Serine Protease HTRA1 as a Novel Target Antigen in Primary Membranous Nephropathy (original) (raw)
Significance Statement
Membranous nephropathy (MN) is a glomerular disease that often leads to nephrotic syndrome and is caused by autoantibodies that target podocyte proteins. Recent work has identified new targets in this disease, although many cases remain untyped, making clinical monitoring difficult. A combination of conventional and more novel techniques reveals a new target podocyte antigen in MN, serine protease HTRA1. Fourteen patients were identified in a demographic group with mean age of 67.3 years. The titer of circulating anti-HTRA1 antibodies appears to correlate with the disease course, suggesting serial monitoring could facilitate diagnostic and therapeutic decisions. Study of the function of HTRA1 may provide important clues to podocyte biology and the underlying pathogenesis of this type of MN.
Keywords: membranous nephropathy, podocyte, nephrotic syndrome
Visual Abstract

Abstract
Background
Identification of target antigens PLA2R, THSD7A, NELL1, or Semaphorin-3B can explain the majority of cases of primary membranous nephropathy (MN). However, target antigens remain unidentified in 15%–20% of patients.
Methods
A multipronged approach, using traditional and modern technologies, converged on a novel target antigen, and capitalized on the temporal variation in autoantibody titer for biomarker discovery. Immunoblotting of human glomerular proteins followed by differential immunoprecipitation and mass spectrometric analysis was complemented by laser-capture microdissection followed by mass spectrometry, elution of immune complexes from renal biopsy specimen tissue, and autoimmune profiling on a protein fragment microarray.
Results
These approaches identified serine protease HTRA1 as a novel podocyte antigen in a subset of patients with primary MN. Sera from two patients reacted by immunoblotting with a 51-kD protein within glomerular extract and with recombinant human HTRA1, under reducing and nonreducing conditions. Longitudinal serum samples from these patients seemed to correlate with clinical disease activity. As in PLA2R- and THSD7A- associated MN, anti-HTRA1 antibodies were predominantly IgG4, suggesting a primary etiology. Analysis of sera collected during active disease versus remission on protein fragment microarrays detected significantly higher titers of anti-HTRA1 antibody in active disease. HTRA1 was specifically detected within immune deposits of HTRA1-associated MN in 14 patients identified among three cohorts. Screening of 118 “quadruple-negative” (PLA2R-, THSD7A-, NELL1-, EXT2-negative) patients in a large repository of MN biopsy specimens revealed a prevalence of 4.2%.
Conclusions
Conventional and more modern techniques converged to identify serine protease HTRA1 as a target antigen in MN.
Primary membranous nephropathy (MN) is one of the most common causes of nephrotic syndrome and a significant cause of ESKD.1 MN is an autoimmune disease that targets the glomeruli and is characterized by deposition of immune complexes and complement along the outer aspect of the glomerular basement membrane (GBM). Previous discoveries in MN include the discovery of phospholipase A2 receptor (PLA2R) and thrombospondin type 1 domain–containing 7A (THSD7A) as target antigens in approximately 80%–85% of patients with primary MN.2,3 However, target antigens in the remaining 15%–20% of patients had remained unidentified until recently.4–7 The identification of causal antigens and autoantibodies has allowed diagnosis of initial disease, or relapse, without requiring invasive kidney biopsies, and has been instrumental for monitoring response to therapy, allowing tailoring of immunosuppressive therapy, and defining treatment duration.
It has become more challenging to identify novel antigens using conventional techniques, such as screening for common bands by Western blotting (WB) with MN serum samples.8 This is likely due to the infrequency of the remaining “idiopathic” MN population recognizing any one single antigen and technical limitations inherent to these traditional methods. Newer technologies, such as protein G tissue immunoprecipitation, laser-capture microdissection (LCM) followed by mass spectrometry (MS), or protein arrays, serve as adjunctive techniques for identifying unknown antigens or autoantibodies, and have already proved their utility in MN and other glomerular diseases.5–7,9
LCM followed by MS proteomic analysis is a powerful methodology that allows the selective isolation of glomeruli from kidney tissue to look for enriched proteins, and has been used in amyloidosis, C3 glomerulonephritis, and now in MN. LCM-MS has been used in subtypes of primary and secondary MN to identify the exostosin 1 (EXT1) and EXT2 complex in a subset of class V membranous lupus nephritis,7 neural EGF-like 1 (NELL1) in primary MN,5 and Semaphorin-3B in a type of MN enriched in the pediatric population.6 Protein G tissue immunoprecipitation followed by MS analysis has recently emerged as a novel method to isolate IgG-containing immune complexes from frozen tissue and to identify the specific antigens isolated from the complexes.10,11 Moreover, autoimmune profiling on protein microarrays may be used for broadscale analyses of autoantibody responses and the identification of novel immune targets.9,12–18
In this article, we combine the more conventional WB and immunoprecipitation approaches used for the identification of PLA2R and THSD7A with newer laser-capture proteomics and autoimmune profiling techniques to converge on a novel autoantigen in MN. Using this combination of approaches, we have identified the secreted 51-kD serine protease HTRA1 (also known as high-temperature requirement A serine peptidase 1), as a novel podocyte-expressed antigen in primary MN.
Methods
Human serum and biopsy specimens for this study were obtained from four sites (see Supplemental Figure 1), with institutional review board approval at each site. The first cohort comprises patients with biopsy sample–proved MN who were prospectively recruited at University of Utah, starting in 2018. Serum was obtained at the time of enrollment before immunosuppressive therapy, and longitudinal serum samples were collected from the patients with PLA2R-negative MN at intervals between 1 and 3 months. Renal tissue from 17 patients who were PLA2R negative, with no clinical history or histologic features of lupus, was sent to Arkana Laboratories (Little Rock, AK) for MS and comprises the Utah discovery cohort. Two patients were ultimately found to have HTRA1-associated MN by MS (MN01 and MN03, which also exhibited reactive sera). Of the remaining patients, one each was found to be associated with THSD7A, EXT1/2, and Semaphorin-3B. Archived biopsy samples dating back to 2005 were included in this study and comprised the Utah validation cohort. A waiver of consent was obtained for historical samples.
A second cohort comprised a renal biopsy series from Arkana Laboratories, representing consecutive patients (_n_=349) with MN over a 9-month period, with sufficient biopsy tissue remaining for antigen characterization. The specifics of this cohort, which included 266 patients considered to have primary MN and 83 patients with pure class V (membranous) lupus nephritis, have been recently described.10 Immunostaining for antigens known at that time revealed that 181 specimens were PLA2R positive (51.9%), ten were THSD7A positive (2.9%), 33 were EXT2 positive (9.5%), and six were NELL1 positive (1.7%). Of the remaining 119 patients with unknown antigen, one had insufficient tissue for further staining, and thus 118 individuals negative for all four tissue antigens were assayed for the presence of HTRA1 within deposits.
A third cohort was derived from the Oregon Health and Science University renal tissue archives. The Oregon validation cohort included 35 PLA2R-negative MN kidney tissue samples, obtained from biopsies performed between 2016 and 2019. Specimens from patients with a clinical history of lupus or histologic features of lupus (proliferation or full-house staining) were excluded.
Selected serum samples representing cases of known MN subtypes and other glomerular disease sera from Boston Medical Center were included as controls.
Schematics of the cohorts and methods are shown as Supplemental Figures 1 and 2, and further information is available in the Cohorts and Methods sections of the Supplemental Methods.
Western Blotting
WB was performed according to published methods,2 with additional details available in the Supplemental Methods. Bio-Rad 4%–20% gradient gels were loaded with human glomerular extract or with recombinant human HTRA1 (catalog number, RP-77538; Thermo Fisher). Patient serum was diluted 1:25 in blocking solution and incubated overnight at 4°C, followed by incubation with sheep antibodies against the four human IgG subclasses, used as recommended by the manufacturer (The Binding Site), and then incubation with peroxidase-conjugated anti-sheep IgG at 1:10,000. HTRA1 was detected using a rabbit polyclonal antibody (provided by Moran Eye Center, University of Utah) at 1:500, followed by peroxidase-conjugated anti-rabbit IgG at 1:5000 (Jackson ImmunoResearch, West Grove, PA) secondary antibodies. Blots were incubated as directed with SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Scientific, Waltham, MA), and imaged using a Kodak X-OMAT film developer or a gel imaging system (Fuji).2,3
Immunoprecipitation and Isolation of the Candidate Antigen
Equal amounts (200 _µ_l) of a protein extract prepared from human glomeruli2 were incubated overnight at 4°C with a standard volume (75 _µ_l) of serum from the index patient MN01 when nephrotic versus in remission, from a patient with PLA2R-associated MN, and from a healthy control. IgG4-antigen complexes were immunoprecipitated using CaptureSelect Anti-Human IgG4 beads after incubation for 1 hour and washed five times with Tris-buffered saline. To isolate the approximately 50 kD candidate antigen band (as assessed on WB) for MS analysis, the immunoprecipitates were boiled in 2× gel loading buffer, under nonreducing conditions, and gel electrophoresed on a 4%–20% gradient gel flanked by protein marker lanes. Gel strips, approximately 2 mm wide, were excised from the 50-kD region and transferred to sterile tubes. These were sent for in-gel trypsin digestion and MS analysis, as described below. The remainder of the gel was stained with Coomassie Brilliant Blue to assess protein loading.
Mass Spectrometric Analysis of Gel Bands
As described previously,19 the gel bands were digested at 37°C overnight with 60 ng of Pierce Trypsin Protease, MS Grade (Thermo Scientific), in 75 _µ_l of 50 mM triethylammonium bicarbonate, pH 8.5. After digestion, the samples were acidified with formic acid, and peptides were extracted from the gel pieces with acetonitrile. The extracts were dried in a SpeedVac, the residue was dissolved in 20 _µ_l of 2% vol/vol acetonitrile/0.1% vol/vol formic acid, and filtered through a 0.45-_µ_m regenerated cellulose syringe filter (Thermo Scientific). The tryptic peptides were analyzed by an UltiMate 3000 NanoLC and Q Exactive HF Orbitrap MS system (Thermo Scientific).
Peptides were separated on an in-house–packed C18 analytic column (particle size, 3.6 _µ_m, 100 _µ_m i.d.×135 mm) with a 100 minute, 2.5%–45% acetonitrile gradient using a binary solvent system (solvent A, 0.1% formic acid; solvent B, 100% acetonitrile with 0.1% formic acid). Eluate from the column was directly ionized by a nanospray source and analyzed by mass spectrometer in data-dependent acquisition mode. The mass resolution was set to 120,000 and 15,000 for MS and tandem MS (MS/MS), respectively. Automatic gain control (AGC), isolation window, and offset were set to 3e6, 2 m/Z, and 0.5 m/Z. Other settings included the spray voltage at 1.65 kV, capillary temperature at 250°C, and S-Lens radio frequency at 60%.
The acquired data were processed by Proteome Discoverer (version 2.3.0.523; Thermo Scientific) with the Sequest HT search engine, using the SwissProt HumanReviewed190212. fasta file appended with the cRAP190304.fasta file to exclude known affinity purification contaminant proteins. The static modification cysteine carbamidomethylation (+57 Da) and the dynamic modifications methionine oxidation (+15.99 Da) and acetyl (+42.01 Da at protein amino-termini [N-termini]) were applied in the search. The match tolerances were set to 10 ppm and 0.02 Da for precursor and fragment ions, respectively. The target-decoy PSM validator node in PD version 2.3 was used to estimate the false discovery rates (FDRs) for peptide identifications. High-confidence peptide sequence assignments were used at the ≤1% FDR. Protein identifications were based on one peptide or greater. Relative quantification was based on unique plus razor peptides, and the protein abundances were normalized to the same total peptide amount per channel and scaled, so that the average abundance per protein and peptide is 100.
Laser-Capture Microdissection and Preparation for MS
Renal biopsy tissue from formalin-fixed, paraffin-embedded (FFPE) tissue was cut at a thickness of 10 _µ_m onto Leica PET membrane frame slides (four 10-_µ_m sections per case, with ≥20 glomeruli each). These slides were then deparaffinized and stained with Mayer hematoxylin for 20 seconds. The glomeruli were microdissected into microcentrifuge tubes in PBS using a Leica DM6000B microscope. The microdissected glomeruli were lysed in 2% SDS and 0.1 M dithiothreitol at 99°C for 1 hour and processed by filter-assisted sample preparation. The clarified lysate was transferred onto Vivacon 500 concentrators with a mol wt cutoff of 30 kD (Sartorius, Gottingen, Germany). SDS was removed by repeat washes with 8 M urea in 0.1 M Tris/chloride, pH 8.5. The samples were then alkylated with 0.05 M iodoacetamide. Proteins were digested with trypsin (sequencing grade; Promega) at a 40:1 wt/wt ratio at 37°C for 16 hours. Peptides were collected by centrifugation and desalted on C18 stage tips (Thermo Scientific).20
Protein G Tissue Immunoprecipitation
IgG coimmunoprecipitation was performed from protein lysates prepared from remnants of frozen kidney biopsy tissue. To do this, optimal cutting temperature–frozen tissue from archived renal biopsy specimens was thawed, washed four times in PBS, and lysed by mechanical disruption of tissue cores in Pierce IP lysis buffer by bead beating. Bead beating was performed three times at 2 minutes per cycle. The samples were centrifuged and the supernatant protein extracts were incubated with 50 _µ_l of protein G magnetic Dynabeads (Invitrogen, Carlsbad, CA), at room temperature (RT) for 1 hour, with shaking. The beads were washed four times with PBS to reduce nonspecific binding interactions. Proteins were digested from the beads using trypsin before mass spectrophotometric analysis.
MS Proteomic Analysis of Glomeruli/Tissue Immunoprecipitates
For data-dependent acquisitions, digested peptides were analyzed by NanoLC-MS/MS using a Thermo Orbitrap Fusion Lumos mass spectrometer. The peptides were loaded onto a reverse-phase trap column (IntegraFrit, New Objective, Littleton, MA) containing 2.5 _μ_m Waters XSelect CSH resin coupled to a 150 mm ×0.075 mm analytical column containing the same reverse-phase resin as used in the trap. A nanoAcquity UPLC system (Waters Corporation, Milford, MA) was then used to generate a 60-minute gradient of the buffer A/B ratio from 98:2 to 60:40 (buffer A, 0.1% formic acid with 0.5% acetonitrile; buffer B, 0.1% formic acid with 99.9% acetonitrile). Peptides were eluted from the column with an integrated spray tip (PicoFrit, New Objective) and ionized by electrospray (2.0 kV) followed by MS/MS analysis using higher-energy, collision-induced dissociation. Survey scans of peptide precursors were performed at 240,000 resolution (at 400 m/z) with a 5×105 ion count target. MS/MS was performed by isolation at 1.6 Th with the quadrupole, higher-energy, collision-induced dissociation fragmentation with normalized collision energy of 30 eV, and rapid-scan MS analysis in the ion trap. The obtained MS/MS data were searched against the most recent Uniprot human database containing both the Swiss-Prot and the TREMBL entries using MaxQuant. Visualization of data was performed using Scaffold version 4.6. The FDR was set at 1% for the peptide-to-spectrum matches. Normalized intensity-based absolute quantification (iBAQ) values from MaxQuant were used for quantitation. iBAQ distributions for each sample were adjusted to control for differences in loading. iBAQ values equal to zero were removed from the dataset. For statistical hypothesis testing, a two-sample t test was performed for each protein using normalized iBAQ values for the two groups. If a protein was only detected in one group, a one-sample t test was performed, using the smallest-detected iBAQ value as the null hypothesis.
For data-independent acquisition (DIA), tryptic peptides were separated by reverse-phase, XSelect CSH C18, 2.5-_μ_m resin (Waters) on an in-line, 150×0.075 mm column using an UltiMate 3000 RSLCnano system (Thermo Scientific). Peptides were eluted using a 60-minute gradient of the buffer A/B ratio from 97:3 to 60:40 (buffer A, 0.1% formic acid with 0.5% acetonitrile; buffer B, 0.1% formic acid and 99.9% acetonitrile). Eluted peptides were ionized by electrospray (2.15 kV) followed by MS analysis on an Orbitrap Exploris 480 mass spectrometer (Thermo Scientific). To assemble a chromatogram library, six gas-phase fractions were acquired on the Orbitrap Exploris with 4 m/z DIA spectra (4 m/z precursor isolation windows at 30,000 resolution, normalized AGC target of 100%, maximum injection time of 66 ms) using a staggered-window pattern from narrow mass ranges using optimized window placements.21 Precursor spectra were acquired after each DIA duty cycle, spanning the m/z range of the gas-phase fraction (i.e., 496–602 m/z, 60,000 resolution, normalized AGC target of 100%, maximum injection time of 50 ms). For wide-window acquisitions, the Orbitrap Exploris was configured to acquire a precursor scan (385–1015 m/z, 60,000 resolution, normalized AGC target of 100%, maximum injection time of 50 ms) followed by 50×12 m/z DIA spectra (12 m/z precursor isolation windows at 15,000 resolution, normalized AGC target of 100%, maximum injection time of 33 ms) using a staggered-window pattern with optimized window placements. Precursor spectra were acquired after each DIA duty cycle. Gas-phase fractionated files were searched against a predicted spectral library generated by Prosit to obtain an empirically corrected chromatogram library. The wide-window acquisitions were then searched against the empirically corrected library using EncyclopeDIA and ScaffoldDIA (Proteome Software).22,23
Immunodetection of Tissue Antigens
Immunofluorescence
FFPE sections, cut at 3 _μ_m, were deparaffinized, and antigen retrieval was performed by incubation at 99°C. The sections were reacted with mouse monoclonal anti-HTRA1 antibody (1:50, clone 275603; R & D Systems), followed by a Rhodamine Red-X AffiniPure Goat Anti-Mouse IgG, which was solid-phase adsorbed to ensure minimal cross reaction with human IgG (1:100; Jackson Immunoresearch). Each case was run with positive and negative controls. The stain was evaluated by standard immunofluorescence microscopy. The stain was judged to be positive if there was granular capillary loop staining in the glomeruli, and negative if there was no capillary loop staining in glomeruli. Negative controls were performed to ensure antibody specificity by omitting primary antibodies. For identification of control MN cases, immunofluorescence was performed for PLA2R (PLA2R rabbit polyclonal antibody, catalog number HPA012657; Sigma), THSD7A (THSD7A rabbit polyclonal antibody, catalog number AMAB91234; Atlas Antibodies), and EXT1 (EXT1 rabbit polyclonal antibody, catalog number PA5-60699; Invitrogen).
Primary Podocyte Staining
Cells were cultured at a low density in a six-well format for 24 hours using a humidified incubator at 37°C with 5% carbon dioxide. Immunofluorescence analysis of primary podocytes (Novabiosis) was performed using a HTRA1 polyclonal antibody (product number PA5-11412; Thermo Fisher) at a dilution of 1:10–50, followed by a fluor-conjugated goat anti-rabbit secondary antibody (green).
Immunohistochemistry
HTRA1 was detected in human kidney tissue by incubating mouse anti-human HTRA1 at 1:100 (MAB29161, clone number 275603; R&D Systems) overnight at 4°C in blocking buffer (PBS containing 1 mg/ml BSA, and 0.1% [vol/vol] Triton X-100) after deparaffinization of 3- to 4-_µ_m tissue sections and antigen retrieval. Slides were heated to 110°C for 15 minutes in a decloaking chamber (Biocare Medical), using Diva Decloaker (pH 6.2; Biocare Medical) as the antigen retrieval buffer, followed by anti-mouse IgG (1:10,000, Mouse-on-Farma HRP) for 30 minutes at RT. Alternatively, another anti-HTRA1 rabbit polyclonal antibody (PA511412; Thermo Fisher) was used at a dilution of 1:170 (10 _μ_g/ml) in Monet Blue (Biocare Medical) for 60 minutes, followed by anti-rabbit IgG (1:10,000, Rabbit-on-Farma HRP) for 30 minutes (Supplemental Table 1).
Autoimmune Profiling Using Protein Fragment Microarray Platform
Large-scale, high-density, recombinant protein fragment microarrays were used to profile the autoantibody repertoire in plasma/serum samples from patients with MN and from healthy controls. The protein fragment microarrays (SciLifeLab, Stockholm, Sweden) cover 94% of the gene-centric human proteome and are composed of 42,000 protein fragments, produced by The Human Protein Atlas Project, with a mean length of 80 amino acids, which have been selected on the basis of sequence uniqueness.24 Reactive serum samples with distinctive WB bands (e.g., MN01) were applied in those microarrays to more precisely specify potential autoantigens. The samples were diluted 1:100 in assay buffer (0.1% PBS with Tween 20, 3% BSA, 5% milk, supplemented with 160 _μ_g/ml His6ABP) and incubated in the assay buffer for 15 minutes before transfer to the arrays. The samples were incubated on the arrays for 1 hour at RT on the bench before washing twice for 5 minutes in 0.01% PBS with Tween 20. The arrays were then incubated with hen anti-His6ABP, at a dilution of 1:20,000 (hen anti-His6ABP, concentration 25 mg/ml, was produced within The Human Protein Atlas Project), for 1 hour on a shaker. The detection antibodies anti-human IgG (H+L) Alexa Fluor 647 (2 mg/ml, catalog number A21445; Life Technology) and goat anti-chicken IgY Alexa Fluor 555 (catalog number A-21437; Invitrogen) were diluted 1:10,000 in 0.01% PBS with Tween 20 and incubated on the arrays for 1 hour at RT before washing in 0.01% PBS with Tween 20. The arrays were scanned using a CapitalBio LuxScan HT24 at a resolution of 10 _µ_m. The images were analyzed using the GenePix Pro 5.1 image analysis program. R was used for the statistical analysis.
The concentration of the antibody is expressed using SD. The “raw” unit is the median fluorescence intensity (see Supplemental Table 2).
Autoimmune Profiling Prioritizing Algorithms
A prioritization algorithm was implemented to identify candidate protein antigens in MN. Proteins with autoantibody reactivity that were enriched in urinary podocyte exosomes and had previously characterized murine podocyte analogues were identified.25,26 Proteins meeting these criteria were then sorted by autoantibody signal intensity, as quantified by SD. We used Python 3.6 software for coding. The data and code are available at https://github.com/ehlin92/membranous_nephropathy_HTRA1 (Supplemental Tables 3 and 4).
Electron Microscopy Sample Preparation for Gold Immunolabeling
Whole, wild-type, mouse kidney was immersed directly in fixative containing 2% paraformaldehyde and 0.2% glutaraldehyde in 0.1 M phosphate buffer, pH 7.0. Small cubes of edges, of about 3 mm, were trimmed from the cortex with a razor blade and stored in fresh fixative at RT overnight. To isolate glomeruli, the specimen blocks were transferred to 0.1 M phosphate buffer containing 0.15% glycine, and individual glomeruli were dissected out under the dissecting microscope, using fine-pointed forceps. The isolated glomeruli were pelleted with centrifugation and further processed for either pre- or postembedding immunocytochemistry (see Supplemental Methods).
Statistical Methods
To summarize the data in the participant table (Table 8), because of the small number (_n_=13) of observations, we used the sample mean and evaluated its 95% confidence interval using the bootstrap resampling method.27 For urine protein-creatinine (Cr) ratio levels, where data indicated only that the levels were in the nephrotic range, we assumed the value was 3.5 g/g Cr, which is the lower end of the standard range indicating proteinuria.
Statistical hypothesis testing of MS DIA data were performed using the moderated t test from Limma.28 The statistical test was performed on protein quantities, which are obtained by summing peptide intensities.
Results
Our groups have been interested in identifying techniques to more rapidly identify novel antigens in cases of MN of unclear etiology and have focused on adult subjects with MN who are negative for PLA2R-, NELL1-, and THSD7A. One method is to screen for such patients using serum samples from the time of severe nephrosis (diagnosis or relapse) and remission, and then to assess the differential reactivity of these serial samples by performing WB against human glomerular proteins. The presence of a reactive band at a time of clinical activity that specifically disappears during remission could suggest the presence of relevant anti-glomerular antibodies involved in disease pathogenesis. Our discovery cohort included an index patient (MN01), who is a 75-year-old White female; an 87-year-old White female (MN03); and a 68-year-old Hispanic male (MN02). They all represent patients with biopsy specimen–proven MN that were negative for PLA2R and THSD7A in the absence of any apparent secondary causes for their disease.
Detection and Identification of a Novel Candidate Antigen
The use of longitudinal serum samples from our index patient with MN to examine reactivity by WB human glomerular proteins allowed the identification of an IgG4 band migrating just below 50 kD. This 50-kD protein was present during nephrosis and absent during remission (Figure 1A). This band, representing a putative target glomerular antigen, was not detected in the serum of a patient with anti-PLA2R antibodies (lane 4) or from healthy controls (not shown).
Figure 1.

Identification of a 50-kDa target antigen present within human glomerular protein extract. Identical nitrocellulose membrane strips of transferred human glomerular protein extract were cut from the original membrane, separately Western blotted with patient serum samples (top labels) and reassembled prior to imaging. (A) Longitudinal serum samples from our index patient with MN (MN01) used for WB of human glomerular protein extract. The 50-kD band was detected with serum sampled at time of a relapse of nephrotic syndrome (lane 1), but it declined and disappeared (lanes 2–3) as the patient achieved clinical remission. This band, representing a putative target glomerular antigen, was not detected by the serum of a patient with anti-PLA2R antibodies (lane 4; the large 180-kD band is PLA2R) or by healthy controls (not shown). (B) WB of human glomerular protein extract using rabbit anti-human HTRA1 (leftmost lane) or with individual MN serum samples and detected with appropriate secondary antibodies. Rabbit anti-HTRA1 strongly recognizes a band migrating to just below 50 kD (arrow), which is identical in size to the band recognized by the index patient (MN01) and another patient (MN03). Additional sera from patients with uncharacterized MN (MN-A, -B, -C, and -D) or known PLA2R antibody–associated MN (PLA2R-Ab+) fail to recognize the approximately 50 kD band. Anti-Hu, anti-human; anti-Rb, anti-rabbit.
Knowing there was differential IgG4 reactivity with the putative glomerular antigen, we performed IgG4-specific immunoprecipitation using samples from a time point when the patient was nephrotic in comparison with one when the patient was in remission, in addition to control sera. The IgG4-specific immunoprecipitates were separated by gel electrophoresis, under nonreducing conditions, and 50-kD gel regions were separately excised, subjected to tryptic digestion, and analyzed by MS. The potential candidates (Supplemental Table 5) were selected on the basis of presence of peptide spectra in the immunoprecipitated sample at the time of nephrosis, but absence at the time of remission and absence from the control samples.
The leading candidate from this analysis was a 51-kD protein known as serine protease HTRA1. A rabbit polyclonal antibody to human HTRA1 (generated by New England Peptide against full-length, inactive HTRA1) identified a band by WB that migrated in the identical position as the band identified by the sera of both our index patient with MN and another patient (MN03) with this subtype of MN (Figure 1B). These sera did not react with recombinant vitamin D–binding protein, the other similarly sized candidate identified by MS (Supplemental Table 5, Supplemental Figure 3, B and C).
Characteristics of Anti-HTRA1 Antibodies and Association with Disease Course
Recombinant human HTRA1 from a commercial source was immunoblotted with serum from patients with MN and controls. Serum samples from the index patient, at the time when she had active MN, recognized both recombinant HTRA1 and the presumed native HTRA1 band from glomerular extract, both of which migrated to exactly the same position (Figure 2A). There were additional bands recognized in the lane containing the recombinant protein, which likely represent oligomers (upper bands) and proteolytic fragments of HTRA1 (lower bands), on the basis of the known characteristics of this protein.29 Of note, serum from the point at which the index patient was in partial clinical remission reacted weakly with recombinant HTRA1, but not with human glomerular extract (Figure 2A). In contrast, sera from patients with MN who had anti-PLA2R or anti-THSD7A antibodies, and sera from healthy controls, did not recognize HTRA1 (Figure 2A).
Figure 2.
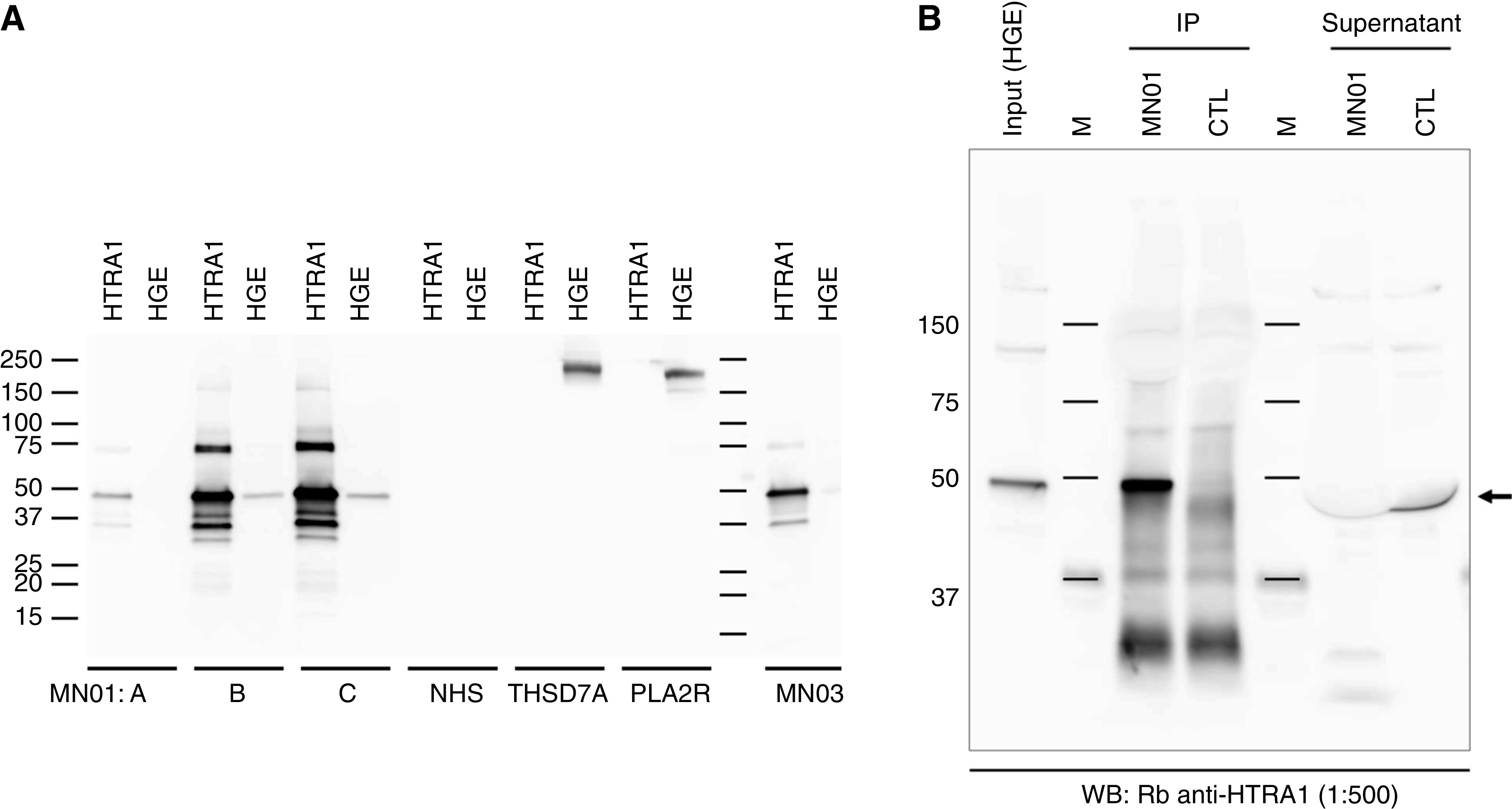
Western blot detection and immunoprecipitation of glomerular proteins with sera from patients with HTRA1-associated MN and controls. (A) Paired lanes of recombinant HTRA1 and human glomerular extract (HGE) individually underwent WB with patient serum (represented by black bars and labels at bottom) and were detected for IgG4. Serial samples (A–C) from index patient MN01 showed increased reactivity for the recombinant and native protein over time. In addition, serum from MN03 is clearly shown to react with recombinant HTRA1. In contrast, there was no reaction in the approximately 50 kD region when recombinant HTRA1 or HGE was blotted with normal control human serum (NHS) or from patients with known THSD7A- or PLA2R-associated MN. The 250- and 180-kD bands demarcate the position of THSD7A and PLA2R, respectively. (B) Serum from the index patient (MN01) or a healthy control (CTL) was used to immunoprecipitate the antigen from HGE. The HGE input, the immunoprecipitates (IPs), and the residual HGE supernatant post-IP were gel electrophoresed and immunoblotted with a rabbit (Rb) antibody to human HTRA1. HTRA1 was identified in the IP from MN01 (corresponding to the size in the input lane) and was largely depleted from the supernatant. In contrast, CTL serum did not IP HTRA1 and the protein was retained in the supernatant (arrow; band is displaced downward due to large albumin band above). M, marker.
We confirmed that the band recognized in the human glomerular extract was indeed native HTRA1 by performing immunoprecipitation from glomerular extract with serum from the index patient followed by WB of the immunoprecipitate with rabbit anti-HTRA1. This serum could precipitate a protein that was reactive with rabbit anti-HTRA1 and could deplete the input human glomerular extract of this band (Figure 2B). In contrast, serum from healthy controls was neither able to immunoprecipitate HTRA1 nor deplete human glomerular extract of this protein.
Human autoantibody reactivity with PLA2R and THSD7A is abolished under reducing immunoblot conditions when disulfide bonds are reduced.2,3 Because HTRA1 also has a number of disulfide bonds in its N-terminal domains we asked if there was also a reduction sensitivity to the epitopes in HTRA1. Reactive serum samples from the patients with MN retained reactivity to HTRA1, both the native protein from human glomerular extract and the recombinant protein, when assayed under reducing conditions (Figure 3). The equivalent detection of the protein in nonreducing and reducing conditions suggests the conformation of the epitope(s) is not dependent on the presence of disulfide bonds.
Figure 3.
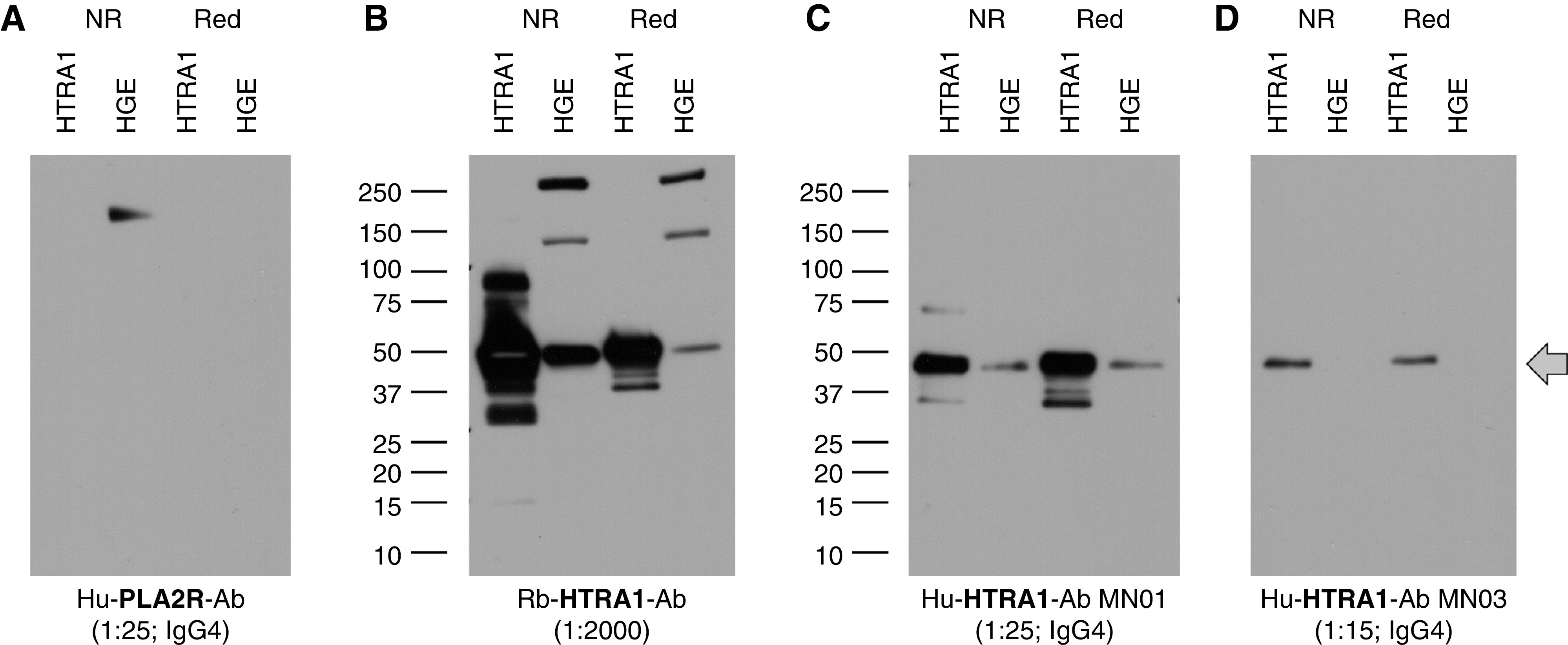
Characterization of antibody reactivity with reduced and non-reduced forms of native and recombinant HTRA1. WB of recombinant HTRA1 or native human glomerular extract (HGE) within HGE electrophoresed under reducing and nonreducing conditions. (A) WB with PLA2R antibody (PLA2R-Ab)–containing serum shows loss of reactivity to PLA2R (180-kD band) under reducing (Red) conditions. (B) The polyclonal anti-HTRA1 antibody (HTRA1-Ab) detects recombinant and native HTRA1 under both reducing and nonreducing (NR) conditions. (C) Serum from the index patient MN01 reacts with recombinant and native HTRA1 under both reducing and nonreducing conditions. (D) The weaker-titer MN03 serum also recognizes recombinant HTRA1 under both conditions. The arrow demarcates the position of the HTRA1 monomer. Higher and lower bands in (B) are likely oligomers and proteolytic fragments, respectively. Hu, human; Rb, rabbit.
The anti-HTRA1 antibodies in our index patient were primarily of the IgG4 subclass, followed by IgG3 and IgG1 (Supplemental Figure 3), consistent with other forms of primary, autoimmune MN. We assessed the reactivity of all available serum samples from our index patient and MN03 against recombinant HTRA1 by WB to correlate the strength of the signal on WB (as a surrogate for titer) with clinical features, such as proteinuria and serum albumin (Figure 4). Periods of strong reactivity appeared to correlate with periods of low or decreasing serum albumin and high or increasing proteinuria, likely signifying disease activity, whereas attenuation of the signal was associated with disease remission. Although such clinical correlation could only be shown in two patients due to sample availability, these findings support the role of circulating anti-HTRA1 as a potential biomarker for disease activity.
Figure 4.
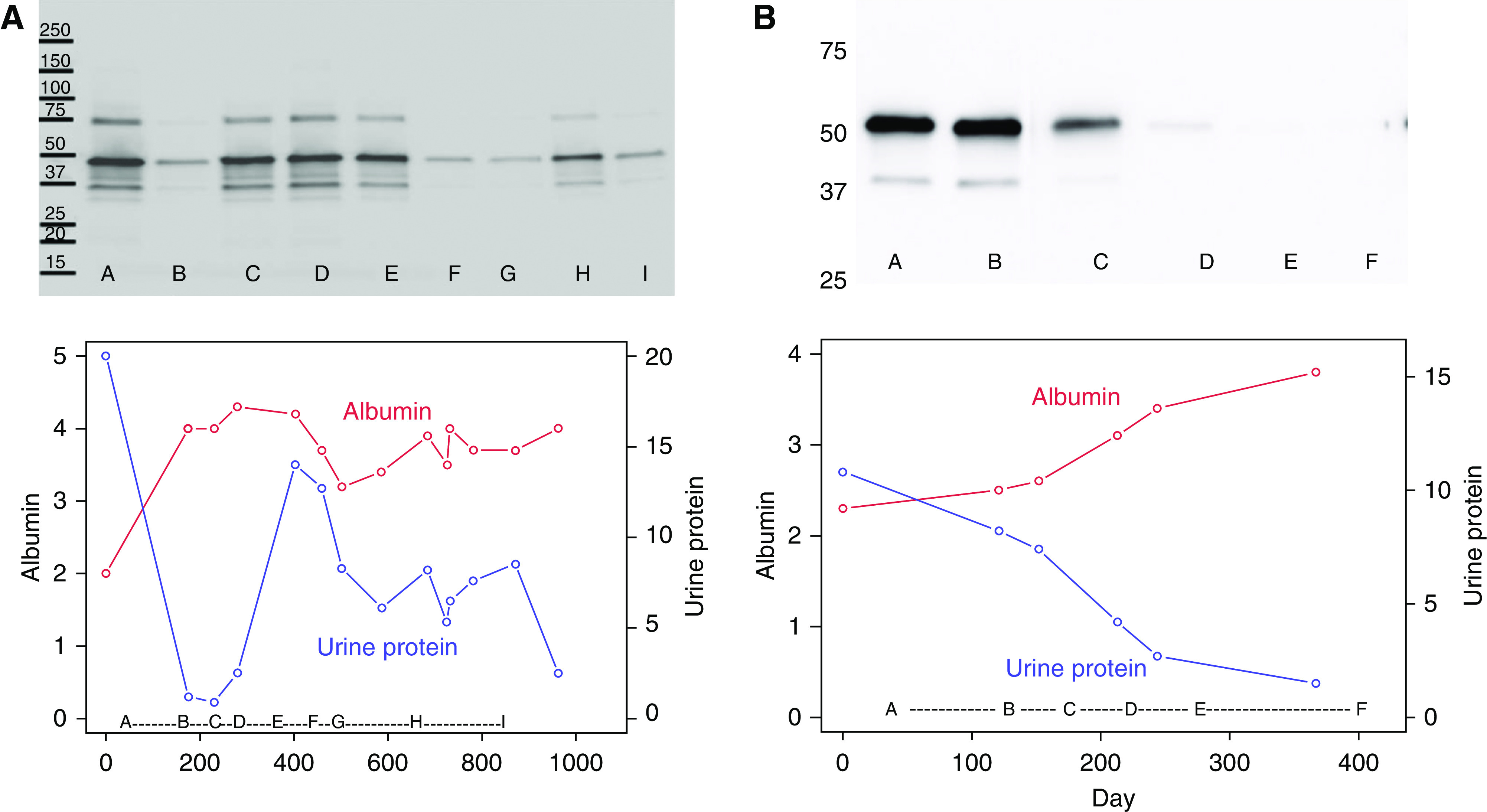
Anti-HTRA1 antibody response in relation to clinical disease parameters in two patients with HTRA1-associated MN. Longitudinal serum samples from (A) the index patient MN01 and (B) MN03 were used to immunoblot equal amounts of recombinant HTRA1 and demonstrate the changes in anti-HTRA1 titer over time. Identical amounts of recombinant HTRA1 were gel electrophoresed and transferred to a nitrocellulose membrane, and after visualization of the protein bands with Ponceau S, the individual lanes were manually cut into strips with scissors. Each strip was incubated with a standard dilution of patient serum from different time points (letters at bottom of images and graphs) then detected for human IgG4. These images represent digital merges of the chemiluminescence image captures (dark bands at 50 kDa. Please see Supplemental Figure 3 and the detailed methods in the Supplemental Material. Urine protein-Cr ratio (blue) and serum albumin (red) are shown over the same time periods.
Autoimmune Profiling of the Serum of the Index Case
A separate, but related, line of investigation was simultaneously pursued in which we used high-density, whole-proteome microarrays of human protein fragments (SciLifeLab) to assess differences in the autoantibody repertoire present in the reactive (relapse) sample from our index patient versus the minimally reactive remission sample. When the results were filtered according to differential reactivity between the two samples, protein size, and known expression by podocytes, HTRA1 again emerged as a leading candidate, with a difference in mean fluorescence intensity of 6.9 SD (Supplemental Figure 4) which was supported by preliminary results of an ELISA in development (Supplemental Figure 5). Of note, the particular protein fragment from the microarray that was differentially recognized by the relapse versus remission sample is located in the carboxy-terminal (C-terminal) PDZ domain of HTRA1 (Supplemental Table 6). This domain lacks disulfide bonds and, therefore, the presence of an epitope in this region would be consistent with our previous WB findings that human anti-HTRA1 reactivity is maintained under reducing conditions.
Tissue Immunoprecipitation and MS Identification of HTRA1
In another, independent line of investigation, biopsy specimens from patients with MN known to be negative for PLA2R, THSD7A, and EXT1/EXT2 were investigated by LCM of glomeruli from FFPE kidney biopsy tissue followed by MS analysis to identify accumulation of putative antigens. Additionally, immunoprecipitation of antibody-antigen immune complexes was performed from optimal cutting temperature–frozen tissue, followed by MS analysis.10 In three patients, the leading candidate antigen was HTRA1 (Figure 5, Supplemental Figure 6).
Figure 5.
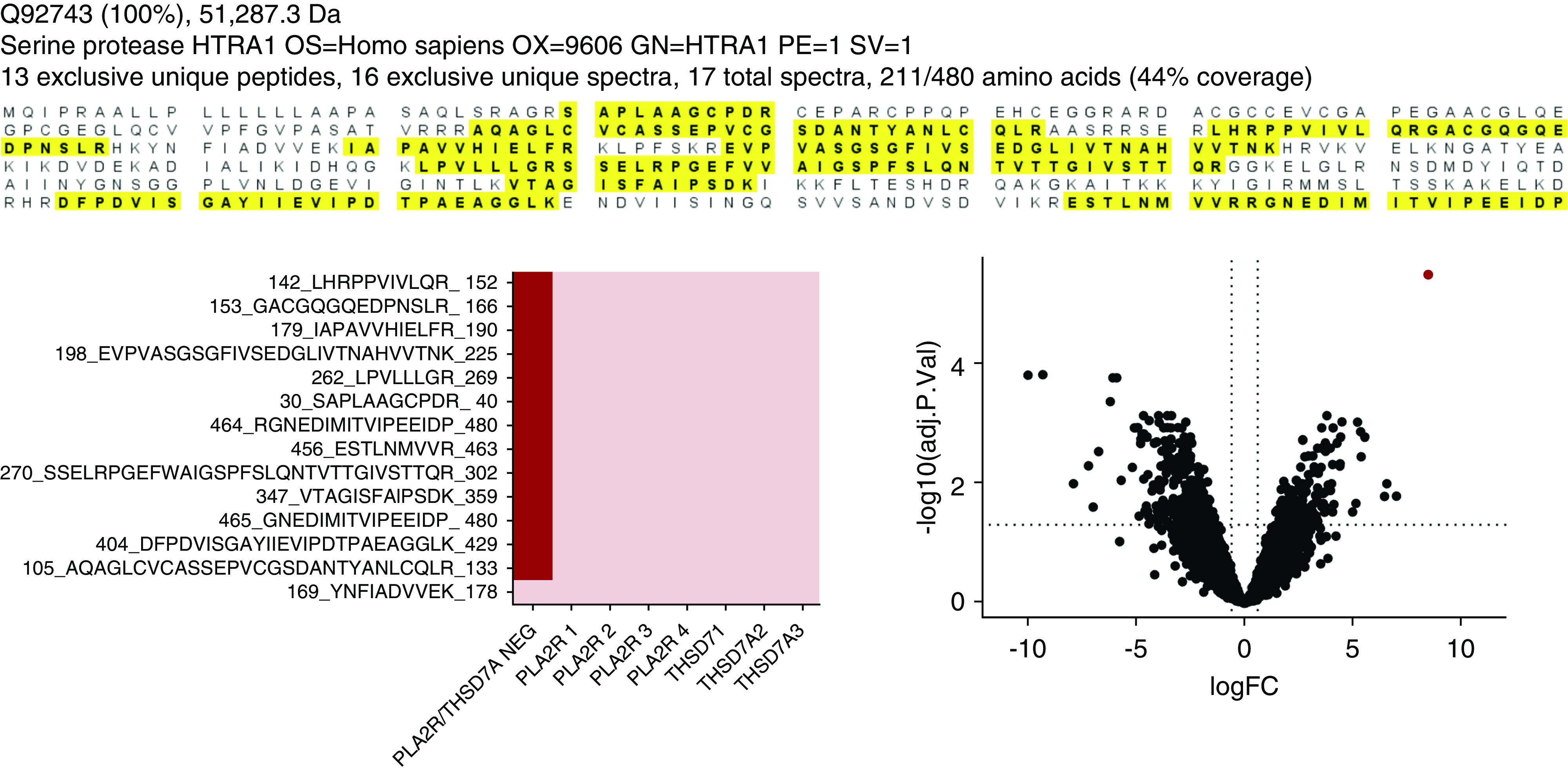
Ig capture from frozen kidney biopsy specimens followed by MS identifies serine protease HTRA1 as a target antigen. (A) Sequence coverage maps representing peptides detected from MN01 showing the extensive coverage by MS/MS. Peptide sequences detected are highlighted in bold letters over yellow background. (B) Heat map of 13 peptides quantified from the same patients. These peptides were specific to HTRA1-associated MN and were not present in four patients with PLA2R-associated MN and three patients with THSD7A-associated MN included as controls. (C) The volcano plot showing fold-change (FC) and P value of proteins quantified in HTRA1 versus non-HTRA1 cases. The serine protease HTRA1 (red dot) is a potential protein of interest, with the highest fold-change and lowest P value relative to other quantified proteins. Adj. P. Val, adjusted P value; neg, negative; OS, organism name; OX, organism identifier; GN, gene name; PE, protein existence; SV, sequence version.
Localization of HTRA1 in MN and Normal Kidney Tissue
The paradigm for immune complex formation in MN involves the accumulation of antigen with Ig within immune complexes after shedding (or secretion) of the antigen from the podocyte. Staining for HTRA1 alone via immunofluorescence and immunohistochemistry showed granular peripheral capillary wall staining for HTRA1 in cases of HTRA1-associated MN (Figure 6, A and C, Supplemental Figures 7 and 8), but weaker cell body staining in cases of other subtypes of MN, such as PLA2R-associated MN (Figure 6, B and D). Specificity of HTRA1 staining was demonstrated by the lack of granular HTRA1 deposits in other forms of MN (Figure 6, F–I) or in other proteinuric disease states, such as FSGS, minimal change disease, fibrillary glomerulopathy, and diabetic glomerulopathy (Supplemental Figure 9). HTRA1 and IgG colocalize within the immune deposits in this subtype of MN but not in other types of MN (Figure 7, Supplemental Figure 10). Confocal imaging of kidney biopsy cryosections demonstrated 93% colocalization (averaged over nine glomeruli) of IgG with the antigen HTRA1 in a patient with HTRA1-associated MN, as opposed to 3.6% in a patient with PLA2R- associated MN (_P_=3.46505×10−17; Supplemental Figure 11, Supplemental Table 7).
Figure 6.
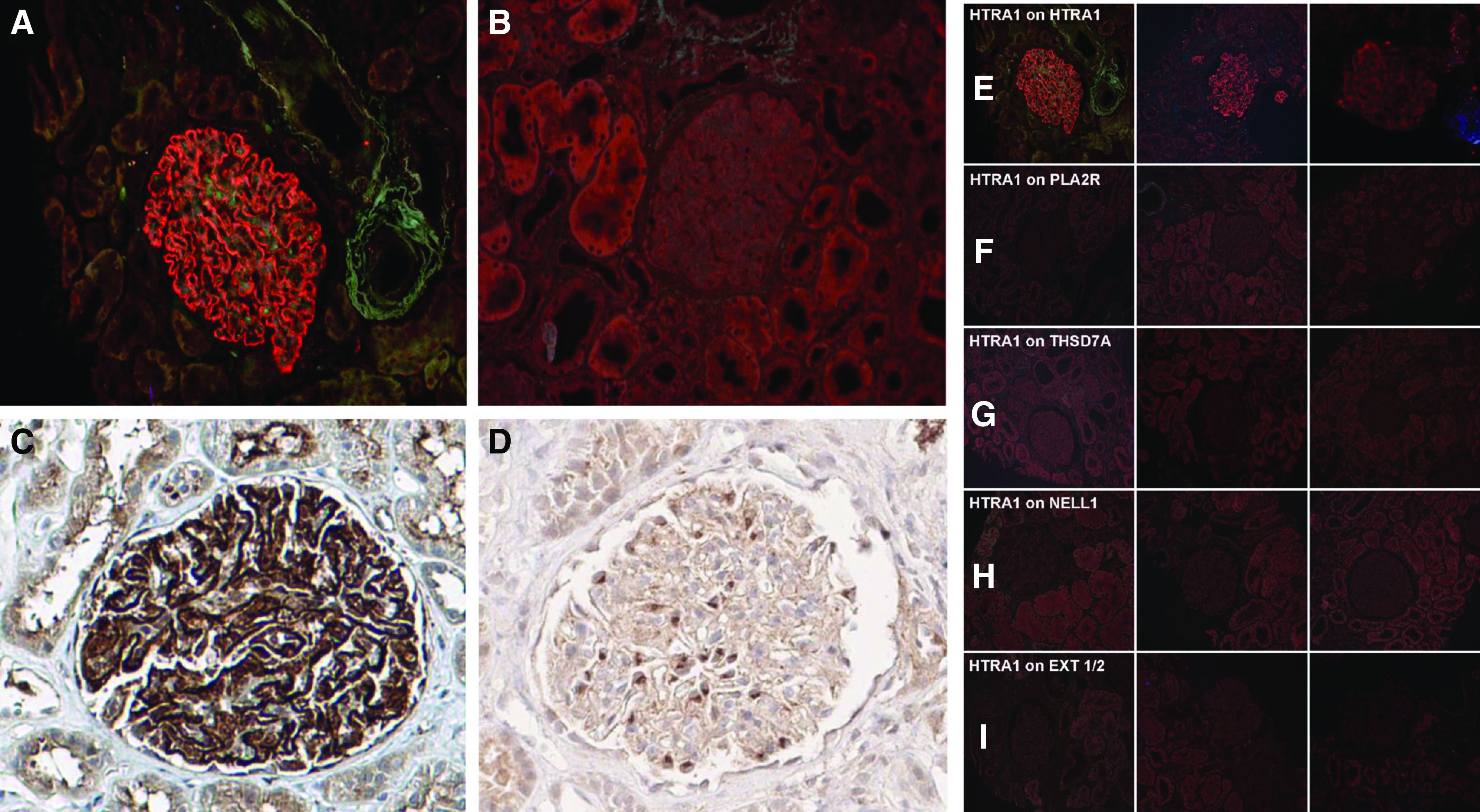
Immunofluorescence and immunohistochemical staining in HTRA1-associated MN patients and controls. Immunofluorescence microscopy of MN biopsy tissue (MN03) demonstrates a strong, granular, peripheral capillary loop staining pattern in (A) HTRA1-associated MN but not in (B) PLA2R-associated MN. (C) Immunohistochemical staining similarly showed strong capillary wall staining for HTRA1 in a peripheral capillary loop pattern, (D) but much weaker staining, consistent with low-level baseline podocyte expression, was seen in PLA2R-associated MN. (E–I) HTRA1 staining is uniquely present in (E) HTRA1-associated MN, but not within other forms of MN of known type, including (F) PLA2R-associated MN, (G) THSD7A-associated MN, (H) NELL1-associated MN (H), and (I) EXT1/EXT2-associated MN. A representative glomerulus from three biopsy specimens is shown within each row.
Figure 7.

Dual immunofluorescence staining for IgG (green) and HTRA1 (red) shows colocalization (right panels, yellow) in HTRA1-associated MN (MN02) but not in PLA2R-associated MN.
The bulk of the published literature suggests that HTRA1 is expressed by the podocyte within the glomerulus, including by induced podocytes in vitro.26,30–32 Human immortalized podocytes express mRNA for HTRA1 at a moderate level (unpublished transcriptional microarray data) and at the protein level (Figure 8A). Both immortalized and primary human podocytes express HTRA1 in a perinuclear punctate or reticular pattern by immunofluorescence staining (Figure 8B and data not shown). Immunohistochemical staining of normal human kidney tissue for HTRA1 demonstrates moderate staining of the podocyte cell bodies (Figure 8C). We next investigated localization of HTRA1 in normal mouse kidney at the ultrastructural level with gold-conjugated antibodies. HTRA1 was found in vesicular structures in the foot processes, within the subpodocyte space between foot processes (Supplemental Figure 12), and occasionally within the GBM (data not shown), supportive of its role as a secreted protein.
Figure 8.
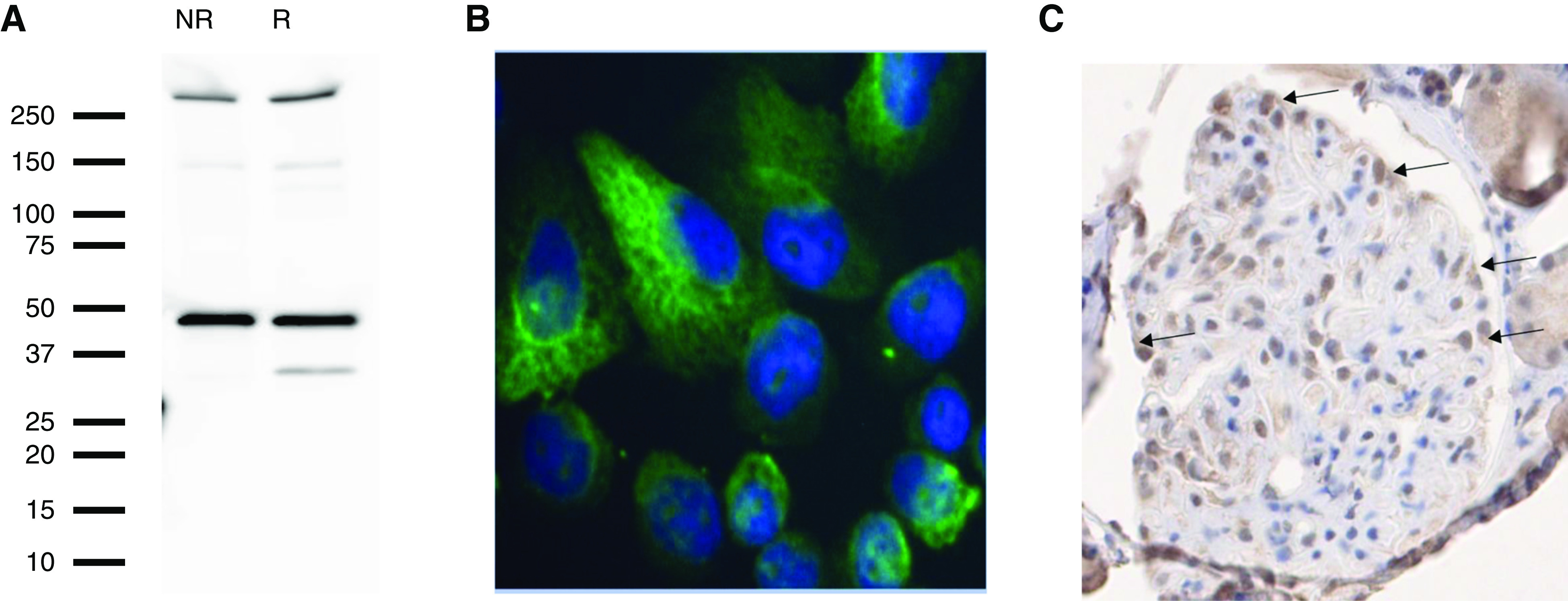
HTRA1 is a podocyte-expressed protein. (A) Protein extracts of differentiated, immortalized human podocytes were immunoblotted with rabbit anti-HTRA1 and they exhibited reactivity with HTRA1 in both the nonreduced (NR) and reduced (R) state. (B) Immunofluorescence imaging of primary podocytes using rabbit anti-HTRA1 revealed cellular expression of the HTRA1 (green) in a reticular and perinuclear pattern (nuclei in blue via 4′,6-diamidino-2-phenylindole staining). Original magnification, ×400. (C) Immunohistochemical staining of normal kidney tissue with rabbit anti-HTRA1 shows positive HTRA1 staining of podocyte cell bodies (arrows).
On the basis of these data, we feel the most likely source of HTRA1 in this subtype of MN is locally from the podocyte. However, we also excluded the possibility of circulating immune complexes containing HTRA1 and could not detect circulating immune complexes of HTRA1–IgG via protein G immunoprecipitation of IgG from relevant serum samples (data not shown).
Screening/Validation in Additional Biopsy Cohorts
We used the large 9-month consecutive case series of MN available at Arkana Laboratories10 to assess the prevalence of HTRA1-associated MN. Out of the 349 patients with MN, 181 were PLA2R positive (51.9%), ten were THSD7A positive (2.9%), 33 were EXT2 positive (9.5%), and six were NELL1 positive (1.7%). No individual was positive for more than one of these existing biomarkers. Screening of the 118 “quadruple-negative” (i.e., PLA2R-, THSD7A-, NELL1-, and EXT2-negative) biopsy specimens for HTRA1 identified five positive patients. Thus, we estimate that HTRA1 is a biomarker for 1.43% (95% CI, 0.19% to 2.68%) of patients with MN, and that it explains 4.24% (95% CI, 0.6% to 7.87%) of the patients unexplained by existing biomarkers. We combined the Oregon and Utah patients with PLA2R-negative MN (_n_=180) and estimated the frequency of HTRA1-associated MN to be 3.3% (95% CI, 0.7% to 6%), which may be a more representative prevalence in institutions that test only for PLA2R.
We next assessed the specificity of the HTRA1 tissue staining by assessing biopsy tissue of other patients with MN who were positive for other antigens. Not all of the 230 samples that had previously stained positive for an existing marker had sufficient remaining tissue for HTRA1 testing. However, we stained 146 PLA2R-positive, 38 lupus-associated MN, and three NELL1-positive samples for HTRA1, and all 187 were found to be negative for HTRA1 within deposits. This is consistent with the hypothesis that HTRA1 and the existing markers are mutually exclusive and HTRA1-associated MN is a unique and specific entity.
Demographic, Clinical, and Biopsy Specimen Features of HTRA1-Associated MN
With the addition of additional cohorts from Utah and Oregon, HTRA1-associated MN was confirmed in a total of 14 patients from our three cohorts (Supplemental Figure 1). Summary statistics are shown in Supplemental Tables 8 and 9 and are presented as mean (95% CI). The mean age was 67.3 years (95% CI, 58.7 to 75.9). Mean serum Cr was 1.57 mg/dl (95% CI, 1.04 to 2.1 mg/dl), and the mean urine protein-Cr ratio was 9.17 g/g (95% CI, 6.56 to 11.78 g/g). Twelve patients did not have any evidence of malignancy or infection. One patient was positive for ANCA-associated vasculitis with concurrent crescentic lesions, consistent with the clinical presentation of rapidly progressive GN. One patient had been diagnosed with stage IV small cell lung cancer (SCLC) 2 years before the kidney biopsy, with no proteinuria at that time. There was no reported evidence of cancer when nephrotic syndrome developed, although, given the well-known poor outcome and treatment resistance of SCLC, the presence of malignancy cannot be excluded.
The kidney biopsy specimens of all patients with HTRA1–associated MN showed thickened capillary loops on light microscopy, granular IgG and C3 staining along the capillary wall on immunofluorescence microscopy, and subepithelial deposits on electron microscopy. Of the 14 patients, 12 had a diffuse pattern of subepithelial deposits (80%), whereas a segmental distribution to the deposits was found in the remaining two patients. Consistent with PLA2R- and THS7A-associated MN, IgG4 was the dominant subclass in seven out of eight patients in which staining for IgG subclasses was performed. One patient showed IgG2 as the predominant subclass. All patients exhibited polyclonal staining for both κ and λ light chains. Electron microscopy showed subepithelial deposits with no subendothelial or tubuloreticular inclusions. Occasional mesangial deposits were reported in two out of 14 patients, and C1q was positive in the patient with SCLC and did not show full-house staining (Supplemental Table 9).
Discussion
We used a multipronged approach—which included immunoblotting and differential immunoprecipitation of human glomerular proteins using nephrotic versus remission serum samples, LCM and protein G tissue immunoprecipitation/MS, and autoimmune profiling—to identify serine protease HTRA1 as a novel podocyte antigen in a subset of patients with primary MN. The candidate antigen was initially detected as a discrete band in human glomerular protein extract detected in serum at a time of disease activity, but not during remission. Immunoprecipitation of this candidate target antigen, using serum from these two time points, followed by MS, suggested HTRA1 as the putative target antigen, which was confirmed with the recombinant human HTRA1 protein. LCM and immunoprecipitation of glomerular IgG-antigen complexes, followed by MS, also identified HTRA1, which was uniquely enriched in the glomeruli in this subclass of MN. Immunofluorescence staining of biopsy tissue for HTRA1 demonstrates localization to immune deposits only in this type of MN, and not in cases of PLA2R- or THSD7A-associated MN. Analysis of three separate US cohorts of MN suggests that HTRA1-associated MN may explain 1%–2% of all suspected primary MN cases, and 3%–4% of cases with unknown antigen association.
Circulating antibodies against HTRA1 appeared to correlate with disease activity. Analysis of longitudinal samples from our index case (MN01) and MN03 showed that circulating antibodies to HTRA1 are present in the nephrotic state, and then decline with remission of disease. Similar to PLA2R- and THSD7A-associated MN, antibodies were predominantly IgG4. Autoantibodies against HTRA1 reacted with recombinant HTRA1 protein, both in the native state and in reducing conditions, suggesting the epitope was not sensitive to reducing conditions and was less likely to be in the disulfide bridge–rich N-terminus. Our autoimmune profiling results suggest autoantibodies may target a region in the C-terminal region of the molecule.
HTRA1-associated MN is a distinct subtype of MN that appears to have a mean age of onset that occurs later than what is more commonly seen in primary MN (e.g., early 50s for PLA2R- associated MN). The male-female ratio was 4:3, and there was a predilection to White race in our small and geographically limited cohort. The degree of proteinuria can be within the severe nephrotic range, with five patients presenting with >8 g/g Cr of urinary protein. Clinically, there is usually an absence of clear secondary causes, such as malignancy, infections, or systemic autoimmune diseases; however, we recognize the incomplete clinical dataset from our small cohort is a major limitation in fully characterizing the clinical phenotype of HTRA1-associated MN. This is supported by biopsy specimen features showing predominance of IgG4, absence of C1q or a full-house pattern of staining by immunofluorescence, and a lack of subendothelial deposits or endothelial tubuloreticular inclusions by electron microscopy. The peripheral capillary wall and subepithelial pattern of HTRA1 staining is more suggestive of a mechanism of immune complex formation related to shedding or secretion from the podocyte, similar to PLA2R- and THSD7A-associated MN, versus the antigens or immune complexes circulating in plasma becoming trapped in the glomerulus.
HTRA1 is a member of the serine protease family.33,34 It has nonspecific tissue distribution, with variable expression, and increased levels in the placenta.35 It is a very well conserved protein among different species.36,37 It is composed of four distinct protein domains: IGF-binding domain, a kazal domain, a trypsin-like peptidase domain, and a PDZ domain.34 Certain mammalian species (e.g., rabbit, cow) lack the Mac25 region (IGFBP and kazal domain; Supplemental Figure 13). Although the role of HTRA1 in kidney disease has not been well investigated, HTRA1 has been studied extensively in other fields, such as cancer,38–40 age-related macular degeneration,41,42 Alzheimer disease,43 and rheumatoid arthritis.33 Its specific role in progression of these diseases has been linked to its ability to directly degrade cartilage and stimulate matrix metalloproteinase production by synovial fibroblasts in arthritic disorders,42,44–48 and a proposed tumor suppressor role in malignancy, because loss of HTRA1 expression contributes to the aggressiveness, metastatic ability, and chemoresistance of certain tumors.39,40
A bacterial form of the HTRA protein found in the cell wall of Orientia tsutsugamushi is known to be antigenic, and this 47-kD bacterial protein has been used in a vaccine developed against scrub typhus, which is caused by this bacteria.49,50 In one study, a significant number of patients developed antibodies against recombinant human HTRA1, raising concern about safety of this vaccine given its antigenicity and potential mimicry mechanism triggering autoimmune process, because there is 35% identity and 55% homology with the human protein across the protease and C-terminal region.49,50 To our knowledge, none of those patients showed features of MN. We were able to test a serum sample from our index patient by PCR and IgG for scrub typhus at the Centers for Disease Control and Prevention laboratory, and the testing was negative for any active or recent infection. A more dedicated study to explore this possibility is needed in areas where the disease is endemic.
Recent advances in proteomics and single-cell sequencing have generated large datasets that allow identification of novel, podocyte-enriched proteins.25,31 It is clear from the aggregate data that HTRA1 is expressed by the podocyte and can be found in the extracellular environment, because it was identified as an element of the glomerular matrisome51–53 and was present in a urinary podocyte exosome fraction.25 Similar to PLA2R and THSD7A, HTRA1 transcripts are present in pluripotent stem cells induced to differentiate toward a podocyte lineage.30 HTRA1 was suggested, on the basis of single-cell transcriptome analysis, to be a marker of mature podocytes and mapped to pathways related to podocyte intercellular adhesion and slit diaphragm structure and function. Older transcriptional data described a 30-fold enrichment of HTRA1 in podocytes versus other glomerular cell types.54 Studies of other, nonglomerular human cell types demonstrated that HTRA1 can be expressed on the cell membrane or, alternatively, linked to the cytoskeleton in colocalization with the microtubules.36,55 Whether or not this is true in podocytes remains to be investigated.
A prerequisite for circulating antibodies to target a podocyte protein is the availability of exposed humoral epitope(s) in the extracellular compartment. HTRA1 has a signal peptide and enters the secretory pathway, possibly being secreted in conjunction with extracellular matrix molecules, many of which serve as a substrate for its protease activity.56 HTRA1 has been shown to localize to the GBM by attaching to collagen fibrils in the extracellular space and deposit between the GBM and podocytes.51,52,57 HTRA1 can undergo conformational changes and adopt different tertiary structures that result in differences in sequestration and activity.57 This evidence supports the secretion of HTRA1 and a potential role in cleavage, modification, and/or turnover of matrix molecules in the GBM. The ability of podocytes to secrete vesicles containing HTRA1 provides another potential route for engagement of HTRA1 with the immune system.
This report demonstrates the convergence of several independent lines of investigation culminating in the identification of serine protease HTRA1 as a novel, podocyte-expressed antigen in MN. This subtype of MN represents approximately 4.2% of cases of previously uncharacterized MN; however, because of its relative rarity (1%–2% of all MN), this subtype would have been difficult to study in detail using only the traditional approaches used in the identification of PLA2R and THSD7A. Our work illustrates the importance of using a variety of proteomic and antibody profiling techniques along with large biopsy specimen repositories and publicly available datasets to help identify the remaining rare MN antigens. We envision that similar protocols can help to find additional autoantigens in MN and in other rare autoimmune diseases.
Disclosures
J. Klein reports serving as a scientific advisor for, or member of, Akebia Therapeutics, Inc. B. Williams reports being an inventor on a patent application related to HTRA1 in the field of macular degeneration. The patent is owned by the University of Utah. All remaining authors have nothing to disclose.
Funding
This project was funded by the National Kidney Foundation of Utah & Idaho award number 51006060 (grant title, “Novel Antibodies in Membranous Nephropathy”). Institutional support for L. H. Beck’s work on this project was provided through the Glomerular Disease Center at Boston Medical Center.
Supplementary Material
Supplemental Figure 3
Supplemental Data
Supplemental Data
Acknowledgments
The authors would like to thank the patients with MN who contributed samples to this study (especially RN and DE for their continued participation) and to New England Donor Services and the families who consented to the use of donor kidneys for research purposes. The authors would like to thank Dr. Gregory Hagman and Dr. Paul Bernstein from the Moran Eye Center, Dr. Vipul Chitalia and Dr. David Salant from Boston Medical Center, and Dr. Martin Gregory and Dr. Alfred Cheung from University of Utah Health for their insightful comments and suggestions on this work. The authors would like to thank Dr. Fuad Shihab and Ms. Deen Vetterli from NKF Utah Idaho/NKF for the continuous support.
L. F. Al-Rabadi and L. H. Beck designed the study; C. Trivin-Avillach, N. Hayashi, B. Williams, C. Zou, T. Cummins, C. Herzog, A. Storey, R. Sjoberg, T. Yang, L. F. Al-Rabadi, and L. H. Beck carried out the experiments; J. Chien, R. Edmondson, M. Merchant, J. Arthur, J. Klein, C. Larsen, M. P. Revelo, F. Clayton, J. Abraham, N. Ramkumar, I. Kawalit, E. Lin, L. F. Al-Rabadi, and L. H. Beck. analyzed the data; C. Trivin-Avillach, N. Hayashi, T. Caza, C. Larsen, L. F. Al-Rabadi, and L. H. Beck made the figures; E. Lin performed the algorithm coding; T. Caza, F. Clayton, M. P. Revelo, and C. Larsen performed the immunofluorescence and immunohistochemistry studies; L. F. Al-Rabadi and L. H. Beck drafted and revised the manuscript; and all authors approved the final version of the manuscript.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2020101395/-/DCSupplemental.
Supplemental Figure 1. Schematic representation of the cohorts.
Supplemental Figure 2. Schematic representation of the methods.
Supplemental Figure 3. Western blot: IgG subclasses, controls, Vit D binding protein and original Fig 4B.
Supplemental Figure 4. Figures derived from whole protein fragment array analysis.
Supplemental Figure 5. Recombinant HTRA1 ELISA.
Supplemental Figure 6. Mass spectrometry data from protein G tissue IP.
Supplemental Figure 7. Immunofluorescence imaging of the positive HTRA1 cases.
Supplemental Figure 8. Low-power immunofluorescence.
Supplemental Figure 9. Specificity of immunofluorescence imaging.
Supplemental Figure 10. Additional confocal imaging.
Supplemental Figure 11. Quantification of co-localization of HTRA1 with IgG on biopsy.
Supplemental Figure 12. Gold immunolabeling.
Supplemental Figure 13. The domain structure of HTRA1.
Supplemental Table 1. Commercial anti-HTRA1 antibodies used.
Supplemental Table 2. Autoimmune profiling data.
Supplemental Table 3. Top podocyte proteins from autoimmune profiling.
Supplemental Table 4. Candidate antigens (strong reactive signal) present in urinary exosome.
Supplemental Table 5. Mass spectrometry findings from 50 kDa gel region.
Supplemental Table 6. HTRA1 protein fragments expressed by autoimmune profiling.
Supplemental Table 7. HTRA1 and IgG co-localization statistics.
Supplemental Table 8. Baseline serum creatinine and urine protein values.
Supplemental Table 9. (A and B) Patient characteristics and biopsy findings.
References
- 1.McGrogan A, Franssen CF, de Vries CS: The incidence of primary glomerulonephritis worldwide: A systematic review of the literature. Nephrol Dial Transplant 26: 414–430, 2011 [DOI] [PubMed] [Google Scholar]
- 2.Beck LH Jr., Bonegio RG, Lambeau G, Beck DM, Powell DW, Cummins TD, et al.: M-type phospholipase A2 receptor as target antigen in idiopathic membranous nephropathy. N Engl J Med 361: 11–21, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tomas NM, Beck LH Jr., Meyer-Schwesinger C, Seitz-Polski B, Ma H, Zahner G, et al.: Thrombospondin type-1 domain-containing 7A in idiopathic membranous nephropathy. N Engl J Med 371: 2277–2287, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iwakura T, Ohashi N, Kato A, Baba S, Yasuda H: Prevalence of enhanced granular expression of thrombospondin type-1 domain-containing 7A in the glomeruli of Japanese patients with idiopathic membranous nephropathy. PLoS One 10: e0138841, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sethi S, Debiec H, Madden B, Charlesworth MC, Morelle J, Gross L, et al.: Neural epidermal growth factor-like 1 protein (NELL-1) associated membranous nephropathy. Kidney Int 97: 163–174, 2020 [DOI] [PubMed] [Google Scholar]
- 6.Sethi S, Debiec H, Madden B, Vivarelli M, Charlesworth MC, Ravindran A, et al.: Semaphorin 3B-associated membranous nephropathy is a distinct type of disease predominantly present in pediatric patients. Kidney Int 98: 1253–1264, 2020 [DOI] [PubMed] [Google Scholar]
- 7.Sethi S, Madden BJ, Debiec H, Charlesworth MC, Gross L, Ravindran A, et al.: Exostosin 1/exostosin 2-associated membranous nephropathy. J Am Soc Nephrol 30: 1123–1136, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Beck LH Jr.: PLA2R and THSD7A: Disparate paths to the same disease? J Am Soc Nephrol 28: 2579–2589, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Delville M, Sigdel TK, Wei C, Li J, Hsieh SC, Fornoni A, et al.: A circulating antibody panel for pretransplant prediction of FSGS recurrence after kidney transplantation. Sci Transl Med 6: 256ra136, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caza TN, Hassen SI, Dvanajscak Z, Kuperman M, Edmondson R, Herzog C, et al.: NELL1 is a target antigen in malignancy-associated membranous nephropathy. Kidney Int 99: 967–976, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Caza TN, Hassen SI, Kuperman M, Sharma SG, Dvanajscak Z, Arthur J, et al.: Neural cell adhesion molecule 1 is a novel autoantigen in membranous lupus nephritis [published online ahead of print October 9, 2020]. Kidney Int 10.1016/j.kint.2020.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zandian A, Forsström B, Häggmark-Månberg A, Schwenk JM, Uhlén M, Nilsson P, et al.: Whole-proteome peptide microarrays for profiling autoantibody repertoires within multiple sclerosis and narcolepsy. J Proteome Res 16: 1300–1314, 2017 [DOI] [PubMed] [Google Scholar]
- 13.Choung RS, Marietta EV, Van Dyke CT, Brantner TL, Rajasekaran J, Pasricha PJ, et al.: Determination of B-cell epitopes in patients with celiac disease: Peptide microarrays. PLoS One 11: e0147777, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hecker M, Lorenz P, Steinbeck F, Hong L, Riemekasten G, Li Y, et al.: Computational analysis of high-density peptide microarray data with application from systemic sclerosis to multiple sclerosis. Autoimmun Rev 11: 180–190, 2012 [DOI] [PubMed] [Google Scholar]
- 15.Sjoberg R, Mattsson C, Andersson E, Hellstrom C, Uhlen M, Schwenk JM, et al.: Exploration of high-density protein microarrays for antibody validation and autoimmunity profiling. Nt Biotechnol 33: 582–592, 2016 [DOI] [PubMed] [Google Scholar]
- 16.Häggmark-Månberg A, Zandian A, Forsström B, Khademi M, Lima Bomfim I, Hellström C, et al.: Autoantibody targets in vaccine-associated narcolepsy. Autoimmunity 49: 421–433, 2016 [DOI] [PubMed] [Google Scholar]
- 17.Li Y, Li CQ, Guo SJ, Guo W, Jiang HW, Li HC, et al.: Longitudinal serum autoantibody repertoire profiling identifies surgery-associated biomarkers in lung adenocarcinoma. EBioMedicine 53: 102674, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poulsen TBG, Damgaard D, Jørgensen MM, Senolt L, Blackburn JM, Nielsen CH, et al.: Identification of novel native autoantigens in rheumatoid arthritis. Biomedicines 8: 141, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Larsen CP, Trivin-Avillach C, Coles P, Collins AB, Merchant M, Ma H, et al.: LDL receptor-related protein 2 (megalin) as a target antigen in human kidney anti-brush border antibody disease. J Am Soc Nephrol 29: 644–653, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sethi S, Vrana JA, Theis JD, Dogan A: Mass spectrometry based proteomics in the diagnosis of kidney disease. Curr Opin Nephrol Hypertens 22: 273–280, 2013 [DOI] [PubMed] [Google Scholar]
- 21.Searle BC, Pino LK, Egertson JD, Ting YS, Lawrence RT, MacLean BX, et al.: Chromatogram libraries improve peptide detection and quantification by data independent acquisition mass spectrometry. Nat Commun 9: 5128, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gessulat S, Schmidt T, Zolg DP, Samaras P, Schnatbaum K, Zerweck J, et al.: Prosit: Proteome-wide prediction of peptide tandem mass spectra by deep learning. Nat Methods 16: 509–518, 2019 [DOI] [PubMed] [Google Scholar]
- 23.Searle BC, Swearingen KE, Barnes CA, Schmidt T, Gessulat S, Küster B, et al.: Generating high quality libraries for DIA MS with empirically corrected peptide predictions. Nat Commun 11: 1548, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al.: Proteomics. Tissue-based map of the human proteome. Science 347: 1260419, 2015 [DOI] [PubMed] [Google Scholar]
- 25.Prunotto M, Farina A, Lane L, Pernin A, Schifferli J, Hochstrasser DF, et al.: Proteomic analysis of podocyte exosome-enriched fraction from normal human urine. J Proteomics 82: 193–229, 2013 [DOI] [PubMed] [Google Scholar]
- 26.Rinschen MM, Gödel M, Grahammer F, Zschiedrich S, Helmstädter M, Kretz O, et al.: A multi-layered quantitative in vivo expression atlas of the podocyte unravels kidney disease candidate genes. Cell Rep 23: 2495–2508, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Effron B: Nonparametric estimate of standard error: The jackknife, the bootstrap and other resampling methods. Biometrika 68: 589–599, 1981 [Google Scholar]
- 28.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 3: Article3, 2004 [DOI] [PubMed] [Google Scholar]
- 29.Jo H, Patterson V, Stoessel S, Kuan CY, Hoh J: Protoporphyrins enhance oligomerization and enzymatic activity of HtrA1 serine protease. PLoS One 9: e115362, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshimura Y, Taguchi A, Tanigawa S, Yatsuda J, Kamba T, Takahashi S, et al.: Manipulation of nephron-patterning signals enables selective induction of podocytes from human pluripotent stem cells. J Am Soc Nephrol 30: 304–321, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rinschen MM, Huesgen PF, Koch RE: The podocyte protease web: Uncovering the gatekeepers of glomerular disease. Am J Physiol Renal Physiol 315: F1812–F1816, 2018 [DOI] [PubMed] [Google Scholar]
- 32.Menon R, Otto EA, Kokoruda A, Zhou J, Zhang Z, Yoon E, et al.: Single-cell analysis of progenitor cell dynamics and lineage specification in the human fetal kidney. Development 145: dev164038, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu SI, Carozza M, Klein M, Nantermet P, Luk D, Crowl RM: Human HtrA, an evolutionarily conserved serine protease identified as a differentially expressed gene product in osteoarthritic cartilage. J Biol Chem 273: 34406–34412, 1998 [DOI] [PubMed] [Google Scholar]
- 34.Eigenbrot C, Ultsch M, Lipari MT, Moran P, Lin SJ, Ganesan R, et al.: Structural and functional analysis of HtrA1 and its subdomains. Structure 20: 1040–1050, 2012 [DOI] [PubMed] [Google Scholar]
- 35.Nie G, Hale K, Li Y, Manuelpillai U, Wallace EM, Salamonsen LA: Distinct expression and localization of serine protease HtrA1 in human endometrium and first-trimester placenta. Dev Dyn 235: 3448–3455, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Chien J, Ota T, Aletti G, Shridhar R, Boccellino M, Quagliuolo L, et al.: Serine protease HtrA1 associates with microtubules and inhibits cell migration. Mol Cell Biol 29: 4177–4187, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Canfield AE, Hadfield KD, Rock CF, Wylie EC, Wilkinson FL: HtrA1: A novel regulator of physiological and pathological matrix mineralization? Biochem Soc Trans 35: 669–671, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Chien J, Staub J, Hu SI, Erickson-Johnson MR, Couch FJ, Smith DI, et al.: A candidate tumor suppressor HtrA1 is downregulated in ovarian cancer. Oncogene 23: 1636–1644, 2004 [DOI] [PubMed] [Google Scholar]
- 39.Mullany SA, Moslemi-Kebria M, Rattan R, Khurana A, Clayton A, Ota T, et al.: Expression and functional significance of HtrA1 loss in endometrial cancer. Clin Cancer Res 17: 427–436, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chien J, Aletti G, Baldi A, Catalano V, Muretto P, Keeney GL, et al.: Serine protease HtrA1 modulates chemotherapy-induced cytotoxicity. J Clin Invest 116: 1994–2004, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Deangelis MM, Ji F, Adams S, Morrison MA, Harring AJ, Sweeney MO, et al.: Alleles in the HtrA serine peptidase 1 gene alter the risk of neovascular age-related macular degeneration. Ophthalmology 115: 1209–1215.e7, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.An E, Sen S, Park SK, Gordish-Dressman H, Hathout Y: Identification of novel substrates for the serine protease HTRA1 in the human RPE secretome. Invest Ophthalmol Vis Sci 51: 3379–3386, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grau S, Baldi A, Bussani R, Tian X, Stefanescu R, Przybylski M, et al.: Implications of the serine protease HtrA1 in amyloid precursor protein processing. Proc Natl Acad Sci U S A 102: 6021–6026, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsuchiya A, Yano M, Tocharus J, Kojima H, Fukumoto M, Kawaichi M, et al.: Expression of mouse HtrA1 serine protease in normal bone and cartilage and its upregulation in joint cartilage damaged by experimental arthritis. Bone 37: 323–336, 2005 [DOI] [PubMed] [Google Scholar]
- 45.Polur I, Lee PL, Servais JM, Xu L, Li Y: Role of HTRA1, a serine protease, in the progression of articular cartilage degeneration. Histol Histopathol 25: 599–608, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vierkotten S, Muether PS, Fauser S: Overexpression of HTRA1 leads to ultrastructural changes in the elastic layer of Bruch’s membrane via cleavage of extracellular matrix components. PLoS One 6: e22959, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hoy B, Lower M, Weydig C, Carra G, Tegtmeyer N, Geppert T, et al.: Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep 11: 798–804, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang L, Lim SL, Du HJ, Zhang M, Kozak I, Hannum G, et al.: High temperature requirement factor A1 (HTRA1) gene regulates angiogenesis through transforming growth factor-β family member growth differentiation factor 6. J Biol Chem 287: 1520–1526, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen HW, Zhang Z, Huber E, Chao CC, Wang H, Dasch GA, et al.: Identification of cross-reactive epitopes on the conserved 47-kilodalton antigen of Orientia tsutsugamushi and human serine protease. Infect Immun 77: 2311–2319, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Choi S, Jeong HJ, Hwang KJ, Gill B, Ju YR, Lee YS, et al.: A recombinant 47-kDa outer membrane protein induces an immune response against Orientia tsutsugamushi strain boryong. Am J Trop Med Hyg 97: 30–37, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hale LJ, Howden SE, Phipson B, Lonsdale A, Er PX, Ghobrial I, et al.: 3D organoid-derived human glomeruli for personalised podocyte disease modelling and drug screening. Nat Commun 9: 5167, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Byron A, Randles MJ, Humphries JD, Mironov A, Hamidi H, Harris S, et al.: Glomerular cell cross-talk influences composition and assembly of extracellular matrix. J Am Soc Nephrol 25: 953–966, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Merchant ML, Barati MT, Caster DJ, Hata JL, Hobeika L, Coventry S, et al.: Proteomic analysis identifies distinct glomerular extracellular matrix in collapsing focal segmental glomerulosclerosis. J Am Soc Nephrol 31: 1883–1904, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nyström J, Fierlbeck W, Granqvist A, Kulak SC, Ballermann BJ: A human glomerular SAGE transcriptome database. BMC Nephrol 10: 13, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Runyon ST, Zhang Y, Appleton BA, Sazinsky SL, Wu P, Pan B, et al.: Structural and functional analysis of the PDZ domains of human HtrA1 and HtrA3. Protein Sci 16: 2454–2471, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fresquet M, Jowitt TA, McKenzie EA, Ball MD, Randles MJ, Lennon R, et al.: PLA2R binds to the annexin A2-S100A10 complex in human podocytes. Sci Rep 7: 6876, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Globus O, Evron T, Caspi M, Siman-Tov R, Rosin-Arbesfeld R: High-temperature requirement A1 (Htra1) – a novel regulator of canonical Wnt signaling. Sci Rep 7: 17995, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 3
Supplemental Data
Supplemental Data