A Human Pleiotropic Multiorgan Condition Caused by Deficient Wnt Secretion (original) (raw)
. Author manuscript; available in PMC: 2022 Apr 19.
Published in final edited form as: N Engl J Med. 2021 Sep 30;385(14):1292–1301. doi: 10.1056/NEJMoa2033911
Abstract
BACKGROUND
Structural birth defects occur in approximately 3% of live births; most such defects lack defined genetic or environmental causes. Despite advances in surgical approaches, pharmacologic prevention remains largely out of reach.
METHODS
We queried worldwide databases of 20,248 families that included children with neurodevelopmental disorders and that were enriched for parental consanguinity. Approximately one third of affected children in these families presented with structural birth defects or microcephaly. We performed exome or genome sequencing of samples obtained from the children, their parents, or both to identify genes with biallelic pathogenic or likely pathogenic mutations present in more than one family. After identifying disease-causing variants, we generated two mouse models, each with a pathogenic variant “knocked in,” to study mechanisms and test candidate treatments. We administered a small-molecule Wnt agonist to pregnant animals and assessed their offspring.
RESULTS
We identified homozygous mutations in WLS, which encodes the Wnt ligand secretion mediator (also known as Wntless or WLS) in 10 affected persons from 5 unrelated families. (The Wnt ligand secretion mediator is essential for the secretion of all Wnt proteins.) Patients had multiorgan defects, including microcephaly and facial dysmorphism as well as foot syndactyly, renal agenesis, alopecia, iris coloboma, and heart defects. The mutations affected WLS protein stability and Wnt signaling. Knock-in mice showed tissue and cell vulnerability consistent with Wnt-signaling intensity and individual and collective functions of Wnts in embryogenesis. Administration of a pharmacologic Wnt agonist partially restored embryonic development.
CONCLUSIONS
Genetic variations affecting a central Wnt regulator caused syndromic structural birth defects. Results from mouse models suggest that what we have named Zaki syndrome is a potentially preventable disorder. (Funded by the National Institutes of Health and others.)
STRUCTURAL BIRTH DEFECTS ARE A CAUSE of substantial morbidity and mortality among infants, observed in 1 in 33 births and accounting for 20.6% of infant deaths.1,2 Such defects result from complex interactions among the genome, the epigenome, and the environment.3 Despite the widespread prevalence of structural birth defects, only a minority of such events can be traced to environmental or genetic causes; in most, causes remain unexplained.4 Structural birth defects commonly involve the neural tube, kidney, heart, facies, and limbs, with 20 to 30% of patients having multiorgan defects.5 Identifying causal factors is especially challenging for pleiotropic conditions that have disparate or partially penetrant phenotypes. Most structural birth defects occur in early embryogenesis, and treatment generally addresses symptoms or involves surgery rather than being preventive.
Wnt signaling controls both development and homeostasis, and its disruption is implicated in a wide range of human diseases.6–10 The human genome encodes 19 Wnt ligands, which have unique and yet partially overlapping expression and functions.7,11,12 Perhaps that explains why loss-of-function mutations in only 8 of the 19 Wnt ligands are associated with distinct conditions (Table S1 in Supplementary Appendix 1, available with the full text of this article at NEJM.org).6,8 Because previous studies focused on loss of individual Wnts, genetic redundancies remain mostly unexplored. Secretion of all Wnts requires the multitransmembrane protein receptor Wntless (WLS).13–15 WLS associates with Wnts in the endoplasmic reticulum after Wnt palmitoleation by an acyl transferase (encoded by the gene PORCN).11,16,17 WLS–Wnt complexes are then transported to the cell membrane, where Wnts (molecular “cargo”) are released into the extracellular milieu (Fig. S1A in Supplementary Appendix 1).18
From a total of 20,248 families, approximately one third of which included family members with structural birth defects, we evaluated 5 families with similar pleiotropic conditions using linkage analysis and exome sequencing. After identifying partial loss-of-function mutations in WLS, we studied knock-in mice to determine the effect of patient variants on Wnt signaling, tissue and cell vulnerability, and potential treatment feasibility.
METHODS
PATIENTS
Five siblings from Family 1 (Patients F1-IV:1 through F1-IV:5) were referred for intellectual disability and dysmorphic features (Fig. 1, and Fig. S1 in Supplementary Appendix 1). The parents are healthy first-degree cousins from the United Arab Emirates. The presence of similar phenotypes in all five siblings and the absence of unaffected siblings were considered unusual but not impossible for a recessive condition (Fig. 1A). All the children were born after uneventful pregnancies (Table 1 and Supplementary Appendix 2. Delayed global milestones, short stature, and microcephaly were evident by 2 years of age, with head circumference ranging from 2.6 to 5.9 SD below the mean and height ranging from 0.3 to 3.4 SD below the mean. Magnetic resonance imaging (MRI) of the brain was performed in one child (F1-IV:4) and showed cerebellar vermis hypoplasia and an enlarged fourth ventricle (Fig. 1B). Dysmorphisms, including cupped ears, sparse eyebrows, and partial toe syndactyly, were extremely similar among siblings. Findings on extensive biochemical assessments were normal.
Figure 1.
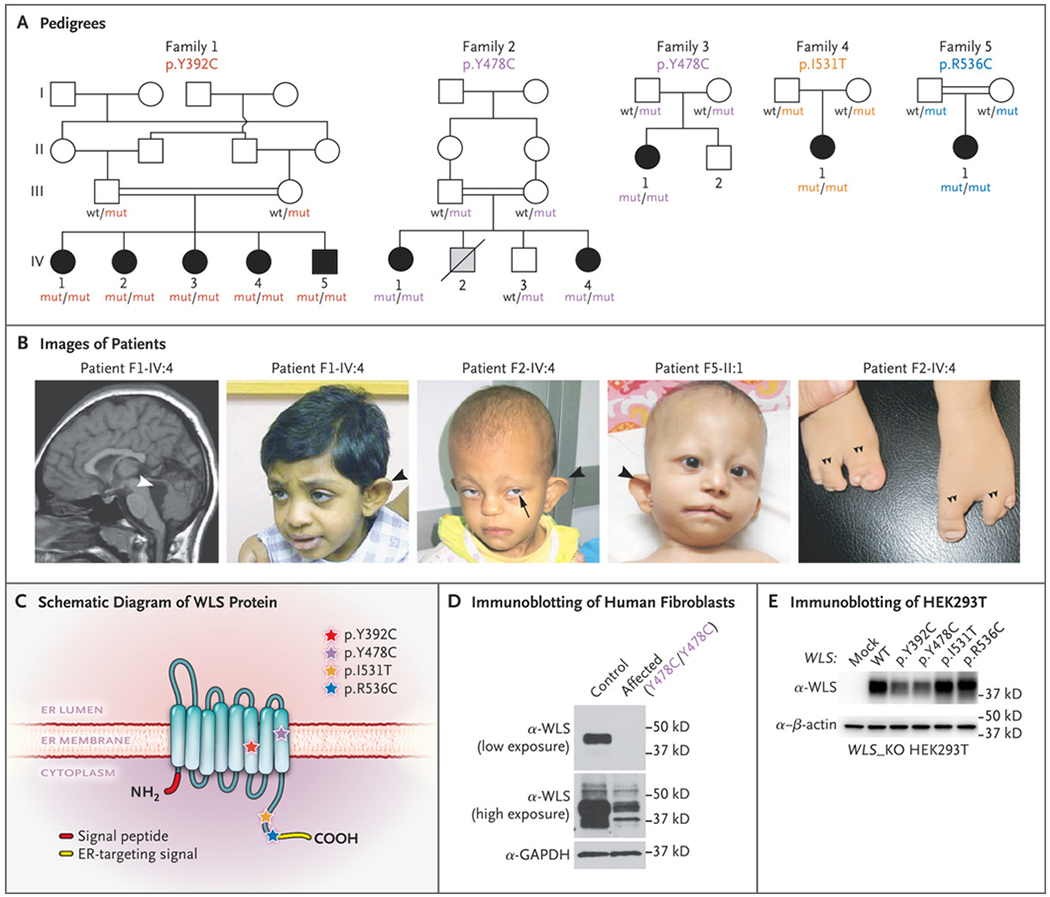
Homozygous Missense Mutations in WLS Leading to a Pleiotropic Recognizable Syndrome. Panel A shows pedigrees of five families with affected persons showing recessive inheritance. Homozygous WLS mutations and genotypes of available family members are indicated. Squares indicate male family members, circles female family members, double bars consanguinity, open symbols unaffected, black symbols affected, gray symbol likely affected, and diagonal slash deceased. The term wt denotes wild type, and mut mutant. Panel B shows images of affected persons. At left, sagittal T1-weighted magnetic resonance imaging of the brain in Patient F1-IV:4 shows reduced brain volume and an enlarged fourth ventricle (white arrowhead). In the three middle images, dysmorphic features include abnormal outer ears (black arrowhead), wide mouth, iris coloboma (Patient F2-IV:4, black arrow), and sparse scalp hair and eyebrows. At right, a “claw” malformation of the feet (double arrowheads) was observed in Patient F2-IV:4. We propose that this distinctive clinical presentation represents Zaki syndrome. Panel C shows the topologic features of WLS within the endoplasmic reticulum (ER) membrane. Patient mutations are represented with stars. Y392 and Y478 locate at the sixth and eighth transmembrane domain, respectively. I531 and R536 locate in the C-terminal tail, just before or within the ER-targeting signal, respectively. Panel D shows immunoblots of endogenous WLS protein in control and patient (F3-II:1) primary dermal fibroblasts. GAPDH denotes glyceraldehyde-3-phosphate dehydrogenase. Panel E shows immunoblots of overexpressed, untagged WLS in WLS knockout (KO) HEK293T cells. The p.Y392C and p.Y478C mutations lead to lower WLS protein levels than wild-type WLS.
Table 1.
Summary of Major Phenotypes in Patients.*
| Phenotypes in Patients with WLS Mutations | Wnt Mutations with Similar Defects |
|---|---|
| Facial dysmorphism† | WNT5A 19 |
| Iris coloboma and microcornea | NA |
| Cupped or abnormal ears† | NA |
| Sparse or no eyebrows | NA |
| Median pseudocleft lip | NA |
| Wide mouth† | WNT5A 19 |
| Microcephaly† | NA |
| Sparse scalp hair | NA |
| Foot syndactyly | WNT10B 20 |
| Renal agenesis and hydronephrosis | WNT4 21 |
| Heart defects | NA |
| Patent ductus arteriosus | NA |
| Patent foramen ovale | NA |
| Short stature | WNT5A 19 |
Two patients from Family 2 (F2-IV:1 and F2-IV:4) were referred for the same issues as those in Family 1. The parents are healthy first-degree cousins from Egypt. The affected children were born after uncomplicated pregnancies and showed delayed global milestones, short stature, and microcephaly (head circumference, 4.1 to 5.8 SD below the mean). Visual impairment, iris coloboma, cupped ears, and sparse hair were noted in both. Ectrodactyly (claw-shaped feet with polysyndactyly) was observed in the younger child (F2-IV:4) (Fig. 1B). MRI of the brain showed corpus callosum hypoplasia in both children and cerebellar vermis hypoplasia in the younger child.
Three patients (F3-II:1, F4-II:1, and F5-II:1) were single affected children from separate families of diverse ancestries (Fig. 1 and Supplementary Appendix 2). Bilateral iris coloboma and congenital diaphragmatic hernia resulting in surgery at birth were evident in the child from Family 3. The affected child in Family 4 had hydronephrosis. MRI of the brain showed corpus callosum hypoplasia, cerebellar vermis hypoplasia, and an enlarged fourth ventricle. Social and speech development were delayed in the affected children in Families 3 and 4 but normal in the child in Family 5. Microcephaly and short stature were seen in all affected patients for whom data were available; data on head circumference were not available for the affected child in Family 4.
OVERSIGHT AND LABORATORY STUDIES
Written informed consent was obtained with the use of consent forms from the University of California, San Diego, and the Genome Institute of Singapore. We performed genomewide singlenucleotide polymorphism genotyping to localize the causative locus in Family 1. We sequenced the exomes of two affected members from Families 1 and 2 and of the father, mother, and affected child from Families 3, 4, and 5. Details of the laboratory procedures, bioinformatics pipelines, and variant prioritization are provided in Supplementary Appendix 1.
We obtained primary dermal fibroblasts from the affected member of Family 3 and generated induced pluripotent stem cells (iPSCs) and then embryoid bodies (three-dimensional aggregates of iPSCs, differentiated through exposure to a defined cell medium); we compared these embryoid bodies with embryoid bodies derived from two unrelated healthy persons. We generated two independent knock-in mouse lines, one with the mutation observed in Family 1 and one with the mutation observed in Families 2 and 3, and bred each line for homozygosity of the relevant mutation. The mouse line carrying the p.Y478C allele was intercrossed with the TCF/Lef:H2B-GFP Wnt reporter line to permit assay of Wnt signaling. Full details regarding the generation of iPSCs, embryoid bodies, and transgenic mice as well as Wnt agonist treatment are provided in Supplementary Appendix 1.
STATISTICAL ANALYSIS
Continuous variables are presented as means with standard deviations. Comparisons were performed with parametric tests for normally distributed data or nonparametric tests when this criterion was not met. For multiple comparisons, adjusted P values were calculated after Bonferroni or šídák correction. Details of the statistical methods used for individual assays are provided in Supplementary Appendix 1.
RESULTS
IdeNTIfICATION Of WLS VARIANTS
Linkage analysis in Family 1, in which there were five affected siblings, revealed a single locus on chromosome 1 (Fig. S1B in Supplementary Appendix 1). Filtering of the exome sequences identified a homozygous p.Y392C missense variant in WLS (ClinVar accession number, SUB9531807) (Fig. 1 and Supplementary Appendixes 2 and 3), which segregated with the phenotypes and was located in the linkage locus. This WLS variant altered a residue conserved in all vertebrates (Fig. S1C and S1D in Supplementary Appendix 1) and was absent in the Genome Aggregation Database of healthy controls.22
In the four other families with similar phenotypes, we identified homozygous WLS mutations likely to impair function (Fig. 1, Fig. S1 in Supplementary Appendix 1, and Supplementary Appendixes 2 and 3). In Families 2 and 3, exome sequencing identified an identical WLS p.Y478C variant (ClinVar accession number, SUB9531807). Families 4 and 5 had independent missense mutations — p.I531T and p.R536C, respectively (ClinVar accession number, SUB9531807). Homozygous variants fell within regions of homozygosity in Families 3 and 4, which suggests distant consanguinity (Supplementary Appendix 4). The implicated variants altered conserved residues either in the transmembrane (p.Y392C and p.Y478C) region of WLS or adjacent to or within the C-terminal endoplasmic reticulum–targeting signal motif (Fig. 1C, and Fig. S2A in Supplementary Appendix 1), which met American College of Medical Genetics and Genomics criteria for moderate pathogenicity.23
To evaluate the WLS protein, we analyzed cultured primary dermal fibroblasts by means of Western blot testing and found that levels of the protein were more than 90% lower in patient cells (F3-II:1) than in the cells from an unrelated healthy control (Fig. 1D). Ectopic WLS expression from constructs harboring distinct patient mutations in _WLS_-deleted HEK293T cells showed protein levels were reduced by half for the two transmembrane mutations but not the two C-terminal mutations (Fig. 1E, and Fig. S2B, S2C, and S2D in Supplementary Appendix 1). To confirm protein reduction, we generated two homozygous knock-in mouse lines (p.Y392C and p. Y478C). Cultures of mouse embryonic fibroblasts showed that levels of WLS protein were reduced by approximately 40% in both lines (Fig. S2E and S2F in Supplementary Appendix 1).
EFFECT OF MUTATIONS ON WNT
We next assessed the effect of patient mutations on the secretion of WNT3A and WNT5A in WLS knockout HEK293T cells. Although intracellular Wnt levels were similar across cells, regardless of whether they harbored a WLS mutation, levels of secreted Wnts were reduced by 60 to 80% in cells carrying a mutation (Fig. 2A and 2B, and Fig. S3A and S3B in Supplementary Appendix 1), findings that were confirmed in both patient fibroblasts and mouse embryonic fibroblasts (Fig. S3C through S3H in Supplementary Appendix 1).
Figure 2.
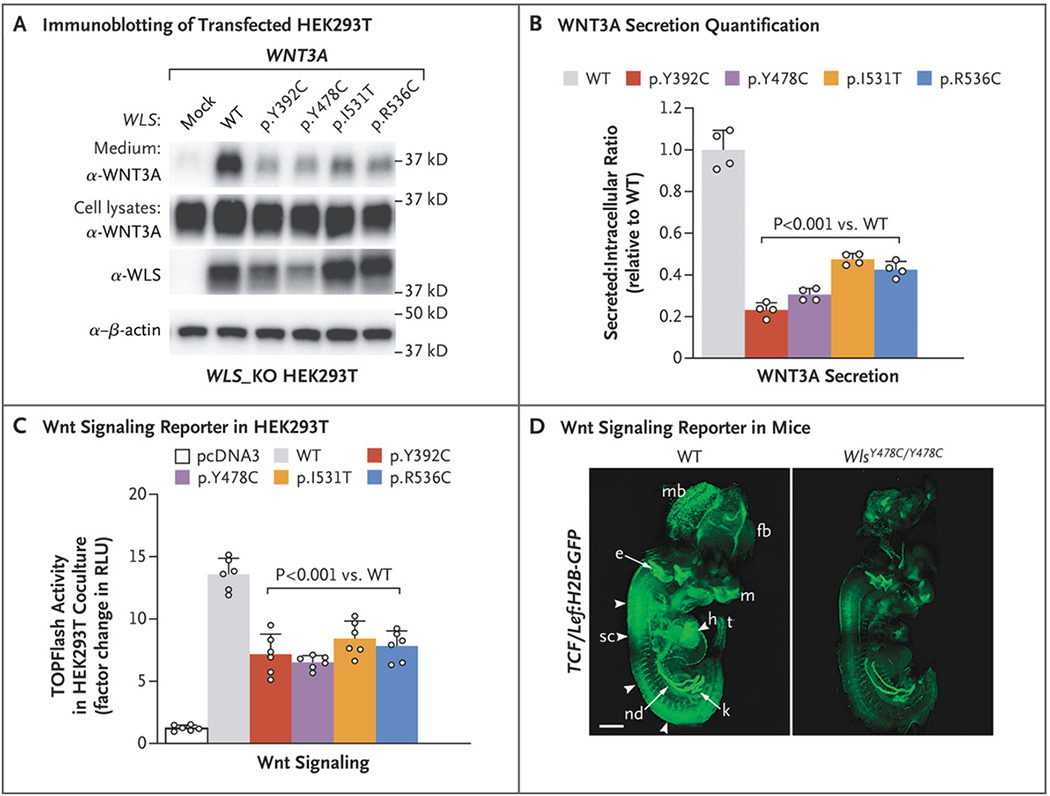
Effect of WLS Mutations on Wnt Secretion and Signaling. Panel A shows Western blot analysis of WNT3A in culture medium and lysates of WLS knockout (WLS_KO) HEK293T cells transfected with WNT3A and pcDNA3 or WLS. Panel B shows the ratio of immunoblots of secreted WNT3A to intracellular WNT3A, normalized to wild-type (WT) WLS. Panel C shows a Super TOPFlash assay from WLS_KO HEK293T cells transfected with Wnt3a and WT or mutant WLS plasmids cocultured with control HEK293T cells transfected with Super TOPFlash and Renilla reporter plasmids. RLU denotes relative light unit. The values in Panels B and C are means calculated by one-way analysis of variance; T bars indicate standard deviations. Panel D shows whole TCF/Lef:H2B-GFP transgenic mouse embryos at embryonic day 12.5 imaged with a light-sheet microscope, revealing globally lower Wnt signaling in WlsY478C/Y478C embryos than in controls. Arrowheads indicate the spinal cord. The term e denotes ear, fb forebrain, h heart, k kidney, m mouth, mb midbrain, nd nephric duct, sc spinal cord, and t tail. The scale bar indicates 1 mm.
We next assessed the ability of these cells to stimulate Wnt signaling in adjacent cells by coculturing them with cells expressing the TOPFlash Wnt reporter.24 We observed a 50 to 60% reduction in the luciferase signal in these cells when cocultured with cells expressing mutant WLS, which indicates decreased Wnt signaling (Fig. 2C).
The effects of WLS p.Y478C on Wnt signaling in vivo were assessed by crossing the Wls p.Y478C mouse line with the TCF/Lef:H2B-GFP reporter line,25 which resulted in progeny in which green fluorescent protein intensity is a surrogate for Wnt activity. In homozygous mutants, we observed globally reduced Wnt reporter activity across the body, most notably in facial structures, the heart, the spinal region, the tail bud, the metanephros, and neural tissues (Fig. 2D, and Fig. S3I and S3J in Supplementary Appendix 1) — many of the same tissues affected in patients.
STRUCTURAL DEFECTS IN WLS KNOCK-IN MICE
Heterozygous Wls p.Y392C and p.Y478C knock-in mice were healthy and viable. In contrast, homozygous mutant mice showed perinatal lethality and had phenotypes similar to those of our patients or those previously ascribed to Wnt functions (Table S1 in Supplementary Appendix 1). Nearly all mutants in both lines showed defects in caudal neural tube closure, with severely reduced or absent tail buds and reduced numbers of caudal vertebrae (Fig. 3A, and Fig. S4A through S4E in Supplementary Appendix 1). Cystic medullary hydronephrosis was observed in both lines (Fig. 3B), a phenotype seen in the affected child in Family 4. We also found reduced numbers of forelimb and hindlimb digits in mutant mice (Fig. 3C, and Fig. S4F and S4G in Supplementary Appendix 1), similar to the foot syndactyly observed in patients. Mutants in both lines also showed a notably reduced brain size, with a severely defective hippocampal dentate gyrus, a decreased number of cortical neurons, and “cobblestone”-like penetration of neurons through the disrupted pial basement membrane (Fig. 3D and 3E, and Figs. S5 and S6 in Supplementary Appendix 1). Together, the findings in knock-in mice recapitulated the major defects in patients.
Figure 3.
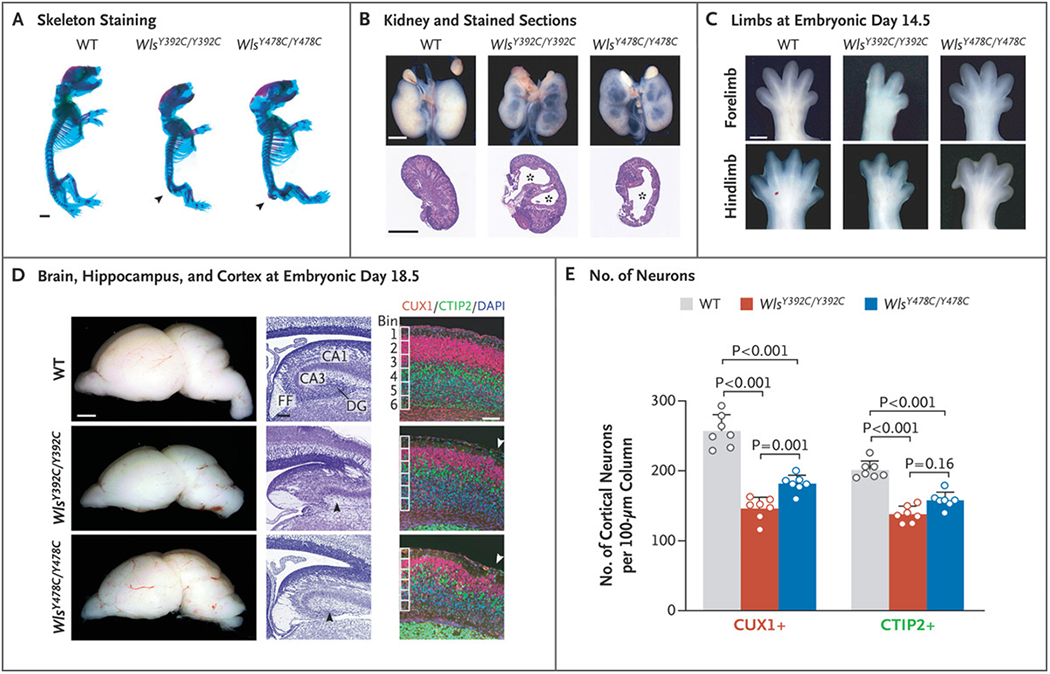
A Spectrum of Developmental Defects in Wls Knock-in Mouse Embryos. Panel A shows mouse skeletons at embryonic day 18.5 stained with Alcian blue (cartilage) and Alizarin red (bone). Wls knock-in embryos had defective caudal vertebrae (arrowhead). Panel B shows whole kidneys (top) and sections stained with hematoxylin and eosin (bottom) from WT, WlsY392C/Y392C, and WlsY478C/Y478C embryos with cysts (asterisk) at embryonic day 18.5. Panel C shows representative images of forelimbs and hindlimbs with reduced digit number in WlsY392C/Y392C embryos at embryonic day 14.5. Panel D shows images of whole brains and sections of such brains at embryonic day 18.5. The left column shows whole brains. In the middle column, Nissl-stained sections of brains show the regions of the hippocampus. Mutants showed defective dentate gyrus (DG) (black arrowhead). CA1 denotes cornu ammonis subfield 1, CA3 cornu ammonis subfield 3, and FF fimbria—fornix. In the right column, magnification of mouse cortex stained for CUX1 and CTIP2 shows defective cortical lamination and ectopias (arrowhead) in Wls mutants. DAPI denotes 4’,6-diamidino-2-phenylindole. Panel E shows quantification of cortical CUX1+ and CTIP2+ neurons in WT and Wls mutant embryos at embryonic day 18.5. Data are for 4 mice per genotype. Values are means calculated with a two-tailed, unpaired t-test, and T bars indicate standard deviations. Scale bars indicate 50 μm in the middle and right columns in Panel D and 1 mm elsewhere.
Next, we investigated cellular defects underlying the microcephaly phenotype, present in all our patients but not previously linked to defective Wnt signaling in humans. Using single-nuclei RNA sequencing and immunohistochemical assays, we observed a striking reduction of neurogenesis in the brains of embryonic mutant mice. This was associated with a defective cell cycle of intermediate neural progenitors, which stalled at the S and G2/M phases (Figs. S7, S8, and S9 in Supplementary Appendix 1), findings consistent with the role of Wnt signaling in cell cycle and proliferation.10,26
PHARMACOLOGIC RESCUE
Given the globally reduced Wnt signaling in mutant mouse embryos, we assessed the ability of the Wnt agonist CHIR99021 to prevent WLS related disease.27,28 iPSC-derived embryoid bodies from patient cells have emerged as a clinically relevant model of the pregastrulation embryonic stage.29 We generated embryoid bodies from the affected child in Family 3 and compared them with those derived from two healthy unrelated controls. Although mutant iPSCs were healthy, embryoid bodies were reduced in size by 50% (Fig. 4A and 4B). The addition of CHIR99021 to the culture led to restoration of the size of the embryoid bodies, which suggests at least partial restoration of critical Wnt-mediated effects.
Figure 4.
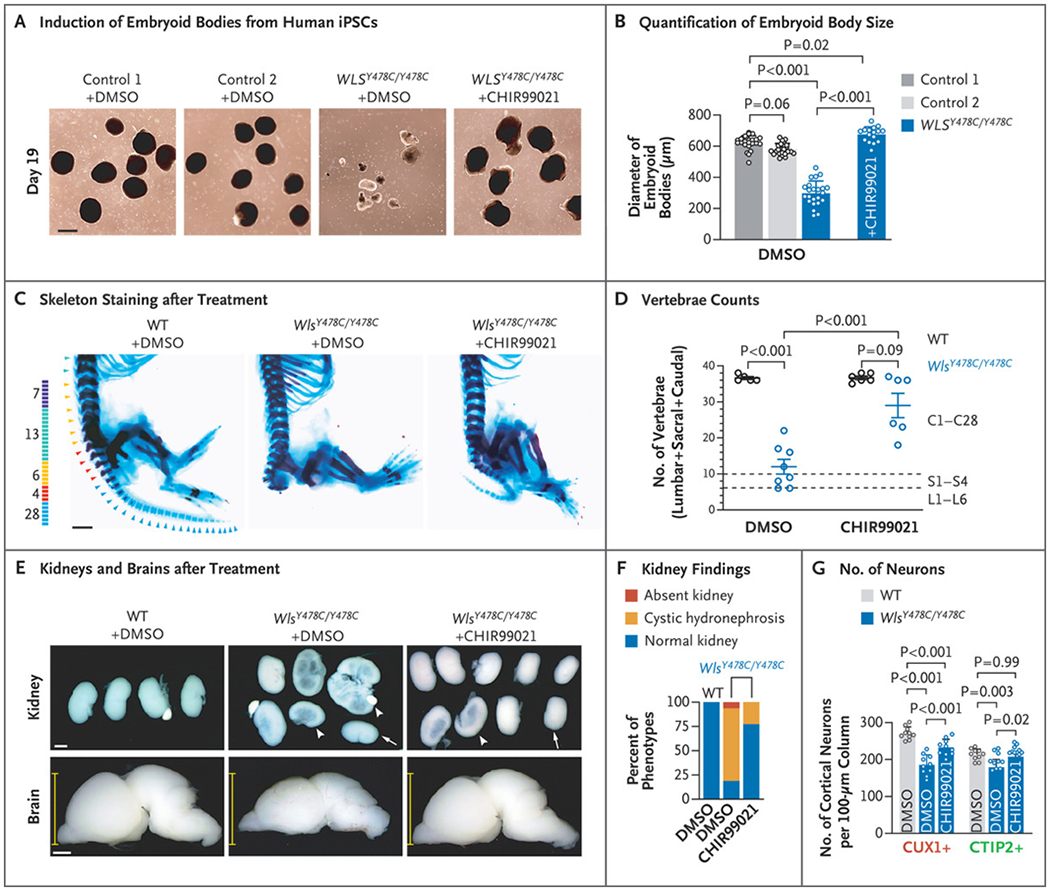
Partial Rescue of Defects in Wls Knock-in Embryos by Wnt Agonist Treatment. Panel A shows the induced embryoid bodies from human induced pluripotent stem cells (iPSCs) at day 19. Panel B shows the quantification of the diameters of embryoid bodies derived from control and patient iPSCs, treated with dimethylsulfoxide (DMSO) or 2.5 μM of CHIR99021. Data are for 23 embryoid bodies from control and patient iPSCs treated with DMSO and 16 embryoid bodies from patient iPSCs treated with CHIR99021. Panel C shows the skeletons of embryos at embryonic day 18.5 stained with Alcian blue (cartilage) and Alizarin red (bone). Panel D shows the quantification of vertebrae, indicating rescued sacral and caudal vertebrae in Wls mutants by CHIR99021. Mice generally have 7 cervical, 13 thoracic, 6 lumbar, 4 sacral, and 28 caudal vertebrae. All embryos had a normal number of cervical and thoracic vertebrae, and the sums of lumbar, sacral, and caudal vertebrae were calculated. Data are for 5 WT mice treated with DMSO, 8 Wls mutants treated with DMSO, 6 WT mice treated with CHIR99021, and 6 Wls mutants treated with CHIR99021. Panel E shows images of whole kidneys (top) and brains (bottom) in embryos at embryonic day 18.5 from dams treated with DMSO or CHIR99021. Arrows indicate small kidneys, and arrowheads cystic kidneys with hydronephrosis. Each yellow line indicates a caliper for dorsoventral axis thickness. Panel F shows the percent of mutants with kidney phenotypes. Data are for 10 WT mice treated with DMSO, 16 mutants treated with DMSO, and 22 Wls mutants treated with CHIR99021. Panel G shows quantification of CUX1+ and CTIP2+ cortical neurons. A total of 4 WT mice and 5 Wls mutants were included for each group, with the left and right sides of the brain measured separately. In Panels B, D, and G, values are means calculated by one-way analysis of variance, and T bars or I bars indicate standard deviations. Scale bars indicate 500 μm in Panel A and 1 mm in Panels C and E.
This observation prompted us to assess the effect of CHIR99021 on mouse embryogenesis. We administered intraperitoneal CHIR99021 (in dimethylsulfoxide) twice daily to dams of each Wls knock-in line, starting at embryonic day 4.5 and continuing until sacrifice at embryonic day 12.5. We observed near-complete rescue of tail growth in the p.Y478C line (Fig. S10A and S10B in Supplementary Appendix 1). In contrast, the p.Y392C line showed only partial rescue, with approximately 25% of treated embryos showing a diminutive tail as compared with absent tail bud in all dimethylsulfoxide-treated mutants. This finding is consistent with a more severe vertebral defect in the p.Y392C line.
To assess potential rescue at later stages, we extended the administration of CHIR99021 and control dimethylsulfoxide only to embryonic day 18.5, focusing on the p.Y478C mouse line. We found that both tail length and the number of caudal vertebrae were increased in the mice receiving CHIR99021 (Fig. 4C and 4D, and Fig. S10C and S10D in Supplementary Appendix 1). Moreover, the incidence of hydronephrosis was reduced from 75.0% to 22.7% (Fig. 4E and 4F). Partial rescue of brain size was also observed, with substantial increases in the dorsoventral axis width (by 16.4%), cortical thickness (by 14.3%), and the number of cortical neurons (by 25.4% for CUX1+ and by 16.0% for CTIP2+) and hippocampal dentate gyrus cells (by 150.3%) (Fig. 4E and 4G, and Fig. S10E through S10I in Supplementary Appendix 1), each approaching sizes or cell numbers seen in controls. Altogether, these results suggest that treatment of the mutant mice with the Wnt agonist CHIR99021 can partially rescue pleiotropic developmental defects.
DISCUSSION
In this study, we explored genetic causes and mechanisms leading to structural birth defects, focusing on a pleiotropic multiorgan condition. We were aided by Family 1, which showed consistency in phenotype among siblings and helped to reveal the genetic cause. Through a worldwide recruitment of patients sharing damaging WLS mutations, we uncovered overlapping but not identical features among 10 patients, which suggests a distinct and recognizable disorder that we term Zaki syndrome (Fig. S11 in Supplementary Appendix 1). The clinical differences among families may represent genetic modifiers or environmental factors.
Our genetic findings provide evidence of impaired WLS function as a cause for this new condition. Although studies of patients’ cells confirmed defects in Wnt signaling and Wnt-dependent defects in embryoid bodies, introducing these mutations into mice confirmed these findings. We showed global reduction of Wnt signaling in knock-in mice on the basis of reporter activity. Mutant intermediate neural progenitors had a lengthened cell cycle as a likely mechanism, leading to the microcephaly seen in patients. A previous study showed that mice with conditionally deleted Wls in the epidermis had a hair-growth defect,30 similar to that observed in the affected children. Moreover, mutations in PORCN, which is required for Wnt lipid modification, cause ectrodactyly,31,32 a phenotype that we observed in the younger affected child in Family 2.
Some of the features of this new condition implicate specific Wnts. For instance, Wnt7b controls morphogenesis of the renal medulla,33 and Wnt3a and Wnt5a control somite and tail-bud morphogenesis,34 both affected in Wls knock-in mice. Similarly, WNT10B is mutated in patients with split-foot malformations,20 a phenotype seen in one of our patients. Rather than affecting a single Wnt, WLS is required for secretion of all Wnts, so the range of phenotypes probably reflects global or partial depletion across all early Wnt signaling.
Our findings suggest that pharmacologic intervention could be considered for some structural birth defects during gestation. In this study, the availability of CHIR99021 as a potent and cell-permeable Wnt agonist provided the opportunity to assess the degree to which the disease was a result of general depletion of Wnt signaling. CHIR99021 promotes the accumulation of beta-catenin, thus enhancing Wnt-mediated transcriptional signaling.35 CHIR99021 rescued diminutive size in patient iPSC-derived embryoid bodies, consistent with the roles of Wnt in maintaining cell “stemness.”36 Administration to pregnant dams also achieved substantial rescue of the brain, vertebrae, tail, and kidney phenotypes in the mutant embryos, without notable toxic effects in the pregnant dams. The safety of proposed therapies should be examined carefully in humans to rule out potential risks to mother and child.
We did not evaluate postnatal use of CHIR99021, but we do not rule out potential benefit, because we suspect that most future diagnoses of this condition will be made after rather than before birth. Differentiating outright structural defects, such as digit malformations, from defects such as microcephaly, short stature, and kidney defects, for which there is the potential to intervene postnatally, will be important. Development of preclinical models for the treatment of amenable structural defects could represent future studies within pediatrics.
Supplementary Material
Supplement 1
Supplement 2
Supplement 3
Supplement 4
Acknowledgments
Supported by the National Institutes of Health (R01NS048453, R01NS09800, and P01-HD104436), the Simons Foundation Autism Research Initiative (51486313), the Yale Center for Mendelian Genomics (U54HG006504), the Broad Institute Center for Mendelian Genomics (UM1HG008900), the Center for Inherited Disease Research (N01 268201700006I-0-26800029), the Howard Hughes Medical Institute, the Rady Children’s Institute for Genomic Medicine, a National Medical Research Council Open Fund–Young Individual Research Grant (OF-YIRG, #OFYIRG18May-0053; and OF-IRG, #OFIRG20Nov-0057), the National Research Foundation (Singapore), the Branco Weiss Foundation (Switzerland), the European Molecular Biology Organization Young Investigator program, and a use-inspired basic research grant from the Agency for Science, Technology, and Research in Singapore.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
We thank the staff of the University of California, San Diego, Mouse Transgenic Core, Neuroscience Microscopy Core, and IGM (Institute for Genomic Medicine) Genomics Core for their help; Lu Qiao for homozygosity analysis; and the families for their participation.
Appendix
The authors’ affiliations are as follows:the Rady Children’s Institute for Genomic Medicine, San Diego (G.C., C.C., Z.L., L.W., T.M., N.J., X.Y., J.M.-V., V.S., P.A., N.L., J.G.G.), and the University of California, San Diego, La Jolla (G.C., C.C., Z.L., L.W., T.M., N.J., X.Y., J.M.-V., V.S., P.A., N.L., K.W., J.G.G.) — both in California; Xuanwu Hospital, Capital Medical University, Beijing (G.C.); the Genome Institute of Singapore (E.S.-R., M.K., V.W., B.R.) and the Institute of Molecular and Cellular Biology (B.R.), Agency for Science, Technology, and Research, and the Program in Cancer and Stem Cell Biology, Duke–NUS (National University of Singapore) Medical School (J.Y., D.M.V.) — all in Singapore; the Medical Genetics Department, Koç University School of Medicine, Istanbul, Turkey (B.R.); the Department of Pediatrics, Duke University, Durham, NC (D.M.V.); the Department of Physiology and Cellular Biophysics, Columbia University Irving Medical Center (R.N., F.M.), and the Departments of Pediatrics and Medicine, Columbia University (R.H., W.K.C.) — both in New York; Centogene, Rostock, Germany (R.M., A.M.B.-A.); DNA Laboratório e Genética Médica, Salvador, Brazil (M.B.P.T., P.M.L.P.); the Institute of Medical Genetics and Genomics, Sir Ganga Ram Hospital, New Delhi, India (R.D.P.); Johns Hopkins Aramco Healthcare, Dhahran, Saudi Arabia (N.A.-S.); and the Clinical Genetics Department, National Research Center, Cairo (M.S.Z.).
Footnotes
The authors’ affiliations are listed in the Appendix.
Contributor Information
Guoliang Chai, Rady Children’s Institute for Genomic Medicine, San Diego, California; University of California, San Diego, La Jolla, California; Xuanwu Hospital, Capital Medical University, Beijing, Singapore
Emmanuelle Szenker-Ravi, Genome Institute of Singapore, Singapore
Changuk Chung, Rady Children’s Institute for Genomic Medicine, San Diego, California; University of California, San Diego, La Jolla, California
Zhen Li, Rady Children’s Institute for Genomic Medicine, San Diego, California; University of California, San Diego, La Jolla, California
Lu Wang, Rady Children’s Institute for Genomic Medicine, San Diego, California; University of California, San Diego, La Jolla, California
Muznah Khatoo, Genome Institute of Singapore, Singapore
Trevor Marshall, Rady Children’s Institute for Genomic Medicine, San Diego, California; University of California, San Diego, La Jolla, California
Nan Jiang, Rady Children’s Institute for Genomic Medicine, San Diego, California; University of California, San Diego, La Jolla, California
Xiaoxu Yang, Rady Children’s Institute for Genomic Medicine, San Diego, California; University of California, San Diego, La Jolla, California
Jennifer McEvoy-Venneri, Rady Children’s Institute for Genomic Medicine, San Diego, California; University of California, San Diego, La Jolla, California
Valentina Stanley, Rady Children’s Institute for Genomic Medicine, San Diego, California; University of California, San Diego, La Jolla, California
Paula Anzenberg, Rady Children’s Institute for Genomic Medicine, San Diego, California; University of California, San Diego, La Jolla, California
Nhi Lang, Rady Children’s Institute for Genomic Medicine, San Diego, California; University of California, San Diego, La Jolla, California, California
Vanessa Wazny, Genome Institute of Singapore, Singapore
Jia Yu, Agency for Science, Technology, and Research, and the Program in Cancer and Stem Cell Biology, Duke—NUS (National University of Singapore) Medical School, Singapore
David M. Virshup, Agency for Science, Technology, and Research, and the Program in Cancer and Stem Cell Biology, Duke-NUS (National University of Singapore) Medical School, Singapore; Department of Pediatrics, Duke University, Durham, NC
Rie Nygaard, Department of Physiology and Cellular Biophysics, Columbia University Irving Medical Center, New York
Filippo Mancia, Department of Physiology and Cellular Biophysics, Columbia University Irving Medical Center, New York
Rijad Merdzanic, Centogene, Rostock, Germany
Maria B.P. Toralles, DNA Laboratório e Genética Médica, Salvador, Brazil
Paula M.L. Pitanga, DNA Laboratorio e Genética Médica, Salvador, Brazil
Ratna D. Puri, Institute of Medical Genetics and Genomics, Sir Ganga Ram Hospital, New Delhi, India
Rebecca Hernan, Departments of Pediatrics and Medicine, Columbia University, New York
Wendy K. Chung, Departments of Pediatrics and Medicine, Columbia University, New York
Aida M. Bertoli-Avella, Centogene, Rostock, Germany
Nouriya Al-Sannaa, Johns Hopkins Aramco Healthcare, Dhahran, Saudi Arabia
Maha S. Zaki, Clinical Genetics Department, National Research Center, Cairo
Karl Willert, University of California, San Diego, La Jolla, California
Bruno Reversade, Genome Institute of Singapore, Singapore; Institute of Molecular and Cellular Biology, Singapore; Medical Genetics Department, Koç University School of Medicine, Istanbul, Turkey
Joseph G. Gleeson, Rady Children’s Institute for Genomic Medicine, San Diego, California; University of California, San Diego, La Jolla, California
REFERENCES
- 1.Mai CT, Isenburg JL, Canfield MA, et al. National population-based estimates for major birth defects, 2010-2014. Birth Defects Res 2019;111:1420–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almli LM, Ely DM, Ailes EC, et al. Infant mortality attributable to birth defects — United States, 2003-2017. MMWR Morb Mortal Wkly Rep 2020;69:25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hobbs CA, Chowdhury S, Cleves MA, et al. Genetic epidemiology and nonsyndromic structural birth defects: from candidate genes to epigenetics. JAMA Pediatr 2014;168:371–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu X-Y, Phung MT, Shaw CA, et al. Genomic imbalances in neonates with birth defects: high detection rates by using chromosomal microarray analysis. Pediatrics 2008;122:1310–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Agopian AJ, Evans JA, Lupo PJ. Analytic methods for evaluating patterns of multiple congenital anomalies in birth defect registries. Birth Defects Res 2018; 110:5–11. [DOI] [PubMed] [Google Scholar]
- 6.Nusse R, Clevers H. Wnt/_β_-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017;169:985–99. [DOI] [PubMed] [Google Scholar]
- 7.Ng LF, Kaur P, Bunnag N, et al. WNT signaling in disease. Cells 2019;8:826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clevers H, Nusse R. Wnt/_β_-catenin signaling and disease. Cell 2012;149:1192–205. [DOI] [PubMed] [Google Scholar]
- 9.Zhan T, Rindtorff N, Boutros M. Wnt signaling in cancer. Oncogene 2017;36:1461–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrison-Uy SJ, Pleasure SJ. Wnt signaling and forebrain development. Cold Spring Harb Perspect Biol 2012;4(7):a008094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willert K, Brown JD, Danenberg E, et al. Wnt proteins are lipid-modified and can act as stem cell growth factors. Nature 2003;423:448–52. [DOI] [PubMed] [Google Scholar]
- 12.Steinhart Z, Angers S. Wnt signaling in development and tissue homeostasis. Development 2018;145:dev146589. [DOI] [PubMed] [Google Scholar]
- 13.Das S, Yu S, Sakamori R, Stypulkowski E, Gao N. Wntless in Wnt secretion: molecular, cellular and genetic aspects. Front Biol (Beijing) 2012;7:587–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bänziger C, Soldini D, Schütt C, Zipperlen P, Hausmann G, Basler K. Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell 2006;125:509–22. [DOI] [PubMed] [Google Scholar]
- 15.Bartscherer K, Pelte N, Ingelfinger D, Boutros M. Secretion of Wnt ligands requires Evi, a conserved transmembrane protein. Cell 2006;125:523–33. [DOI] [PubMed] [Google Scholar]
- 16.Kadowaki T, Wilder E, Klingensmith J, Zachary K, Perrimon N. The segment polarity gene porcupine encodes a putative multitransmembrane protein involved in Wingless processing. Genes Dev 1996; 10:3116–28. [DOI] [PubMed] [Google Scholar]
- 17.Nygaard R, Yu J, Kim J, et al. Structural basis of WLS/Evi-mediated Wnt transport and secretion. Cell 2021;184(1):194.e14–206.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yu J, Chia J, Canning CA, Jones CM, Bard FA, Virshup DM. WLS retrograde transport to the endoplasmic reticulum during Wnt secretion. Dev Cell 2014;29:277–91. [DOI] [PubMed] [Google Scholar]
- 19.Person AD, Beiraghi S, Sieben CM, et al. WNT5A mutations in patients with autosomal dominant Robinow syndrome. Dev Dyn 2010;239:327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ugur SA, Tolun A. Homozygous WNT10b mutation and complex inheritance in split-hand/foot malformation. Hum Mol Genet 2008;17:2644–53. [DOI] [PubMed] [Google Scholar]
- 21.Biason-Lauber A, Konrad D, Navratil F, Schoenle EJA. A WNT4 mutation associated with müllerian-duct regression and virilization in a 46,XX woman. N Engl J Med 2004;351:792–8. [DOI] [PubMed] [Google Scholar]
- 22.Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581:434–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Molenaar M, van de Wetering M, Oosterwegel M, et al. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell 1996;86:391–9. [DOI] [PubMed] [Google Scholar]
- 25.Ferrer-Vaquer A, Piliszek A, Tian G, Aho RJ, Dufort D, Hadjantonakis A-K. A sensitive and bright single-cell resolution live imaging reporter of Wnt/_β_-catenin signaling in the mouse. BMC Dev Biol 2010;10:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Niehrs C, Acebron SP. Mitotic and mitogenic Wnt signalling. EMBO J 2012;31:2705–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Belinson H, Nakatani J, Babineau BA, et al. Prenatal _β_-catenin/Brn2/Tbr2 transcriptional cascade regulates adult social and stereotypic behaviors. Mol Psychiatry 2016;21:1417–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.An WF, Germain AR, Bishop JA, et al. Discovery of potent and highly selective inhibitors of GSK3b. Probe reports from the NIH Molecular Libraries program. Bethesda, MD: National Center for Biotechnology Information, 2010. [PubMed] [Google Scholar]
- 29.Nickolls AR, Lee MM, Zukosky K, Mallon BS, Bönnemann CG. Human embryoid bodies as a 3D tissue model of the extracellular matrix and _α_-dystroglycan-opathies. Dis Model Mech 2020;13(6):dmm042986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Augustin I, Gross J, Baumann D, et al. Loss of epidermal Evi/Wls results in a phenotype resembling psoriasiform dermatitis. J Exp Med 2013;210:1761–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grzeschik KH, Bornholdt D, Oeffner F, et al. Deficiency of PORCN, a regulator of Wnt signaling, is associated with focal dermal hypoplasia. Nat Genet 2007;39:833–5. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Sutton VR, Peraza-Llanes JO, et al. Mutations in X-linked PORCN, a putative regulator of Wnt signaling, cause focal dermal hypoplasia. Nat Genet 2007;39:836–8. [DOI] [PubMed] [Google Scholar]
- 33.Yu J, Carroll TJ, Rajagopal J, Kobayashi A, Ren Q, McMahon APA. A Wnt7b-dependent pathway regulates the orientation of epithelial cell division and establishes the cortico-medullary axis of the mammalian kidney. Development 2009;136:161–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takada S, Stark KL, Shea MJ, Vassileva G, McMahon JA, McMahon AP. Wnt-3a regulates somite and tailbud formation in the mouse embryo. Genes Dev 1994;8:174–89. [DOI] [PubMed] [Google Scholar]
- 35.Wu D, Pan W. GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci 2010;35:161–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu Y, Ai Z, Yao K, et al. CHIR99021 promotes self-renewal of mouse embryonic stem cells by modulation of proteinencoding gene and long intergenic noncoding RNA expression. Exp Cell Res 2013;319:2684–99. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement 1
Supplement 2
Supplement 3
Supplement 4