The Electron Bifurcating FixABCX Protein Complex from Azotobacter vinelandii: Generation of Low-Potential Reducing Equivalents for Nitrogenase Catalysis (original) (raw)
. Author manuscript; available in PMC: 2020 Nov 4.
Published in final edited form as: Biochemistry. 2017 Aug 3;56(32):4177–4190. doi: 10.1021/acs.biochem.7b00389
Abstract
The biological reduction of dinitrogen (N2) to ammonia (NH3) by nitrogenase is an energetically demanding reaction that requires low-potential electrons and ATP; however, pathways used to deliver the electrons from central metabolism to the reductants of nitrogenase, ferredoxin or flavodoxin, remain unknown for many diazotrophic microbes. The FixABCX protein complex has been proposed to reduce flavodoxin or ferredoxin using NADH as the electron donor in a process known as electron bifurcation. Herein, the FixABCX complex from Azotobacter vinelandii was purified and demonstrated to catalyze an electron bifurcation reaction: oxidation of NADH (Em= −320 mV) coupled to reduction of flavodoxin semiquinone (Em= −460 mV) and reduction of coenzyme Q (Em= +10 mV). Knocking out fix genes rendered Δ_rnf A. vinelandii_ cells unable to fix dinitrogen, confirming that the FixABCX system provides another electron delivery route to nitrogenase. Characterization of the purified FixABCX complex revealed the presence of flavin and iron-sulfur cofactors confirmed by native mass spectrometry, electron paramagnetic resonance spectroscopy, and transient absorption spectroscopy. Transient absorption spectroscopy further established the presence of a short-lived flavin semiquinone radical, suggesting that a thermodynamically unstable flavin semiquinone may participate as an intermediate in electron transfer to flavodoxin. A structural model of FixABCX, generated using chemical cross-linking in conjunction with homology modeling, revealed plausible electron transfer pathways to both high- and low-potential acceptors. Overall, this study informs on a mechanism for electron bifurcation, offering insight into a unique method for delivery of low-potential electrons required for energy-intensive biochemical conversions.
Keywords: Azotobacter vinelandii, Electron bifurcation, Electron transfer, FixABCX, Nitrogenase, Rnf
Graphical Abstract
Introduction
Biological dinitrogen (N2) fixation is performed by diazotrophic microbes, which harbor the enzyme nitrogenase. This enzyme converts N2 into bioavailable ammonia (NH3) (Eq. 1) and accounts for at least half of the production of fixed nitrogen on Earth.1–3
| N2+8H++16MgATP+8e−→2NH3+H2+16MgADP+16Pi | (Eq. 1) |
|---|
As summarized by Eq. 1, the biological reduction of N2 is an energy-demanding reaction, requiring both ATP and low reduction-potential electrons. These electrons are provided by small redox proteins, ferredoxin (Fd) and flavodoxin (Fld), which serve as direct electron donors to the iron (Fe) protein of nitrogenase.4–10 The redox active iron-sulfur cluster(s) of Fd typically access one redox couple (FdOx/Red) with midpoint reduction potentials (Em) ranging from 0 to −645 mV.4,11 Two redox couples of the flavin in Fld are accessible, including the oxidized quinone/semiquinone (FldOx/Sq) and semiquinone/hydroquinone (FldSq/Hq) couples.5,9,12 In general, the Em of the FldOx/Sq couple ranges from −50 to −250 mV and the FldSq/Hq couple from −370 to −500 mV.4,7 Only the FldSq/Hq couple of Fld has enough driving force to donate electrons to nitrogenase. While much is known about other aspects of biological nitrogen fixation, pathways for delivery of the low-potential reducing equivalents for Fd and Fld reduction are not well understood for many diazotrophs.4 It has been shown that the nitrogen-fixing organism Klebsiella pneumoniae uses the anaerobic oxidation of pyruvate to reduce Fld,13,14 and it was proposed that other microbes likely use energy associated with the proton motive force to drive reduction of a low-potential electron donor.15,16 Recently, a new mechanism for generating reductant for nitrogen fixation was put forward. Flavin-based electron bifurcation (FBEB), considered a third fundamental form of energy conservation, couples exergonic and endergonic electron transfer reactions to limit free energy loss in biological systems.17–19 FBEB exploits a favorable electron transfer event in order to drive a thermodynamically unfavorable reaction without the use of ATP or an electrochemical gradient.17–19 Several bifurcating complexes, all of which contain a flavin as the proposed site of bifurcation, have been identified in anaerobic bacteria and archaea.17,18 While these enzymes catalyze a variety of reactions, all characterized thus far use Fld/Fd as an electron donor or acceptor.20–24 For example, the electron transferring flavoprotein/butyryl-coenzyme A (Etf-Bcd) bifurcating system uses the electron donor NADH (Em= −320 mV) to reduce Fld (EmSq/Hq= −430 mV) or Fd (Em= −405 mV). This thermodynamically unfavorable reaction is achieved by coupling it to an exergonic one, in this case, the reduction of crotonyl-CoA (Em= −10 mV).17,22,23 The bifurcation of electrons to both a high- and a low-potential acceptor results in an overall thermodynamically favorable reaction.17,22,23,25
Homologs of bifurcating electron transfer flavoproteins (Etfs), known as FixAB, have been found in physiologically and phylogenetically distinct nitrogen fixing organisms such as Azotobacter vinelandii,26,27 Rhodopseudomonas palustris,28 Rhodospirillum rubrum,29 and Sinorhizobium meliloti.30,31 Previous studies demonstrated that disrupting the Fix system in R. palustris, R. rubrum, and S. meliloti completely abolishes or significantly impairs their ability to grow under nitrogen fixing conditions, suggesting that Fix may serve as a source of electrons to nitrogenase.28–30 Given the homology between FixAB and known bifurcating Etfs, it was hypothesized that the Fix system uses electron bifurcation to generate low-potential reducing equivalents for nitrogenase.20
While the FixABCX complex is clearly linked to nitrogen fixation in many diazotrophs, its specific role has not been firmly established, nor has its ability to generate reductant for nitrogenase via electron bifurcation been demonstrated. Here, we report electron bifurcation by the FixABCX complex from the obligate aerobe A. vinelandii and characterize the Fix proteins using advanced biochemical and spectroscopic tools. Further, we provide evidence that FixABCX provides electrons for nitrogenase in A. vinelandii cells. Overall, this work establishes a new pathway for the generation of low-potential reductant required by nitrogenase and elucidates a mechanism by which biology can overcome thermodynamic barriers to accomplish a difficult reductive reaction.
Materials and methods
Construction of A. vinelandii Δfix mutant
A. vinelandii strain DJ was the wild type strain used for physiological studies and mutant construction. Strains UW195 (Δrnf1) and UW207 (Δrnf2) were kindly donated by Luis Rubio.32 The Δfix mutant was generated by gene disruption with an antibiotic resistance cassette as shown in Figure S1 using primers in Table S1. Briefly, two 1.2 kb DNA fragments that included 0.3 kb of fixA (Avin_10520) and 0.1 kb of fixC (Avin_10540) respectively were obtained by PCR and cloned sequentially in pT7–7 ampicillin resistant vector using _Nde_I/_BamH_I and _BamH_I/_Hind_III as restriction cloning sites. The kanamycin resistance (KmR) gene (aph), was isolated as a 1.5-kb _BamH_I fragment from mini-Tn533 and inserted between the 1.2 kb upstream and downstream regions previously cloned in pT7–7 vector using _BamH_I restriction cloning site. The final construct was transformed into A. vinelandii strain DJ as described previously.34,35 KmR transformants were screened for sensitivity to ampicillin (AmpS); AmpS derivatives were assumed to have arisen from a double crossover recombination event in which the wild-type fixABC genes were replaced by the _aph_-containing cassette (Figure S1). This replacement was confirmed by PCR and sequencing (Figure S1, Table S1). Burk’s media36 with no nitrogen source or supplemented with ammonium acetate (1 g/L) was used for physiological analyses of A. vinelandii DJ and Δrnf and Δfix mutants.
Homologous overexpression of FixABCX
The fix genes of A. vinelandii were homologously overexpressed by placing them under control of the nifH promoter, which is used for the transcription of genes associated with the molybdenum-dependent nitrogenase (Figure S2). The 4.3 kb fix operon, which includes six genes (fixfd, Avin_10510; fixA, Avin_10520; fixB, Avin_10530; fixC, Avin_10540; fixX, Avin_10550; ORF6, Avin_10560) (Figure S2), was PCR amplified using primers specified in Table S2. The PCR product was digested with _Xba_I and _BamH_I and inserted into a slightly modified pUC19 vector for blue-white screening. Using _Nde_I and _BamH_I restriction sites, the fix genes were then cloned into a vector built specifically to support the insertion of genes behind the nifH promoter.37 The fix operon was inserted between two segments of DNA from the _nif_-region of A. vinelandii. The flanking regions served as sites for homologous recombination within the chromosome, allowing replacement of nifHD with fix genes (Figure S2). The presence of a streptomycin resistance gene (SmR) between the flanking regions provided a selectable marker upon transformation of the plasmid into _A. vinelandi_i.34 The proper insertion of the fix genes into the final plasmid and the chromosome of A. vinelandii were confirmed by PCR and sequencing (Table S2).
Growth of recombinant A. vinelandii and purification of the FixABCX complex
Recombinant A. vinelandii was grown in a 100 L fermenter (New Brunswick BioFlo 610, Hauppauge, NY) at Utah State University’ Synthetic Biomanufacturing Institute. Cells were grown in Burk’s medium supplemented with 10 mM urea as a nitrogen source.36 In the presence of urea, the nifH promoter is repressed. When the cells reached an OD600nm of ~1.8, they were harvested in a stacked disk centrifuge (TSE 10, GEA Westfalia, Northvale, NJ) at 14,200 × g and resuspended in Burk’s medium with no source of fixed nitrogen. Upon removal of fixed nitrogen source, the nifH promoter is derepressed and gene expression occurs. A. vinelandii was derepressed for 5 h to achieve optimal expression of Fix proteins. Because nif genes were replaced with fix genes, Fix rather than nitrogenase was expressed. Following the derepression, cells were harvested and stored at −80°C until further use. Fermenter conditions were maintained as follows throughout the growth and derepression: 30°C, pH 7, 20% dissolved oxygen, and agitation at ~200 rpm.
All purification steps were performed anaerobically under an argon atmosphere. 100 g of wet cell paste was resuspended in 50 mM HEPES, pH 8, 150 mM NaCl, 1 mM dithiothreitol (DTT) and 2 mg DNase at a cell to buffer ratio of 1:5. Flavin adenine dinucleotide (FAD) (0.25 mM) was also added to the lysis buffer as it increased the flavin cofactor occupancy of the Fix complex. Lysis was achieved with a French pressure cell (SLM Aminco FA-078, Aminco, Rochester, NY) at 200 MPa. The cell extract was centrifuged at 8,000 × g for 15 min to remove cell debris. A second centrifugation was then conducted at 50,000 × g for 2 h to obtain the cell membrane fraction. The membranes were solubilized for 1 h at 4°C in 50 mM HEPES, pH 8, 150 mM NaCl, 1% (w/v) n-dodecyl β-D-maltoside (DDM), and 1 mM DTT at a pellet to buffer ratio of approximately 1:6. The solubilized membrane was obtained by centrifuging at 50,000 × g for 1 h and then diluted 3-fold in Buffer A (50 mM HEPES, pH 8, 0.02% (w/v) DDM, and 1mM DTT) to reduce the salt and detergent concentration before loading onto a 100 ml Q-Sepharose column. The Q-Sepharose column was prewashed with 2 column volumes of Buffer B (50 mM HEPES, pH 8, 1 M NaCl, 0.02% (w/v) DDM, and 1 mM DTT) and then equilibrated with 2 column volumes of Buffer A. Once the protein was loaded, unbound proteins were removed with 2 column volumes of Buffer A, followed by elution of bound proteins with a salt gradient of 15–60% (5 column volumes). FixABCX eluted between 32–36% NaCl. Fractions were pooled and diluted to less than 100 mM NaCl for concentration on a 15 ml Q-Sepharose column pre-washed and equilibrated as described above. After loading, the column was washed with 2 column volumes of Buffer A and bound protein eluted with 500 mM NaCl. The resultant concentrated fraction was loaded onto a Sephacryl-200 column equilibrated with 50 mM HEPES, pH 8, 150 mM NaCl, 0.02% (w/v) DDM, and 1 mM DTT. Fractions containing FixABCX were pooled and concentrated using an Amicon (EMD Millipore, Billerica, MA) concentrator with a 100 kDa cutoff membrane and stored in liquid nitrogen. The purity of FixABCX was determined using SDS-PAGE and protein concentration was measured using the DC Protein Assay (Bio-Rad, Hercules, CA).
Heterologous overexpression and purification of R. palustris FixAB
R. palustris FixAB was co-expressed in E. coli strain NiCo21(DE3) (New England Biolabs, Ipswich, MA) transformed with plasmids based on pMCSG28 and pMCSG21 (DNASU Plasmid Repository, Arizona State University) modified to carry the fixA gene with a C-terminal His-tag and the fixB gene with an N-terminal His-tag, respectively. Cells were grown in terrific broth (TB) supplemented with riboflavin (20 mg/L) and MgSO4 (2 mM) along with carbenicillin (100 μg/mL) and spectinomycin (100 μg/ml) at 37°C with shaking at 200 RPM to an OD600 of ~2. After fully cooling the culture to 18–20°C, fix gene expression was induced with β-D-1-thiogalactopyranoside (IPTG) (0.1 mM) and cells were grown for an additional 12 hours at this lower temperature. Cells were harvested by centrifugation at 11,899 × g at 4°C for 6 min, and the pellet was stored at −80°C.
The cell pellet was suspended in BugBuster (80 ml) (EMD Millipore, Billerica, MA) containing 4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF) (1 mM), Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (1 mM), FAD (1 mM), benzonase nuclease (20 μL) and rLysozyme (2 μL) (EMD Millipore, Billerica, MA), and further incubated at 4°C for 2 hours with stirring. After centrifugation at 20,000 × g for 30 min at 4°C, the supernatant was filtered through a 0.22 μm syringe filter. The resulting protein solution was mixed with 3 mL pre-equilibrated Ni-NTA resin and incubated overnight at 4°C with stirring. The next day, the mixture was transferred to a column at 4°C. After collecting the flow-through, the column was washed with TPGT buffer (20 mM Tris, pH 7.8, 500 mM KCl, 10% (w/v) glycerol and 1 mM TCEP) containing 20 mM, 40 mM and then 50 mM imidazole in sequence using 20, 2 and 2 column volumes for each, respectively. Finally, the column was developed with 2 column volumes of TPGT buffer containing 100 mM imidazole and the eluate was collected in different fractions. After SDS-PAGE analysis, imidazole was removed from the pooled pure fractions by passage over a 10DG column (Bio-Rad Laboratories, Hercules, CA) equilibrated with BPGT buffer (20 mM Bis-Tris propane, pH 9.0, 200 mM KCl, 10% (w/v) glycerol and 1 mM TCEP). Any apo-flavin sites were then reconstituted by overnight incubation of the protein in 1 mM FAD at 4°C overnight. Excess flavin was removed by gel filtration on a 10DG column (see above) prior to prompt use or flash freezing in liquid nitrogen and storage at −80°C.
Heterologous overexpression and purification of R. castenholzii FixX
R. castenholzii FixX was overexpressed in E. coli using a pCDFDuet-1 vector modified to include a C-terminal strep-tag. Transformed cells were grown in Luria-Bertani (LB) broth containing streptomycin (50 mg/ml) at 37°C and 250 RPM to an OD600 of 0.4–0.5. To induce expression of the _fix_X gene, IPTG (1.5 mM) was added to the cell culture. Ammonium Fe(III) citrate (4 mM), L-cysteine (2 mM), and sodium fumarate (10 mM) were also added. Ammonium Fe(III) citrate and cysteine increased iron-sulfur cluster occupancy and sodium fumarate served (10 mM) as an electron acceptor during anaerobic metabolism. The flasks were sealed with rubber stoppers containing cannulas sparging argon into the cell suspension and suited with exhaust tubes flowing into a water trap. Cells were incubated for 4–6 h at room temperature and then anaerobically transferred to centrifugation bottles in a Coy chamber (Coy Laboratories, Grass Lake, MI) under a nitrogen atmosphere. Cells were harvested at 5,000 × g for 10 min.
All purification steps were performed anaerobically under a nitrogen or argon atmosphere. Wet cell pellets were suspended in lysis buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1 mM sodium dithionite (Na-dithionite), 5% glycerol, 1 μL/mL supernatant of a super saturated phenylmethylsulfonyl fluoride (PMSF) solution, 1 mg/10 mL DNAse, and 5 mg/10 mL lysozyme) at a cell to buffer ratio of 1:5. Cells were lysed using a cell bomb (Parr Bomb Instrument Co., Moline, IL) by slowly increasing the pressure to 1500 PSI and equilibrating for 30 min before collecting the lysate. This process was repeated and the supernatant collected by centrifugation at 18,000 × g for 20 min. FixX was purified using a single step affinity column purification using Strep-Tactin Superflow Plus (Qiagen, Hilden, Germany) resin pre-equilibrated with wash buffer containing 50 mM Tris-HCl, pH 8, 150 mM NaCl, and 1 mM Na-dithionite. Once the protein was loaded, the column was washed until the baseline absorbance returned. The protein was then eluted with 50 mM Tris-HCl, pH 8, 150 mM NaCl, 1 mM Na-dithionite, and 2.5 mM D-desthiobiotin as a brown band and anaerobically concentrated using an Amicon (EMD Millipore, Billerica, MA) centrifugal filter with a membrane molecular mass cutoff of 10 kDa. The concentrated protein was snap frozen with liquid nitrogen for further analysis.
Heterologous overexpression and purification of R. rubrum FixAB
FixAB from Rhodospirillum rubrum was overexpressed and purified as described previously.26 Briefly, E. coli BL21 cells containing the plasmid pET101/d-TOPO_::fixAB_26 were cultured anaerobically in LB supplemented with 2.8 mM glucose,17 mM KH2PO4,72 mM K2HPO4 and 15 mg/L riboflavin at 20°C until early exponential growth phase. At this point gene expression was induced with the addition of 0.5 M IPTG, and cells were cultured under the same conditions for an additional 12 h. Harvested cells were resuspended in degassed lysis buffer (20 mM Tris-Cl, pH 7.6, 10 mM imidazole, 500 mM NaCl, 5 mg/ml lysozyme, and 1 mg/ml Dnase I) and submitted to three freeze/thaw cycles. The lysate was added to a nickel column (Qiagen, Hilden, Germany) that had been equilibrated with degassed lysis buffer. The column was washed with degassed lysis buffer, and FixAB was eluted into anaerobic vials using a 10–300 mM imidazole gradient in degassed 20 mM Tris-HCl, pH 7.6, and 500 mM NaCl. Yellow fractions indicating the presence of FixAB were subjected to buffer exchange using a PD-10 column (GE-Healthcare, Little Chalfont, UK) and stored in desalting buffer (20 mM Tris-HCl, pH7.6, and 100 mM NaCl). Purified FixAB was incubated overnight at 4°C with 1 mM FAD in desalting buffer to reconstitute FAD cofactors lost during purification.38,39 Unbound FAD was rinsed by concentrating the samples with Amicon membrane (cutoff 30 kDa) followed by dilution with desalting buffer containing 0.5 μM FAD. Reconstituted samples were stored at −20°C. Purity and identity of the fractions were assessed with SDS-PAGE and mass spectrometry, respectively.
Dynamic light scattering
Dynamic light scattering (DLS) was performed on DynaPro NanoStar (Wyatt Technology, Santa Barbara, CA) to determine the size distribution and hydrodynamic radii of species in solution. Samples were filtered through a 0.1 μm syringe filter prior to the experiments. DLS was measured aerobically using 10 μL of buffer as the control and also 1 mg/mL enzyme using a disposable cyclic olefin copolymer cuvette at 25°C.
Protein identification, chemical cross-linking, and model construction
Protein identification from gel bands and solution digestion was performed according to standard protocols recommended by the manufacturers using a trypsin (Promega, Madison, WI) protease:complex ratio of 1:50–1:100 overnight and pepsin (Sigma, St. Louis, MO) protease:complex ratio of 1:10 for 60 s. Proteins were identified as described5 using a maXis Impact UHR-QTOF instrument (Bruker Daltonics, Billerica, MA) interfaced with a Dionex 3000 nano-uHPLC (Thermo-Fisher, Waltham, MA) followed by data analysis in Peptide Shaker.40 Chemical cross-linking was performed using 20 μg of the FixABCX complex and 1 mM bis(sulfosuccinimidyl)suberate (BS3) (Thermo-Fisher, Waltham, MA) in 50mM HEPES, pH 7.2, 150mM NaCl buffer at room temperature for 1 h. The reaction was quenched by addition of 120 mM Tris (final concentration). The resulting mixture was separated by SDS-PAGE (4–20% linear gradient gel, Bio-Rad, Hercules, CA) and stained with Coomassie Brilliant Blue (Thermo-Fisher, Waltham, MA). Protein bands of interest were excised from the gel and digested with trypsin as described above. For cross-link mapping, a Spectrum Identification Machine, SIM, was used.41 Precursor and fragment ion tolerances were set to ±20ppm. Intact protein analysis was performed as described previously using a Bruker Micro-TOF mass spectrometer (Bruker Daltonics, Billerica, MA).42 Non-covalent mass spectrometry experiments were conducted on SYNAPT G2-Si instrument (Waters, Milford, MA) in a similar fashion as described.43 Briefly, the FixABCX complex sample was buffer exchanged to 200 mM ammonium acetate, pH 7 (Sigma, St. Louis, MO) using 3 kDa molecular mass cutoff spin filters (Pall Corporation, Port Washington, NY) and infused from in-house prepared gold-coated borosilicate glass capillaries to electrospray source at protein concentration of 2–3 μM and the rate around 90 nl/min. The instrument was tuned to enhance performance in the high mass-to-charge range. Settings were as follows: source temperature 30°C, capillary voltage 1.7 kV, trap bias voltage 16 V and argon pressure in collision cell (trap) 7 ml/min. Transfer collision energy was held at 10 V while trap energy varied between 10–200 V. Data analysis was performed in MassLynx software version 4.1 (Waters, Milford, MA).
Protein homology models were generated by Phyre2,44 and energy minimized models were docked using ClusPro2 with restrictions derived from chemical cross-linking experiments.45–48 The flavin and iron sulfur cluster co-factors were added using SwissDock49,50 for individual subunits and eventually added as rigid bodies to the final FixABCX complex model. Molecular graphics were created using the UCSF Chimera package.51
Electron paramagnetic resonance spectroscopy
Electron paramagnetic resonance (EPR) spectra were recorded with a Bruker E-500 spectrometer (X-band, 9.38 Ghz) equipped with a SHQ resonator (Bruker, Billerica, MA), an in-cavity cryogen free VT system (ColdEdge Technologies, Allentown, PA) and MercuryiTC temperature controller (Oxford Instruments, Abingdon, UK). Spin quantifications were determined by double integration of the spectra after manual baseline subtraction in the OriginPro software package and referenced to copper-triethylamine standards (75–125 μM) measured at the same conditions. To assist with spectral deconvolution and assignment of _g_-factors (±0.003), computer simulations of the experimental spectra were carried out in MatLab using the EasySpin package and ‘esfit’ fitting function incorporating _g_-strain to replicate line broadening.
Electron bifurcation assays
FixABCX electron bifurcation activity was measured anaerobically in a UV-Vis spectrophotometer (Varian Cary 50 Bio, Agilent Technologies, Santa Clara, CA) using quartz cuvettes (_d_= 1 cm). All assays were carried out in 50 mM HEPES, pH 7.5, 10% glycerol and 0.02% DDM and contained the following: 0.8 μM FixABCX (1.7 nmol flavin/nmol FixABCX and 9.1 nmol Fe/nmol FixABCX), 85 μM FldSq, 200 μM NADH, and 300 μM Coenzyme Q1 (CoQ1). NADH, FldSq, and FldOx were monitored at 340 nm (ε= 6.2 mM−1 cm−1), 580 nm (ε= 5.7 mM−1 cm−1), and 450 nm (ε= 11.3 mM−1 cm−1), respectively.12,52 The Fld used in the assays as the low-potential electron acceptor was purified in the hydroquinone state as previously described.5 FldHq was exposed to oxygen for a short period of time, upon which the majority (>80%) of the FldHq converted to the semiquinone form. The protein was then degassed with argon, and the absorbance of the semiquinone species was measured at 580 nm. The concentration was then calculated using the extinction coefficient (ε= 5.7 mM−1 cm−1).
Thermodynamic calculations
Standard reduction midpoint potentials of the NAD+/NADH (Em= −320 mV), CoQ/CoQH2 (Em= +10 mV), FldOx/Sq (Em= −180 mV), FldSq/Hq (Em= −460 mV) half reactions were converted to standard Gibbs free energies (ΔG°’) with the equation,
where n= electrons (mol), F= Faraday constant (96,485.34 J/V·mol) (Table S5). The standard Gibbs free energy of reaction (ΔGrxn°'; J/mol) can be calculated for the reaction of interest using the equation,
| ΔGrxn°'=∑|vi|ΔG(products)°'−∑|vi|ΔG(reactants)°' | (Eq. 4) | | -------------------------------------------------- | ------- |
where |𝑣𝑖| = stoichiometric coefficient, 𝑖 = product or reactant, ΔG(productsorreactants)°′= standard Gibbs free energy of the products and reactants (J/mol) calculated from equation 3.
Transient absorption spectroscopy
The ultrafast (100 fs to 5.1 ns) transient absorption spectroscopy (TAS) spectrometer employed in this study uses an amplified 4W Ti:sapphire laser (Libra, Coherent, 800 nm, 1 kHz, 100 fs pulse width), and the Helios spectrometer (Ultrafast Systems LLC, Sarasota, FL). A fraction of the 800 nm Libra output was frequency-doubled in beta barium borate (BBO) to produce the desired pump wavelength (480 nm for the data described here) for sample excitation, which was then directed into the Helios. The pump pulses were passed through a depolarizer and chopped by a synchronized chopper to 500 Hz before reaching the sample. The pump pulse energy was 1.1 μJ per pulse at the sample. Another fraction of the 800 nm Libra output was guided directly into the Helios for generation of the probe. Within the spectrometer, a white light continuum of wavelengths including 340–800 nm was generated using a 2 mm thick CaF2 crystal. This beam was split into a probe and a reference beam. The probe beam was focused into the sample where it was overlapped with the pump beam. The transmitted probe and reference beams were then focused into optical fibers coupled to multichannel spectrometers with CMOS sensors with 1 kHz detection rates. The reference signal is used to correct the probe signal for pulse-to-pulse fluctuations in the white-light continuum. The time delay between the pump and probe pulses was controlled by a motorized delay stage. For all transient absorption measurements, the sample was made in an Mbraun glove-box (N2 atmosphere), sealed in a 2 mm quartz cuvette and constantly stirred to prevent photodegradation. R. rubrum FixAB and A. vinelandii FixABCX concentrations were approximately 150 μM and 43 μM, respectively, and they were measured in their as-isolated state (mostly oxidized). The R. rubrum dimer contained >0.7 nmol flavin/nmol FixAB, and the A. vinelandii FixABCX complex contained 1.7 nmol flavin/nmol FixABCX. For the purpose of this study, light initiated the formation of the semiquinone intermediates for each flavin through generation of FAD excited state and electron donation from nearby protein residues or the other flavins. Qualitatively, this experiment shows which type of semiquinone is formed for a particular FAD site and suggests how thermodynamically stable that intermediate is, based on its lifetime. All experiments were conducted at room temperature. The change in absorbance signal (ΔA) was calculated from the intensities of signals detected after sequential probe pulses with and without the pump pulse excitation. The data collection (350 pump shots per time point) was carried out three consecutive times and then averaged. The experiment was repeated three times for A. vinelandii FixABCX and once for R. rubrum FixAB. Data were corrected for spectral chirp using SurfaceXplorer (Ultrafast Systems LLC, Sarasota, FL). ASQ signals were fit in Igor Pro with a double exponential function. The 550 nm emissive feature in R. rubrum FixAB is due to stimulated emission.53
Results and discussion
Effect of fix and rnf deletions on nitrogen fixation in A. vinelandii
To determine whether the Fix system is associated with diazotrophic growth in A. vinelandii, as in R. palustris, R. rubrum, and S. meliloti,28,29,31 a deletion mutant lacking fixABC was generated (Figure S1). This mutant, unlike those of R. palustris, R. rubrum, and S. meliloti exhibited equally robust growth on solid media with and without added fixed nitrogen (Figure 1). This nitrogen fixing (Nif+) phenotype was also observed when the A. vinelandii fix mutant expressed the vanadium and iron-only alternative nitrogenases or when cultured under low aeration conditions (data not shown). Although the results appeared to indicate that Fix is not associated with nitrogen fixation in A. vinelandii and were consistent with a previous study by Wientjens,27 we also tested the possibility of redundancy,54 thinking that an alternative complex could participate in balancing the chemical energy and reductant pools in the absence of Fix, masking the effect of the ΔfixABC mutation.
Figure 1.

Phenotype of A. vinelandii DJ wt (A), Δfix (B), Δrnf1 (C), ΔfixΔrnf1 (D), Δrnf2 (E), and ΔfixΔrnf2 (F) strains. Wild type and mutant strains were cultivated in Burk’s medium supplemented with ammonium acetate (+N) or with no added fixed nitrogen (−N). All samples were grown aerobically.
Rnf (_Rhodobacter n_itrogen _f_ixation) is a membrane-bound complex found in some diazotrophs that, like Fix, has been hypothesized to generate reductant in the form of Fd or Fld for nitrogen fixation.16,55,56 Nitrogen-fixing organisms that have genes coding for Rnf typically do not have genes coding for Fix and vice versa. Interestingly, A. vinelandii is one of the few known diazotrophs with genes coding for both complexes.57 The physiological implications of having both of Rnf and Fix are not altogether clear, but given that A. vinelandii fixes nitrogen under highly oxic conditions, the combination of Rnf and Fix could confer the ability to fine-tune the redox status of the cell to a high degree under a wide range of oxygen tensions.
It has been suggested that Rnf uses the energy of the proton motive force to generate reduced Fd/Fld15,16 while Fix uses electron bifurcation.18,20 Neither the Rnf nor Fix complex, however, has previously been directly implicated in electron delivery to nitrogenase. Curatti et al. showed that A. vinelandii possesses two Rnf complexes, Rnf1 associated with nitrogen fixation and Rnf2, which is expressed constitutively.32 Δrnf1 mutants, although still able to grow diazotrophically (Figure 1), consistently exhibit a long lag in nif gene expression and diazotrophic growth during derepression studies.32 To test whether Rnf and Fix might be compensating for one another, we transformed Δ_rnf_ mutants of A. vinelandii with the Δ_fix_ construct (Figure S1)32 to generate double mutants of A. vinelandii lacking either Rnf1 and Fix, or Rnf2 and Fix. The Δfix-Δrnf1 A. vinelandii strain was able to grow when fixed nitrogen was supplied, but no growth was observed under nitrogen fixing conditions (Figure 1). Therefore, cells lacking both Rnf1 and Fix displayed a non-nitrogen fixing (Nif−) phenotype. Our data provide support for a role of both Fix and Rnf1 in providing reducing equivalents to nitrogenase and indicate that the two complexes have compensatory activities, such that either one can replace the other to ensure electron flow. Furthermore, this is the first study to link the Fix complex to nitrogen fixation in A. vinelandii. The Δfix-Δrnf2 A. vinelandii mutant control strain was able to grow with or without added fixed nitrogen (Figure 1), confirming that Rnf1, but not Rnf2, can support nitrogen fixation.
Characterization of the FixABCX complex
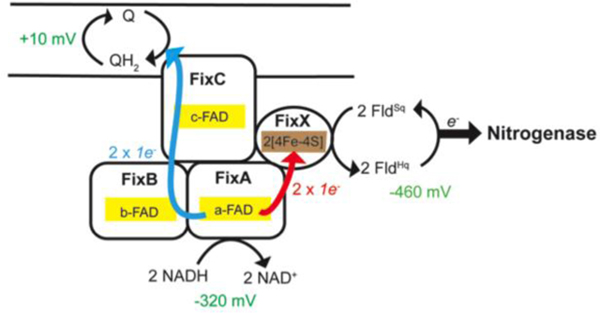
To understand how Fix is able to support nitrogen fixation, we characterized the FixABCX complex and its activity in vitro. The fix operon of A. vinelandii consists of six genes, _fix_Fd (annotated as _fix_P in some literature), _fix_A, _fix_B, _fix_C, _fix_X, and Avin_10560 (designated ORF6) (Figure S2). FixAB is homologous to the previously characterized EtfAB, which is part of the Etf-Bcd bifurcating complex. FixCX is similar to Etf ubiquinone oxidoreductase (Etf-QO), involved in transfer of electrons to the quinone pool in the membrane.29 Based on the similarity to other Etf systems, it is hypothesized that the Fix complex oxidizes NADH and bifurcates one electron to the high-potential quinone pool (exergonic branch) and drives the other electron to a low-potential acceptor in the form of Fd or Fld (endergonic branch) (Figure 2).20
Figure 2.

(A) Proposed mechanism of FixABCX electron bifurcation in A. vinelandii, operating within the framework of the known oxidation-reduction potentials of NADH, Fld/Fd, and CoQ. It is hypothesized that crossed potentials of the bifurcating flavin (a-FAD) promote electron bifurcation based on the appearance of a short-lived ASQ. Once the first electron transfers into the exergonic branch, the second electron, sitting at a very low reduction potential, is thermodynamically unstable and immediately transfers into the endergonic branch composed of the low potential iron-sulfur clusters of FixX. The exergonic branch is set up to ensure electron delivery to c-FAD where electrons accumulate before being transferred to CoQ. The specific oxidation-reduction potential levels are qualitative unless otherwise noted. (B) Electron transfer pathways illustrated in FixABCX structural model generated by docking (ClusPro2) four homology models (Phyre2) (Figure S8).
Most microbes with fix genes only have _fix_ABCX; homologs _fix_Fd and ORF6 have not been described in fix gene clusters of any other species thus far.27,58 Although FixFd and ORF6 were not specifically addressed in this study, it is thought that FixFd, a small iron-sulfur protein,27,59 may serve as a low-potential acceptor in Fix electron bifurcation. ORF6 is described as being ferritin-like, but studies on its function have yet to be conducted.
The fix genes from A. vinelandii were homologously overexpressed under control of the _nif_H promoter (Figure S2), which regulates transcription of genes encoding molybdenum nitrogenase. This approach allows gene(s) of interest to be overexpressed upon removal of a fixed nitrogen source. Based on the hypothesis that the FixABCX complex delivers one electron per bifurcation reaction to the quinone pool (coenzyme Q, CoQ), it was anticipated that the enzyme could be associated with the membrane fraction, as observed for Etf-QO.60,61 Indeed, a previous study found that heterologously overexpressed R. rubrum FixC was localized to the membrane fraction in Escherichia coli.29 Furthermore, an N-terminal lipophilic tail and transmembrane helix were predicted on the FixC subunit based on the amino acid sequence (SACS MEMSAT2 and Phyre 2 prediction software).44,62 Purification attempts for the FixABCX complex confirmed membrane association. After trying a range of NaCl concentrations (0–1 M) and a variety of non-ionic detergents to solubilize the complex, the detergent n-dodecyl β-D-maltoside (DDM) used for many membrane proteins,61,63 was found to be most effective for the isolation of intact FixABCX in a soluble and active form.
Upon extraction of the FixABCX complex from the membrane, the DDM-solubilized fraction was subjected to anion-exchange and size-exclusion chromatography. SDS-PAGE analysis of the purified FixABCX complex revealed four bands corresponding in size to FixA, FixB, FixC, and FixX (Figure 3) and was >80% homogenous (defined by percent band (Image Lab, BioRad)). Mass spectrometry confirmed the identities of the four Fix subunits (Figure S3), and densitometry of the stained gel indicated that the band intensities were consistent with a subunit stoichiometry of 1:1:1:1 for A:B:C:X.
Figure 3.

SDS-PAGE gel of purified (~80%) FixABCX complex from A. vinelandii (extra lanes were removed). All Fix protein bands were verified by mass spectrometry (Figure S3) and according to densitometry, the subunit stoichiometry of FixABCX is 1:1:1:1.
Dynamic light scattering
DLS was used to determine the oligomeric state of the FixABCX complex on the basis of particle size. The complex in buffer containing 0.02% DDM displayed an average hydrodynamic radius of 5.1 ± 0.3 nm, corresponding to an estimated molecular mass of 150 kDa versus the expected 129 kDa for the FixABCX heterotetramer (Figure S4). This slightly larger-than-expected complex is consistent with attachment of DDM to the protein tetramers. While there was also some contribution from larger aggregates, which are very prominent in the DLS output due to their much larger scattering efficiencies (≈12–16 nm), the aggregates comprised <1% of the population (Figure S4). Buffer containing DDM was used as a negative control and produced micelles with a radius of ≈4.3 nm (Figure S4). Overall, the DLS data suggest FixABCX exists as individual heterotetramers in solution.
FixABCX ligand composition and structural model
Multiple electron transfer cofactors, including flavins and iron-sulfur clusters, have been identified in electron bifurcating enzyme complexes, with a single flavin proposed to serve as the site of bifurcation.17,18,20,25 Based on common sequence motifs for ligand binding, FixABCX was predicted to have 3 FAD moieties, one each in FixA, FixB, and FixC, and two [4Fe-4S] clusters in FixX. Native mass spectrometry (MS) was employed to reveal the subunit stoichiometry and ligand composition of the complex.
All complex members were first identified based on fragments produced by two different proteases, trypsin and pepsin. Proteolysis reactions revealed that FixABCX and a small amount of Fld (NifF) purify together. The next step was to determine the molecular masses of the intact proteins. Reverse phase LC-MS confirmed full-length FixA and that FixB and FixC subunits did not contain N-terminal Met residues. The purified FixABCX complex, under anaerobic conditions, was analyzed using native MS conditions by direct infusion on a Synapt G2-Si. Under standard conditions (60V collision energy), the complex dissociated into subcomplexes of FixAB and FixCX. A consistent ion signal for the tetrameric FixABCX complex could not be maintained despite the presence of all four proteins and cofactors in the sample after size-based purification. The FixAB component contained two FAD molecules and closely matched the expected size (71,414.8 Da vs predicted MW = 71,413.7 Da) (Figures 4A, 4B); whereas, the FixCX subcomplex containing FAD and two [4Fe-4S] clusters was slightly heavier than expected (60,310.1 Da vs predicted MW = 59,932.9 Da: FixCX) (Figure 4B). The additional mass of 377.2 Da matches very closely to riboflavin (Rf) (MW = 376.4 Da), raising the intriguing possibility that it may also be associated with the complex (Figure 4B).
Figure 4.
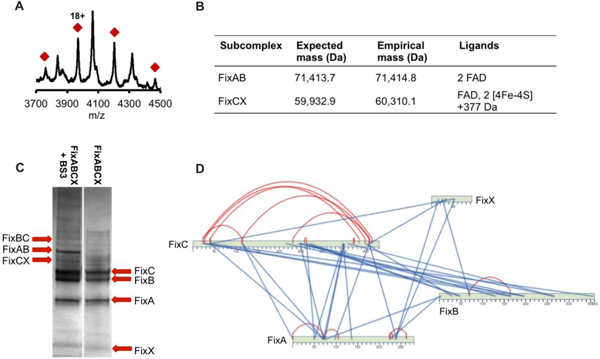
A. vinelandii FixABCX complex composition. (A) Native mass spectrum of FixAB containing two FAD cofactors (71,414.8 Da; calculated MW = 71,413.7 Da) generated during FixABCX complex activation in the gas phase. Red diamonds signify charge state envelope centered around charge 18+. The unmarked masses are the charge state envelop of the molecular chaperone DnaK. (B) Sub-complexes obtained during FixABCX complex activation. (C) SDS-PAGE gel of FixABCX complex (right) cross-linked with BS3 reagent (left). The most distinguished bands in cross-linking reaction, migrating around the 60 kDa, 70 kDa, and 88 kDa molecular weight marker, were identified as FixCX, FixAB and FixBC dimers respectively. (D) Protein-protein interaction map based on highest scoring cross-links (score 7 and higher; red and blue lines indicate intra and inter cross-links, respectively). A complete list of generated cross-links can be found in Table S3.
The UV-visible spectrum of FixABCX revealed a distinct peak at 428 nm, as well as oxidized flavin signatures with shoulders near 460 nm and 365 nm (Figure 5 (Black trace)). After denaturation of FixABCX and removal of the protein by centrifugation, the soluble fraction demonstrated clear signatures of flavins with broad maxima at 370 and 450 nm (Figure 5 (Red trace)). The species responsible for the band at 428 nm is not ausual signature of flavins, and such a band has not been observed in FixAB from other diazotrophs that have been expressed in E. coli. Thus, it islikely that this species is associated with FixC or FixX and may be covalently attached to the protein, as it does not remain in the supernatant when denatured protein is removed. The signatures of the [4Fe-4S] clusters are not evident in as-purified FixABCX, but are not expected to be prominent because of scattering and their broad and relatively weak signals (400 nm-420 nm, ε≈ 2–4 mM−1 cm−1).64
Figure 5.

UV-visible spectrum of the FixABCX complex from A. vinelandii demonstrating the presence of flavin. Black: as-purified FixABCX, red: cofactors released upon denaturation of FixABCX, and blue: flavins as observed in the spectrum of FixAB from R. palustris, vertically offset by −0.05 AU for clarity. The two spectra representing FixABCX were corrected for Raleigh scattering by fitting the baseline to the equation for scattering and then subtracting the fit from the measured spectrum to obtain a corrected spectrum. A. vinelandii FixABCX was prepared in 50 mM Tris-HCl, pH 8, 150 mM NaCl, and 0.02% (w/v) DDM. R. palustris FixAB was prepared in 20 mM Bis-Tris propane, pH 9, 200 mM KCl, 10% (w/v) glycerol and 1 mM tris(2-carboxyethyl)phosphine (TCEP).
Once we had established the subunit stoichiometry and ligand composition of the FixABCX complex, we investigated the quaternary interactions that define the active enzyme. Models for each of the Fix subunits were generated using Phyre244, and the figures of merit are provided in Figures S5–S8. To validate these subunit models and to elucidate protein-protein interactions within the FixABCX complex, chemical cross-linking was used. The bis(sulfosuccinimidyl)suberate (BS3) cross-linking reagent is homobifunctional, reacting with primary amines and hydroxyl groups to form covalent bonds. Cross-linked samples were separated using 1D SDS-PAGE to confirm connections between subunits and to enrich cross-linked species in samples subsequently analyzed by liquid chromatography tandem mass spectrometry (LC-MS/MS) (Figure 4C and Table S3). Overall, the reaction of the FixABCX complex with BS3 reagent produced over 200 connections. For initial model building of the FixABCX complex and to confirm the subunit homology models, a reduced set of cross-links of very high confidence were selected (probability score >7, significance is >3) (Figure 4D).
The resulting model of the quaternary structure provides insightful hypotheses to be tested in next generation of experiments (Figures 2B, S8E). The MS-based model places the cofactors in locations that suggest a pair of possible paths for electron transfer, both emanating from the flavin in FixA. One provides a plausible route to the [4Fe-4S] clusters in FixX and the other can provide a path to the site at which quinone is found to be bound in the functional homolog of FixC, the Etf-QO (Figures 2B, S8E).65 While some of the distances between cofactors are longer than ideal for direct electron transfer, conformational changes could be invoked to bring them sufficiently close,22,39,66 but we also note that the model places conserved Trp and Tyr in locations that could provide electron transfer paths between cofactors (Figure S8E).67–69 Thus, our model provides testable hypotheses for which amino acids are expected to alter complex stability or internal electron transfer upon mutagenesis. These amino acids will be targets of experiments to elucidate fundamental elements of bifurcating activity and test attribution of bifurcating activity to FixABCX.
EPR spectroscopy
FixABCX was analyzed by low-temperature continuous wave X-band EPR spectroscopy to assess its flavin radical content and identity of iron-sulfur clusters. The EPR spectrum of the as-purified enzyme showed a very weak fast relaxing, broad signal that was strongest near 5 K along with a weak radical signal near g = 2 (Figure 6, Table S4). Upon reduction with either NADH or Na-dithionite, the broad signal almost disappeared and was replaced with multiple signals in the S = ½ region indicative of FeS clusters and a flavin radical. Both treatments yielded overlapping rhombic g = 2.07 (g = 2.072, 1.940, 1.895) and axial g = 2.04 (g = 2.041, 1.944, 1.944) signals consistent with the presence of two iron-sulfur clusters, in addition to an isotropic g = 2.0 signal consistent with a flavin radical that was dominant above 50 K (Figure S9). The temperature dependencies of the rhombic g = 2.07 and axial g = 2.04 signals were characteristic of [4Fe-4S] clusters,70 in agreement with the sequence prediction for a pair of [4Fe-4S] clusters. The optimal temperature (Topt) for the rhombic g = 2.07 signal was near 15 K with broadening above 30 K (Figure S9). The axial g = 2.04 signal was faster relaxing with a lower Topt between 5 and 10 K and broadening above 15 K. Also present for the NADH-reduced sample was a highly temperature dependent axial g = 2.03 signal (g = 2.030, 2.00, 2.00) observed at 10 K. The signal appeared to correlate with a slight loss in intensity from the axial g = 2.04 signal compared to the Na-dithionite treatment, suggesting that this feature could arise from the spin interaction between the faster relaxing [4Fe-4S] cluster and a flavin radical.
Figure 6.

EPR spectra of FixABCX from A. vinelandii prepared in 50 mM HEPES pH 7.5, 150 mM NaCl, and 5% glycerol. Black: As-prepared FixABCX (100 μM); Blue: FixABCX (100 μM) reduced with NADH (1 mM); Red: FixABCX (100 μM) reduced with sodium dithionite (10 mM). Simulations of spectra of NADH- and Na-dithionite-reduced FixABCX are shown in lighter colored traces. Microwave frequency, 9.38 GHz; microwave power, 1 mW; modulation frequency, 100 kHz; modulation amplitude, 10.0 G; sample temperature, 10 K.
Further EPR analysis of the FixX subunit alone (from Roseiflexus castenholzii) identified very similar rhombic g = 2.07 and axial g = 2.04 signals indicating that these signals in FixABCX originate from the FixX subunit (Figure S10). Line broadening at the high- and low-field edges of the reduced spectra was observed for both FixABCX and FixX and is indicative of spin coupling between the two [4Fe-4S] clusters.71 For FixABCX, the different relative intensities of the rhombic g = 2.07 and axial g = 2.04 signals suggest that the corresponding [4Fe-4S] clusters have different Em values. Accordingly, for FixABCX both clusters are thought to have Em values below that of Na-dithionite (≤−440 mV vs. SHE, pH 7.5), since the sample appeared to be only partially reduced by Na-dithionite. We hypothesize that the [4Fe-4S] cluster giving rise to the axial g = 2.04 signal has a lower Em, due to its weaker contribution to the spectrum, based on simulations (Table S4).
Altogether, the two [4Fe-4S] cluster EPR signals show striking resemblance to the EPR signals assigned to the two [4Fe-4S] clusters in the NADH-dependent ferredoxin:NADP+ oxidoreductase I (Nfn) bifurcating enzyme, as the signals share similar _g_-values, temperature dependence, and oxidation-reduction properties.25 For the Nfn enzyme, structural and biophysical analyses showed that two [4Fe-4S] clusters in the large subunit form an electron-transfer chain from the flavin site of electron-bifurcation to an external ferredoxin redox partner. Spin-coupling between the two clusters is thought to facilitate electron-transfer between the redox cofactors. It was also found for the Nfn enzyme that one of the [4Fe-4S] clusters had an unusually low Em that creates a thermodynamically favorable electron transfer pathway from a highly unstable semiquinone intermediate formed during electron bifurcation. An analogous model is suggested here for FixABCX where the two low-potential [4Fe-4S] clusters in the FixX subunit would form a thermodynamically favorable pathway for facile electron transfer between the site of bifurcation and Fd/Fld.
Electron bifurcation by the FixABCX complex
Electron bifurcation by the FixABCX complex is proposed to drive the endergonic reduction of FldSq by coupling it to the exergonic reduction of CoQ using NADH as an electron donor (Figures 2, 7A). Electron bifurcation by the FixABCX complex was demonstrated by incubating FixABCX with the electron donor NADH, high potential acceptor coenzyme Q1 (CoQ1), and low-potential acceptor FldSq. CoQ1 served as an amphipathic analog of the physiological acceptor coenzyme Q8 (CoQ8) found in the electron transport chain of A. vinelandii.72,73 FldSq was used as the low-potential acceptor in these assays since FldHq has been shown to be a direct electron donor to nitrogenase.5,6 Similarly, the previously characterized electron bifurcating Etf-Bcd complex could direct electrons to either Fd or Fld,22,23 suggesting that Fld could likely serve as an electron acceptor from FixABCX as well.
Figure 7.

Electron bifurcation by the FixABCX complex of A. vinelandii under anaerobic conditions. (A) Overview of electron bifurcation by the FixABCX complex showing the NADH bifurcating an electron to the high-potential acceptor, CoQ1 and the other to the low-potential acceptor, FldSq. Evidence of electron bifurcation was obtained using UV-visible spectrophotometry. NADH oxidation as well as FldSq reduction and oxidation were monitored over time as signatures of the bifurcation reaction. (B) NADH oxidation at 340 nm, C) FldSq reduction/oxidation at 580 nm, and D) formation of FldOx at 450 nm. Final concentrations of components added to the reactions were as follows: 0.8 μM FixABCX, 85 μM FldSq, 200 μM NADH, and 300 μM CoQ1. Data were normalized and demonstrate the overall change in absorbance.
For the electron bifurcation assay, the oxidation of NADH (340 nm), disappearance of FldSq (580 nm), and formation of FldOx (450 nm) were monitored over time. CoQ1 reduction (λmax= 274 nm) was not recorded, due to significant interference at that wavelength. While the simultaneous presence of several redox active components in the assays posed a challenge, monitoring activities at multiple wavelengths provided evidence of electron bifurcation. Control reactions for NADH oxidation (diaphorase) activity linked to CoQ1 reduction in the absence of FldSq yielded a specific activity of 396 ± 44 nmol/min/mg with a turnover frequency of 51 ± 6 min−1 (Figure 7B (No FldSq). Bifurcation reactions containing the additional component needed for bifurcation (FldSq, in addition to the control reactants FixABCX, NADH, and CoQ1) exhibited 25% greater NADH oxidation activity with a specific activity of 524 ± 21 nmol/min/mg and turnover frequency of 68 ± 3 min−1 (Figure 7B (All components)). It is important to note that the specific activity values are not adjusted for flavin occupancy, since it could not be confirmed how much of the protein was fully occupied (how flavins were distributed among the flavin binding sites).
The Fld-dependent increase in NADH consumption is consistent with bifurcation, and previous studies on the Etf-Bcd bifurcating complex also demonstrated increased NADH consumption in a bifurcating reaction.22,23 Omission of FixABCX or CoQ1 resulted in baseline NADH oxidation activity (Figure 7B). To decipher whether the enhanced consumption of NADH in the presence of FldSq could be attributed to the endergonic reduction of FldSq to FldHq (the bifurcation reaction), disappearance of FldSq was monitored at 580 nm where optical absorption by the hydroquinone species is negligible but absorbance by the semiquinone is strong (ε= 5.7 mM−1 cm−1). Reactions monitoring the disappearance of FldSq were difficult to interpret on their own, due to background activity in the absence of FixABCX (Figure 7C). In vivo, CoQ is in the membrane, whereas, Fld is in the cytoplasm so direct contact between them is diminished and spontaneous electron transfer between the two species is suppressed (Table S5). In vitro experiments however lack this separation, and our controls revealed that FldSq is able to donate electrons to CoQ1 in the absence of FixABCX (Figure 7C). Thus, the FldSq concentration decreased while an increase at 450 nm, characteristic of FldOx, was observed (Figure 7D). This non-enzymatic reduction of CoQ1 by FldSq is consistent with the favorable ΔG° of −36.7 kJ/mol (Table S5). However, the increase in absorbance at 450 nm produced by spontaneous FldSq oxidation enabled us to account for this reaction that detracts from the apparent yield of the bifurcating reaction (Figure 7C (All components)). The FldOx species was not detected in the bifurcating reaction assays indicating that FldHq, the expected product of the bifurcation reaction, was formed instead (Figure 7D). However, we do not expect FldHq to accumulate in the presence of CoQ1 due to the favorability of electron transfer between these two that will return Fld to its semiquinone state and thus conceal the true extent of FldHq production (ΔG°= −154 kJ/mol) (Eq. 2, Table S5).
| NADH+2FIdHq+2CoQ+3H+=2CoQH2+2FIdSq+NAD+ | (Eq. 2) |
|---|
Overall, the absence of FldOx in conjunction with the increased activity of the electron bifurcation reaction in comparison to diaphorase activity, suggest that the FixABCX complex can in fact perform electron bifurcation. To further investigate the flavin cofactors in FixABCX, transient absorption spectroscopy (TAS) was conducted.
Transient absorption spectroscopy
A comparative TAS study between FixAB from R. rubrum and FixABCX from A. vinelandii was performed in order to gain qualitative information on spectral contributions from individual flavins. TAS of the incomplete Fix complex, FixAB, revealed an anionic semiquinone (ASQ) absorption at 365 nm with corresponding oxidized (Ox) flavin bleach at 447 nm. The ASQ absorption decays with two components: a short-lived component with a lifetime of tens of picoseconds and a longer short-lived component with a lifetime of one thousand picoseconds (Figure 8A, (Red trace), Table S6). A similar ASQ signal was also observed in the A. vinelandii FixABCX complex (Figure 8A (Black trace)). This suggests that two ASQ species can be formed in the FixAB unit, consistent with the presence of two flavins. Bifurcating flavins have been shown to exhibit crossed potentials, whereby the EmOx/SQ is at lower potential than the EmSQ/HQ (‘crossed potentials’) with the result that the high-energy SQ intermediate does not appreciably accumulate relative to Ox and HQ.
Figure 8.

Transient absorption spectra of as-prepared R. rubrum FixAB (red traces) and A. vinelandii FixABCX (black traces). (A) Kinetic traces of ASQ signal (dots) at 365 nm. ASQ decay shows half-lives of ~15 ps and ~1000 ps when fit with a double exponential function (solid lines). (B) An NSQ absorption peak (~565–650 nm) is only observed in the A. vinelandii FixABCX complex.
A short-lived ASQ has been observed in the bifurcating Nfn from Pyrococcus furiosus and is proposed to play a central role in bifurcation.19,25 The current observation of relatively short-lived ASQ signals in R. rubrum FixAB and A. vinelandii FixABCX is consistent with the work on Nfn, and suggests that neither flavin significantly stabilizes a one-electron reduced species. Based on comparison with Etf, the flavin in FixA (a-FAD) (analogous to the β-FAD in EtfB) is the proposed site of bifurcation and likely corresponds to the tens of picosecond component in our TAS experiments.22 This assignment then implies that the FixB b-FAD acts as an electron transferring flavin, consistent with a slightly longer-lived ASQ, since accumulation of electrons at this site may congest electron flow and impede bifurcation. This may additionally play a role in gating electron transfer, ensuring that only one electron follows the exergonic path, and thus restricting the lower potential electron to travel down the endergonic branch, similar to the mechanism of electron bifurcation in P. furiosus Nfn.25 Due to the crossed potentials believed to characterize bifurcating flavins, the more negative potential electron of the bifurcating flavin (ASQ to Ox transition) would provide enough driving force to reduce the low-potential iron-sulfur clusters (predicted ≤−440 mV vs. SHE, pH 7.5) in FixX directly. In the complete FixABCX complex from A. vinelandii, an additional peak is observed in the TAS corresponding to a neutral semiquinone (NSQ) and is assigned to the c-FAD in FixC (Figure 8B). We propose that the c-FAD serves to accumulate two electrons from successive bifurcations in order to permit 2-electron reduction of CoQ.
Proposed mechanism for FixABCX electron bifurcation
The data presented herein establish that the FixABCX complex from A. vinelandii can bifurcate electrons from NADH to CoQ1 and FldSq. The biochemical and biophysical evidence is consistent with a proposed mechanism of Fix electron bifurcation initiated at the flavin in FixA, which would accept a pair of electrons from NADH and pass one electron to the quinone pool via the flavins in FixB and FixC, and the other to Fld/Fd via the low potential [4Fe-4S] clusters in FixX, with the energetic cost being paid by favorable transfer of the former electron to the quinone pool (Figure 2).
TAS on the FixABCX complex provides evidence by demonstrating the presence of a short-lived anionic flavin semiquinone consistent with the ability to support rapid and efficient electron transfer. The finding here provides experimental support for the mechanism proposed previously for a homologous bifurcating Etf that assigns the site of bifurcation to the flavin bound by FixA (EtfB).22 This assignment is further supported by the demonstration by Sato et al. that the flavin reduced by NADH is the one bound in FixA (EtfB), but that electron transfer to the flavin in FixB (EtfA) is favorable.39 Thus, upon reduction of the FixA flavin, transfer of an electron through the exergonic branch to CoQ via Fix B and FixC can leave the second electron in a very unstable, highly energetic flavin semiquinone state with a low EmOx/Sq able to drive reduction of Fld/Fd, via FixX (Figure 2).
The mechanism presented here offers a means by which Fd/Fld can be reduced by NADH in a reaction that is thermodynamically favorable overall and identifies a new pathway by which low-potential reducing equivalents can be generated to drive nitrogen fixation.
Supplementary Material
Supplemental Information
Acknowledgments
The authors would like to thank Stefan Nordlund at Stockholm University and Tomas Edgren at Umeå University for the plasmid containing R. rubrum FixAB and their guidance in Fix protein expression and purification and Luis Rubio for the A. vinelandii rnf mutants. The authors would also like to acknowledge Utah State University’s Synthetic Biomanufacturing Institute for conducting large-scale fermentations and Montana State University’s Microfabrication Facility for help in preparation of gold-coated borosilica capillaries for non- covalent mass spectrometry. This work is supported as part of the Biological and Electron Transfer and Catalysis (BETCy) EFRC, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science (DE-SC0012518). The Mass Spectrometry Facility at MSU is supported in part by the Murdock Charitable Trust and an NIH IDEA program grant P20GM103474.
Abbreviations
AEBSF
4-(2-aminoethyl)benzenesulfonyl fluoride hydrochloride
AmpS
ampicillin sensitivity
ASQ
anionic semiquinone
BBO
beta barium borate
BS3
bis(sulfosuccinimidyl)suberate
CoQ
coenzyme Q
CoQ1
coenzyme Q1
CoQ8
coenzyme Q8
DDM
dodecyl maltoside
DLS
dynamic light scattering
DTT
dithiothreitol
Etf
electron transfer flavoprotein
Etf-Bcd
electron transferring flavoprotein/butyryl-coenzyme A
ETF-QO
electron transferring flavoprotein ubiquinone oxidoreductase
Em
midpoint potential
EPR
electron paramagnetic resonance
FAD
flavin adenine dinucleotide
Fd
ferredoxin
Fld
flavodoxin
FldOx
oxidized quinone
FldHq
flavodoxin hydroquinone
FldSq
flavodoxin semiquinone
IPTG
Isopropyl β-D-1-thiogalactopyranoside
KmR
kanamycin resistance
LB
Luria-Bertani
LC-MS/MS
liquid chromatography tandem mass spectrometry
MS
mass spectrometry; Na-dithionite, sodium dithionite
Nif+
nitrogen fixing phenotype
Nif−
non-nitrogen fixing phenotype
PMSF
phenylmethylsulfonyl fluoride
Rf
riboflavin
Rnf
Rhodobacter nitrogen fixation
SmR
streptomycin resistance
TAS
transient absorption spectroscopy
TCEP
tris(2-carboxyethyl)phosphine
TB
terrific broth, Topt, optimal temperature
Footnotes
Supporting information
Primers for Δ_fix_ mutant generation (Table S1), Primers for overexpression of FixABCX (Table S2), Protein-protein interactions captured within FixABCX complex during chemical cross-linking reaction (Table S3), Overview of the EPR signals observed for FixABCX from A. vinelandii (Table S4), Calculated Gibbs free energies of possible reactions catalyzed by the Fix complex and possible non-enzymatic reactions (Table S5), Lifetimes of ASQ species from R. rubrum FixAB and A. vinelandii FixABCX (Table S6), Scheme for Δfix mutant generation (Figure S1), Scheme for overexpression of fix genes in A. vinelandii (Figure S2), Protein identification within FixABCX complex purified from Azotobacter vinelandii (Figure S3), Dynamic light scattering of FixABCX complex (Figure S4), Evaluation of subunit homology models (Figure S5), Predicted structural features in Fix subunits (Figure S6), FixB protein interface (Figure S7), Structural model of FixABCX from A. vinelandii (Figure S8), EPR temperature profiles of FixABCX from A. vinelandii (Figure S9), Comparison of EPR spectra of FixABCX from A. vinelandii and the individual FixX subunit from R. castenholzii (Figure S10).
References
- (1).Kim J, and Rees DC (1994) Nitrogenase and biological nitrogen fixation. Biochemistry 33, 389–397. [DOI] [PubMed] [Google Scholar]
- (2).Burgess BK, and Lowe DJ (1996) Mechanism of molybdenum nitrogenase. Chem. Rev. 96, 2983–3012. [DOI] [PubMed] [Google Scholar]
- (3).Seefeldt LC, Hoffman BM, and Dean DR (2009) Mechanism of Mo-dependent nitrogenase. Annu. Rev. Biochem. 78, 701–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Saeki K. (2004) Electron transport to nitrogenase: Diverse routes for a common destination, in Genetics and Regulation of Nitrogen Fixation in Free-living Bacteria (Klipp W, Masepohl B, Gallon JR, and Newton WE, Eds.), pp 257–290. Kluwer Academic Publishers, Dordrecht. [Google Scholar]
- (5).Yang ZY, Ledbetter R, Shaw S, Pence N, Tokmina-Lukaszewska M, Eilers B, Guo Q, Pokhrel N, Cash VL, Dean DR, Antony E, Bothner B, Peters JW, and Seefeldt LC (2016) Evidence that the Pi release event is the rate-limiting step in the nitrogenase catalytic cycle. Biochemistry 55, 3625–3635. [DOI] [PubMed] [Google Scholar]
- (6).Martin AE, Burgess BK, Iismaa SE, Smartt CT, Jacobson MR, and Dean DR (1989) Construction and characterization of an Azotobacter vinelandii strain with mutations in the genes encoding flavodoxin and ferredoxin I. J. Bacteriol. 171, 3162–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Deistung J, and Thorneley RN (1986) Electron transfer to nitrogenase: Characterization of flavodoxin from Azotobacter chroococcum and comparison of its redox potentials with those of flavodoxins from Azotobacter vinelandii and Klebsiella pneumoniae (nifF-gene product). Biochem. J. 239, 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mortenson LE (1964) Ferredoxin and ATP, requirements for nitrogen fixation in cell-free extracts of Clostridium pasteurianum. Proc. Natl. Acad. Sci. U.S.A. 52, 272–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Yates MG (1972) Electron transport to nitrogenase in Azotobacter chroococcum: Azotobacter flavodoxin hydroquinone as an electron donor. FEBS Lett. 27, 63–67. [DOI] [PubMed] [Google Scholar]
- (10).Thorneley RN, and Deistung J. (1988) Electron-transfer studies involving flavodoxin and a natural redox partner, the iron protein of nitrogenase: Conformational constraints on protein-protein interactions and the kinetics of electron transfer within the protein complex. Biochem. J. 253, 587–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Cammack R. (1992) Iron-sulfur clusters in enzymes: Themes and variations, in Advances in Inorganic Chemistry (Cammack R, Ed.), pp 281–322. Academic Press. [Google Scholar]
- (12).Klugkist J, Voorberg J, Haaker H, and Veeger C. (1986) Characterization of three different flavodoxins from Azotobacter vinelandii. Eur. J. Biochem. 155, 33–40. [DOI] [PubMed] [Google Scholar]
- (13).Ludden PW (1991) Energetics of and sources of energy for biological nitrogen fixation, in Current Topics in Bioenergetics (Lee CP, Ed.), pp 369–390. Academic Press. [Google Scholar]
- (14).Shah VK, Stacey G, and Brill WJ (1983) Electron transport to nitrogenase. Purification and characterization of pyruvate:flavodoxin oxidoreductase. The nifJ gene product. J. Biol. Chem. 258, 12064–12068. [PubMed] [Google Scholar]
- (15).Saeki K, and Kumagai H. (1998) The rnf gene products in Rhodobacter capsulatus play an essential role in nitrogen fixation during anaerobic DMSO-dependent growth in the dark. Arch. Microbiol. 169, 464–467. [DOI] [PubMed] [Google Scholar]
- (16).Schmehl M, Jahn A, Vilsendorf AM zu, Hennecke S, Masepohl B, Schuppler M, Marxer M, Oelze J, and Klipp W. (1993) Identification of a new class of nitrogen fixation genes in Rhodobacter capsalatus: A putative membrane complex involved in electron transport to nitrogenase. Molec. Gen. Genet. 241, 602–615. [DOI] [PubMed] [Google Scholar]
- (17).Buckel W, and Thauer RK (2013) Energy conservation via electron bifurcating ferredoxin reduction and proton/Na+ translocating ferredoxin oxidation. Biochim. Biophys. Acta-Bioenerg 1827, 94–113. [DOI] [PubMed] [Google Scholar]
- (18).Peters JW, Miller AF, Jones AK, King PW, and Adams MW (2016) Electron bifurcation. Curr. Opin. Chem. Biol. 31, 146–152. [DOI] [PubMed] [Google Scholar]
- (19).Nitschke W, and Russell MJ (2012) Redox bifurcations: Mechanisms and importance to life now, and at its origin. Bioessays 34, 106–109. [DOI] [PubMed] [Google Scholar]
- (20).Herrmann G, Jayamani E, Mai G, and Buckel W. (2008) Energy conservation via electron-transferring flavoprotein in anaerobic bacteria. J. Bacteriol. 190, 784–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Li F, Hinderberger J, Seedorf H, Zhang J, Buckel W, and Thauer RK (2008) Coupled rerredoxin and crotonyl coenzyme A (CoA) reduction with NADH catalyzed by the butyryl-CoA dehydrogenase/Etf complex from Clostridium kluyveri. J. Bacteriol. 190, 843–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chowdhury NP, Mowafy AM, Demmer JK, Upadhyay V, Koelzer S, Jayamani E, Kahnt J, Hornung M, Demmer U, Ermler U, and Buckel W. (2014) Studies on the mechanism of electron bifurcation catalyzed by electron transferring flavoprotein (Etf) and butyryl-CoA dehydrogenase (Bcd) of Acidaminococcus fermentans. J. Biol. Chem. 289, 5145–5157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Chowdhury NP, Klomann K, Seubert A, and Buckel W. (2016) Reduction of flavodoxin by electron bifurcation and sodium ion-dependent re-oxidation by NAD+ catalysed by ferredoxin:NAD+reductase (Rnf). J. Biol. Chem. 291, 11993–12002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Bertsch J, Parthasarathy A, Buckel W, and Müller V. (2013) An electron-bifurcating caffeyl-CoA reductase. J. Biol. Chem. 288, 11304–11311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lubner CE, Jennings DP, Mulder DW, Schut GJ, Zadvornyy OA, Hoben J, Tokmina-Lukaszewska M, Berry L, Nguyen D, Lipscomb GL, Bothner B, Jones AK, Miller AF, King PW, Adams MWW, and Peters JW (2017) Mechanistic insights into energy conservation by flavin-based electron bifurcation. Nat. Chem. Biol. 13, 655–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Edgren T. (2006) Electron transport to nitrogenase. Stockholm University, Stockholm, Sweden. [Google Scholar]
- (27).Wientjens R. (1993) The involvement of the fixABCX genes and the respiratory chain in the electron transport to nitrogenase in Azotobacter vinelandii. The Department of Chemistry and Biochemistry, Agricultural University, Wageningen, The Netherlands. [Google Scholar]
- (28).Huang JJ, Heiniger EK, McKinlay JB, and Harwood CS (2010) Production of hydrogen gas from light and the inorganic electron donor thiosulfate by Rhodopseudomonas palustris. Appl. Environ. Microbiol. 76, 7717–7722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Edgren T, and Nordlund S. (2004) The fixABCX genes in Rhodospirillum rubrum encode a putative membrane complex participating in electron transfer to nitrogenase. J. Bacteriol. 186, 2052–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ruvkun GB, Sundaresan V, and Ausubel FM (1982) Directed transposon Tn5 mutagenesis and complementation analysis of Rhizobium meliloti symbiotic nitrogen fixation genes. Cell 29, 551–559. [DOI] [PubMed] [Google Scholar]
- (31).Earl CD, Ronson CW, and Ausubel FM (1987) Genetic and structural analysis of the Rhizobium meliloti fixA, fixB, fixC, and fixX genes. J. Bacteriol. 169, 1127–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Curatti L, Brown CS, Ludden PW, and Rubio LM (2005) Genes required for rapid expression of nitrogenase activity in Azotobacter vinelandii. Proc. Natl. Acad. Sci. U.S.A. 102, 6291–6296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).de Lorenzo V, Herrero M, Jakubzik U, and Timmis KN (1990) Mini-Tn5 transposon derivatives for insertion mutagenesis, promoter probing, and chromosomal insertion of cloned DNA in gram-negative eubacteria. J. Bacteriol. 172, 6568–6572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Page WJ, and von Tigerstrom M. (1979) Optimal conditions for transformation of Azotobacter vinelandii. J. Bacteriol. 139, 1058–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Jacobson MR, Cash VL, Weiss MC, Laird NF, Newton WE, and Dean DR (1989) Biochemical and genetic analysis of the nifUSVWZM cluster from Azotobacter vinelandii. Molec. Gen. Genet. 219, 49–57. [DOI] [PubMed] [Google Scholar]
- (36).Toukdarian A, and Kennedy C. (1986) Regulation of nitrogen metabolism in Azotobacter vinelandii: isolation of ntr and glnA genes and construction of ntr mutants. EMBO J. 5, 399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sarma R, Barney BM, Hamilton TL, Jones A, Seefeldt LC, and Peters JW (2008) Crystal structure of the L protein of Rhodobacter sphaeroides light-independent protochlorophyllide reductase with MgADP bound: a homologue of the nitrogenase Fe protein. Biochemistry 47, 13004–13015. [DOI] [PubMed] [Google Scholar]
- (38).Lewis JA, and Escalante-Semerena JC (2006) The FAD-dependent tricarballylate dehydrogenase (TcuA) enzyme of Salmonella enterica converts tricarballylate into cis-aconitate. J Bacteriol. 188, 5479–5486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Sato K, Nishina Y, and Shiga K. (2013) Interaction between NADH and electron-transferring flavoprotein from Megasphaera elsdenii. J. Biochem. 153, 565–572. [DOI] [PubMed] [Google Scholar]
- (40).Vaudel M, Burkhart JM, Zahedi RP, Oveland E, Berven FS, Sickmann A, Martens L, and Barsnes H. (2015) PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 33, 22–24. [DOI] [PubMed] [Google Scholar]
- (41).Lima DB, de Lima TB, Balbuena TS, Neves-Ferreira AGC, Barbosa VC, Gozzo FC, and Carvalho PC (2015) SIM-XL: A powerful and user-friendly tool for peptide cross-linking analysis. J. Proteomics 129, 51–55. [DOI] [PubMed] [Google Scholar]
- (42).Poudel S, Tokmina-Lukaszewska M, Colman DR, Refai M, Schut GJ, King PW, Maness PC, Adams MWW, Peters JW, Bothner B, and Boyd ES (2016) Unification of [FeFe]-hydrogenases into three structural and functional groups. Biochim. Biophys. Acta 1860, 1910–1921. [DOI] [PubMed] [Google Scholar]
- (43).Luo ML, Jackson RN, Denny SR, Tokmina-Lukaszewska M, Maksimchuk KR, Lin W, Bothner B, Wiedenheft B, and Beisel CL (2016) The CRISPR RNA-guided surveillance complex in Escherichia coli accommodates extended RNA spacers. Nucl. Acids Res. 44, 7385–7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Kelley LA, Mezulis S, Yates CM, Wass MN, and Sternberg MJE (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protocols 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kozakov D, Beglov D, Bohnuud T, Mottarella SE, Xia B, Hall DR, and Vajda S. (2013) How good is automated protein docking? Proteins 81, 2159–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Kozakov D, Brenke R, Comeau SR, and Vajda S. (2006) PIPER: an FFT-based protein docking program with pairwise potentials. Proteins 65, 392–406. [DOI] [PubMed] [Google Scholar]
- (47).Comeau SR, Gatchell DW, Vajda S, and Camacho CJ (2004) ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20, 45–50. [DOI] [PubMed] [Google Scholar]
- (48).Comeau SR, Gatchell DW, Vajda S, and Camacho CJ (2004) ClusPro: a fully automated algorithm for protein-protein docking. Nucl. Acids Res. 32, W96–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Grosdidier A, Zoete V, and Michielin O. (2011) SwissDock, a protein-small molecule docking web service based on EADock DSS. Nucl. Acids Res. 39, W270–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Grosdidier A, Zoete V, and Michielin O. (2011) Fast docking using the CHARMM force field with EADock DSS. J. Comput. Chem. 32, 2149–2159. [DOI] [PubMed] [Google Scholar]
- (51).Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004) UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (52).McComb RB, Bond LW, Burnett RW, Keech RC, and Bowers GN (1976) Determination of the molar absorptivity of NADH. Clin. Chem. 22, 141–150. [PubMed] [Google Scholar]
- (53).Enescu M, Lindqvist L, and Soep B. (1998) Excited-state dynamics of fully reduced flavins and flavoenzymes studied at subpicosecond time resolution. Photochem. Photobiol. 68, 150–156. [DOI] [PubMed] [Google Scholar]
- (54).Wang Z, and Zhang J. (2009) Abundant indispensable redundancies in cellular metabolic networks. Genome Biol. Evol. 1, 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Jeong HS, and Jouanneau Y. (2000) Enhanced nitrogenase activity in strains of Rhodobacter capsulatus that overexpress the rnf genes. J. Bacteriol. 182, 1208–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Sarkar A, Köhler J, Hurek T, and Reinhold-Hurek B. (2012) A novel regulatory role of the Rnf complex of Azoarcus sp. strain BH72. Mol. Microbiol. 83, 408–422. [DOI] [PubMed] [Google Scholar]
- (57).Boyd ES, Costas AMG, Hamilton TL, Mus F, and Peters JW (2015) Evolution of molybdenum nitrogenase during the transition from anaerobic to aerobic metabolism. J. Bacteriol. 197, 1690–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Grechkin Y, Ratner A, Jacob B, Huang J, Williams P, Huntemann M, Anderson I, Mavromatis K, Ivanova NN, and Kyrpides NC (2012) IMG: the Integrated Microbial Genomes database and comparative analysis system. Nucl. Acids Res. 40, D115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Reyntjens B, Jollie DR, Stephens PJ, Gao-Sheridan HS, and Burgess BK (1997) Purification and characterization of a _fix_ABCX-linked 2[4Fe-4S] ferredoxin from Azotobacter vinelandii. J. Biol. Inorg. Chem. 2, 595–602. [Google Scholar]
- (60).Watmough NJ, and Frerman FE (2010) The electron transfer flavoprotein: Ubiquinone oxidoreductases. Biochim. Biophys. Acta- Bioenerg. 1797, 1910–1916. [DOI] [PubMed] [Google Scholar]
- (61).Usselman RJ, Fielding AJ, Frerman FE, Watmough NJ, Eaton GR, and Eaton SS (2008) Impact of mutations on the midpoint potential of the [4Fe-4S]+1,+2 cluster and on catalytic activity in electron transfer flavoprotein-ubiquinone oxidoreductase (ETF-QO). Biochemistry 47, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Jones DT, Taylor WR, and Thornton JM (1994) A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry. 33, 3038–3049. [DOI] [PubMed] [Google Scholar]
- (63).le Maire M, Champeil P, and Møller JV (2000) Interaction of membrane proteins and lipids with solubilizing detergents. Biochim. Biophys. Acta- Biomembr 1508, 86–111. [DOI] [PubMed] [Google Scholar]
- (64).Sweeney WV, and Rabinowitz JC (1980) Proteins Containing 4Fe-4S Clusters: An Overview. Annual Review of Biochemistry 49, 139–161. [DOI] [PubMed] [Google Scholar]
- (65).Zhang J, Frerman FE, and Kim J-JP (2006) Structure of electron transfer flavoprotein-ubiquinone oxidoreductase and electron transfer to the mitochondrial ubiquinone pool. Proc. Natl. Acad. Sci. U.S.A. 103, 16212–16217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Demmer JK, Rupprecht FA, Eisinger ML, Ermler U, and Langer JD (2016) Ligand binding and conformational dynamics in a flavin-based electron-bifurcating enzyme complex revealed by Hydrogen-Deuterium Exchange Mass Spectrometry. FEBS Letters 590, 4472–4479. [DOI] [PubMed] [Google Scholar]
- (67).Dryhurst G, and Elving PJ (1969) Electrochemical oxidation-reduction paths for pyrimidine, cytosine, purine and adenine: Correlation and application. Talanta 16, 855–874. [DOI] [PubMed] [Google Scholar]
- (68).Bonetti C, Stierl M, Mathes T, van Stokkum IHM, Mullen KM, Cohen-Stuart TA, van Grondelle R, Hegemann P, and Kennis JTM (2009) The role of key amino acids in the photoactivation pathway of the Synechocystis Slr1694 BLUF domain. Biochemistry 48, 11458–11469. [DOI] [PubMed] [Google Scholar]
- (69).Gray HB, and Winkler JR (2015) Hole hopping through tyrosine/tryptophan chains protects proteins from oxidative damage. Proc. Natl. Acad. Sci. U.S.A. 112, 10920–10925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Rupp H, Rao KK, Hall DO, and Cammack R. (1978) Electron spin relaxation of iron-sulphur proteins studied by microwave power saturation. Biochim. Biophys. Acta- Protein Structure 537, 255–269. [DOI] [PubMed] [Google Scholar]
- (71).Mathews R, Charlton S, Sands RH, and Palmer G. (1974) On the nature of the spin coupling between the iron-sulfur clusters in the eight-iron ferredoxins. J. Biol. Chem. 249, 4326–4328. [PubMed] [Google Scholar]
- (72).Swank RT, and Burris RH (1969) Restoration by ubiquinone of Azotobacter vinelandii reduced nicotinamide adenine dinucleotide oxidase activity. J. Bacteriol. 98, 311–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Knowles CJ, and Redfearn ER (1969) Cytochrome and ubiquinone patterns during growth of Azotobacter vinelandii. J. Bacteriol. 97, 756–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Information