Efficacy and Safety of Abrocitinib in Combination With Topical Therapy in Adolescents With Moderate-to-Severe Atopic Dermatitis: The JADE TEEN Randomized Clinical Trial (original) (raw)
Key Points
Question
What is the short-term efficacy and safety of oral abrocitinib in adolescents with moderate-to-severe atopic dermatitis?
Findings
In the randomized clinical trial JADE TEEN, a phase 3 study of abrocitinib in combination with topical therapy that included 285 adolescents with moderate-to-severe atopic dermatitis, significantly more adolescents who were treated with abrocitinib compared with placebo achieved an Investigator’s Global Assessment response or clear or almost clear, at least 75% improvement in Eczema Area and Severity Index response, and/or at least 4-point improvement in Peak Pruritus Numerical Rating Scale response. Serious adverse events were reported for fewer than 3% of patients.
Meaning
Oral abrocitinib in combination with topical therapy was effective and well-tolerated in adolescents with moderate-to-severe atopic dermatitis.
Abstract
Importance
Dupilumab subcutaneous injection is approved for treating moderate-to-severe atopic dermatitis (AD) in adolescents, but there has been too little research on an efficacious systemic oral treatment with a favorable benefit-risk profile for adolescents with moderate-to-severe AD.
Objective
To investigate the efficacy and safety of oral abrocitinib plus topical therapy in adolescents with moderate-to-severe AD.
Design, Setting, and Participants
The phase 3, randomized, double-blind, placebo-controlled study JADE TEEN was conducted in countries of the Asia–Pacific region, Europe, and North America in patients aged 12 to 17 years with moderate-to-severe AD and an inadequate response to 4 consecutive weeks or longer of topical medication or a need for systemic therapy for AD. The study was conducted between February 18, 2019, and April 8, 2020. The data were analyzed after study completion.
Interventions
Patients were randomly assigned 1:1:1 to receive once-daily oral abrocitinib, 200 mg or 100 mg, or placebo for 12 weeks in combination with topical therapy.
Main Outcomes and Measures
Coprimary end points were achievement of an Investigator’s Global Assessment (IGA) response of clear (0) or almost clear (1) with improvement of 2 or more grades from baseline (IGA 0/1) and 75% or greater improvement from baseline in Eczema Area and Severity Index (EASI-75) response at week 12. Key secondary end points included 4-point or greater improvement in Peak Pruritus Numerical Rating Scale (PP-NRS4) at week 12. Adverse events (AEs) were monitored.
Results
This study included 285 adolescents with moderate-to-severe AD (145 boys [50.9%] and 140 girls [49.1%]), of whom 160 (56.1%) were White and 94 (33.0%) were Asian; the median age was 15 years (interquartile range 13-17 years). Substantially more patients treated with abrocitinib (200 mg or 100 mg) vs placebo achieved an IGA response of 0/1 (46.2%; 41.6% vs 24.5%; P < .05 for both), EASI-75 (72.0%; 68.5% vs 41.5%; P < .05 for both), and PP-NRS4 (55.4%; 52.6% vs 29.8%; P < .01 for 200 mg vs placebo) at week 12. Adverse events were reported for 59 (62.8%), 54 (56.8%), and 50 (52.1%) patients in the 200 mg, 100 mg, and placebo groups, respectively; nausea was more common with abrocitinib, 200 mg (17 [18.1%]) and 100 mg (7 [7.4%]). Herpes-related AEs were infrequent; 1 (1.1%), 0, and 2 (2.1%) patients had serious AEs.
Conclusions and Relevance
This randomized clinical trial found that oral abrocitinib combined with topical therapy was significantly more effective than placebo with topical therapy in adolescents with moderate-to-severe AD, with an acceptable safety profile.
Trial Registration
ClinicalTrials.gov identifier: NCT03796676
This randomized clinical trial examines the efficacy and safety of abrocitinib plus topical therapy in adolescents with moderate-to-severe atopic dermatitis.
Introduction
Atopic dermatitis (AD) is a chronic, relapsing inflammatory skin condition with immune dysfunction that affects up to 20% of children and adolescents.1,2 Atopic dermatitis affects all skin types and is characterized by pruritus and eczematous lesions.2,3 It adversely affects a patient’s quality of life (QoL), academic performance, and social relationships,4,5,6,7 as well as the QoL of their caregivers.8,9
Dupilumab subcutaneous injection is approved for treating moderate-to-severe AD in adolescents.10 To our knowledge, an efficacious systemic oral treatment that has a favorable benefit-risk profile for adolescents with moderate-to-severe AD remains an unmet need.
Once-daily oral abrocitinib, a Janus kinase (JAK) 1–selective inhibitor that modulates the function of key cytokines that are involved in AD pathogenesis and pruritus,11,12 was effective and well-tolerated in phase 3 monotherapy studies in a combined patient population of adolescents and adults with moderate-to-severe AD (JADE MONO-1 [NCT03349060]; JADE MONO-2 [NCT03575871]).13,14 A separate, phase 3 study (JADE TEEN [NCT03796676]) investigated efficacy and safety of abrocitinib in adolescents with moderate-to-severe AD in combination with medicated topical therapy and evaluated the effect of abrocitinib on the QoL of patients and caregivers. In this article, we report the efficacy and safety of abrocitinib, 200 mg and 100 mg, vs placebo combined with topical therapy in adolescents with moderate-to-severe AD in JADE TEEN.
Methods
Study Design and Participants
JADE TEEN was a multicenter, international, phase 3, randomized, placebo-controlled, parallel-group study conducted between February 18, 2019, and April 8, 2020, in Australia, China, Czech Republic, Germany, Hungary, Italy, Japan, Latvia, Mexico, Poland, Spain, Taiwan, the UK, and the US (Supplement 1 and Supplement 2). Patients were screened within 28 days of the first dose followed by treatment with a study drug for 12 weeks. Eligible patients were aged 12 to 17 years with a body weight of 55 lb or greater (limit determined based on pharmacokinetic modeling) and a confirmed diagnosis of AD according to the diagnostic criteria of Hanifin and Rajka.15 Patients had moderate-to-severe AD (Investigator’s Global Assessment [IGA] score of ≥3; Eczema Area and Severity Index [EASI]16 score of ≥16; affected percentage of body surface area [BSA], ≥10; and Peak Pruritus Numerical Rating Scale [PP-NRS (used with permission of Regeneron Pharmaceuticals and Sanofi SA)] score of ≥417) at baseline. Patients had a documented history of inadequate responses to treatment with medicated topical therapy for AD that was given for 4 consecutive weeks or longer within 6 months before screening, or had been treated with systemic therapy for AD within 6 months before screening or were candidates for systemic therapy for AD. Because JAK inhibition has the potential to increase the risk of varicella-zoster virus infection,18 prior documentation of varicella-zoster virus immunity was required for inclusion. Patients with substantial psychiatric conditions or with current or past medical history of conditions that were associated with thrombocytopenia, coagulopathy, platelet dysfunction, or disseminated herpes were excluded; those with a history of herpes labialis were allowed to participate. Patients were required to wash out prior AD treatments (eg, biologic therapies, including dupilumab, immunosuppressive drugs, and medicated topical therapies) before study initiation. Standardized regimens of nonmedicated topical emollients and medicated topical therapy were required during the study, as detailed in the eMethods in Supplement 3. Patients were permitted to use oral antihistamines.
The study was conducted in compliance with ethical principles from the Declaration of Helsinki and all International Council for Harmonisation Good Clinical Practice Guidelines. All local regulatory requirements were followed. This research was approved by institutional review boards or ethics committees at each study site. An independent data monitoring committee monitored patient safety. All patients or their legal guardian provided written informed consent.
Randomization and Masking
Patients were randomly assigned 1:1:1 to receive oral, once-daily abrocitinib, 200 mg or 100 mg, or placebo with medicated topical therapy for 12 weeks. Randomization was stratified by baseline disease severity (moderate vs severe) and administered via an interactive response technology system. Patients, investigators, and sponsors were masked to the study treatment.
Outcome Measures
Coprimary, multiplicity-controlled efficacy end points were the proportion of patients who achieved an IGA response (clear [0] or almost clear [1] and improvement of ≥2 grades from baseline) and/or EASI-75 response (≥75% improvement from baseline) at week 12. Key secondary, multiplicity-controlled efficacy end points were the proportion of patients who achieved an improvement of 4 points or more in PP-NRS score (PP-NRS4) from baseline at weeks 2, 4, and 12 and a change from baseline in Pruritus and Symptoms Assessment for AD (PSAAD)19 total score at week 12. The PP-NRS score was recorded daily from days 1 to 15 and at weeks 4, 8, and 12; daily PP-NRS values were used directly rather than averaged weekly. The PSAAD score was recorded daily. Both end points were recorded by patients using a handheld device. Secondary and other efficacy end points that were not controlled for multiplicity were the proportion of patients who achieved IGA and EASI-75 responses at weeks 2, 4, and 8 and PP-NRS4 response at week 8; time to achieve a PP-NRS4 response; proportion of patients who achieved an EASI-50 response (≥50% improvement from baseline), EASI-90 response (≥90% improvement from baseline), EASI-100 response (100% improvement from baseline), Scoring of AD index (SCORAD)–50 response (≥50% improvement from baseline), SCORAD-75 response (≥75% improvement from baseline); and change from baseline in SCORAD subjective assessments of itch and sleep loss at weeks 2, 4, 8, and 12.20 Patient-reported outcomes included change from baseline at week 12 in the Children’s Dermatology Life Quality Index (CDLQI),21 Patient-Oriented Eczema Measure (POEM),22 and Dermatitis Family Impact (DFI).23 Incidence of adverse events (AEs), serious AEs, and AEs that led to discontinuation were recorded. Vital signs, 12-lead electrocardiogram, serum chemistry/hematology, high-sensitivity C-reactive protein levels, and urinalysis were assessed at baseline and weeks 2, 4, 8, and 12. A lipid panel was conducted at baseline and weeks 4 and 12.
Statistical Analysis
Sample size calculation was based on the Fisher exact test for comparing 2 proportions (eMethods in Supplement 3). Familywise type 1 error rates for testing coprimary and key secondary end points were strictly controlled at 5% using a sequential, Bonferroni-based procedure (eMethods and eFigure 1 in Supplement 3). Testing of all other secondary end points was performed at a nominal 5% significance level and was not controlled for multiplicity. For all binary end points, patients who permanently discontinued the study were defined as nonresponders at all visits after discontinuation. Statistical analyses were conducted using SAS, version 9.4 (SAS Institute). Additional details of the statistical analyses are presented in the eMethods in Supplement 3.
Results
Patients
In total, 273 patients completed JADE TEEN (Figure 1). The median (interquartile range) age was 15.0 (13.0-17.0) years (Table 1). A total of 160 (56.1%) were White, 94 (33.0%) were Asian, and 17 (6.0%) were Black. Seventy-six (26.7%) identified as Hispanic or Latino. Prior medication use is shown in Table 1. Most patients had moderate disease (175 [61.4%]) per their IGA score.
Figure 1. Patient Disposition in JADE TEEN.

AE indicates adverse event; FAS, full analysis set; SAF, safety analysis set.
aTwo patients randomly assigned to receive abrocitinib, 200 mg, were not treated and were not included in the analysis sets.
Table 1. Demographic and Baseline Characteristics.
| Characteristics | No. (%) | |||
|---|---|---|---|---|
| Placebo (n = 96) | Abrocitinib | All (n = 285) | ||
| 100 mg (n = 95) | 200 mg (n = 94) | |||
| Age, median (IQR), y | 14.0 (13.5-16.5) | 16.0 (14.0-17.0) | 15.0 (13.0-16.0) | 15.0 (13.0-17.0) |
| Age group, y | ||||
| 12-17a | 95 (99.0) | 95 (100.0) | 94 (100.0) | 284 (99.6) |
| ≥18b | 1 (1.0) | 0 | 0 | 1 (0.4) |
| Female | 52 (54.2) | 50 (52.6) | 38 (40.4) | 140 (49.1) |
| Male | 44 (45.8) | 45 (47.4) | 56 (59.6) | 145 (50.9) |
| Race | ||||
| White | 56 (58.3) | 52 (54.7) | 52 (55.3) | 160 (56.1) |
| Asian | 32 (33.3) | 31 (32.6) | 31 (33.0) | 94 (33.0) |
| Black or African American | 3 (3.1) | 9 (9.5) | 5 (5.3) | 17 (6.0) |
| Multiracial | 1 (1.0) | 0 | 1 (1.1) | 2 (0.7) |
| Otherc | 2 (2.0) | 3 (3.2) | 5 (5.4) | 10 (3.5) |
| Not reported | 2 (2.1) | 0 | 0 | 2 (0.7) |
| Ethnicity | ||||
| Not Hispanic or Latino | 65 (67.7) | 63 (66.3) | 69 (73.4) | 197 (69.1) |
| Hispanic or Latino | 25 (26.0) | 26 (27.4) | 25 (26.6) | 76 (26.7) |
| Not reported | 6 (6.3) | 6 (6.3) | 0 | 12 (4.2) |
| Disease duration, mean (SD), y | 10.5 (4.8) | 9.8 (5.4) | 9.7 (5.3) | 10.0 (5.2) |
| Prior medication for ADd | ||||
| Any prior medication | 95 (99.0) | 95 (100.0) | 92 (97.9) | 282 (98.9) |
| Topical agents alonee | 71 (74.0) | 68 (71.6) | 70 (74.5) | 209 (73.3) |
| Systemic ± topical agentsf | 24 (25.0) | 27 (28.4) | 22 (23.4) | 73 (25.6) |
| Dupilumab | 1 (1.0) | 1 (1.1) | 1 (1.1) | 3 (1.1) |
| IGA score | ||||
| 3 | 57 (59.4) | 57 (60.0) | 61 (64.9) | 175 (61.4) |
| 4 | 39 (40.6) | 38 (40.0) | 33 (35.1) | 110 (38.6) |
| EASI score, mean (SD) | 29.2 (12.7) | 31.0 (12.8) | 29.5 (12.2) | 29.9 (12.5) |
| BSA affected, mean (SD), % | 45.8 (22.4) | 51.2 (21.7) | 48.7 (21.7) | 48.6 (22.0) |
| PP-NRS total score, mean (SD) | 7.2 (1.7) | 7.0 (1.8) | 6.8 (2.0) | 7.0 (1.8) |
| PSAAD | ||||
| No. of patients | 95 | 95 | 93 | 283 |
| Total score, mean (SD) | 5.0 (2.4) | 4.9 (2.1) | 4.8 (2.3) | 4.9 (2.3) |
| SCORAD | ||||
| No. of patients | 96 | 95 | 93 | 284 |
| Total score, mean (SD) | 68.5 (13.4) | 67.6 (13.5) | 66.2 (13.3) | 67.5 (13.4) |
| SCORAD Sleep Loss VAS | ||||
| No. of patients | 96 | 95 | 93 | 284 |
| Total score, mean (SD) | 5.7 (2.9) | 5.3 (2.9) | 5.6 (2.9) | 5.5 (2.9) |
| CDLQI | ||||
| No. of patients | 96 | 95 | 94 | 285 |
| Total score, mean (SD) | 14.0 (6.7) | 14.3 (6.1) | 13.6 (7.0) | 14.0 (6.6) |
| POEM | ||||
| No. of patients | 95 | 95 | 94 | 284 |
| Total score, mean (SD) | 19.8 (5.9) | 19.5 (6.4) | 19.2 (6.2) | 19.5 (6.2) |
Efficacy
At week 12, IGA responses occurred in 43 (46.2%), 37 (41.6%), and 23 (24.5%) patients in the abrocitinib 200-mg, abrocitinib 100-mg, and placebo groups, respectively (eTable in Supplement 3; Figure 2A). Differences in IGA response rates at week 12 vs placebo for abrocitinib, 200 mg and 100 mg, were 20.6% (95% CI, 7.3%-33.9%; P < .05) and 16.7% (95% CI, 3.5%-29.9%; P < .05) (eTable in Supplement 3). At week 12, EASI-75 responses occurred in 67 (72.0%), 61 (68.5%), and 39 (41.5%) patients in the abrocitinib, 200 mg, 100 mg, and placebo groups, respectively (eTable in Supplement 3; Figure 2B). Differences in EASI-75 response rates vs placebo for abrocitinib, 200 mg and 100 mg, were 29.4% (95% CI, 16.3%-42.5%; P < .05) and 26.5% (95% CI, 13.1%-39.8%; P < .05) (eTable in Supplement 3). The proportion of patients who achieved IGA and EASI-75 responses was higher with abrocitinib treatment at each time assessed compared with placebo. Sensitivity analyses performed for coprimary end points using the per-protocol analysis set at week 12 (eMethods in Supplement 3) yielded similar results.
Figure 2. Proportion of Patients Who Achieved Investigator’s Global Assessment (IGA) Response and Eczema Area and Severity Index (EASI-75) Response.

Error bars represent 95% CIs. Conclusion of statistical significance was controlled for multiplicity at week 12. P values for other comparisons were not controlled for multiplicity.
a_P_ < .05 vs placebo.
b_P_ < .0001 vs placebo.
The proportions of patients who achieved EASI-50 and EASI-90 responses were higher for the abrocitinib groups than for placebo from weeks 2 to 12 (eTable and eFigure 2 in Supplement 3). The least-squares mean percentage change from baseline in EASI at week 12 was −80.6 (95% CI, −86.5 to −74.8), −77.3 (95% CI, −83.1 to −71.5), and −63.7 (95% CI, −69.5 to −57.9) in the abrocitinib 200-mg, abrocitinib 100-mg, and placebo groups, respectively.
The proportion of patients who achieved a PP-NRS4 response was significantly higher for the abrocitinib groups than placebo (eTable in Supplement 3; Figure 3A). Differences in PP-NRS scores between the abrocitinib groups vs placebo were observed within 2 days of dose 1 (Figure 3B). The median time to a PP-NRS4 response was 29.0 (95% CI, 15.0-61.0) days, 70.0 (95% CI, 30.0-85.0) days, and 90.0 (95% CI, 62.0-not evaluable) in the abrocitinib 200-mg, abrocitinib 100-mg, and placebo groups, respectively (Figure 3C). Decreases from baseline in PSAAD scores were greater for each abrocitinib group vs placebo at all times assessed (eTable and eFigure 3 in Supplement 2). For the proportion of patients who achieved SCORAD-50, SCORAD-75, and a SCORAD sleep loss visual analog scale score of less than 2, responses were higher for the abrocitinib groups than placebo (eTable and eFigure 4 in Supplement 3).
Figure 3. Peak Pruritus Numerical Rating Scale (PP-NRS) Outcomes.
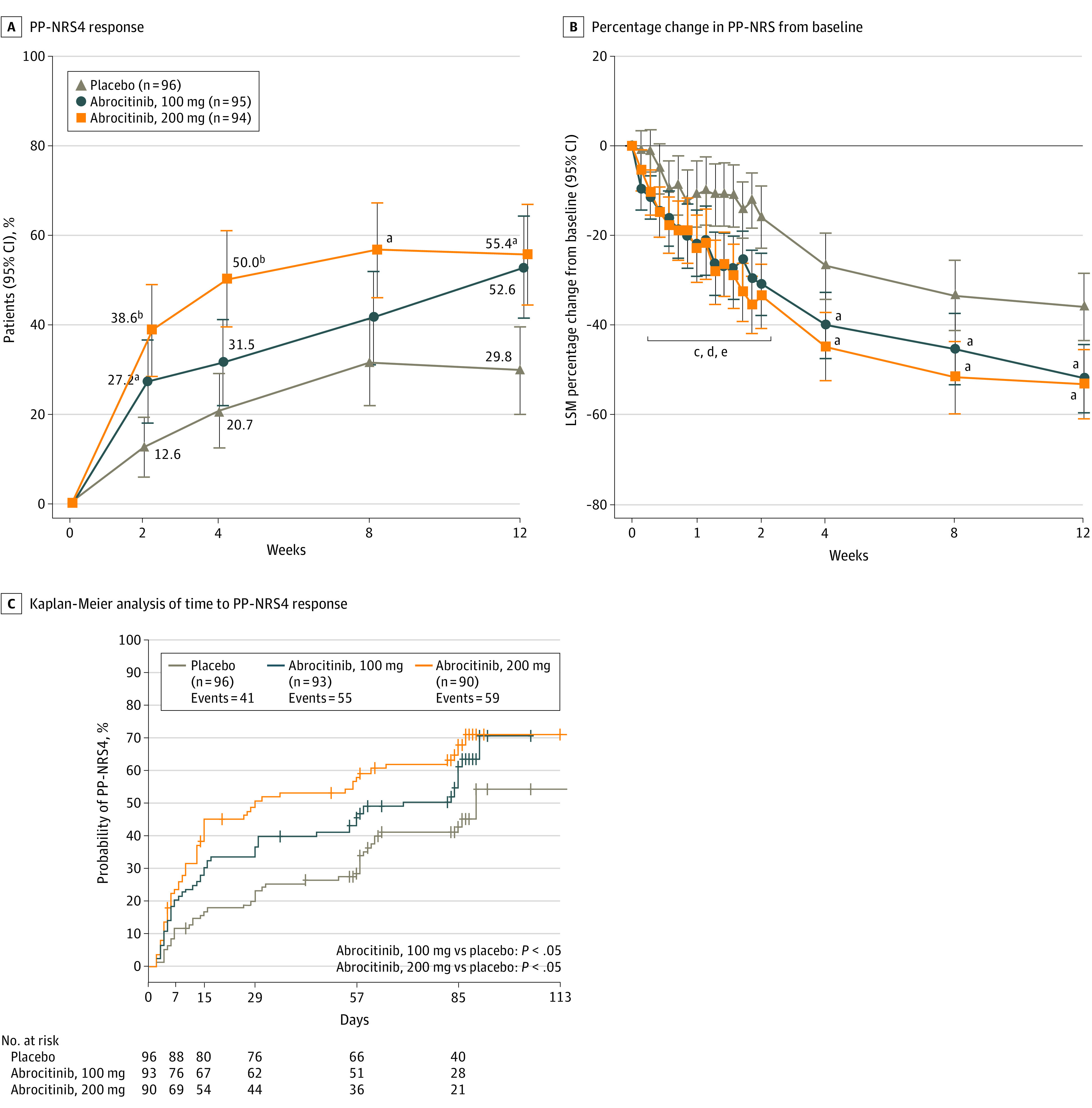
A, Proportion of patients who achieved at least 4-point improvement from baseline in PP-NRS (PP-NRS4). B, Least-squares mean (LSM) percentage change from baseline in PP-NRS scores. C, Kaplan-Meier analysis of time to achieve at least 4-point improvement from baseline in PP-NRS. Error bars in panels A and B represent 95% CIs. Conclusion of statistical significance was controlled for multiplicity at weeks 2, 4, and 12. P values for other comparisons were not controlled for multiplicity. P values shown in panel C are from log-rank tests for median time to response.
a_P_ < .05 vs placebo.
b_P_ < .001 vs placebo.
c_P_ < .05 for abrocitinib, 100 mg, vs placebo at days 3, 4, 6, and 8 to 15.
d_P_ < .05 for abrocitinib, 200 mg, vs placebo at days 3, 4, 6, 8 to 12, and 15.
e_P_ < .001 for abrocitinib, 200 mg, vs placebo at days 13 and 14.
Patient-Reported Outcomes
From weeks 2 to 12, adolescents reported improvement in QoL, with greater improvements in CDLQI scores for the abrocitinib groups than placebo. Improvement in disease severity measured by POEM was greater for both abrocitinib doses compared with placebo from weeks 2 to 12. Caregivers of adolescents reported improvement in QoL, with greater improvements in DFI scores for abrocitinib groups than placebo (eTable and eFigure 5 in Supplement 3).
Safety
Overall, 59 (62.8%), 54 (56.8%), and 50 (52.1%) patients in the abrocitinib 200-mg, abrocitinib 100-mg, and placebo groups, respectively, experienced treatment-emergent adverse events (TEAEs; Table 2); of these, 2 (2.1%), 0, and 2 (2.1%), respectively, were severe. Most frequently reported TEAEs were nausea in the abrocitinib, 200 mg, group (17 [18.1%]), upper respiratory tract infection in the abrocitinib, 100 mg, group (9 [9.5%]), and upper respiratory tract infection in the placebo group (10 [10.4%]). The median (interquartile range) time to onset of nausea was 2.0 (1.0-5.0), 4.0 (1.0-25.0), and 1.0 (1.0-1.0) days in the abrocitinib 200-mg, abrocitinib 100-mg, and placebo groups, respectively; the median time to resolution was 13.0 (95% CI, 3.0-87.0), 23.0 (95% CI, 1.0-86.0), and 1.0 (95% CI, NE-NE) days. Most nausea TEAEs were mild; however, there were 2 severe cases in the abrocitinib, 200 mg, group.
Table 2. Summary of Adverse Events.
| Event | No. (%) | ||
|---|---|---|---|
| Placebo (n = 96) | Abrocitinib | ||
| 100 mg (n = 95) | 200 mg (n = 94) | ||
| TEAEs of any causality | 50 (52.1) | 54 (56.8) | 59 (62.8) |
| Serious AEs of any causality | 2 (2.1) | 0 | 1 (1.1) |
| Severe AEs of any causality | 2 (2.1) | 0 | 2 (2.1) |
| TEAEs of any causality that led to treatment discontinuation | 2 (2.1) | 1 (1.1) | 2 (2.1) |
| Deaths | 0 | 0 | 0 |
| Most frequently reported TEAEs of any causality (≥3% in any treatment group) | |||
| Nausea | 1 (1.0) | 7 (7.4) | 17 (18.1) |
| Upper respiratory tract infection | 10 (10.4) | 9 (9.5) | 10 (10.6) |
| Headache | 7 (7.3) | 5 (5.3) | 8 (8.5) |
| Nasopharyngitis | 9 (9.4) | 8 (8.4) | 8 (8.5) |
| Dizziness | 1 (1.0) | 0 | 6 (6.4) |
| Acne | 1 (1.0) | 3 (3.2) | 5 (5.3) |
| Vomiting | 0 | 4 (4.2) | 5 (5.3) |
| Abdominal pain upper | 0 | 0 | 4 (4.3) |
| Blood creatine phosphokinase increased | 0 | 4 (4.2) | 4 (4.3) |
| Abdominal pain | 1 (1.0) | 1 (1.1) | 3 (3.2) |
| Pharyngitis | 3 (3.1) | 5 (5.3) | 3 (3.2) |
| Sinusitis | 0 | 0 | 3 (3.2) |
| Folliculitis | 1 (1.0) | 7 (7.4) | 2 (2.1) |
| Influenza | 1 (1.0) | 4 (4.2) | 2 (2.1) |
| Atopic dermatitis | 3 (3.1) | 2 (2.1) | 1 (1.1) |
| Cough | 2 (2.1) | 4 (4.2) | 1 (1.1) |
| Pyrexia | 4 (4.2) | 3 (3.2) | 1 (1.1) |
| Rhinorrhea | 3 (3.1) | 1 (1.1) | 0 |
| TEAEs of special interest | |||
| Herpes zoster | 0 | 1 (1.1) | 0 |
| Herpes simplex | 0 | 0 | 1 (1.1) |
| Oral herpes | 0 | 1 (1.1) | 2 (2.1) |
| Eczema herpeticum | 0 | 1 (1.1) | 0 |
| Conjunctivitis | 1 (1.0) | 0 | 0 |
Serious AEs were reported for 1 (1.1%) and 2 (2.1%) patients in the abrocitinib 200-mg and placebo groups, respectively. There were no reports of serious AEs in the abrocitinib, 100 mg, group (Table 2). One patient in the abrocitinib, 200 mg, group reported a serious AE of anxiety. One patient in the placebo group developed a serious AE of angioedema due to allergy; another experienced a serious AE of worsening of AD. These events did not result in dosing changes and were not considered treatment-related per investigators.
Two (2.1%), 1 (1.1%), and 2 (2.1%) patients in the abrocitinib 200-mg, abrocitinib 100-mg, and placebo groups, respectively, discontinued treatment because of experiencing AEs (Table 2). One patient in the abrocitinib, 200 mg, group developed headache, and another developed gastroesophageal reflux, nausea, and vomiting; all were considered related to treatment. One patient in the abrocitinib, 100 mg, group developed a gastrointestinal infection, which was not considered treatment related. One patient in the placebo group developed an upper respiratory tract infection, and another developed a wound abscess; neither was considered treatment related.
Regarding AEs of special interest, cutaneous herpes zoster was reported for 1 patient (1.1%) in the abrocitinib, 100 mg, group (Table 2). Herpes simplex was reported for 1 patient (1.1%) in the abrocitinib, 200 mg, group. Oral herpes was reported for 2 (2.1%) and 1 (1.1%) patients in the abrocitinib 200-mg and abrocitinib 100-mg groups, respectively. Eczema herpeticum was reported for 1 patient (1.1%) in the abrocitinib, 100 mg group. Conjunctivitis was reported for 1 patient (1.0%) in the placebo group. Acne was reported for 5 (5.3%), 3 (3.2%), and 1 (1.0%) patients in the abrocitinib 200-mg, abrocitinib 100-mg, and placebo groups, respectively. There were no cases of serious infection, venous thromboembolism, cancers, major adverse cardiovascular events, or deaths (Table 2).
There were modest increases in total cholesterol and high- and low-density lipoprotein cholesterol levels for both abrocitinib doses vs placebo that plateaued at week 4. Among the abrocitinib 200-mg, abrocitinib 100-mg, and placebo groups, elevated (>1.2 × upper limit of normal) low-density lipoprotein cholesterol levels occurred in 3 (3.2%), 2 (2.1%), and 2 (2.1%) patients, respectively.
Dose-related decreases in median platelet cell counts were observed in patients who were treated with abrocitinib, with a nadir at week 4 (median platelet count, 229 × 103/μL and 246 × 103/μL [to convert to × 109/L, multiply by 1] in the 200 mg and 100 mg groups, respectively) and a return toward baseline values thereafter (eFigure 6 in Supplement 3). No patients discontinued participation because of changes in platelet cell counts or bleeding disorders. There were no clinically significant changes in hemoglobin levels or neutrophil or lymphocyte cell counts. One patient met prespecified discontinuation criteria for elevated aspartate aminotransferase levels. All other observed changes in clinical laboratory values, electrocardiographic measurements, and vital signs did not lead to study discontinuation.
Discussion
In this phase 3 study, abrocitinib combined with medicated topical therapy in adolescents with moderate-to-severe AD resulted in significant improvement in AD signs and symptoms compared with placebo, with improvements in the QoL of patients and caregivers. More patients who were treated with abrocitinib achieved IGA, EASI-75, and PP-NRS4 responses at week 2 compared with placebo, illustrating early onset of activity. Larger mean percentage reductions in PP-NRS scores were observed with abrocitinib vs placebo within 2 days of treatment initiation, indicating fast relief of pruritus with abrocitinib. Improvement in signs and symptoms of AD could explain the improvement in the sleep of adolescents and potentially contribute to reported QoL improvement in patients and caregivers.
Both abrocitinib doses resulted in statistically significant IGA and EASI-75 response rates compared with placebo. The difference in IGA and EASI-75 response rates between the abrocitinib 200-mg and abrocitinib 100-mg group doses at week 12 were numerically smaller vs those at weeks 2, 4, and 8. In contrast, a clearer abrocitinib dose-response effect was observed in the phase 3 monotherapy studies JADE MONO-1 and JADE MONO-2 and phase 3 JADE COMPARE (abrocitinib plus medicated topical therapy) across the times assessed, with higher response rates observed with abrocitinib, 200 mg, than abrocitinib, 100 mg.13,14,24 Still, treatment effects of both abrocitinib doses were clinically meaningful as monotherapy or with topical therapy in these phase 3 studies of patients with moderate-to-severe AD.
Most patients who were included in JADE MONO-1 and JADE MONO-2 were adults 18 years or older (78% and 90%, respectively), although many adolescents were included (84 in JADE MONO-1 and 40 in JADE MONO-2). JADE MONO-1 and JADE MONO-2 yielded lower placebo response rates and a significantly higher efficacy of abrocitinib vs placebo than observed in JADE TEEN. The higher placebo IGA and EASI-75 response rates observed in JADE TEEN vs JADE MONO-1 and JADE MONO-2 were likely related to the use of concomitant topical therapy. Differences in demographic characteristics and other baseline characteristics may have also contributed to a higher placebo response. It is unknown to what extent elevated placebo response rates due to concomitant topical therapy in JADE TEEN contributed to a reduced dose response. Preliminary evidence of abrocitinib efficacy in the adolescent subgroups of JADE MONO-1 and JADE MONO-2 was demonstrated based on IGA and EASI-75 response rates at week 12. In the adolescent subgroup of JADE MONO-1 (n = 84), IGA response rates at week 12 were 27.3% in the 200 mg group, 26.5% in the 100 mg group, and 12.5% in the placebo group; EASI-75 response rates at week 12 were 54.5% in the 200 mg group, 44.1% in the 100 mg group, and 12.5% in the placebo group. In the adolescent subgroup of JADE MONO-2 (n = 40), IGA response rates at week 12 were 40.0% in the 200 mg group, 12.5% in the 100 mg group, and 0% in the placebo group; EASI-75 response rates were 60.0% in the 200 mg group, 43.8% in the 100 mg group, and 0% in the placebo group.
The safety of abrocitinib demonstrated in this study suggests that combining abrocitinib with topical therapies does not adversely affect the benefit-risk profile in adolescents. Incidence of serious AEs, severe AEs, and AEs that led to study discontinuation were similar or lower in the abrocitinib groups than placebo. There was a higher incidence of nausea in the abrocitinib, 200 mg, group. The incidence of herpes infections was very low, but more cases occurred in the abrocitinib than placebo groups. Although adolescents are at high risk of acne, very few cases were reported as an AE (more among those treated with abrocitinib than placebo). Dose-related decreases in median platelet cell counts were observed, with a nadir at week 4 and platelet cell count stabilization by week 12. Platelet cell count changes were not associated with bleeding disorders. The results of a systems model of the effect of JAK inhibition on platelet homeostasis suggest that abrocitinib affects platelet progenitor production,25 which results in the observed changes in platelet cell counts. This effect has been observed in other abrocitinib studies.13,14
Limitations
Limitations include the 12-week duration; studies are needed to address long-term efficacy and safety of abrocitinib in adolescents. This study was not powered to compare the 200-mg and 100-mg abrocitinib doses. Concomitant topical therapy may have reduced a potential dose-response effect between the 100-mg and 200-mg doses of abrocitinib.
Conclusions
Considering all data presented, the study results suggest that abrocitinib, 200 mg or 100 mg, combined with medicated topical therapy was efficacious and well-tolerated in adolescents with moderate-to-severe AD, with a favorable benefit-risk profile.
Supplement 1.
Trial protocol
Supplement 2.
Statistical analysis plan
Supplement 3.
eMethods.
eTable. Summary of Efficacy End Points and Patient-Reported Outcomes
eFigure 1. Bonferroni-Based Procedure for Testing the Coprimary and Key Secondary End Points
eFigure 2. Proportions of Patients Who Achieved (A) EASI-50 and (B) EASI-90 Responses
eFigure 3. Change From Baseline in PSAAD Score
eFigure 4. SCORing Atopic Dermatitis Scale Outcomes
eFigure 5. Patient-Reported Outcomes
eFigure 6. Median Absolute Platelet Count
Supplement 4.
Data sharing statement
References
- 1.Williams H, Robertson C, Stewart A, et al. Worldwide variations in the prevalence of symptoms of atopic eczema in the International Study of Asthma and Allergies in Childhood. J Allergy Clin Immunol. 1999;103(1 Pt 1):125-138. doi: 10.1016/S0091-6749(99)70536-1 [DOI] [PubMed] [Google Scholar]
- 2.Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD. Atopic dermatitis. Nat Rev Dis Primers. 2018;4(1):1. doi: 10.1038/s41572-018-0001-z [DOI] [PubMed] [Google Scholar]
- 3.Suárez-Fariñas M, Tintle SJ, Shemer A, et al. Nonlesional atopic dermatitis skin is characterized by broad terminal differentiation defects and variable immune abnormalities. J Allergy Clin Immunol. 2011;127(4):954-64.e1, 4. doi: 10.1016/j.jaci.2010.12.1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boguniewicz M, Fonacier L, Guttman-Yassky E, Ong PY, Silverberg J, Farrar JR. Atopic dermatitis yardstick: practical recommendations for an evolving therapeutic landscape. Ann Allergy Asthma Immunol. 2018;120(1):10-22.e2. doi: 10.1016/j.anai.2017.10.039 [DOI] [PubMed] [Google Scholar]
- 5.Newton L, DeLozier AM, Griffiths PC, et al. Exploring content and psychometric validity of newly developed assessment tools for itch and skin pain in atopic dermatitis. J Patient Rep Outcomes. 2019;3(1):42. doi: 10.1186/s41687-019-0128-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ezzedine K, Shourick J, Merhand S, Sampogna F, Taïeb C. Impact of atopic dermatitis in adolescents and their parents: a French study. Acta Derm Venereol. 2020;100(17):adv00294. doi: 10.2340/00015555-3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng MS, Tan S, Chan NH, Foong AY, Koh MJ. Effect of atopic dermatitis on quality of life and its psychosocial impact in Asian adolescents. Australas J Dermatol. 2018;59(2):e114-e117. doi: 10.1111/ajd.12632 [DOI] [PubMed] [Google Scholar]
- 8.Marciniak J, Reich A, Szepietowski JC. Quality of life of parents of children with atopic dermatitis. Acta Derm Venereol. 2017;97(6):711-714. doi: 10.2340/00015555-2633 [DOI] [PubMed] [Google Scholar]
- 9.Ricci G, Bellini F, Dondi A, Patrizi A, Pession A. Atopic dermatitis in adolescence. Dermatol Reports. 2011;4(1):e1. doi: 10.4081/dr.2012.e1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dupixent. Package insert. Regeneron Pharmaceuticals Inc; 2019.
- 11.Szilveszter KP, Németh T, Mócsai A. Tyrosine kinases in autoimmune and inflammatory skin diseases. Front Immunol. 2019;10:1862. doi: 10.3389/fimmu.2019.01862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vazquez ML, Kaila N, Strohbach JW, et al. Identification of N-cis-3-[methyl(7H-pyrrolo[2,3-d]pyrimidin-4-yl)amino]cyclobutylpropane-1-sulfonamide (PF-04965842): a selective JAK1 clinical candidate for the treatment of autoimmune diseases. J Med Chem. 2018;61(3):1130-1152. doi: 10.1021/acs.jmedchem.7b01598 [DOI] [PubMed] [Google Scholar]
- 13.Silverberg JI, Simpson EL, Thyssen JP, et al. Efficacy and safety of abrocitinib in patients with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2020;156(8):863-873. doi: 10.1001/jamadermatol.2020.1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simpson EL, Sinclair R, Forman S, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396(10246):255-266. doi: 10.1016/S0140-6736(20)30732-7 [DOI] [PubMed] [Google Scholar]
- 15.Hanifin JM, Rajka G. Diagnostic features of atopic dermatitis. Acta Derm Venereol. 1980;60(92):44-47. https://www.medicaljournals.se/acta/content_files/files/pdf/60/92/924447.pdf [Google Scholar]
- 16.Hanifin JM, Thurston M, Omoto M, Cherill R, Tofte SJ, Graeber M; EASI Evaluator Group . The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. Exp Dermatol. 2001;10(1):11-18. doi: 10.1034/j.1600-0625.2001.100102.x [DOI] [PubMed] [Google Scholar]
- 17.Yosipovitch G, Reaney M, Mastey V, et al. Peak pruritus numerical rating scale: psychometric validation and responder definition for assessing itch in moderate-to-severe atopic dermatitis. Br J Dermatol. 2019;181(4):761-769. doi: 10.1111/bjd.17744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sunzini F, McInnes I, Siebert S. JAK inhibitors and infections risk: focus on herpes zoster. Ther Adv Musculoskelet Dis. 2020;12:X20936059. doi: 10.1177/1759720X20936059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hall R, Lebwohl MG, Bushmakin AG, et al. Development and content validation of pruritus and symptoms assessment for atopic dermatitis (PSAAD) in adolescents and adults with moderate-to-severe AD. Dermatol Ther (Heidelb). 2021;11(1):221-233. doi: 10.1007/s13555-020-00474-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Severity scoring of atopic dermatitis: the SCORAD index. consensus report of the European Task Force on Atopic Dermatitis. Dermatology. 1993;186(1):23-31. doi: 10.1159/000247298 [DOI] [PubMed] [Google Scholar]
- 21.Lewis-Jones MS, Finlay AY. The Children’s Dermatology Life Quality Index (CDLQI): initial validation and practical use. Br J Dermatol. 1995;132(6):942-949. doi: 10.1111/j.1365-2133.1995.tb16953.x [DOI] [PubMed] [Google Scholar]
- 22.Charman CR, Venn AJ, Williams HC. The patient-oriented eczema measure: development and initial validation of a new tool for measuring atopic eczema severity from the patients’ perspective. Arch Dermatol. 2004;140(12):1513-1519. doi: 10.1001/archderm.140.12.1513 [DOI] [PubMed] [Google Scholar]
- 23.Lawson V, Lewis-Jones MS, Finlay AY, Reid P, Owens RG. The family impact of childhood atopic dermatitis: the Dermatitis Family Impact Questionnaire. Br J Dermatol. 1998;138(1):107-113. doi: 10.1046/j.1365-2133.1998.02034.x [DOI] [PubMed] [Google Scholar]
- 24.Bieber T, Simpson EL, Silverberg JI, et al. ; JADE COMPARE Investigators . Abrocitinib versus placebo or dupilumab for atopic dermatitis. N Engl J Med. 2021;384(12):1101-1112. doi: 10.1056/NEJMoa2019380 [DOI] [PubMed] [Google Scholar]
- 25.Koride S, Nayak S, Banfield C, Peterson MC. Evaluating the role of Janus kinase pathways in platelet homeostasis using a systems modeling approach. CPT Pharmacometrics Syst Pharmacol. 2019;8(7):478-488. doi: 10.1002/psp4.12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement 1.
Trial protocol
Supplement 2.
Statistical analysis plan
Supplement 3.
eMethods.
eTable. Summary of Efficacy End Points and Patient-Reported Outcomes
eFigure 1. Bonferroni-Based Procedure for Testing the Coprimary and Key Secondary End Points
eFigure 2. Proportions of Patients Who Achieved (A) EASI-50 and (B) EASI-90 Responses
eFigure 3. Change From Baseline in PSAAD Score
eFigure 4. SCORing Atopic Dermatitis Scale Outcomes
eFigure 5. Patient-Reported Outcomes
eFigure 6. Median Absolute Platelet Count
Supplement 4.
Data sharing statement