cells (N2A) following JEV infection was analyzed by cDNA microarray followed by real-time RT-PCR. Eighty mRNA transcripts belonging to various functional classes exhibited significant alterations in gene expression. There was considerable induction of genes involved in apoptosis and anti-viral response. Modified levels of several transcripts involved in proinflammatory and anti-inflammatory processes exemplified the balance between opposing forces during JEV pathogenesis. Other genes displaying altered transcription included those associated with host translation, cellular metabolism, cell cycle, signal transduction, transcriptional regulation, protein trafficking, neurotransmitters, neuron maturation, protein modulators, ER stress and cytoskeletal proteins. The infection of neurons results in the synthesis of proinflammatory chemokines, which are early important mediators of leukocyte recruitment to sites of viral infection. Our results clearly suggest the implication of chemokines in neuropathogenesis of JEV infection leading to neurological sequelae. Pro- and anti-inflammatory agents targeted against chemokines such as CXCL10 may provide possible therapeutic modalities that can mitigate the morbidity associated with JEV infection of the CNS.
Keywords: Mouse neuroblastoma cells, Japanese encephalitis, Chemokine, Microarray, Gene expression
Japanese encephalitis has become a major cause of mortality and morbidity in wide areas of south and southeast Asia and kept in a zoonotic transmission cycle between pigs or birds and mosquitoes (Burke et al., 2001, Solomon et al., 2003, Tsai, 2000). JEV spreads to dead-end hosts, including humans, through the bite of JEV-infected mosquitoes and causes infection of the central nervous system, with a high mortality rate (Parida et al., 2006a). JEV targets the CNS, clinically manifesting with fever, headache, vomiting, signs of meningeal irritation, and altered consciousness, leading to high mortality and neurological sequelae in some of those who survive (Kumar et al., 1990). After entry into the host, JEV generates a rapid inflammatory response, including peripheral neutrophil leucocytosis or infiltration of neutrophils in extraneural tissue. Clinically, the infection of JEV results in increased levels of cytokines such as macrophage-derived chemotactic factor, TNF, and interleukin-8 (IL-8) in the serum and cerebrospinal fluid (Khanna et al., 1991, Ravi et al., 1997, Singh et al., 2000). The increased levels of inflammatory mediators appear to play a protective role or initiate an irreversible immune response leading to cell death. Despite the fact that Japanese encephalitis is a major disease affecting the tropical world, little is known of its pathogenesis due, partly, to the lack of a suitable animal model and the complex cell interactions in infected individuals. JEV tends to cause a neurotrophic infection, attacking neural rather than non-neural tissues in humans. Experimental studies in rats have demonstrated that neuron cells, especially developing neurons are the major target cells for infection. The role that neurons play in the induction of the immune response following CNS viral infection is poorly understood, largely owing to the belief that these cells are immunologically quiescent. In order to understand the nature and consequence at the transcriptional level, we have carried out the microarray analysis of mock-infected and JE virus-infected murine neuroblastoma cells. The microarray data was validated with qRT-PCR experiments and time point analysis was done to demonstrate the induction of chemokine by JEV-infected neurons as an early defense mechanism. We employed mouse neuronal cells (N2A) as a model system because of the permissiveness of these cells to JEV infection (Murali-Krishna et al., 1995, Yang et al., 2004, Lin et al., 2004, Hong-Lin et al., 2002). In this study, we demonstrated that virus infection of neuronal cells results in the strong induction of proinflammatory chemokines along with robust anti-viral response. The replicating virus within the infected host neurons alters normal gene expression profiles, which can be of significance in JEV pathogenesis at the molecular level.
A neurovirulent JEV strain, JE S982, was employed throughout this study. The propagation of virus was carried out in C6/36 cells utilizing RPMI-1640 medium containing 10% fetal bovine serum. To determine virus titers, culture media were harvested for plaque-forming assays using Porcine stable Kidney cells. The mouse neuroblastoma, N2A cell line was maintained in antibiotic-free minimum essential medium (MEM) with Earle's salts supplemented with 2 mM l-glutamine and 10% fetal bovine serum.
For viral infection, monolayers of the mouse neuroblastoma cells grown in 6- or 12-well plates were exposed to either live JE Virus (multiplicity of infection MOI = 5) or mock-infected for 1 h, at 37 °C. The unbound virus was removed from cells by gentle washing with phosphate-buffered saline (PBS), and then the cells were cultured in medium at 37 °C. The quantification of the viral load in harvested infected culture fluid was determined by one-step single tube SYBR Green I mediated JEV Env gene-specific real-time RT-PCR and RT-LAMP (Santhosh et al., 2007, Parida et al., 2006b). Total RNA was extracted from uninfected or JEV-infected cells at 36 h post-infection using the Qiagen (Valencia, CA) RNAEasy Mini kit. RNA quality and integrity was assessed using RNA 6000 Nano Lab Chip on the 2100 Bioanalyzer (Agilent, Palo Alto, CA). Agilent mouse whole genome array (4× 44k; G 4122F) was used and processed for labeling and hybridization as per manufacturer's protocol.
Hybridized arrays were scanned at 5 μm resolution on an Agilent DNA Microarray Scanner, Model G2565BA. Data extraction from images was done using Feature Extraction software of Agilent. The microarray experiment was repeated once. Feature extracted data was analyzed using GeneSpring Gx v7.3.1 software from Agilent. Normalization of the data was done using per spot per chip intensity dependent lowess normalization. Further quality control of normalized data was done using correlation based condition tree to eliminate experimental error. One folds and above differentially regulated genes was filtered from the data. Differentially regulated genes were clustered using gene tree to identify significant gene expression patterns. Ontology based biological analysis was done using Gene Ontology browser in GeneSpring Gx.
Genes with significant transcriptional changes known to be associated with biological significance were selected for further analysis. RT2 Profiler PCR Array of Mouse Inflammatory Cytokines and Receptors (PAMM-011A Superarray Bioscience Corporation, USA) was used for qRT-PCR studies. The housekeeping genes were used for standardization of the initial RNA content of a sample. The result for an individual sample was expressed as the mean expression level of a specific gene. The relative expression between each infected sample and the uninfected control was then calculated and expressed as fold change.
The real-time RT-PCR was carried out for the time point analysis of chemokines using gene-specific primers for CXCL9, CXCL10, and CXCL11 using Quanti Tect primer assay kit (Qiagen, Germany) and Quanti Fast one-step RT-PCR kit (Qiagen, Germany) to validate the secretion of chemokines by neurons itself as preliminary anti-viral response. Reactions were run on Stratagene Mx 3005p system. The threshold cycle (C t) of gene of interest and housekeeping gene (HK) and the difference between their C t values (C t) were determined. Relative changes of gene expression were calculated by the following formula (Livak and Schmittgen, 2001), and the data are represented as fold upregulation/downregulation.fold change=2−ΔΔCt,where ΔΔ_C_ t = (C t of gene of interest, treated – C t of HK gene, treated) − (C t of gene of interest, control − Ct of HK gene, control), C t is the threshold cycle number and HK is the house keeping gene.
The time point for microarray and PCR array study consisted of only 36 hpi of mock-infected (control) and JE virus-infected samples. For microarray and PCR array, two replicates each (control and infected) were used and the experiment is repeated once. The data of Table 1, Table 2, Table 3, Table 4were analyzed by _t_-test and _p_-value of ≤0.05 was considered significant. Data of time course study on chemokine synthesis was analyzed by one-way ANOVA followed Dunnet's test for comparison between control and treatment groups. The level of significance was set at p ≤ 0.05. All experiments were repeated at least thrice.
Table 1.
List of upregulated genes involved in pathogenesis of mouse neuroblastoma cells (N2A) infected with Japanese encephalitis virus.
| Accession no. | Gene name | Gene description | Fold change | _p_-Value |
|---|---|---|---|---|
| NM_021274 | Cxcl10 | Chemokine (C–X–C motif) ligand 10 | 7.71 | 0.019 |
| NM_013653 | Ccl5 | Chemokine (C–C motif) ligand 5 | 6.10 | 0.041 |
| NM_021384 | Rsad2 | Viral hemorrhagic septicemia virus (VHSV) induced gene 1 | 5.11 | 0.049 |
| NM_023386 | Rtp4 | Receptor transporting protein 4 | 4.80 | 0.01 |
| NM_015783 | G1p2 | Interferon, alpha-inducible protein | 4.25 | 0.008 |
| NM_011909 | Usp18 | Ubiquitin specific protease 18 | 4.17 | 0.011 |
| NM_011854 | Oasl2 | 2′–5′ oligoadenylate synthetase-like 2 | 4.06 | 0.025 |
| NM_145211 | Oas1a | 2′–5′ oligoadenylate synthetase 1A | 3.39 | 0.092 |
| NM_010501 | Ifit3 | Interferon-induced protein with tetratricopeptide repeats 3 | 3.38 | 0.07 |
| NM_145209 | Oasl1 | 2′–5′ oligoadenylate synthetase-like 1 | 2.85 | 0.035 |
| NM_007498 | Atf3 | Activating transcription factor 3 | 2.71 | 0.033 |
| NM_019440 | Iigp2 | Interferon-g induced GTPase | 2.51 | 0.046 |
| XM_488522 | Parp14 | Poly(ADP-ribose) polymerase family, member 14 | 2.39 | 0.004 |
| NM_173368 | Chd6 | Chromodomain helicase DNA binding protein 6 | 2.27 | 0.084 |
| NM_029720 | Creld2 | Cysteine-rich with EGF-like domains 2 | 2.22 | 0.023 |
| NM_022324 | Sdf2l1 | Stromal cell-derived factor 2-like 1 | 2.21 | 0.005 |
| NM_008326 | Irgm | Interferon inducible protein 1 | 2.16 | 0.001 |
| NM_173743 | 2310016F22Rik | Hypothetical protein LOC71898 | 2.15 | 0.011 |
| NM_010800 | Bhlhb8 | Muscle, intestine and stomach expression 1 | 2.12 | 0.04 |
| NM_008332 | Ifit2 | Interferon-induced protein with tetratricopeptide repeats 2 | 2.11 | 0.011 |
| NM_013760 | Dnajb9 | DnaJ (Hsp40) homolog, subfamily B, member 9 | 2.10 | 3.96E-04 |
| NM_020583 | Isg20 | Interferon-stimulated protein | 2.02 | 0.031 |
| NM_172715 | A230097K15Rik | Hypothetical protein LOC231510 | 1.95 | 0.007 |
| NM_007837 | Ddit3 | DNA-damage inducible transcript 3 | 1.93 | 0.004 |
| NM_197986 | 1110007F12Rik | Hypothetical protein LOC68487 | 1.90 | 0.085 |
| NM_019717 | Arl6ip2 | ADP-ribosylation factor-like 6 interacting protein 2 isoform 1 | 1.90 | 0.126 |
| NM_022310 | Hspa5 | Heat shock 70 kDa protein 5 (glucose-regulated protein) | 1.86 | 0.076 |
| NM_175397 | 5830484A20Rik | Sp110 nuclear body protein | 1.86 | 0.066 |
| NM_013760 | Dnajb9 | DnaJ (Hsp40) homolog, subfamily B, member 9 | 1.81 | 0.008 |
| NM_008326 | Irgm | Interferon inducible protein 1 | 1.80 | 0.030 |
| NM_009283 | Stat1 | Signal transducer and activator of transcription 1 | 1.79 | 0.031 |
| NM_008929 | Dnajc3 | DnaJ (Hsp40) homolog, subfamily C, member 3 | 1.75 | 0.040 |
| NM_008654 | Myd116 | Myeloid differentiation primary response gene 116 | 1.75 | 0.088 |
| NM_030150 | D11Lgp2e | DNA segment, Chr 11, Lothar Hennighausen 2, expressed | 1.75 | 0.009 |
| NM_012024 | Ppp2r5e | Epsilon isoform of regulatory subunit B56, protein phosphatase 2A | 1.72 | 0.097 |
| NM_013642 | Dusp1 | Dual specificity phosphatase 1 | 1.71 | 0.185 |
| NM_008929 | Dnajc3 | DnaJ (Hsp40) homolog, subfamily C, member 3 | 1.71 | 0.106 |
| NM_013760 | Dnajb9 | DnaJ (Hsp40) homolog, subfamily B, member 9 | 1.69 | 0.002 |
| NM_027057 | Wdfy1 | WD repeat and FYVE domain containing 1 | 1.64 | 0.030 |
| NM_011990 | Slc7a11 | Solute carrier family 7 (cationic amino acid transporter, y+ system), member 11 | 1.63 | 0.111 |
| NM_022331 | Herpud1 | Homocysteine-inducible, endoplasmic reticulum stress-inducible, ubiquitin-like domain member 1 | 1.62 | 0.021 |
| NM_018738 | Igtp | Interferon gamma induced GTPase | 1.60 | 0.017 |
| NM_013606 | Mx2 | Myxovirus (influenza virus) resistance 2 | 1.60 | 0.002 |
Table 2.
List of downregulated genes involved in pathogenesis of mouse neuroblastoma cells (N2A) infected with Japanese encephalitis virus.
| Accession no. | Gene name | Gene description | Fold change | _p_-Value |
|---|---|---|---|---|
| NM_133743 | Lypd3 | GPI-anchored metastasis-associated protein homolog | −0.60 | 0.123 |
| NM_010317 | Gng4 | Guanine nucleotide binding protein (G protein), gamma 4 subunit | −0.60 | 0.107 |
| NM_025681 | Lix1 | Limb expression 1 homolog | −0.60 | 0.099 |
| XM_001005167 | Synpo2 | −0.60 | 0.098 | |
| NM_010826 | Mrvi1 | MRV integration site 1 isoform a | −0.60 | 0.093 |
| NM_027820 | 8430429K09Rik | Hypothetical protein LOC71523 | −0.60 | 0.112 |
| AK080781 | −0.60 | 0.105 | ||
| NM_144556 | Lgi4 | Leucine-rich repeat LGI family, member 4 | −0.60 | 0.127 |
| 4933400F03Rik | −0.60 | 0.153 | ||
| NM_010585 | Itpr1 | Inositol 1,4,5-triphosphate receptor 1 | −0.60 | 0.096 |
| XM_622260 | AI427138 | −0.60 | 0.149 | |
| NM_028133 | Egln3 | EGL nine homolog 3 | −0.60 | 0.130 |
| NM_028940 | 4933402J24Rik | Hypothetical protein LOC74438 | −0.61 | 0.095 |
| NM_053139 | Pcdhb14 | Protocadherin beta 14 | −0.61 | 0.143 |
| NM_138661 | Pcdha9 | Protocadherin alpha 9 | −0.61 | 0.036 |
| NM_011373 | St6galnac4 | ST6 (alpha-N-acetyl-neuraminyl-2,3-beta-galactosyl-1,3)-N-acetylgalactosaminide alpha-2,6-sialyltransferase 4 | −0.61 | 0.112 |
| 8430408O14 | −0.61 | 0.102 | ||
| NM_011867 | Slc26a4 | Pendrin | −0.61 | 0.096 |
| XM_125817 | 1300007O09Rik | −0.61 | 0.102 | |
| NM_133683 | Tmem19 | Transmembrane protein 19 | −0.62 | 0.096 |
| XM_204152 | Frmpd1 | −0.62 | 0.099 | |
| NM_001029889 | Gm608 | Hypothetical protein LOC207806 | −0.62 | 0.108 |
| NM_053146 | Pcdhb21 | Protocadherin beta 21 | −0.62 | 0.931 |
| NM_008666 | Myt1l | Myelin transcription factor 1-like | −0.63 | 0.088 |
| NM_009824 | Cbfa2t3h | Core-binding factor, runt domain, alpha subunit 2, translocated to, 3 homolog | −0.63 | 0.099 |
| 6330439K17Rik | −0.63 | 0.095 | ||
| Gm288 | −0.63 | 0.156 | ||
| NM_175383 | B3gnt6 | Beta-1,3-N-acetylglucosaminyltransferase bGnT-6 | −0.63 | 0.091 |
| NM_009211 | Smarcc1 | SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily c, member 1 | −0.63 | 0.092 |
| XM_485743 | D630002J15Rik | −0.63 | 0.095 | |
| NM_178444 | Egfl7 | Vascular endothelial statin isoform 1 precursor | −0.63 | 0.095 |
| NM_030137 | Cstad | CSA-conditional, T cell activation-dependent protein | −0.63 | 0.092 |
| NM_207298 | Ceecam1 | Cerebral endothelial cell adhesion molecule 1 | −0.63 | 0.109 |
| NM_198119 | Lrrc24 | Leucine-rich repeat containing 24 | −0.64 | 0.087 |
| C130057N11Rik | −0.64 | 0.142 | ||
| NM_027865 | Tmem25 | Transmembrane protein 25 | −0.64 | 0.107 |
| NM_025823 | Pcyox1 | Prenylcysteine oxidase 1 | −0.64 | 0.089 |
| NM_012014 | Gprin1 | G protein-regulated inducer of neurite outgrowth 1 | −0.64 | 0.089 |
| XM_485800 | Aak1 | −0.64 | 0.105 | |
| XM_981846 | Col22a1 | −0.64 | 0.097 | |
| NM_001039079 | Prkcz | Protein kinase C, zeta isoform b | −0.64 | 0.040 |
| NM_025285 | Stmn2 | Stathmin-like 2 | −0.65 | 0.092 |
| NM_020043 | Nope | Neighbor of Punc e11 protein | −0.65 | 0.106 |
| XM_283153 | Polr3g | −0.65 | 0.101 | |
| C230014O12Rik | −0.65 | 0.131 | ||
| XM_130797 | Tnik | −0.65 | 0.103 | |
| NM_013884 | Cspg5 | Chondroitin sulfate proteoglycan 5 | −0.65 | 0.104 |
| NM_172861 | Slc7a14 | Solute carrier family 7, member 14 | −0.65 | 0.097 |
Table 3.
Significant pathways of the upregulated genes at 36 h post-infection with JE virus.
| Pathways | No. of genes | _p_-Value |
|---|---|---|
| Protein export | 2 | 5.46E−06 |
| Toll-like receptor signaling pathway | 7 | 1.88E−05 |
| Circadian rhythm | 5 | 3.22E−05 |
| SNARE interactions in vesicular transport | 5 | 0.000157 |
| MAPK signaling pathway | 14 | 0.0005 |
| Alanine and aspartate metabolism | 3 | 0.002 |
| Aminoacyl-tRNA synthetases | 3 | 0.002 |
| Antigen processing and presentation | 4 | 0.004 |
| One carbon pool by folate | 2 | 0.005 |
| Jak-STAT signaling pathway | 5 | 0.008 |
| Methane metabolism | 1 | 0.023 |
| Novobiocin biosynthesis | 1 | 0.035 |
| N-glycan biosynthesis | 2 | 0.048 |
| Parkinson's disease | 2 | 0.050 |
| Glycosylphosphatidylinositol (GPI)-anchor biosynthesis | 1 | 0.053 |
| Retinol metabolism | 1 | 0.058 |
Table 4.
Significant pathways of the downregulated genes at 36 h post-infection with JE virus.
| Pathway | No. of genes | _p_-Value |
|---|---|---|
| Biosynthesis of steroids | 5 | 0.0006 |
| Androgen and estrogen metabolism | 1 | 0.013 |
| ECM-receptor interaction | 4 | 0.015 |
| Glycan structures – biosynthesis 2 | 2 | 0.019 |
| Diterpenoid biosynthesis | 1 | 0.021 |
| Focal adhesion | 6 | 0.024 |
| Alkaloid biosynthesis I | 1 | 0.031 |
Viral replication in N2A cells infected with JE virus JaOAr S982 for 96 h duration was determined by plaque assay of the virus released in the cell culture medium (Fig. 1). The results showed that new extracellular viral progeny reached maximum at 72 h. After 72 h, a degree of cell death becomes apparent and the experiment was terminated at 96 h (Fig. 2). Transcript analysis on 36 h post-infection was thus selected to track early changes in gene expression in JE virus-infected cells. A total of 497 genes were found to be significantly upregulated after JEV infection and 223 genes were downregulated (Table 1, Table 2). Some of the pathways which show upregulated gene expression are toll-like receptor signaling, MAPK, JAK-STAT, SNARE reactions in vesicular transport, antigen processing and presentation (Table 3). Some of the significantly downregulated pathways are biosynthesis of steroids, androgen and estrogen metabolism, ECM interaction, focal adhesion, etc. (Table 4). Other downregulated genes are making clusters in CNS signature like neuron maturation, nerve ensheathment, neurophysiological process, transmission of nerve impulse, ionic insulation of neurons, myelination, transport, localization and neuron projection (data not shown). Other differentially regulated genes that were significantly changed include those associated with cell signaling, lipid metabolism, cell cycle and vesicular transport.
Fig. 1.
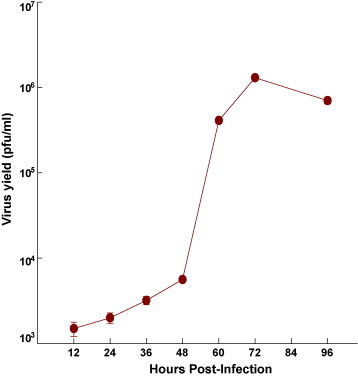
Kinetics of JEV growth in Mouse N2A cells. Monolayers of N2A cells were infected with the JE S982 strain of JEV at an MOI of ∼5 and incubated at 37 °C. At various time intervals, samples were removed and virus titre was assayed. Values are mean ± SE of three replicates each.
Fig. 2.
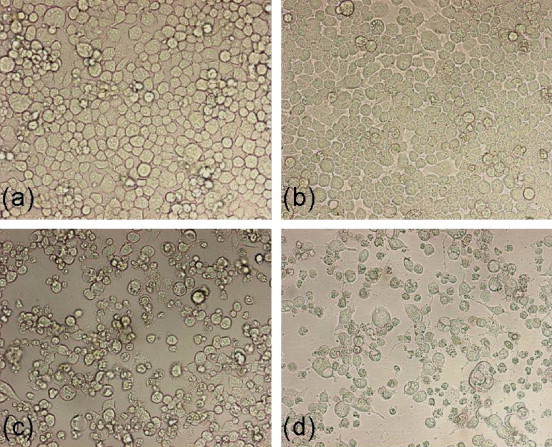
Morphological pattern of neuroblastoma cells infected with JEV. Monolayers of the mouse neuroblastoma cells were adsorbed with either live JE virus (multiplicity of infection (MOI) ∼5) or mock-infected for 1 h, at 37 °C. (a) N2A cells mock-infected at 36 hpi; (b) N2A cells infected with JEV at 36 hpi; (c) N2A cells infected with JEV at 72 hpi; (d) N2A cells infected with JEV at 96 hpi.
In order to verify the reproducibility of the microarray results, the real-time PCR plate assay of mouse inflammatory cytokines and cytokine receptors was carried out. The results showed a set of six genes viz. Itgb, CD40, Tollip, Xcr1, CXCL1 and CXCL10 well corroborated with microarray data. GAPDH, β actin, Gusb, Hsp90 ab1 and Hprt1 were used as housekeeping genes. Out of those six genes, five genes showed congruent and significant differences in expression, while one displayed opposite expression pattern (Table 5). Upregulated genes include CXCL1 (chemokine, C–X–C motif, ligand 1), CXCL10 (chemokine, C–X–C motif, ligand 10), Itgb (integrin beta), and CD 40 (CD40 ligand). downregulated genes include Xcr1 (chemokine, C motif receptor 1). However, the qRT-PCR showed greater dynamism in fold changes than the microarray results because of the greater sensitivity of PCR compared with fluorescent detection in microarray experiment.
Table 5.
Comparison of gene expression changes between microarray and qRT-PCR of some select genes in mouse neuroblastoma cells at 36 h post-infection with JE virus.
| Gene name | Description | Pathway | Microarray | Real-time RT-PCR |
|---|---|---|---|---|
| Itgb | Integrin beta | Focal adhesion, ECM-receptor interaction | +1 | +1.42 |
| CD40 | CD40 ligand | Toll-like receptor signaling | +1.06 | +1.6 |
| Tollip | Toll interacting protein | Toll-like receptor signaling | −0.61 | +1.40 |
| Xcr1 | Chemokine (C motif) receptor 1 | Cytokine–cytokine receptor interaction | −0.3 | −0.5 |
| Cxcl1 | Chemokine (C–X–C motif) ligand 1 | Cytokine–cytokine receptor interaction | +3.1 | +6.68 |
| Cxcl10 | Chemokine (C–X–C motif) ligand 10 | Toll-Like receptor signaling | +7.7 | +35.7 |
We found marked increase in expression of IP-10/CXCL10, which is known to play important role in the host defense against viral infection (Chen et al., 2006). CXCL10 is found to be a crucial molecule governing the protective response against various diseases like dengue, Trypanosoma cruzi, Klebsiella pneumoniae, rabies virus and corona virus infection of the CNS by enhancing innate immune responses (Hsieh et al., 2006, Hardison et al., 2006). IP-10 is an essential component in host defense by coordinating the trafficking of Th1 T lymphocytes into the CNS in response to viral infection. We demonstrated the strong induction of CXCL10 by infected neurons itself. The qRT-PCR analysis showed sustained induction of CXCL10 in JEV-infected mouse neuroblastoma cells. The results indicate that neurons may be a source of chemokine synthesis for primary anti-viral response in JE infection (Fig. 3B). There is gradual decrease in CXCL10 expression with increased viral load in cells, suggesting its anti-viral role in JE infection. No significant fold change was detected for other chemoattractant like CXCL11 and CXCL9 (Fig. 3A–C).
Fig. 3.
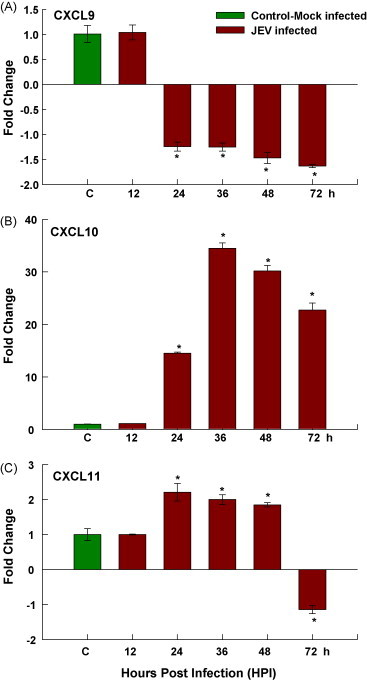
Chemokine expression by JEV-infected mouse neuroblastoma. N2A cells were infected with JEV, at least three replicates per time point were collected at different time intervals, and analysis was done with real-time RT-PCR. The expression of (A) CXCL9, (B) CXCL10 and (C) CXCL11 are shown. Values are mean ± SE of three replicates each. *Significantly different from control at p < 0.05 by Dunnet's test.
Array data also showed Ccl-5 upregulation in JE infection indicating virus modulation of the host machinery in the initial stages of infection. Ccl5–Ccr5 interaction provides anti-apoptotic signals for macrophage survival during viral infection. Ccl5-responsive genes comprise a significant number of enzymes, transcription factors, and miscellaneous molecules involved in neuronal survival and differentiation, including neurite outgrowth and synaptogenesis (Valerio et al., 2004). There was significant upregulation of a number of ubiquitin–proteasome system related genes such as Usp18 during JE virus replication but it was unclear if this response was anti-viral or if the JE virus utilized components of the ubiquitin–proteasome system for its replication. We found significant upregulation in anti-viral response genes like IFN induced Isg15, Viperin, Mx 2, anti-viral genes like oligoadenylate synthetase, Parp14, Irgm, transcription factor-like STAT and ER stress regulated genes like Herpud1, ATF6 and XBP1. The marked elevation in expression of these genes at 36 h post-infection suggests a highly notable anti-viral response to JEV pathogenesis.
Downregulated genes were mainly from focal adhesion and CNS signature, as detailed and vast study is required to elaborate the importance and involvement of these significantly downregulated genes in JEV pathogenesis. Genes like Lypd3, Lgi4 are involved in protein binding, neuron maturation, myelination, etc. The real-time RT-PCR validated genes elucidate the varied transcriptional responses of JE virus-infected neuroblastoma cells. Together with other identified differentially expressed genes, these transcripts provide a better understanding of the pathogenesis of JEV at a transcriptomic level, particularly the molecular events that underpin host defense mechanism against JEV infection.
The results of the present study with mouse neuronal cells clearly show that many genes and host response pathways were upregulated during JE infection. Specific components of the response to virus such as Viperin, G1p2, Ifit3, Atf3, Irgm and CXCL10 have been implicated in JE infection for the first time to the best of our knowledge. Future research is required to explore the mechanism of JEV modulation of these genes to evade the host defense response. Our study offers an overview of the cascade of changes in host cellular expression culminating from infection with JEV. The counterbalancing of several anti-inflammatory and proinflammatory pathways together with the variable expression of apoptosis-related genes is a significant finding of the present study. The involvement of these genes indicated modulation of initial host cell response and balance between cell proliferation and cell death there by enabling virus multiplication. The emerging picture from this study implicates a central role for the immune response in the pathobiology of JE infection. It will be interesting to compare the host response to different JEV isolates with inactivated preparations of the virus.
The present study implies that neurons play an important role in their own defense against viral infections. Although this challenges the long-held belief that neurons are immunologically quiescent, an improved understanding of the proinflammatory effects responsible for immune-mediated control of viral infection and neuronal injury during JEV infection is an essential step for developing strategies for limiting the severity of CNS disease.
Acknowledgements
We thank Dr. R. Vijayaraghavan, Director, DRDE for offering all facilities and support required for this study. Mr. Nimesh Gupta is recipient of DRDO Senior research fellowship.
References
- Burke D.S., Monath T.P., Flaviviruses ., Knipe D.M., Howley P.M., Griffin D.E., Lamb R.A., Martin M.A., Roizman B., Straus S.E., editors. 4th ed. vol. 1. Lippincott Williams & Wilkins; Philadelphia: 2001. pp. 1043–1125. (Fields Virology). [Google Scholar]
- Chen J.P., Lu H.L., Lai S.L., Campanella G.S., Sung J.M., Lu M.Y., Wu-Hsieh B.A., Lin Y.L., Lane T.E., Luster A.D., Liao F. Dengue virus induces expression of CXC chemokine ligand 10/IFN-gamma-inducible protein 10, which competitively inhibits viral binding to cell surface heparan sulfate. J. Immunol. 2006;177:3185–3192. doi: 10.4049/jimmunol.177.5.3185. [DOI] [PubMed] [Google Scholar]
- Hardison J.L., Wrightsman R.A., Carpenter P.M., Lane T.E., Manning J.E. The chemokines CXCL9 and CXCL10 promote a protective immune response but do not contribute to cardiac inflammation following infection with Trypanosoma cruzi. Infect. Immun. 2006;74:125–134. doi: 10.1128/IAI.74.1.125-134.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong-Lin Su, Ching-Len Liao, Lin Yi-Ling. Japanese encephalitis virus infection initiates endoplasmic reticulum stress and an unfolded protein response. J. Virol. 2002:4162–4171. doi: 10.1128/JVI.76.9.4162-4171.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh M.F., Lai S.L., Chen J.P., Sung J.M., Lin Y.L., Wu-Hsieh B.A., Gerard C., Luster A., Liao F. Both CXCR3 and CXCL10/IFN-inducible protein 10 are required for resistance to primary infection by dengue virus. J. Immunol. 2006;177:1855–1863. doi: 10.4049/jimmunol.177.3.1855. [DOI] [PubMed] [Google Scholar]
- Khanna N., Agnihotri M., Mathur A., Chaturvedi U.C. Neutrophil chemotactic factor produced by Japanese encephalitis virus stimulated macrophages. Clin. Exp. Immunol. 1991;86:299–303. doi: 10.1111/j.1365-2249.1991.tb05813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R., Mathur A., Kumar A., Sethi G.D., Sharma S., Chaturvedi U.C. Virological investigations of acute encephalopathy in India. Arch. Dis. Child. 1990;65:1227–1230. doi: 10.1136/adc.65.11.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R.J., Liao C.L., Lin Y.L. Replication-incompetent virions of Japanese encephalitis virus trigger neuronal cell death by oxidative stress in a culture system. J. Gen. Virol. 2004;85:521–533. doi: 10.1099/vir.0.19496-0. [DOI] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D. Analysis of relative gene expression data using real- time quantitative PCR and the 2−ΔΔCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Murali-Krishna K., Ravi V., Manjunath R. Japanese encephalitis virus infection of mouse cell lines: ability to prime mice for generation of virus specific cytotoxic T lymphocytes and differences in CTL recognisable viral determinants. Arch. Virol. 1995;140:127–143. doi: 10.1007/BF01309728. [DOI] [PubMed] [Google Scholar]
- Parida M.M., Dash P.K., Tripathi N.K., Ambuj, Santhosh S.R., Saxena P.S., Agarwal S., Sahni A.K., Singh S.P., Rathi A.K., Bhargava R., Abhyankar A., Verma S.K., Rao P.V.L., Sekhar K. Japanese encephalitis outbreak, India, 2005. Emerg. Infect. Dis. 2006;12:1427–1430. doi: 10.3201/eid1209.060200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parida M.M., Santhosh S.R., Dash P.K., Tripathi N.K., Saxena P., Ambuj, Sahni A.K., Rao P.V.L., Morita K. Development and evaluation of reverse transcription Loop mediated isothermal amplification assay for rapid and real-time detection of Japanese encephalitis virus. J. Clin. Microbiol. 2006;44(11):4172–4178. doi: 10.1128/JCM.01487-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravi V., Parida S., Desai A., Chandramuki A., Gourie-Devi M., Grau G.E. Correlation of tumor necrosis factor levels in the serum and cerebrospinal fluid with clinical outcome in Japanese encephalitis patients. J. Med. Virol. 1997;51:132–136. [PubMed] [Google Scholar]
- Santhosh S.R., Parida M.M., Dash P.K., Pateriya A., Pattnaik B., Pradhan H.K., Tripathi N.K., Ambuj S., Gupta N., Saxena P., Rao P.V.L. Development and evaluation of SYBR Green I-based one-step real time RT-PCR assay for detection and quantification of Japanese encephalitis virus. J. Virol Methods. 2007;143:73–80. doi: 10.1016/j.jviromet.2007.02.011. [DOI] [PubMed] [Google Scholar]
- Singh A., Kulshreshtha R., Mathur A. Secretion of the chemokine interleukin-8 during Japanese encephalitis virus infection. J. Med. Microbiol. 2000;49:607–612. doi: 10.1099/0022-1317-49-7-607. [DOI] [PubMed] [Google Scholar]
- Solomon T., Ni H., Beasley D.W., Ekkelenkamp M., Cardosa M.J., Barrett A.D. Origin and evolution of Japanese encephalitis virus in southeast Asia. J. Virol. 2003;77:3091–3098. doi: 10.1128/JVI.77.5.3091-3098.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai T.F. New initiatives for the control of Japanese encephalitis by vaccination: minutes of a WHO/CVI meeting, Bangkok, Thailand. Vaccine. 2000;26:1–25. doi: 10.1016/s0264-410x(00)00037-2. [DOI] [PubMed] [Google Scholar]
- Valerio A., Ferrario M., Martinez F., Locati O.M., Ghisi V., Bresciani L.G., Mantovani A., Spano A.P. Gene expression profile activated by the chemokine CCL5/RANTES in human neuronal cells. J. Neurosci. Res. 2004;78:371–382. doi: 10.1002/jnr.20250. [DOI] [PubMed] [Google Scholar]
- Yang K.D., Yeh W.T., Chen R.F., Chuon H.L., Tsai H.P., Yao C.W., Shaio M.F. A model to study neurotropism and persistency of Japanese encephalitis virus infection in human neuroblastoma cells and leukocytes. J. Gen. Virol. 2004;85:635–642. doi: 10.1099/vir.0.19426-0. [DOI] [PubMed] [Google Scholar]