Host Pathways of Hemostasis that Regulate Group A Streptococcus pyogenes Pathogenicity (original) (raw)
. Author manuscript; available in PMC: 2020 Nov 17.
Abstract
A hallmark feature of severe Group A Streptococcus pyogenes (GAS) infection is dysregulated hemostasis. Hemostasis is the primary pathway for regulating blood flow through events that contribute towards clot formation and its dissolution. However, a number of studies have identified components of hemostasis in regulating survival and dissemination of GAS. Several proteins have been identified on the surface of GAS and they serve to either facilitate invasion to host distal sites or regulate inflammatory responses to the pathogen. GAS M-protein, a surface-exposed virulence factor, appears to be a major target for interactions with host hemostasis proteins. These interactions mediate biochemical events both on the surface of GAS and in the solution when M-protein is released into the surrounding environment through shedding or regulated proteolytic processes that dictate the fate of this pathogen. A thorough understanding of the mechanisms associated with these interactions could lead to novel approaches for altering the course of GAS pathogenicity.
Keywords: Group A Streptococcus, hemostasis, inflammation, pyogenes, pathogen, M-protein
1. INTRODUCTION
Group A Streptococcus pyogenes (GAS) causes >700 million cases of human pharyngeal and dermal infections worldwide/year, ranging from simple antibiotic-sensitive pharyngitis and impetigo/dermatitis to severe antibiotic-resistant forms of disseminated infectious diseases, e.g., necrotizing fasciitis and streptococcal toxic shock syndrome. In addition, post-infection non-pyogenic sequelae, e.g., rheumatic fever and glomerulonephritis, can occur. Due to the existence and continuous emergence of highly virulent strains of GAS, identification, characterization, and regulation of GAS virulence factors are topics of high interest. While there are >250 serotypes of GAS, among the most prevalent is a Pattern D skin trophic group, which conscripts components of the human innate immune system, including hemostasis and complement factors, in order to survive and disseminate in the host. Traditionally, the hemostasis system has been exclusively associated with mechanisms involved in regulating blood flow. Only within the past few decades, a role for components of this system has been identified as participants in other biological processes. Most of these advances have been made from the utilization of isolated components of this pathway in in vitro cell biology and biochemical studies as well as in vivo models utilizing gene-altered mice. Understanding the relationships between hemostasis and GAS in survival and dissemination of this bacteria can lead to novel therapeutic approaches in arresting the pathogenic processes of GAS infection.
2. GROUP A STREPTOCOCCUS PYOGENES
GAS is a common commensal bacterium of humans that typically colonizes the skin and mucosal surfaces, often leading to self-limiting skin and respiratory tract infections, e.g., impetigo and pharyngitis, that are sensitive to antibiotics. However, when treatment is delayed or absent, GAS infections can result in post-infection pyogenic or nonpyogenic sequelae, some of which evoke autoimmune responses that lead to glomerulonephritis and rheumatic heart disease [1]. In the most severe infections, when GAS breaches the epithelial barrier and invades blood and other deep tissue sites, life-threatening complications, e.g., necrotizing fasciitis and toxic septic shock, can occur [2]. The ~250 M-protein (M-Prt)-based serotypes of GAS that have been identified to date, possess highly evolved mechanisms to combat host innate immune defenses, and this human-selective microorganism continuously evolves to adjust to the more general defenses employed by the host that are genetically less focused on a battle with a single microorganism. GAS is genetically well constructed to combat host defenses. While this Gram+ microbe does not possess a protective outer membrane, it does contain a thick peptidoglycan (PGN)-rich semi-porous cell wall (CW) layer around its cytoplasmic membrane (CM). This CW is coated by a nonimmunogenic hyaluronic acid (HA) capsule. The (CW+HA) provides structural strength to GAS and presents a barrier to osmotic lysis. Importantly, the CW is also a matrix with functional groups to which proteins and carbohydrate can covalently tether.
The surface of GAS is studded by projections that extend beyond the HA capsule, which consists of four major types, viz.:(1) sortase A-dependent CW covalently-anchored LPXTG proteins, e.g., M-protein (M-Prt) [3], as well as glycopolymers, e.g., CW-bound teichoic acid, that serve as adhesins and also stabilize the Gram+ cell surface [4]; (2) non-covalently-bound glycolytic enzymes normally found in the cytosol, e.g., enolase (Eno) and GAPDH, provide ATP at the GAS surface, as well as serve as receptors for host proteins [5, 6]; (3) CM-bound moieties, e.g., leucine-rich repeat (LRR) lipoproteins (Lp) [7, 8], which are inserted in the CM through their N-terminal lipid components and bind GAS to host cell extracellular matrix (ECM) components [9]; (4) pathogen-associated molecular patterns (PAMP)s, e.g., CW PGN and lipotechoic acid (LTA), the latter also anchored in the CM by its N-terminal lipid moiety [10], that provides strength to the CW [11]. Some LTA is released from the CM and appears both in the cell media and on the cell surface where it is bound to positively charged proteins, e.g., M-Prt [12, 13]. This cell surface LTA provides hydrophobicity and functions in biofilm formation, as well as in adhesion [10, 14]. LTA can also engage CD14 and toll-like receptor (TLR)-2, thus upregulating inflammatory mediators [15, 16]. To colonize within various host niches, GAS employs ~30 one-component (OC) and ~13 two-component (TC) regulatory systems to sense, respond, and accommodate gene transcription to local environmental changes [17]. Control of human plasminogen/human plasmin (hPg/hPm) binding to GAS must be regulated since uncontrolled protease activity is harmful to the bacteria as they encounter disparate environments during different stages of infection. As a relevant example, M-Prt expression is positively controlled by the OC transcriptional regulator, Mga, which is most active during the bacterial growth phase. Intracellular Mga responds to the environment of GAS, likely by sensing metabolites in growing cells, e.g., glucose, and regulates ~10% of the GAS genome [18]. TC transcriptional regulatory systems normally consist of an extracellular environmental sensor (S) and intracellular responder (R) proteins. The most widely studied TC system is the control of virulence (Cov) RS system, which regulates ~18% of GAS transcription [19, 20]. An example of an important gene regulated by CovRS is the major secreted cysteine proteinase, streptococcal pyrogenic exotoxin B (SpeB). Overall, many GAS strains engage the hemostasis system to assist in its survival, and the human host also employs hemostasis as an innate immune defense.
3. EFFECTS OF COMPONENTS OF HEMOSTASIS ON GAS
Hemostasis, a component of human innate immunity, combines pathways of coagulation (intrinsic and extrinsic), anticoagulation, fibrinolysis, and platelet activation, to seize bleeding and maintain blood flow within the vessels. After infection, many components of hemostasis, in collaboration with inflammatory systems, can also be exploited by the pathogen for survival benefits of the microbe.
3.1. Coagulation and GAS Infection
The contact activation pathway (intrinsic coagulation pathway) consists of three serine proteases, Factor (F) XIIa, FXIa, and plasma kallikrein, as well as a non-enzymatic cofactor, high molecular weight kininogen (HMWK). Activation of this pathway is initiated by FXII binding to a negatively charged surface, which results in the autoactivation of FXII to FXIIa. FXIIa then activates prekallikrein and the clotting factor FXI, in combination with HMWK. Kallikrein cleaves HMWK which generates bradykinin (BK), a vasoactive proinflammatory peptide [21, 22]. Further proteolytic processing, i.e., neutrophil elastase, of HMWK generates antimicrobial peptides (AMPs) which assist in host defense responses against invading pathogens (Fig. 1).
Fig. (1).
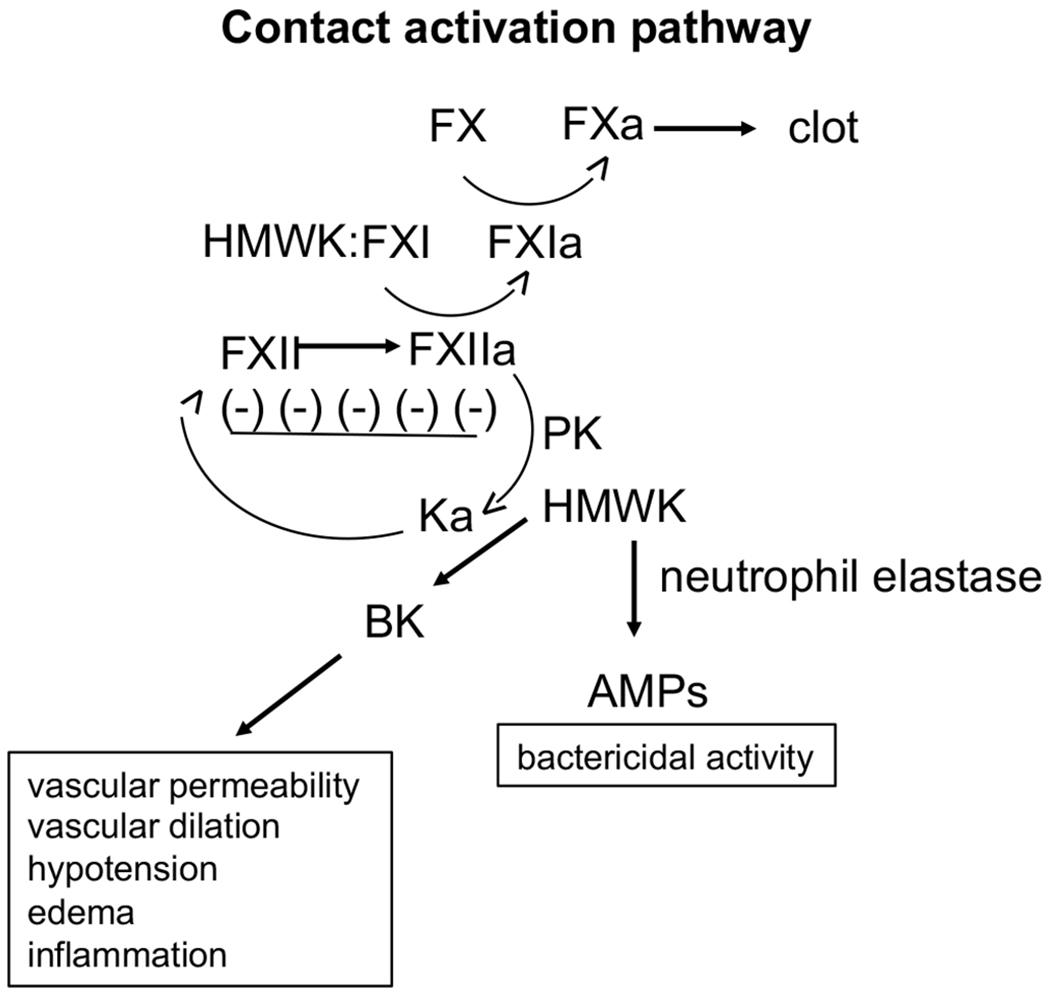
The contact activation system (intrinsic coagulation pathway) consists of three serine proteases, FXII, FXI, and plasma kallirein (Ka), as well as a non-enzymatic cofactor, high molecular weight kininogen (HMWK). Activation of this pathway is initiated by FXII binding to a charged surface, which results in autoactivation of FXII. FXIIa then activates prekallikrein (PK) and FXI in complex with HMWK. Ka cleaves HMWK, generating bradykinin (BK), a vasoactive proinflammatory peptide, and kininogen can be further proteolytically degraded to generate anti-microbial peptides (AMPs).
GAS, specifically the cell wall PAMPs, via toll-like receptors (TLR)s, produce inflammatory mediators (IM) that originate from various host cells, e.g., monocytes [23–25]. This results in activation of the extrinsic pathway of hemostasis by the expression of Tissue Factor (TF) on monocytes and liberation of TF-containing microparticles [26], culminating in the upregulation of coagulation proteases, e.g., activated FXa and thrombin, which also enhance IM generation through binding to cellular protease-activated receptors (PARs) [27, 28]. Activation of endothelial cells (EC)s occurs during this process, which further upregulates TF [29] and plasminogen activated inhibitor (PAI)-1 [30], and downregulates anticoagulant activated Protein C (aPC) [31], which in combination enhances further FXa/thrombin expression. Stressed ECs, and other cell types, also release their intracellular contents (DAMPs), further stimulating inflammation [32]. This cycle of coagulation-inflammation is propagated until systemic inflammation, DIC, and eventually, lethal organ failure can occur.
Thrombin also originates from the intrinsic coagulation system, and is important in severe GAS infections [33–35]. It has been shown that coagulation FXII interacts with the GAS cell surface [36], where it is capable of a low degree of activation to FXIIa [37, 38]. Both FXI and PK (substrates of FXIIa) are bound to the GAS surface through HMWK, which in turn is bound to GAS by some M-Prts [39]. Ka [40], and/or a bacterial secreted protease, SpeB [41], converts HK to bradykinin (BK). The activation of Factor XII and Factor XI, critical components of the contact activation system, has also been shown to occur in the presence of GAS. Clinically, low levels of Factor XII and prolonged aPTTs are seen in patients with septic shock, an end-stage event of severe GAS infection [42].
3.2. Fibrinolysis and GAS Infection
Human fibrin (hFn), a consequence of thrombin/FXIIIa generation, serves other roles, e.g., hFn engulfs and confines the bacteria, thus hindering bacterial dissemination [43]. GAS responds by secreting a protein, streptokinase (SK), which is highly specific for activation of the human fibrinolytic system. This leads to the degradation of hFn, thus liberating the bacteria (Fig. 2). The human fibrinolytic system, consisting of hPg, hPm, fibrinogen (hFg), and activators and inhibitors of fibrinolysis, regulates hFn formation, which is important for a variety of normal processes [44–46], e.g., thrombotic stroke prevention, that require control of microthrombosis [47–49]. hPg is an 810-amino acid residue single-chain modular protein consisting of a 19-residue signal peptide, followed by five consecutive ~80 residue kringle (K) domains, which function to bind fibrinolytic effectors, e.g., Cl-, and lysine [50, 51], and a cryptic serine protease chain. hPg is activated by cleavage of the R580-V581 peptide bond (numbering from methionine 1 of the signal peptide) forming hPm, which contains two peptide chains, viz., the five-kringle nonprotease heavy chain (HC; E20-R580) double disulfide-linked to a trypsin-like serine protease light chain (LC; V581-N810) [52]. The essential features for GAS needed to enlist the host fibrinolytic system in its virulence are first to specifically and functionally interact with hPg and then to activate hPg on its surface to hPm. Many skin trophic strains of GAS can directly, and with very high affinity, bind hPg and hPm through the lysine binding site (LBS) of the K2 domain of hPg/hPm (K2hPg) and a small N-terminal a1a2 module of the M-Prt, plasminogen binding Group A streptococcal M-like protein (PAM) [53–56]. SK is arranged into two clusters, SK1 and SK2, while SK2 is subdivided into subtypes SK2a and SK2b. SK2b is selectively generated in skin-trophic Pattern D GAS strains that express hPg binding PAM. Studies have indicated that SK2b-mediated activation of hPg is enhanced by its interaction with the surface-expressed PAM and hPm that is generated remains associated with the cell surface which protects it from interacting with Pm inhibitors, i.e., α2-antiplasmin [57, 58] (Fig. 3A). Thus, a stable proteolytic surface is formed on GAS that provides the bacteria with a potent weapon against host innate immunity. Other strains of GAS with nasopharynx trophicity contain surface M-Prts, i.e., serotype M1, that first binds hFg through the D-domain of hFg and the B-domain of M-Prts [59]. hPg then binds to GAS via the bound hFg, through the E domain of hFg/hFn and the K1/K4/K5 domains of hPg/hPm (Fig. 3B). Other less virulent strains of GAS do not tightly bind to hPg or hFg. hPm bound to GAS functions in its defense against several host innate immune responses. For example, Pm allows GAS to penetrate the epithelial cell (EpC) and endothelial cell (EC) tight junctions (TJ), thus aiding the spread of the bacteria [60, 61]. hPm can also activate pro-metalloproteinases (pMMP) to active MMPs, which degrade the ECM, as well as EpC and EC TJs, thus allowing GAS dissemination [62]. In addition, hPm engages human cell integrin α5β1 and rearranges the cell cytoskeleton, promoting bacterial invasion of cells [63], thus allowing GAS to persist intracellularly. Lastly, mechanisms exist whereby hPm interferes with complement C3b deposition [64, 65], thereby attenuating opsonophagocytosis of GAS by phagocytic cells.
Fig. (2).
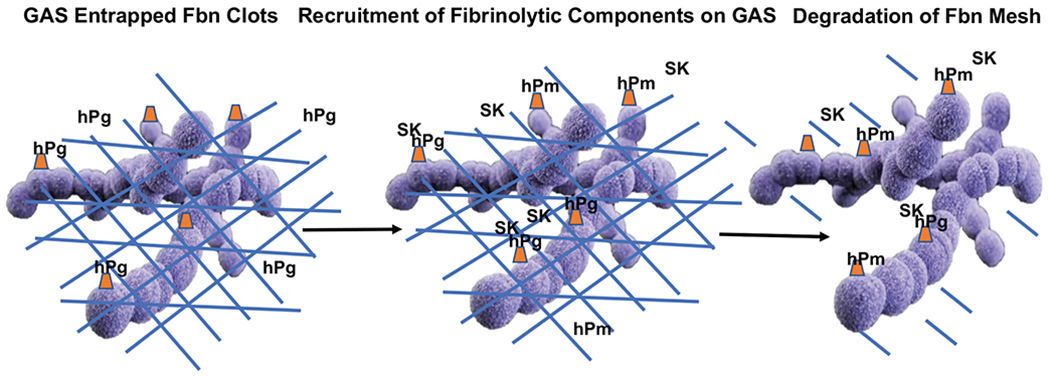
In an attempt to inhibit the dissemination of GAS in host tissue, activation of the host coagulation cascade occurs which results in engulfing GAS in a fibrin mesh (blue hash markings). GAS responds to this entrapment by binding host plasminogen (Pg) to M-Prts (orange triangles), directly or indirectly, exposed on the surface of GAS. GAS-derived SK is then released and surface-bound Pg is activated to plasmin (Pm) which facilitates in the degradation of fibrin and the release of GAS.
Fig. (3).
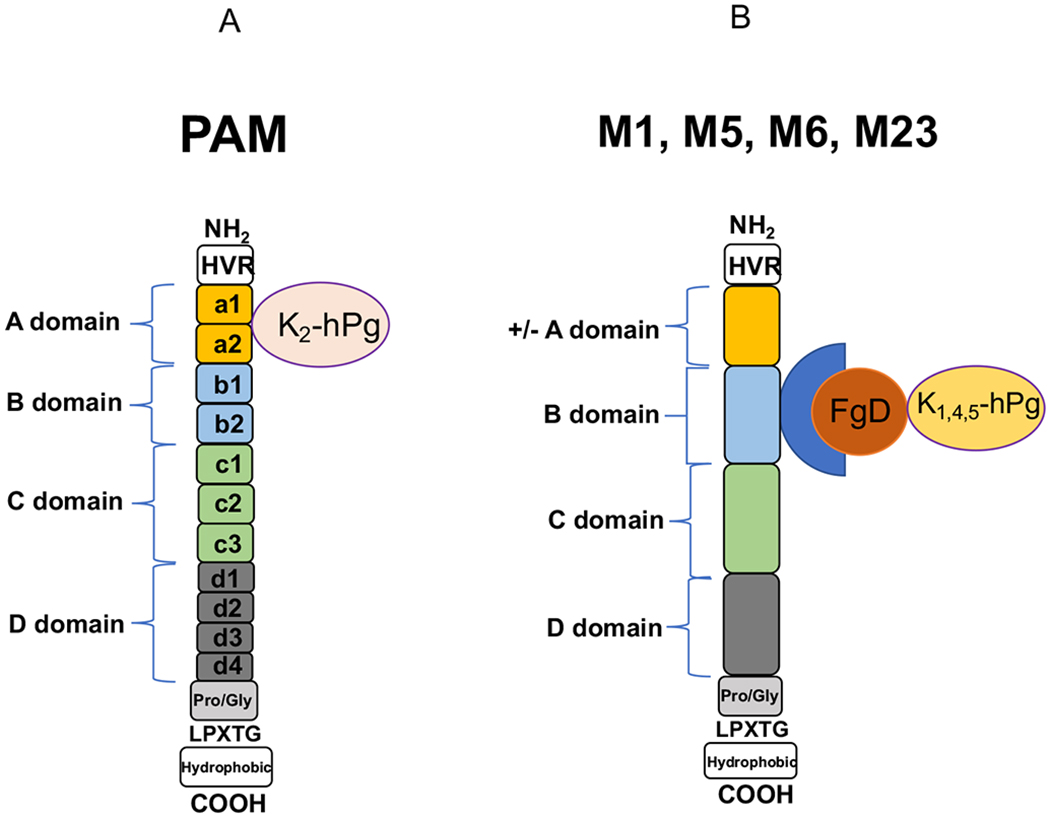
Plasminogen assembly on plasminogen binding Group A streptococcal M-like protein (PAM) and M proteins from GAS occurs either directly (A) through interaction with the a1a2 repeats of PAM through the lysine binding site of Kringle (K2) of human plasminogen (hPg) or indirectly (B) through the B domain of M-Prts interacting with the D domain of fibrinogen (Fg). Fg, through its E domain, then interacts with K1, 4, 5 of hPg. GAS expressing PAM is skin trophic while GAS expressing M-Prts, e.g., M1, that bind Fg are nasopharynx trophic. HVR = hypervariable region; LPXTG = Sortase A recognition site.
In an attempt to mimic the human condition during GAS infection in an animal model, a murine model was developed [66]. Since mouse Pg (mPg) binds weakly to PAM and is not activated by the GAS plasminogen activator, SK, a model of GAS infection was developed in which mice express hPg, through a hPg transgene. The transgene is directed for expression in the liver by the insertion of albumin gene regulatory sequences upstream of the hPg gene. When challenged with a GAS infection, these mice (expressing ~17% hPg) demonstrated a lower survival rate than WT mice (~80% mortality utilizing AP53 GAS). Additionally, genetically altered bacteria that do not express SK or PAM result in enhanced survival in these hPg expressing mice. However, depleting host fibrinogen Fg in these mice leads to enhanced mortality. The results from this study indicate that a major mechanism for bacterial invasion and dissemination is SK-mediated host Pg activation allowing for degradation of fibrin barriers.
3.3. Platelets and GAS Infection
Previous studies have demonstrated that platelets, major components of a thrombus, can interact with GAS leading to host tissue injury and inflammation [67–69]. Dysregulated hemostasis is correlated with severe GAS infections [2]. During infection, M1 protein is released from the bacteria, through both proteolytic processing and shedding, which binds plasma Fg [70]. The Fg receptors of platelets interact with these M1/Fg complexes on the platelet surface. Additionally, the presence of antibodies to M1 protein on these complexes allows for the activation of platelets through interaction with Fc receptors on platelets [69]. The ultimate result is the formation of platelet aggregates and microthrombi. Quantitative mass spectrometry from GAS infections identified M1 in plasma in association with Fg, IgG3, and complement component C1q [71]. Through flow cytometry, these components were also identified on the platelet surface with resultant complement activation. Monocyte-mediated M1-activated phagocytosis was also enhanced demonstrating a novel mechanism for platelet activation with resultant platelet consumption, a hallmark feature of sepsis. Clinically, platelet aggregates, IgG, and M1 have been identified in biopsy samples from patients with S. pyogenes toxic shock syndrome [69]. Additionally, tissue biopsies at the site of S. pyogenes infection show colocalization of bacteria and platelets in microthrombi, indicating that platelet/bacteria interaction and platelet activation may occur at the primary site of invasion during the early stages of infection [72]. Murine studies using a GAS model of sepsis (AP1 clinical isolate) demonstrated an increase in platelet-neutrophil complexes and plasma levels of CD62P (P-selectin, a marker of platelet activation) during bacterial dissemination in the development of sepsis which is decreased in the late stages of the disease. This correlated with organ damage and accumulation of platelets in the liver.
3.4. Regulating GAS Pathogenicity Targeting GAS Components and Host Hemostasis
It has been reported that natural GAS infections result in the development of protective antibodies. It is not surprising that children are more susceptible to the incidence of symptomatic infection which declines as a result of aging. These protective antibodies have been derived from exposure to sequences within the M-protein and other conserved antigens of GAS. Currently, there is no licensed vaccine for GAS which may be the result of these vaccines generating antibodies that induce an autoimmune effect towards host tissue, i.e., ARF. Additionally, the complexity of the disease in terms of the variety of emm types, host location of the infection, and prevalence of disease burden based on geographic location also contribute to the lack of development of an effective vaccine. Many approaches at developing a vaccine have targeted epitopes within the M-Prt that elicit a protective (opsonic) effect without human tissue cross- reactivity [73]. Some approaches have involved generating vaccines that contain a fusion protein with 4-30 N-terminal peptides of M-Prt from different serotypes in association with a carrier protein [74, 75].
In contrast to focusing on the N-terminal sequences of M-Prt, other approaches involve targeting conserved sequences within the M-Prt, i.e., the C-domain. These approaches include utilizing the entire C-domain [76] or 12 amino acid minimal B-cell or B-cell/T cell epitopes within this domain. In some cases, these peptides have been conjugated to the cholera toxin B subunit [76–78]. An epitope within the C-domain (P145) was found to be recognized by individuals in a highly endemic area and these antibodies have been shown to have opsonic activity [79–81]. Further characterization of this epitope determined that a 12 amino acid sequence within the P145 sequence (J8i) is a B cell epitope and does not stimulate T cell responses [82, 83]. However, due to structural restrains of this small peptide, it was found to be poorly immunogenic. This peptide was further modified (J8) and conjugated to diphtheria toxin to generate an immunogenic reagent (J8-DT) in mice [84]. This complex was able to induce B cell memory cells, protect mice from GAS challenge [85], and also prevent pyoderma during GAS skin infection [86]. The antibodies induced by J8-DT did not cross-react with human tissue [81]. Additionally, a related peptide, J14, was able to inhibit throat colonization after an intranasal challenge with GAS [87, 88].
Interestingly, J8-DT was shown to be ineffective against GAS containing mutations in CovR/S two-component regulatory system. Mutations in CovR/S lead to changes in the expression of virulence factors, some of which alter the immune response. These strains degrade CXC chemokines and therefore inhibit neutrophil chemotaxis. A vaccine was then developed that recognized two components of GAS, the J8-DT and an inactive streptococcal CXC protease, Streptococcus pyogenes cell envelope proteinase (SpyCEP). This vaccine protected against pyoderma and bacteremia with 100-1000-fold reduction in bacterial burden after challenge. [86]. However, given the amount of effort in understanding the biochemistry of critical surface virulence proteins of GAS, there are no licensed vaccines to combat GAS infection.
With regard to host hemostasis, it is known that a hallmark feature of severe GAS infection is dysregulated hemostasis and clearly demonstrates that hemostasis plays a major role in GAS infection and invasion in host. Mice with genetic alterations of components of hemostasis have largely assisted in determining key players during disease progression [66, 89]. One of these studies looked at the effects of both plasma and platelet Factor V, a major cofactor in prothrombin activation, on infection [89]. Relative to plasma FV, platelet FV has enhanced procoagulant properties and is more resistant to degradation by activated Protein C. The results from these studies demonstrated a unique role of platelet FV in regulating GAS infection and supports a critical role of thrombin generation in host defense. Additionally, while humans expressing a mutant FV Leiden (resistant to degradation by the anticoagulant protein, Protein C) were resistant to severe sepsis, mice carrying this mutation had no survival advantage when infected with GAS [89–91].
Fibrin has been shown to associate with GAS and inhibit its dissemination in the host. However, fibrin can also assist GAS by acting as a shield against host innate immune responses [92–97]. Although this has only been demonstrated in Staphylococcus aureus infection, which expresses a coagulase that forms a complex with host thrombin (staphylothrombin). Similar observations have not been observed in GAS infections. Other studies have shown that fibrinogen deficient (FG−/−) mice are more susceptible to GAS infection than wild type mice [89]. It has been shown that Fg can also function as a modulator of leukocyte function by interacting with the leukocyte integrin receptor, MAC-1 [98, 99]. In an infection model using mice expressing a Fg mutant (Fibgamma (390-396A)) that can no longer interact with MAC-1, but still has procoagulant activity, demonstrated a compromised host inflammatory response but a delayed and less severe mortality than observed in FG−/− mice [99]. This indicates that these two functions of Fg are important for regulating host responses to infection. It is known that components of the contact activation pathway can bind to GAS. Recent studies in mice have demonstrated that domain-5 of HMWK can bind to GAS, inhibit the contact activation pathway, and protect mice from developing lung pathology [100]. Therefore, blocking bacteria/host interactions, specifically those interactions involving bacteria and host hemostasis components, may be a reasonable approach towards developing new therapies for arresting GAS invasion.
CONCLUSION AND AUTHORS INSIGHT ON THE TOPIC
Hemostasis has traditionally been viewed simply as a method for regulating bleeding. However, through a number of in vitro studies using purified components of hemostasis and the development of mice deficient for or expressing mutant forms of these proteins, a role of hemostasis has been identified in a number of physiological and pathophysiological processes. Not surprisingly, hemostasis also plays a role in regulating GAS infection by utilizing components of this pathway to protect itself from host immune responses and to invade the surrounding barrier tissue. Recent studies have begun to unravel the mechanisms associated with these functions by identifying specific interactions between host and bacteria. Therefore, elucidation of these critical interactions may serve to identify new modalities of arresting bacterial infection and invasion.
ACKNOWLEDGEMENTS
FUNDING
This work was supported by a grant from the National Institute of Health (NHLBI) HL013423 to FJC and VAP.
Footnotes
CONSENT FOR PUBLICATION
Not applicable.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- [1].Cunningham MW. Pathogenesis of group a streptococcal infections. Clin Microbiol Rev 2000; 13(3): 470–511. [ 10.1128/CMR.13.3.470] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Walker MJ, Barnett TC, McArthur JD, et al. Disease manifestations and pathogenic mechanisms of Group A Streptococcus. Clin Microbiol Rev 2014; 27(2): 264–301. [ 10.1128/CMR.00101-13] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Navarre WW, Schneewind O. Proteolytic cleavage and cell wall anchoring at the LPXTG motif of surface proteins in gram-positive bacteria. 1994; 14: 115–21. [ 10.1111/j.1365-2958.1994.tb01271.x] [DOI] [PubMed] [Google Scholar]
- [4].Swoboda JG, Campbell J, Meredith TC, Walker S. Wall teichoic acid function, biosynthesis, and inhibition. ChemBioChem 2010; 11(1): 35–45. [ 10.1002/cbic.200900557] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Winram SB, Lottenberg R. The plasmin-binding protein Plr of group A streptococci is identified as glyceraldehyde-3-phosphate dehydrogenase. Microbiology 1996; 142(Pt 8): 2311–20. [ 10.1099/13500872-142-8-2311] [DOI] [PubMed] [Google Scholar]
- [6].Pancholi V, Fischetti VA. alpha-enolase, a novel strong plasmin(ogen) binding protein on the surface of pathogenic streptococci. J Biol Chem 1998; 273(23): 14503–15. [ 10.1074/jbc.273.23.14503] [DOI] [PubMed] [Google Scholar]
- [7].Sutcliffe IC, Russell RR. Lipoproteins of gram-positive bacteria. J Bacteriol 1995; 177(5): 1123–8. [ 10.1128/jb.177.5.1123-1128.1995] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Biagini M, Garibaldi M, Aprea S, et al. The human pathogen Streptococcus pyogenes releases lipoproteins as lipoprotein-rich membrane vesicles. Mol Cell Proteomics 2015; 14(8): 2138–49. [ 10.1074/mcp.M114.045880] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bober M, Morgelin M, Olin AI, von Pawel-Rammingen U, Collin M. The membrane bound LRR lipoprotein Slr, and the cell wall-anchored M1 protein from Streptococcus pyogenes both interact with type I collagen. PLoS One 2011; 6(5) e20345 [ 10.1371/journal.pone.0020345] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Beachey EH, Simpson WA. The adherence of group A streptococci to oropharyngeal cells: the lipoteichoic acid adhesin and fibronectin receptor. Infection 1982; 10(2): 107–11. [ 10.1007/BF01816738] [DOI] [PubMed] [Google Scholar]
- [11].Morath S, von Aulock S, Hartung T. Structure/function relationships of lipoteichoic acids. J Endotoxin Res 2005; 11(6): 348–56. [ 10.1177/09680519050110061001] [DOI] [PubMed] [Google Scholar]
- [12].Courtney HS, Ofek I, Penfound T, et al. Relationship between expression of the family of M proteins and lipoteichoic acid to hydrophobicity and biofilm formation in Streptococcus pyogenes. PLoS One 2009; 4(1) e4166 [ 10.1371/journal.pone.0004166] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ofek I, Simpson WA, Beachey EH. Formation of molecular complexes between a structurally defined M protein and acylated or deacylated lipoteichoic acid of Streptococcus pyogenes. J Bacteriol 1982; 149(2): 426–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Simpson WA, Beachey EH. Adherence of group A streptococci to fibronectin on oral epithelial cells. Infect Immun 1983; 39(1): 275–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Farnell MB, Crippen TL, He H, Swaggerty CL, Kogut MH. Oxidative burst mediated by toll like receptors (TLR) and CD14 on avian heterophils stimulated with bacterial toll agonists. Dev Comp Immunol 2003; 27(5): 423–9. [ 10.1016/S0145-305X(02)00115-5] [DOI] [PubMed] [Google Scholar]
- [16].Thapa S, Nagy E, Abdul-Careem MF. In ovo delivery of Toll-like receptor 2 ligand, lipoteichoic acid induces pro-inflammatory mediators reducing post-hatch infectious laryngotracheitis virus infection. Vet Immunol Immunopathol 2015; 164(3-4): 170–8. [ 10.1016/j.vetimm.2015.02.006] [DOI] [PubMed] [Google Scholar]
- [17].Vega LA, Malke H, McIver KS. Virulence-related transcriptional regulators of Streptococcus pyogenes Pulmonary Fibrosis. New York: Marcel Dekker; 2016; pp. 135–71. [Google Scholar]
- [18].Hondorp ER, McIver KS. The Mga virulence regulon: infection where the grass is greener. Mol Microbiol 2007; 66(5): 1056–65. [ 10.1111/j.1365-2958.2007.06006.x] [DOI] [PubMed] [Google Scholar]
- [19].Churchward G The two faces of Janus: virulence gene regulation by CovR/S in group A streptococci. Mol Microbiol 2007; 64(1): 34–41. [ 10.1111/j.1365-2958.2007.05649.x] [DOI] [PubMed] [Google Scholar]
- [20].Bao YJ, Liang Z, Mayfield JA, Lee SW, Ploplis VA, Castellino FJ. CovRS regulated transcriptome analysis of a hypervirulent M23 strain of Group A Streptococcus pyogenes provides new insights on virulence determinants. J Bacteriol 2015; 197(19): 3191–205. [ 10.1128/JB.00511-15] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wu Y Contact pathway of coagulation and inflammation. Thromb J 2015; 13: 17 [ 10.1186/s12959-015-0048-y] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Golias C, Charalabopoulos A, Stagikas D, Charalabopoulos K, Batistatou A. The kinin system – bradykinin: biological effects and clinical implications 2007; 11: 124–8. [PMC free article] [PubMed] [Google Scholar]
- [23].Sriskandan S, Cohen J. Gram-positive sepsis. Mechanisms and differences from gram-negative sepsis. Infect Dis Clin North Am 1999; 13(2): 397–412. [ 10.1016/S0891-5520(05)70082-9] [DOI] [PubMed] [Google Scholar]
- [24].Oehmcke S, Morgelin M, Malmstrom J, et al. Stimulation of blood mononuclear cells with bacterial virulence factors leads to the release of pro-coagulant and pro-inflammatory microparticles. Cell Microbiol 2012; 14(1): 107–19. [ 10.1111/j.1462-5822.2011.01705.x] [DOI] [PubMed] [Google Scholar]
- [25].Soult MC, Dobrydneva Y, Wahab KH, Britt LD, Sullivan CJ. Outer membrane vesicles alter inflammation and coagulation mediators. J Surg Res 2014; 192(1): 134–42. [ 10.1016/j.jss.2014.05.007] [DOI] [PubMed] [Google Scholar]
- [26].Mackman N Role of tissue factor in hemostasis and thrombosis. Blood Cells Mol Dis 2006; 36(2): 104–7. [ 10.1016/j.bcmd.2005.12.008] [DOI] [PubMed] [Google Scholar]
- [27].Coughlin SR. Protease-activated receptors in vascular biology. Thromb Haemost 2001; 86(1): 298–307. [PubMed] [Google Scholar]
- [28].Coughlin SR. How the protease thrombin talks to cells. Proc Natl Acad Sci USA 1999; 96(20): 11023–7. [ 10.1073/pnas.96.20.11023] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Lopes-Bezerra LM, Filler SG. Endothelial cells, tissue factor and infectious diseases. Braz J Med Biol Res 2003; 36(8): 987–91. [ 10.1590/S0100-879X2003000800004] [DOI] [PubMed] [Google Scholar]
- [30].Biemond BJ, Levi M, Ten Cate H, et al. Plasminogen activator and plasminogen activator inhibitor I release during experimental endotoxaemia in chimpanzees: effect of interventions in the cytokine and coagulation cascades. Clin Sci (Lond) 1995; 88(5): 587–94. [ 10.1042/cs0880587] [DOI] [PubMed] [Google Scholar]
- [31].Faust SN, Levin M, Harrison OB, et al. Dysfunction of endothelial protein C activation in severe meningococcal sepsis. N Engl J Med 2001; 345(6): 408–16. [ 10.1056/NEJM200108093450603] [DOI] [PubMed] [Google Scholar]
- [32].Yang X, Li L, Liu J, Lv B, Chen F. Extracellular histones induce tissue factor expression in vascular endothelial cells via TLR and activation of NF-κB and AP-1. Thromb Res 2016; 137: 211–8. [ 10.1016/j.thromres.2015.10.012] [DOI] [PubMed] [Google Scholar]
- [33].DeLa Cadena RA, Laskin KJ, Pixley RA, et al. Role of kallikrein-kinin system in pathogenesis of bacterial cell wall-induced inflammation. Am J Physiol 1991; 260(2 Pt 1): G213–9. [DOI] [PubMed] [Google Scholar]
- [34].Sriskandan S, Kemball-Cook G, Moyes D, Canvin J, Tuddenham E, Cohen J. Contact activation in shock caused by invasive group A Streptococcus pyogenes. Crit Care Med 2000; 28(11): 3684–91. [ 10.1097/00003246-200011000-00025] [DOI] [PubMed] [Google Scholar]
- [35].Oehmcke S, Herwald H. Contact system activation in severe infectious diseases. J Mol Med (Berl) 2010; 88(2): 121–6. [ 10.1007/s00109-009-0564-y] [DOI] [PubMed] [Google Scholar]
- [36].Herwald H, Mörgelin M, Dahlbäck B, Björck L. Interactions between surface proteins of Streptococcus pyogenes and coagulation factors modulate clotting of human plasma. J Thromb Haemost 2003; 1(2): 284–91.[ 10.1046/j.1538-7836.2003.00105.x] [DOI] [PubMed] [Google Scholar]
- [37].Miller G, Silverberg M, Kaplan AP. Autoactivatability of human Hageman factor (factor XII). Biochem Biophys Res Commun 1980; 92(3): 803–10. [ 10.1016/0006-291X(80)90774-3] [DOI] [PubMed] [Google Scholar]
- [38].Silverberg M, Diehl SV. The autoactivation of factor XII (Hageman factor) induced by low-Mr heparin and dextran sulphate. The effect of the Mr of the activating polyanion. Biochem J 1987; 248(3): 715–20. [ 10.1042/bj2480715] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ben Nasr AB, Herwald H, Müller-Esterl W, Björck L. Human kininogens interact with M protein, a bacterial surface protein and virulence determinant. Biochem J 1995; 305(Pt 1): 173–80. [ 10.1042/bj3050173] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Kaplan AP, Joseph K, Shibayama Y, et al. Bradykinin formation. Plasma and tissue pathways and cellular interactions. Clin Rev Allergy Immunol 1998; 16(4): 403–29. [ 10.1007/BF02737659] [DOI] [PubMed] [Google Scholar]
- [41].Herwald H, Collin M, Müller-Esterl W, Björck L. Streptococcal cysteine proteinase releases kinins: a virulence mechanism. J Exp Med 1996; 184(2): 665–73. [ 10.1084/jem.184.2.665] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Smith-Erichsen N, Aasen AO, Gallimore MJ, Amundsen E. Studies of components of the coagulation systems in normal individuals and septic shock patients. Circ Shock 1982; 9(5): 491–7. [PubMed] [Google Scholar]
- [43].Matsuda Y, Osaki T, Hashii T, Koshiba T, Kawabata S. A cysteine-rich protein from an arthropod stabilizes clotting mesh and immobilizes bacteria at injury sites. J Biol Chem 2007; 282(46): 33545–52. [ 10.1074/jbc.M705854200] [DOI] [PubMed] [Google Scholar]
- [44].Ploplis VA, Carmeliet P, Vazirzadeh S, et al. Effects of disruption of the plasminogen gene on thrombosis, growth, and health in mice. Circulation 1995; 92(9): 2585–93. [ 10.1161/01.CIR.92.9.2585] [DOI] [PubMed] [Google Scholar]
- [45].Ploplis VA, Castellino FJ. Gene targeting of components of the fibrinolytic system. Methods 2000; 21: 103–10. [ 10.1006/meth.2000.0981] [DOI] [PubMed] [Google Scholar]
- [46].Ploplis VA, Castellino FJ. Gene targeting of components of the fibrinolytic system. Thromb Haemost 2002; 87(1): 22–31. [ 10.1055/s-0037-1612938] [DOI] [PubMed] [Google Scholar]
- [47].Dressler DK. Death by clot: acute coronary syndromes, ischemic stroke, pulmonary embolism, and disseminated intravascular coagulation. AACN Adv Crit Care 2009; 20(2): 166–76. [ 10.1097/NCI.0b013e3181a0b5e8] [DOI] [PubMed] [Google Scholar]
- [48].Fisher MJ. Brain regulation of thrombosis and hemostasis. From Theory to practice. Stroke 2013; 44:3275–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]; Int J Immunopharmacol 2019; 72: 473–8. [DOI] [PubMed] [Google Scholar]
- [49].Li X, Zhu Z, Gao S, et al. Inhibition of fibrin formation reduces neuroinflammation and improves long-term outcome after intracerebral hemorrhage. Int Immunopharmacol 2019; 72: 473–8. [ 10.1016/j.intimp.2019.04.029] [DOI] [PubMed] [Google Scholar]
- [50].McCance SG, Castellino FJ. Contributions of individual kringle domains toward maintenance of the chloride-induced tight conformation of human glutamic acid-1 plasminogen. Biochemistry 1995; 34(29): 9581–6. [ 10.1021/bi00029a035] [DOI] [PubMed] [Google Scholar]
- [51].Urano T, Chibber BAK, Castellino FJ. The reciprocal effects of epsilon-aminohexanoic acid and chloride ion on the activation of human [Glu1]plasminogen by human urokinase. Proc Natl Acad Sci USA 1987; 84(12): 4031–4. [ 10.1073/pnas.84.12.4031] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Robbins KC, Summaria L, Hsieh B, Shah RJ. The peptide chains of human plasmin. Mechanism of activation of human plasminogen to plasmin. J Biol Chem 1967; 242(10): 2333–42. [PubMed] [Google Scholar]
- [53].Wistedt AC, Kotarsky H, Marti D, et al. Kringle 2 mediates high affinity binding of plasminogen to an internal sequence in streptococcal surface protein PAM. J Biol Chem 1998; 273(38): 24420–4. [ 10.1074/jbc.273.38.24420] [DOI] [PubMed] [Google Scholar]
- [54].Rios-Steiner JL, Schenone M, Mochalkin I, Tulinsky A, Castellino FJ. Structure and binding determinants of the recombinant kringle-2 domain of human plasminogen to an internal peptide from a group A Streptococcal surface protein. J Mol Biol 2001; 308(4): 705–19. [ 10.1006/jmbi.2001.4646] [DOI] [PubMed] [Google Scholar]
- [55].Fu Q, Figuera-Losada M, Ploplis VA, et al. The lack of binding of VEK-30, an internal peptide from the group A streptococcal M-like protein, PAM, to murine plasminogen is due to two amino acid replacements in the plasminogen kringle-2 domain. J Biol Chem 2008; 283(3): 1580–7. [ 10.1074/jbc.M705063200] [DOI] [PubMed] [Google Scholar]
- [56].Wang M, Zajicek J, Geiger JH, Prorok M, Castellino FJ. Solution structure of the complex of VEK-30 and plasminogen kringle 2. J Struct Biol 2010; 169(3): 349–59. [ 10.1016/j.jsb.2009.09.011] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chandrahas V, Glinton K, Liang Z, Donahue DL, Ploplis VA, Castellino FJ. Direct host plasminogen binding to bacterial surface M-protein pattern D strains of Streptococcus pyogenes is required for activation by natural coinherited SK2b protein. J Biol Chem 2015; 290(30): 18833–42. [ 10.1074/jbc.M115.655365] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hall SW, Humphries JE, Gonias SL. Inhibition of cell surface receptor-bound plasmin by alpha 2-antiplasmin and alpha 2-macroglobulin. J Biol Chem 1991; 266(19): 12329–36. [PubMed] [Google Scholar]
- [59].Macheboeuf P, Buffalo C, Fu CY, et al. Streptococcal M1 protein constructs a pathological host fibrinogen network. Nature 2011; 472(7341): 64–8. [ 10.1038/nature09967] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sumitomo T, Nakata M, Higashino M, Yamaguchi M, Kawabata S, Group A. Group A Streptococcus exploits human plasminogen for bacterial translocation across epithelial barrier via tricellular tight junctions. Sci Rep 2016; 7: 20069 [ 10.1038/srep20069] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Magalhães V, Andrade EB, Alves J, et al. Group B Streptococcus hijacks the host plasminogen system to promote brain endothelial cell invasion. PLoS One 2013; 8(5) e63244 [ 10.1371/journal.pone.0063244] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Lijnen HR. Plasmin and matrix metalloproteinases in vascular remodeling. Thromb Haemost 2001; 86(1): 324–33. [ 10.1055/s-0037-1616230] [DOI] [PubMed] [Google Scholar]
- [63].Siemens N, Patenge N, Otto J, Fiedler T, Kreikemeyer B. Streptococcus pyogenes M49 plasminogen/plasmin binding facilitates keratinocyte invasion via integrin-integrin-linked kinase (ILK) pathways and protects from macrophage killing. J Biol Chem 2011; 286(24): 21612–22. [ 10.1074/jbc.M110.202671] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Agrahari G, Liang Z, Mayfield JA, Balsara RD, Ploplis VA, Castellino FJ. Complement-mediated opsonization of invasive group A Streptococcus pyogenes strain AP53 is regulated by the bacterial two-component cluster of virulence responder/sensor (CovRS) system. J Biol Chem 2013; 288(38): 27494–504. [ 10.1074/jbc.M113.494864] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Agrahari G, Liang Z, Glinton K, Lee SW, Ploplis VA, Castellino FJ. Streptococcus pyogenes employs strain-dependent mechanisms of C3b inactivation to inhibit phagocytosis and killing of bacteria. J Biol Chem 2016; 291(17): 9181–9. [ 10.1074/jbc.M115.704221] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Sun H, Ringdahl U, Homeister JW, et al. Plasminogen is a critical host pathogenicity factor for group A streptococcal infection. Science 2004; 305(5688): 1283–6. [ 10.1126/science.1101245] [DOI] [PubMed] [Google Scholar]
- [67].Kurpiewski GE, Forrester LJ, Campbell BJ, Barrett JT. Platelet aggregation by Streptococcus pyogenes. Infect Immun 1983; 39(2): 704–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Cox D, Kerrigan SW, Watson SP. Platelets and the innate immune system: mechanisms of bacterial-induced platelet activation. J Thromb Haemost 2011; 9(6): 1097–107. [ 10.1111/j.1538-7836.2011.04264.x] [DOI] [PubMed] [Google Scholar]
- [69].Shannon O, Hertzén E, Norrby-Teglund A, Mörgelin M, Sjöbring U, Björck L. Severe streptococcal infection is associated with M protein-induced platelet activation and thrombus formation. Mol Microbiol 2007; 65(5): 1147–57. [ 10.1111/j.1365-2958.2007.05841.x] [DOI] [PubMed] [Google Scholar]
- [70].Berge A, Björck L. Streptococcal cysteine proteinase releases biologically active fragments of streptococcal surface proteins. J Biol Chem 1995; 270(17): 9862–7. [ 10.1074/jbc.270.17.9862] [DOI] [PubMed] [Google Scholar]
- [71].Palm F, Sjöholm K, Malmström J, Shannon O. Complement activation occurs at the surface of platelets activated by streptococcal M1 protein and this results in phagocytosis of platelets. J Immunol 2019; 202(2): 503–13. [ 10.4049/jimmunol.1800897] [DOI] [PubMed] [Google Scholar]
- [72].Hurley SM, Lutay N, Holmqvist B, Shannon O. The dynamics of platelet activation during progression of streptococcal sepsis. PLoS One 2016; 11(9) e0163531 [ 10.1371/journal.pone.0163531] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Cunningham MW. Pathogenesis of group A streptococcal infections. Clin Microbiol Rev 2000; 13(3): 470–511. [ 10.1128/CMR.13.3.470] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Dale JB, Penfound TA, Chiang EY, Walton WJ. New 30-valent M protein-based vaccine evokes cross-opsonic antibodies against nonvaccine serotypes of group A streptococci. Vaccine 2011; 29(46): 8175–8. [ 10.1016/j.vaccine.2011.09.005] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Dale JB, Penfound TA, Tamboura B, et al. Potential coverage of a multivalent M protein-based group A streptococcal vaccine. Vaccine 2013; 31(12): 1576–81. [ 10.1016/j.vaccine.2013.01.019] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Bessen D, Fischetti VA. Influence of intranasal immunization with synthetic peptides corresponding to conserved epitopes of M protein on mucosal colonization by group A streptococci. Infect Immun 1988; 56(10): 2666–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Bessen D, Fischetti VA. Passive acquired mucosal immunity to group A streptococci by secretory immunoglobulin A. J Exp Med 1988; 167(6): 1945–50. [ 10.1084/jem.167.6.1945] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Bessen D, Fischetti VA. Synthetic peptide vaccine against mucosal colonization by group A streptococci. I. Protection against a heterologous M serotype with shared C repeat region epitopes. J Immunol 1990; 145(4): 1251–6. [PubMed] [Google Scholar]
- [79].Pruksakorn S, Currie B, Brandt E, et al. Towards a vaccine for rheumatic fever: identification of a conserved target epitope on M protein of group A streptococci. Lancet 1994; 344(8923): 639–42. [ 10.1016/S0140-6736(94)92083-4] [DOI] [PubMed] [Google Scholar]
- [80].Pruksakorn S, Galbraith A, Houghten RA, Good MF. Conserved T and B cell epitopes on the M protein of group A streptococci. Induction of bactericidal antibodies. J Immunol 1992; 149(8): 2729–35. [PubMed] [Google Scholar]
- [81].Brandt ER, Hayman WA, Currie B, et al. Opsonic human antibodies from an endemic population specific for a conserved epitope on the M protein of group A streptococci. Immunology 1996; 89(3): 331–7. [ 10.1046/j.1365-2567.1996.d01-754.x] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Relf WA, Cooper J, Brandt ER, et al. Mapping a conserved conformational epitope from the M protein of group A streptococci. Pept Res 1996; 9(1): 12–20. [PubMed] [Google Scholar]
- [83].Hayman WA, Brandt ER, Relf WA, Cooper J, Saul A, Good MF. Mapping the minimal murine T cell and B cell epitopes within a peptide vaccine candidate from the conserved region of the M protein of group A streptococcus. Int Immunol 1997; 9(11): 1723–33. [ 10.1093/intimm/9.11.1723] [DOI] [PubMed] [Google Scholar]
- [84].Batzloff MR, Hayman WA, Davies MR, et al. Protection against group A streptococcus by immunization with J8-diphtheria toxoid: contribution of J8- and diphtheria toxoid-specific antibodies to protection. J Infect Dis 2003; 187(10): 1598–608. [ 10.1086/374800] [DOI] [PubMed] [Google Scholar]
- [85].Pandey M, Wykes MN, Hartas J, Good MF, Batzloff MR. Long-term antibody memory induced by synthetic peptide vaccination is protective against Streptococcus pyogenes infection and is independent of memory T cell help. J Immunol 2013; 190(6): 2692–701. [ 10.4049/jimmunol.1202333] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Pandey M, Langshaw E, Hartas J, Lam A, Batzloff MR, Good MF. A synthetic M protein peptide synergizes with a CXC chemokine protease to induce vaccine-mediated protection against virulent streptococcal pyoderma and bacteremia. J Immunol 2015; 194(12): 5915–25. [ 10.4049/jimmunol.1500157] [DOI] [PubMed] [Google Scholar]
- [87].Batzloff MR, Yan H, Davies MR, et al. Toward the development of an antidisease, transmission-blocking intranasal vaccine for group a streptococcus. J Infect Dis 2005; 192(8): 1450–5. [ 10.1086/466528] [DOI] [PubMed] [Google Scholar]
- [88].Batzloff MR, Hartas J, Zeng W, Jackson DC, Good MF. Intranasal vaccination with a lipopeptide containing a conformationally constrained conserved minimal peptide, a universal T cell epitope, and a self-adjuvanting lipid protects mice from group A streptococcus challenge and reduces throat colonization. J Infect Dis 2006; 194(3): 325–30. [ 10.1086/505146] [DOI] [PubMed] [Google Scholar]
- [89].Sun H, Wang X, Degen JL, Ginsburg D. Reduced thrombin generation increases host susceptibility to group A streptococcal infection. Blood 2009; 113(6): 1358–64. [ 10.1182/blood-2008-07-170506] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Yan SB, Nelson DR. Effect of factor V Leiden polymorphism in severe sepsis and on treatment with recombinant human activated protein C. Crit Care Med 2004; 32(5)(Suppl.): S239–46. [ 10.1097/01.CCM.0000126122.34119.D1] [DOI] [PubMed] [Google Scholar]
- [91].Weiler H, Kerlin B, Lytle MC, Factor V. Factor V Leiden polymorphism modifies sepsis outcome: evidence from animal studies. Crit Care Med 2004; 32(5)(Suppl.): S233–8. [ 10.1097/01.CCM.0000126126.79861.08] [DOI] [PubMed] [Google Scholar]
- [92].Smith W, Hale JH, Smith MM. The role of coagulase in staphylococcal infections. Br J Exp Pathol 1947; 28(1): 57–67. [PMC free article] [PubMed] [Google Scholar]
- [93].Cheng AG, McAdow M, Kim HK, Bae T, Missiakas DM, Schneewind O. Contribution of coagulases towards Staphylococcus aureus disease and protective immunity. PLoS Pathog 2010; 6(8) e1001036 [ 10.1371/journal.ppat.1001036] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Thomer L, Emolo C, Thammavongsa V, et al. Antibodies against a secreted product of Staphylococcus aureus trigger phagocytic killing. J Exp Med 2016; 213(3): 293–301. [ 10.1084/jem.20150074] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Panizzi P, Friedrich R, Fuentes-Prior P, Bode W, Bock PE. The staphylocoagulase family of zymogen activator and adhesion proteins. Cell Mol Life Sci 2004; 61(22): 2793–8. [ 10.1007/s00018-004-4285-7] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].McAdow M, Missiakas DM, Schneewind O. Staphylococcus aureus secretes coagulase and von Willebrand factor binding protein to modify the coagulation cascade and establish host infections. J Innate Immun 2012; 4(2): 141–8. [ 10.1159/000333447] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Panizzi P, Nahrendorf M, Figueiredo JL, et al. In vivo detection of Staphylococcus aureus endocarditis by targeting pathogen-specific prothrombin activation. Nat Med 2011; 17(9): 1142–6. [ 10.1038/nm.2423] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Lishko VK, Podolnikova NP, Yakubenko VP, et al. Multiple binding sites in fibrinogen for integrin alphaMbeta2 (Mac-1). J Biol Chem 2004; 279(43): 44897–906. [ 10.1074/jbc.M408012200] [DOI] [PubMed] [Google Scholar]
- [99].Flick MJ, Du X, Witte DP, et al. Leukocyte engagement of fibrinogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest 2004; 113(11): 1596–606. [ 10.1172/JCI20741] [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Oehmcke S, Shannon O, von Köckritz-Blickwede M, et al. Treatment of invasive streptococcal infection with a peptide derived from human high-molecular weight kininogen. Blood 2009; 114(2): 444–51. [ 10.1182/blood-2008-10-182527] [DOI] [PubMed] [Google Scholar]