Single cell analysis of PANoptosome cell death complexes through an expansion microscopy method (original) (raw)
. Author manuscript; available in PMC: 2023 Sep 28.
Published in final edited form as: Cell Mol Life Sci. 2022 Sep 28;79(10):531. doi: 10.1007/s00018-022-04564-z
Abstract
In response to infection or sterile insults, inflammatory programmed cell death is an essential component of the innate immune response to remove infected or damaged cells. PANoptosis is a unique innate immune cell death pathway regulated by multifaceted macromolecular complexes called PANoptosomes, which integrate components from other cell death pathways. Growing evidence shows that PANoptosis can be triggered in many physiological conditions, including viral and bacterial infections, cytokine storms, and cancers. However, PANoptosomes at the single cell level have not yet been fully characterized. Initial investigations have suggested that key pyroptotic, apoptotic, and necroptotic molecules including the inflammasome adaptor protein ASC, apoptotic caspase-8 (CASP8), and necroptotic RIPK3 are conserved components of PANoptosomes. Here, we optimized an immunofluorescence procedure to probe the highly dynamic multiprotein PANoptosome complexes across various innate immune cell death-inducing conditions. We first identified and validated antibodies to stain endogenous mouse ASC, CASP8, and RIPK3, without residual staining in the respective knockout cells. We then assessed the formation of PANoptosomes across innate immune cell death-inducing conditions by monitoring the colocalization of ASC with CASP8 and/or RIPK3. Finally, we established an expansion microscopy procedure using these validated antibodies to image the organization of ASC, CASP8, and RIPK3 within the PANoptosome. This optimized protocol, which can be easily adapted to study other multiprotein complexes and other cell death triggers, provides confirmation of PANoptosome assembly in individual cells and forms the foundation for a deeper molecular understanding of the PANoptosome complex and PANoptosis to facilitate therapeutic targeting.
Keywords: ASC, inflammasome, PANoptosis, PANoptosome, pyroptosis, apoptosis, necroptosis, caspase-8, RIPK3, ASC, caspase-1, innate immunity, infection, inflammation, cell death, influenza, ZBP1, NLRP3, AIM2, HSV-1, KPT-330, IFN, method, protocol
1. INTRODUCTION
The innate immune system is the host’s first line of defense against cellular insults. Pathogen- and damage-associated molecular patterns (PAMPs and DAMPs, respectively) are sensed by innate immune sensors to activate downstream signaling pathways, including the induction of innate immune cell death. While innate immune cell death is important for host defense, aberrant activation can result in inflammation and pathology. Therefore, it is important to understand the molecular mechanisms of innate immune cell death to identify strategies to mitigate disease.
Several innate immune cell death pathways have been described to date; one such pathway is PANoptosis. PANoptosis is a unique innate immune cell death pathway regulated by multifaceted PANoptosome complexes, which integrate components from other cell death pathways such as pyroptosis, apoptosis, and/or necroptosis (1–16). The totality of biological effects observed in PANoptosis cannot be individually accounted for by these other cell death pathways alone. To date, two prototypical PANoptosomes have been biochemically identified: the Z-DNA binding protein 1 (ZBP1)-PANoptosome, which forms in response to influenza A virus (IAV) infection, and the absent in melanoma 2 (AIM2)-PANoptosome, which forms in response to herpes simplex virus 1 (HSV-1) or Francisella novicida infections (2, 6, 12, 14, 17). Additionally, key upstream regulators IRF1, TAK1, and RIPK1 have also been characterized for their role in PANoptosis initiation under various conditions (3, 7, 10, 11, 13). While the upstream sensor is different between PANoptosomes, many of the core proteins are shared, including molecules canonically associated with the pyroptosis, apoptosis, and necroptosis.
Pyroptosis is a lytic innate immune cell death pathway molecularly characterized by caspase-1 (CASP1) activation (18). This pathway is associated with the formation of a multiprotein complex called the inflammasome, which is comprised of a sensor, the adaptor protein ASC, and an inflammatory caspase (19). Inflammasomes can act as integral components of PANoptosomes, and ASC has been identified as a shared molecular feature (6, 12, 14, 17). Alternatively, apoptosis causes morphological membrane blebbing and cell shrinkage (20) and is molecularly controlled by either the formation of an apoptosome containing an adaptor APAF-1 and CASP9 or by the formation of a death-inducing signaling complex (DISC) containing a death receptor and CASP8; both activate the executioner caspases, CASP3 and CASP7 (21–28). Necroptosis is another lytic innate immune cell death pathway initiated by the formation of a necrosome which contains RIPK1 and RIPK3. Necroptosis is executed by RIPK3-mediated activation of pore-forming MLKL downstream of CASP8 inhibition (29–35).
To study cell death complexes and their regulation, microscopy can be an informative tool to assess complex formation and dynamics. Fluorescence microscopy has visualized inflammasomes (36) and necrosomes (37) as well as the colocalization of CASP8 and ASC in single cells (14, 38–40). Biochemical characterizations of ZBP1- and AIM2-PANoptosomes suggest that they both contain ASC as well as CASP8 and RIPK3 (12, 14). However, much remains unknown about the dynamics of PANoptosome assembly, and there is currently no established protocol to investigate the formation of PANoptosome complexes in single cells.
Here, we present an immunofluorescence protocol to assess the dynamics of PANoptosome formation at the single cell level. Given the central roles of ASC, CASP8, and RIPK3 in the biochemical analysis of PANoptosis, we focused on these key molecular pillars of innate immune cell death; their colocalization in a single cell would be indicative of the formation of a PANoptosome. We first screened and validated antibodies and developed a method to stain and visualize the highly dynamic protein complexes induced by IAV (41), HSV-1 (14), the nuclear export inhibitor KPT-330 and interferon-β (IFN-β) (15), and the canonical NLRP3 inflammasome trigger LPS + ATP. Finally, we developed a protocol for expansion microscopy to visualize the molecular organization of PANoptosomes. Overall, these protocols allow for a streamlined process to visualize multiprotein complexes, such as PANoptosomes and inflammasomes, at the single cell level.
2. MATERIALS AND METHODS
2.1. Mice
Wildtype (WT) C57/Bl6, _Casp8_–/–_Ripk3_–/– (42), _Ripk3_–/– (43), _Pycard_–/– (44), _Zbp1_–/– (45), _Aim2_–/– (46), and _Nlrp3_–/– (47) mice have been described previously. All mice were bred at the Animal Resources Center at St. Jude Children’s Research Hospital and maintained under specific pathogen-free conditions and extensively backcrossed to the C57/Bl6 background. Age- and sex-matched 6- to 9-week-old male and female mice were used in this study. Mice were maintained with a 12 h light/dark cycle and were fed standard chow. Studies were conducted under protocols approved by the St. Jude Children’s Research Hospital committee on the Use and Care of Animals.
2.2. Antibodies
Rabbit anti-mASC (AL177, Adipogen), mouse anti-mASC (2EI-7, Millipore), mouse anti-mRIPK3 (B-2, Santacruz), rat anti-mRIPK3 (8G7, Millipore), rat anti-mCASP8 (3B10, Adipogen), rabbit anti-mCASP8 (#4927, CST), goat anti-mouse IgG AlexaPlus-488 (A32723, ThermoFisher), goat anti-rat IgG AlexaPlus-555 (A48263, ThermoFisher), and goat anti-rabbit IgG AlexaPlus-647 (A32733, ThermoFisher) antibodies were used. Antibodies were all used at 1:200 dilution in blocking buffer (10% goat serum in PBS-T).
2.3. Bone marrow-derived macrophage (BMDM) culture and stimulations
Primary BMDMs were grown for 6 days in IMDM (12440–53, Gibco) supplemented with 1% non-essential amino acids (11140–050, Gibco), 10% FBS, 30% L929 conditioned media, and 1% penicillin and streptomycin (15070–63, Gibco). Cells were seeded in 24-well plates containing Poly-L-Lysine coated coverslips (72292–04, Electron Microscopy Science) at a density of 0.5 × 105 cells/well on day 6 and incubated at 37°C, 5% CO2 overnight. Stimulations were performed on day 7.
2.4. In vitro infection
The influenza A virus (A/Puerto Rico/8/34, H1N1 [PR8]) was generated by reverse genetics as previously described (48). The seed virus (P2) was propagated by inoculating into the allantoic cavity of 9- to 11-day old embryonated chicken eggs for two days. The allantoic fluid was collected and filtered through a 0.22 μm filter then aliquoted and placed in −80°C as IAV stocks. Viral titer was measured by a plaque assay in MDCK cells. For infecting BMDMs, IAV stock was diluted with DMEM medium (Sigma, D6171) to MOI = 20, and virus was added onto cells after washing with PBS. After 2 h adsorption, 10% FBS was added into the medium for 10 h before fixing cells.
Human herpes simplex virus 1 (HSV-1) infection was performed similarly to the IAV infection with MOI = 10 for 12 h. Generation of stock HSV-1 (HF strain, ATCC VR-260) was made by infecting Vero cells with MOI = 0.1 for 2 days. Culture supernatant with floating dead cells was collected and underwent two cycles of freeze-thawing before being centrifuged at 3000 × g for 10 min. The supernatant was filtered with a 0.22 μm filter and aliquoted as viral stock in −80°C; the viral titer was measured with a plaque assay in Vero cells.
For stimulation with IFN-β and KPT-330 in combination, cells were washed one time with PBS and stimulated with 50 ng/mL of IFN-β (12400–1, PBL Assay) and 5 mM of KPT-330 (S7252, Selleckchem) together in DMEM medium (11995–065, Gibco) supplemented with 10% FBS and 1% penicillin and streptomycin (15070–63, Gibco) for 24 h.
For canonical inflammasome activation, BMDMs were primed for 4 h with 100 ng/mL ultrapure LPS from Escherichia coli (0111:B4) (tlrl-3pelps, Invivogen) then stimulated for 10 or 30 min (western blotting) or 10 min (microscopy) with 5 mM ATP (10127531001, Roche), unless otherwise indicated. Real-time cell death assays were performed with the IncuCyte S3. After BMDMs were stimulated, 100 nM Sytox Green (Thermo Fisher Scientific) was added. Images were acquired hourly, while at 37°C and 5 % CO2, and image analysis occurred through the use of the software package supplied with the IncuCyte, which counts the number of Sytox Green-positive BMDM nuclei in each frame (49).
After stimulation, cells were fixed by either 4% PFA (15710, Electron Microscopy Sciences; diluted with PBS) at room temperature or 100% methanol (230–4, Honeywell) at −20°C for 10 min. After fixing, cells were washed with PBS three times, then stored in PBS at 4°C for future staining.
2.5. Immunofluorescence staining and expansion microscopy
Prior to staining, PFA-fixed cells were permeabilized by 0.5% Triton-X100 in PBS at room temperature for 10 min and washed three times with PBS. Cells were blocked by 10% normal goat serum (#5425, CST) at room temperature for 1 h and stained by primary antibodies overnight at 4°C. Cells were then washed by PBS-T (PBS with 0.05% Tween-20) three times for 10 min each and stained by secondary antibodies for 2 h at room temperature in the dark. After staining, cells were washed by PBS-T three times for 10 min and kept in PBS. For observation by conventional microscopy, the cells were stained with 1 μg/mL DAPI (4’,6-Diamidino-2-Phenylindole, 40043, Biotium) for 5 min and mounted using mounting medium (P36961, ProLong™ Diamond Antifade Mountant, Invitrogen) on glass slides, then imaged using the Marianas spinning disc confocal system (Intelligent Imaging Innovations) comprised of an inverted AxioObserver Z.1 microscope (Carl Zeiss), CSU-W1 with SoRa (Yokogawa), Prime95B sCMOS camera (Photometrics), 405 nm, 473 nm, 561 nm, 647 nm solid state laser lines (Coherent), and a 1.4NA 100 × oil objective. Images were acquired using Slidebook software with the laser power set as current and the exposure time at 100 ms for each channel. To ensure consistency, the same microscopy parameters were used in all independent experiments. The quantification was done with images obtained with a 60 × objective and counted manually.
For expansion microscopy, the coverslips with stained cells were processed according to published protocols (50). Briefly, the coverslips with stained cells were further incubated with 0.1 mg/mL AcX (A20770, ThermoFisher) in PBS overnight at room temperature and then washed twice with PBS. Cold gelation solution (86 mg/mL sodium acrylate [S03880, Pfaltz & Bauer], 25 mg/mL acrylamide [A9099–25G, Sigma], 1.5 mg/mL N,N’-Methylenebis-acrylamide [146072–100G, Sigma], 2 M NaCl [BP358–10, Fisher Scientific], 100 μg/mL TEMED [T7024, Sigma] and 100 μg/mL APS [BP179–100, Fisher Scientific] in PBS) was added into the ice-cold plate wells containing the coverslips. The coverslips were lifted, inverted, and placed on two coverslip spacers. Forty microliters of gelation solution was added into the space under the cell coverslips to allow gelation in a humid chamber at 37°C for 1 h. The coverslip containing cells together with gel above was then placed into 10 mL digestion buffer (0.5% Triton X-100, 1 mM EDTA [15575020, Thermo Fisher], 50 mM Tris-HCl [BP152–5, Fisher Scientific, stock 1 M pH 8.0], 1 M NaCl, and 8 U/mL proteinase K [P8107S, NEB]) with orbital shaking at 60 rpm overnight at room temperature. The gel was expanded by three changes of water with 20 min incubation intervals with orbital shaking at 60 rpm before imaging with an inverted A1R HD25 point scanning confocal (Nikon) equipped with 488 nm and 561 nm laser lines and a 1.3 NA 40 × oil objective. Images were acquired using NIS Elements software with speed setting as 2 ×, and Nyquist sampling was automatically performed using the software to adjust scan area and pinhole opening. The images were processed in the same software with consistent parameter settings across the WT and knockout controls.
2.6. Immunoblot analysis
For probing caspase activation, cells along with culture supernatants were lysed in caspase lysis buffer (25 mM DTT, 10% NP-40, 1 × protease inhibitors, and 1 × phosphatase inhibitors) and 4 × Laemmli loading buffer (containing SDS and 2-mercaptoethanol). For assessment of signaling activation, supernatants were removed, and the cells were washed once with Dulbecco’s PBS (DPBS) followed by lysis in RIPA buffer and Laemmli loading buffer. Proteins were separated by gel electrophoresis through 8–12% polyacrylamide gels and transferred onto PVDF membranes (IPVH00010, Millipore). Membranes were incubated with 5% skim milk for 1 h at room temperature to block non-specific binding. Then, membranes were incubated at 4°C overnight with the following primary antibodies: caspase-1 (AG-20B-0044-C100, AdipoGen), anti–caspase-8 (CST, #4927), anti-cleaved caspase-8 (CST, #8592), caspase-3 (9662, CST), cleaved caspase-3 (9661, CST), caspase-7 (9492, CST), cleaved caspase-7 (9491, CST), pMLKL (37333, CST), tMLKL (AP14242B, Abgent), GSDMD (ab209845, abcam), GSDME (ab19859, abcam), and β-actin (Proteintech, 66009–1-IG). The next day, the membranes were incubated with the appropriate horseradish peroxidase (HRP)–conjugated anti-mouse (315-035-047, Jackson ImmunoResearch Laboratories) or anti-rabbit (111-035-047, Jackson ImmunoResearch Laboratories) secondary antibodies. Images were acquired using Immobilon® Forte Western HRP Substrate (WBLUF0500, Millipore) on an Amersham Imager.
3. RESULTS
3.1. Staining optimization
PANoptosis has been broadly implicated across the disease spectrum, including in infection, inflammatory diseases, and cancer (2, 3, 5, 7, 8, 11–16, 51–57). While ZBP1- and AIM2-PANoptosomes, along with upstream regulatory molecules such as IRF1, TAK1, and RIPK1, have been characterized as key regulators of PANoptosis under specific conditions (3, 7, 10–14), much remains unknown about the dynamics of PANoptosome assembly. Furthermore, there is a critical need to implement strategies to study dynamic multiprotein complexes like PANoptosomes at the single cell level to understand cell death activation and regulation. To address this gap, we sought to develop an immunofluorescence assay to analyze PANoptosome assembly in single cells. We chose to target conserved PANoptosome proteins that have been identified across triggers (6, 10–12, 14, 15), namely ASC, CASP8, and RIPK3. In addition to being conserved across PANoptosis triggers, this unique combination of proteins allows differentiation between PANoptosomes and other cell death complexes such as necrosomes, where ASC is not expected to colocalize, and apoptosomes, where ASC and RIPK3 are not expected to colocalize.
We first tested commercially available antibodies to stain mouse ASC, CASP8, and RIPK3 in bone marrow-derived macrophages (BMDMs) fixed with paraformaldehyde (PFA) or methanol (MeOH), two of the most popular fixation methods for immunofluorescence. We also selected antibodies from a variety of host species with an aim to identify a combination of three different hosts to develop a primary antibody panel that could be appropriately stained with unique secondary antibodies. Rabbit polyclonal ASC antibody (Adipogen, AL177) and mouse monoclonal ASC antibody (Millipore, 2EI-7) both stained WT cells but not Pycard (the gene encoding ASC)-deficient cells after PFA fixation (Fig. 1A). MeOH fixation resulted in staining of WT cells as well. However, this procedure also resulted in the spontaneous formation of speck-like structures and nuclear staining (Fig. 1A); this spontaneous speck formation is not expected in unstimulated cells. In addition, while a rat antibody to mouse RIPK3 (Millipore, 8G7) showed specific staining irrespective of the fixation method (Fig. 1B), a mouse clone against RIPK3 (Santa Cruz, B-2) stained RIPK3 specifically only in MeOH-fixed cells; this antibody produced non-specific staining in _Ripk3_–/– cells following PFA fixation. For CASP8 staining, two different antibodies against mouse CASP8 (CST, 4927; Adipogen, 3B10) resulted in specific staining in WT cells following either PFA or MeOH fixation. Since deleting caspase-8 causes embryonic lethality (42), _Casp8_–/–_Ripk3_–/– cells were used as the staining control for CASP8. Fluorescence intensity remained low or undetectable in _Casp8_–/–_Ripk3_–/– cells for both antibodies (Fig. 1C), suggesting that both fixation methods are suitable for CASP8 staining. Overall, these data show that to visualize PANoptosome complexes in single cells, the optimal methodology includes the use of PFA fixation with the following antibodies: ASC (Millipore 2EI-7, mouse), RIPK3 (Millipore 8G7, rat), and CASP8 (CST 4927, rabbit). Further, these data underscore the importance of validating antibodies and experimenting to find the appropriate fixation method to obtain reliable immunofluorescence images.
Figure 1. Optimizing conditions for staining mouse ASC, CASP8, and RIPK3.
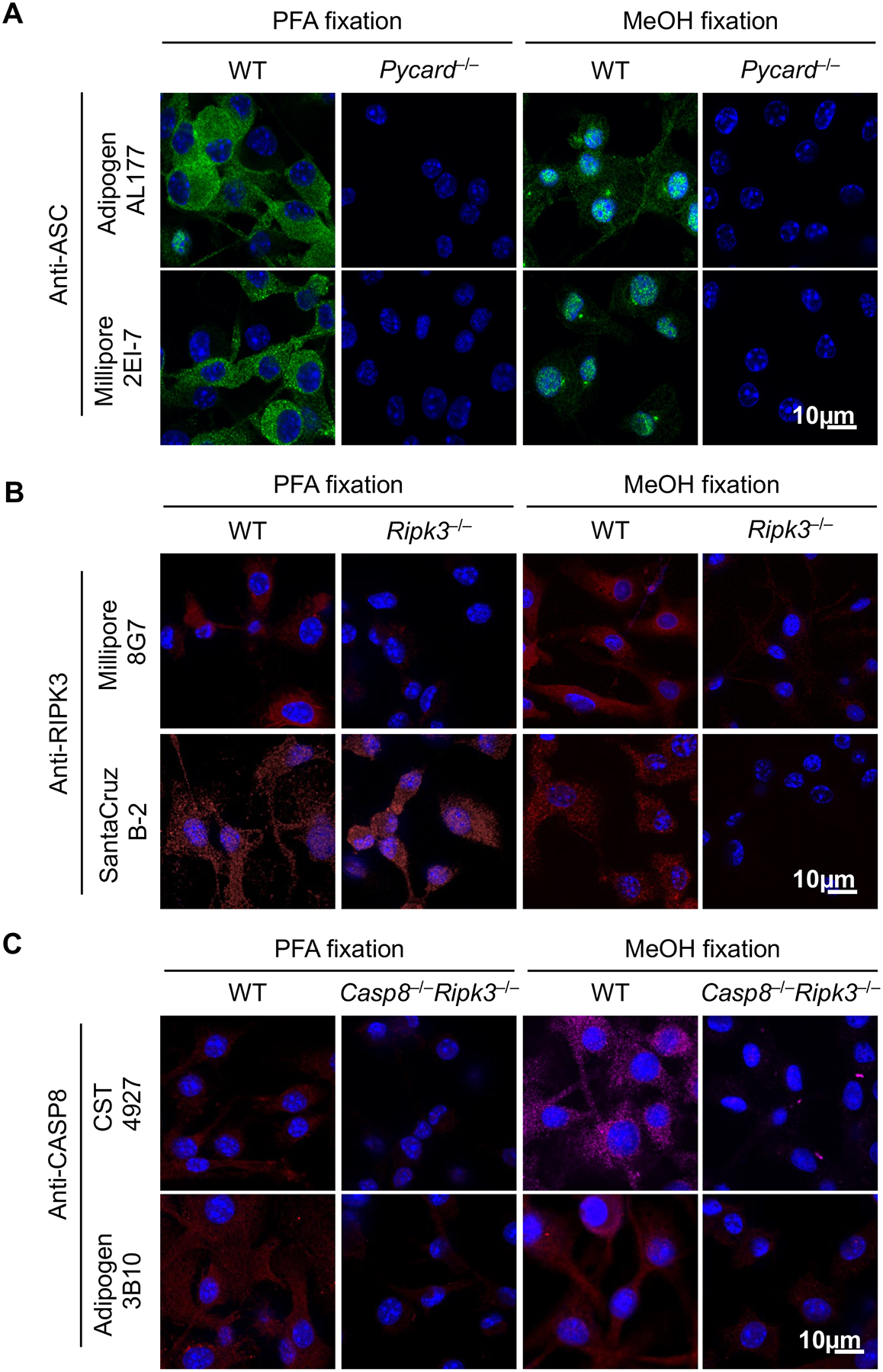
A-C) Bone marrow-derived macrophages (BMDMs) from the indicated genotypes were fixed by 4% paraformaldehyde (PFA) or methanol (MeOH) and stained with indicated primary antibodies to evaluate staining specificity. Images are representative of at least three independent experiments.
3.2. ASC colocalizes with CASP8 and/or RIPK3 under PANoptosis-inducing conditions
Upon stimulations that induce innate immune cell death, ASC assembles into an ASC speck, a condensate that can be seen through microscopy (58). We examined the colocalization of ASC specks with CASP8 and RIPK3 in conditions where PANoptosis has been observed biochemically. IAV was the first trigger formally identified to induce PANoptosis in BMDMs through the formation of a ZBP1-PANoptosome; this complex has been molecularly characterized to include ZBP1, RIPK3, RIPK1, caspase-8, caspase-6, ASC, and NLRP3 (6, 12). We stained ASC, CASP8, and RIPK3 in mock treated (Fig. 2A) or IAV infected BMDMs (Fig. 2B–F). Although ASC specks were frequently observed in WT cells after IAV infection (Fig. 2B–E, Supplemental Figure 1A), we did not observe any ASC specks in ZBP1-deficient cells (Fig. 2F), confirming the critical role of ZBP1 in mediating IAV-induced PANoptosis (2).
Figure 2. ASC specks are heterogenous in response to IAV infection.
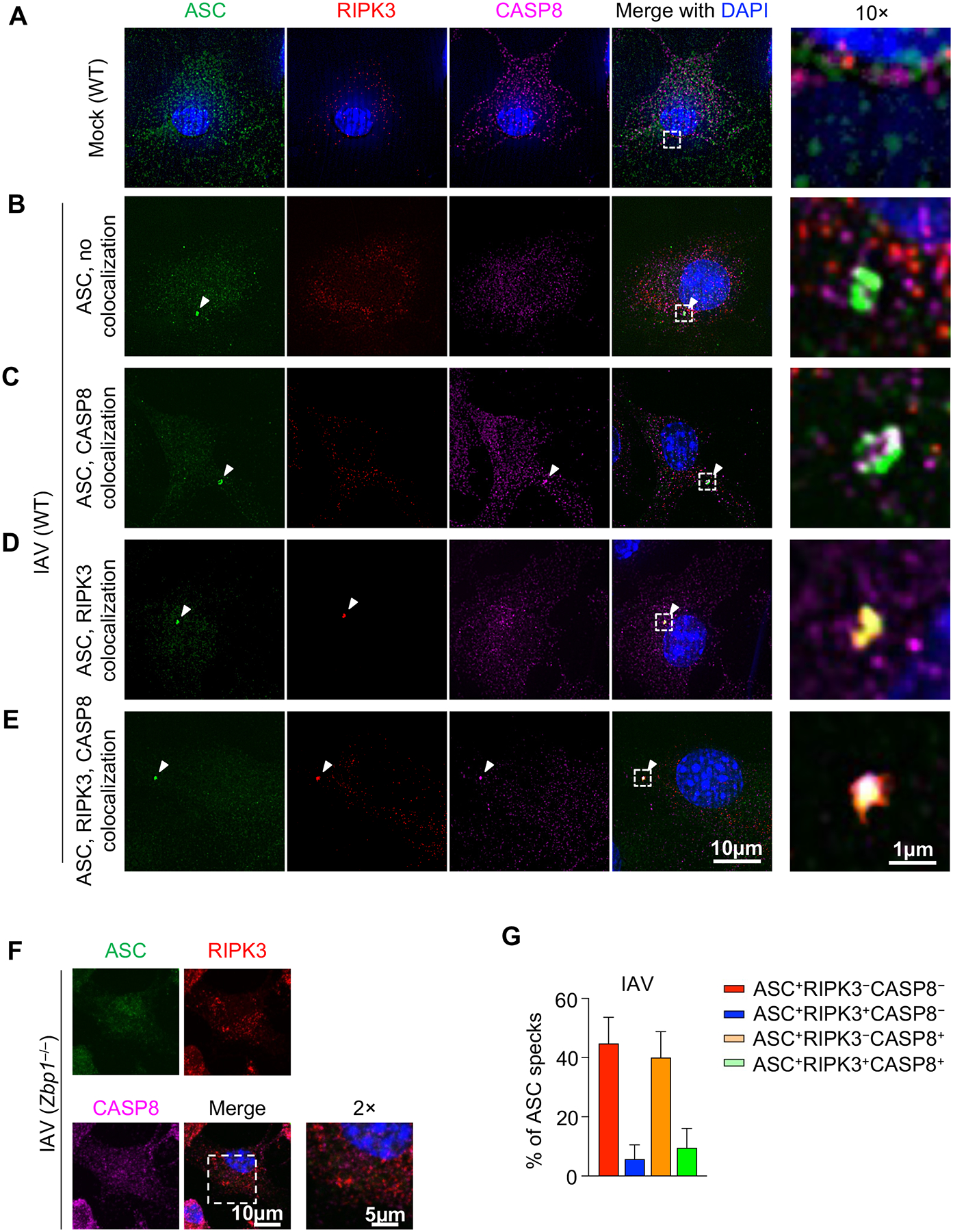
A–E) Wild type (WT) bone marrow-derived macrophages (BMDMs) were mock treated (A) or infected with influenza A virus (IAV) (B–E) and stained for ASC, RIPK3, and CASP8. Different compositions of ASC specks are shown. F) ASC, RIPK3, and CASP8 staining of IAV-infected Zbp1 –/– BMDMs. G) Compositional analysis of ASC specks (n ≥ 100) in IAV-infected WT BMDMs at 9 h and 12 h post-infection (h.p.i.). Mean ± standard error is shown. Images are representative of at least three independent experiments.
The ASC specks induced by IAV were not homogenous, and their molecular composition differed within the same population of cells (Fig. 2B–E). We observed four different types of IAV-induced ASC specks, those without CASP8 or RIPK3 colocalization (Fig. 2B), those colocalizing with only CASP8 (Fig. 2C), those colocalizing with only RIPK3 (Fig. 2D), and those colocalizing with both CASP8 and RIPK3 (Fig. 2E); this triple colocalization is consistent with previous findings (41). We observed that the composition of ASC specks changed at different stages of infection (Fig. 2G), suggesting that the heterogeneity of ASC specks likely reflects the dynamic nature of the PANoptosome as a multiprotein complex. To further confirm the relevance of this colocalization as an indicator of PANoptosis, we also assessed staining in other PANoptosis-inducing conditions. We observed colocalization of ASC, CASP8, and RIPK3 in HSV-1 stimulated WT, but not AIM2-deficient, BMDMs (Supplemental Figure 1B), confirming previous findings (14). Similarly, upon treatment with the ZBP1-PANoptosome–inducing combination of a nuclear export inhibitor, KPT-330, and IFN-β (15), we observed colocalization of ASC, CASP8, and RIPK3 in a ZBP1-dependent manner (Supplemental Figure 1B), consistent with the phenotype of PANoptosis (15). The composition of the ASC specks was also heterogeneous in response to KPT-330 and IFN-β (Supplemental Figure 1C,D), as was seen with IAV infection (Fig. 2G, Supplemental Figure 1A).
3.3. ASC colocalizes with CASP8 and/or RIPK3 under inflammasome-inducing conditions
Colocalization of ASC with CASP8 and RIPK3 was seen in single cells under PANoptotic conditions (Fig. 2B–E; Supplemental Figure 1). Furthermore, inflammasomes can act as integral components of PANoptosomes in response to multiple stimuli (6, 12, 14, 17). Therefore, we also extended our analysis to examine the localization of ASC, CASP8, and RIPK3 in response to a canonical NLRP3 inflammasome trigger. Using the same staining protocol, ASC, CASP8, and RIPK3 were stained in cells treated with LPS priming and ATP stimulation to induce NLRP3 inflammasome activation (Fig. 3A). It is well-established that CASP8 can be a component of ASC specks during inflammasome activation (14, 38–40), and we observed the same (Fig. 3A). However, we also found that some ASC specks colocalized with both CASP8 and RIPK3, and that some ASC specks colocalized with RIPK3 alone (Fig. 3A).
Figure 3. Inflammasome stimulation by LPS + ATP induces formation of ASC specks containing RIPK3 and CASP8.
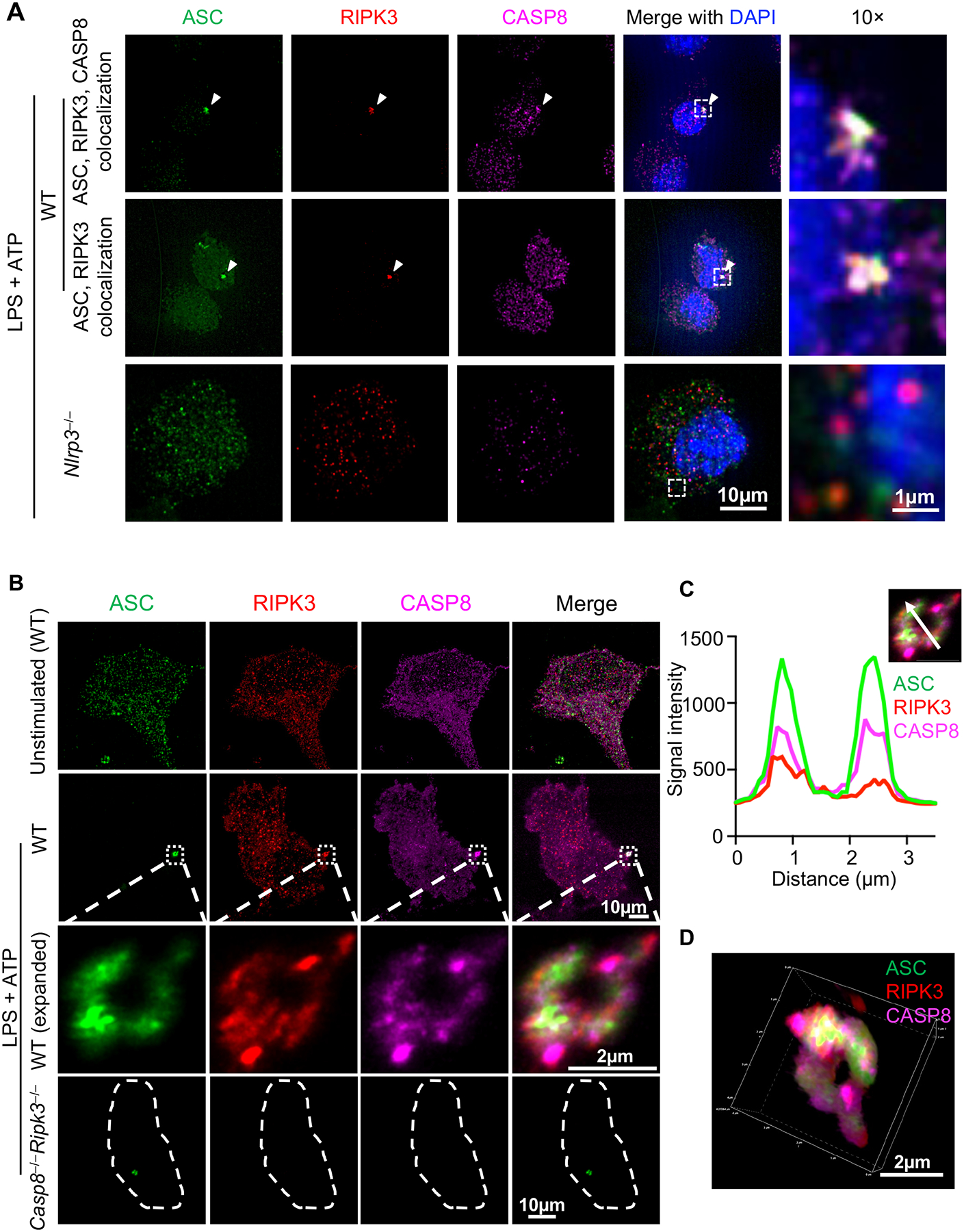
A) Representative images of ASC specks from bone marrow-derived macrophages (BMDMs). Cells of the indicated genotypes were primed with 100 ng/μl LPS for 4 h, then stimulated with 5 mM ATP (i.e., LPS + ATP) for 10 min. B) Expansion microscopy views of a RIPK3/CASP8-containing ring-shaped ASC speck induced by LPS + ATP. C) Fluorescence intensity of ASC, RIPK3, and CASP8 in the ring section along the white arrow. X axis indicates the distance along the white arrow traveling from the base of the arrow to the arrowhead. D) 3D view of the ASC speck in (B). Images are representative of at least three independent experiments.
To further investigate the RIPK3-ASC colocalization, we established a PANoptosome expansion microscopy technique based on previous methods (50); this technique physically expands samples and allows for better resolution than conventional immunofluorescence imaging (59). Although BMDMs are 10–20 μm under unexpanded conditions (Fig. 3A), they were expanded approximately five times (i.e., to 50–100 μm in size) after the expansion technique was applied (Fig. 3B). After immunofluorescence staining and expansion (see Materials and Methods), we identified ASC specks with a ring-like structure (Fig. 3B), which has previously been seen with STORM imaging (60) but was not seen with our conventional confocal microscopy techniques due to resolution limitations (Fig. 3A). Although RIPK3 and CASP8 were distributed throughout the cell, they were also detected in the ASC ring (Fig. 3B–3D). Analysis of the intensity distribution across a section on the speck (white arrow) showed that both RIPK3 and CASP8 colocalized with ASC (Fig. 3C). Neither RIPK3 nor CASP8 staining was detected in ASC specks from _Casp8_−/−_Ripk3_−/− cells (Fig. 3B), confirming the specificity of the staining.
In addition to the ring-shaped ASC specks, we also observed “firework-like” RIPK3 structures after LPS + ATP stimulation (Supplemental Figure 2A); these structures were absent in unstimulated cells (Fig. 3B, unstimulated). In these structures, RIPK3 had a higher signal intensity (Supplemental Figure 2B, i.e., several thousands) when compared with the RIPK3 signal in the ring-shaped structure (Fig. 3C, i.e., several hundreds); the ASC and CASP8 signal intensities were comparable between the two structures. Despite the difference in signaling intensity, ASC, CASP8, and RIPK3 were also colocalized in the “firework-like” speck structures (Supplemental Figure 2B). While we found that more ASC specks were induced by LPS + ATP compared with IAV infection (Supplemental Figure 1A, 2C), we also observed that ASC specks induced by LPS + ATP were similarly heterogeneous, and ASC specks both with and without RIPK3 and CASP8 were observed (Supplemental Figure 2D).
To further understand similarities and differences between LPS + ATP-induced ASC specks and IAV-induced ASC specks, we also performed expansion microscopy on IAV-infected samples. We observed an ASC speck with a core of ASC and CASP8 that was surrounded by RIPK3 in response to IAV infection. Colocalization of RIPK3 and CASP8 in an area without ASC was also found in the same cell (Supplemental Figure 2E). Overall, these observations indicate that the colocalization of RIPK3 and CASP8 with ASC can be triggered by a variety of stimuli and highlight the power of expansion microscopy techniques to further define ASC speck details and heterogeneity.
3.4. LPS + ATP-induced inflammasome activation is independent of RIPK3
Given the observation that RIPK3 localized in ASC specks, we examined whether RIPK3 had any functional role in LPS + ATP-stimulated inflammasome activation and cell death. We first examined the phosphorylation status of RIPK3 during inflammasome induction, as RIPK3 phosphorylation is important for its activation. However, we did not observe phosphorylation upon LPS + ATP stimulation (Supplemental Figure 3A). In addition, RIPK3-deficient BMDMs were not protected from LPS + ATP-induced cell death (Supplemental Figure 3B), had no notable reduction in caspase-1, −7, GSDMD and GSDME cleavage (Supplemental Figure 3C), and released similar amounts of IL-1β and IL-18 compared with WT cells (Supplemental Figure 3D); however, we did note a minor, but consistent, reduction in caspase-3, −8 cleavage (Supplemental Figure 3C). Therefore, although RIPK3 was responding to LPS + ATP stimulation by colocalizing with the ASC speck, there was no detectable function in the downstream inflammasome activation. It is still possible that RIPK3 has other functions that remain to be identified. Alternatively, LPS + ATP-induced changes in RIPK3 may alter its function.
4. DISCUSSION
Our current study provides detailed, validated methods to investigate the presence of PANoptosomes in PANoptosis-inducing conditions such as IAV infection, HSV-1 infection, and KPT-330 + IFN-β treatment. This new method provides further evidence of the presence of PANoptosomes under these conditions. Additionally, on the molecular level, our results contextualize the structural components of PANoptosomes and show, in a single cell, the formation of a protein complex containing CASP8 with the inflammasome component, ASC, and the RHIM-containing protein, RIPK3.
Immunofluorescence staining is one of the most common methods in biomedical research, and antibody specificity for the protein of interest is crucial for reliable data interpretation. Our validation of antibodies against mouse ASC, CASP8, and RIPK3 highlights the need for knockout controls in immunofluorescence staining experiments. The fixation method is also critical, since antibodies may generate nonspecific binding depending on the type of fixation (61). Our validation of reagents and use of appropriate knockout controls identified specific antibody combinations that generate reliable staining of ASC, CASP8, and RIPK3 in a single sample, enabling the study of these proteins in a multiprotein complex such as that formed during PANoptosis.
We found that ASC specks are dynamic and heterogeneous structures that vary in regard to whether or not they contain RIPK3 and CASP8. The dynamics of PANoptosome formation and ASC speck heterogeneity may depend on variations in the expression of specific proteins from cell to cell. The functional consequences of such heterogeneity in the cell fate decision and the dynamics of cell death under physiologically relevant conditions remain unknown. However, future studies to perform and integrate different single cell analyses will be critical to evaluate this possibility and yield informative results. The methods developed here could be further supplemented with live imaging of complex formation under a variety of conditions to address these functional questions. Additionally, upstream regulation of this complex formation remains an open question, and applying the expansion microscopy methods described here in various knockout cells will provide key answers regarding the regulatory factors involved.
The presence of specks containing ASC, RIPK3, and CASP8 in single cells was found not only in response to traditional PANoptosis triggers, such as IAV, but also in response to classical pyroptosis triggers like LPS + ATP stimulation. This suggests that the NLRP3 inflammasome can function as a platform to engage machinery traditionally associated with other cell death pathways. Such versatility has been shown in the context of the AIM2 inflammasome as well (14). These observations suggest that other inflammasomes, such as Pyrin, NLRC4, and NLRP1, may also engage RIPK3 and other cell death molecules to integrate inflammasome-independent functions. Future work will be needed to fully understand the functional relevance of these observations. However, we did find that RIPK3 played a minor role in the activation of some apoptotic caspases in response to the classical pyroptotic trigger LPS + ATP in murine BMDMs, although this did not affect overall cell death. Our results do not rule out the possibility that incorporation of RIPK3 in PANoptosomes changes RIPK3’s functionality (e.g., necroptosis initiation). Specifically, the “firework”-like structure induced by LPS + ATP may represent an aggregate of RIPK3 involved in its functional inactivation. Additional studies will be needed to investigate this possibility.
Using expansion microscopy allows for visualization of the detailed structure of ASC specks. Through this technique, we were able to differentiate multiple ASC speck subtypes, namely “ring-like” and “firework-like” structures of ASC specks. This cannot be achieved with common confocal microscopy techniques, where the details of ASC specks on this scale and resolution are not adequately visualized. These procedures can be implemented in labs where super-resolution microscopy is not accessible, offering broad applicability. The current expansion microscopy method is limited by the use of antibodies, which are approximately 10–15 nm in length. Antibody accessibility has been proposed as a key factor to reliably stain components in dense protein complexes, such as ASC in inflammasomes (60). Therefore, the use of nanobodies (~4 nm in length (62)) in combination with expansion microscopy could be used to further examine the protein distribution of these components in multiprotein complexes.
Overall, the current protocol allows the visualization of the formation of a multiprotein complex containing ASC, CASP8, and RIPK3 in a single cell under multiple innate immune cell death-inducing conditions, providing confirmation of PANoptosome assembly in individual cells. This imaging technique supports the concept in the field that the formation of multiprotein platforms is a common feature of cells undergoing innate immune cell death. The methodology presented here can be applied across research disciplines to examine not only cell death complexes but other cellular processes which result in the formation of multiprotein complexes. This optimized protocol will allow us to gain new insights into PANoptosome formation and regulation, as well as improve understanding of other cellular complexes. Multiprotein complexes, and PANoptosis specifically, are known to be critical across the disease spectrum, and the functions of many innate immune cell death molecules in disease phenotypes have historically been masked by redundancies and synergies between the components of different cell death pathways (2, 3, 5, 7, 8, 11–16, 51–57). Therefore, continued efforts to assess PANoptosomes and the dynamics of their formation in disease will serve as a critical strategy for the identification of therapeutic targets and the development of new treatments.
Supplementary Material
Supp Figure 1. Supplemental Figure 1. ASC, RIPK3, and CASP8 form a complex under PANoptotic conditions.
A) Quantification of the percentage of cells containing ASC specks in IAV-infected wild type (WT) bone marrow-derived macrophages (BMDMs) at 9 h and 12 h post-infection (h.p.i.). B) BMDMs from the indicated genotypes were mock treated, infected with HSV-1, or treated with a KPT-330 + IFN-β and stained for ASC, RIPK3, and CASP8. Representative images of ASC, RIPK3, CASP8-containing specks are shown in WT cells. C) Quantification of the percentage of cells containing ASC specks in WT BMDMs treated with KPT-330 + IFN-β for 24 h. D) Compositional analysis of ASC specks in (C). Mean ± standard error is shown. Images are representative of at least three independent experiments.
Supp Figure 2. Supplemental Figure 2. “Firework”-like RIPK3 structure forms during LPS + ATP stimulation, and ASC and CASP8 form a core surrounded by RIPK3 during IAV infection.
A) Wild type (WT) bone marrow-derived macrophages (BMDMs) were treated with LPS + ATP and stained for ASC, RIPK3, and CASP8. The cells were expanded as described in the methods section. ASC, RIPK3, and CASP8-containing specks with “firework”-like RIPK3 structure are shown. B) Fluorescence intensity of ASC, RIPK3, and CASP8 across the “firework” section along the white arrow. X axis indicates the distance along the white arrow traveling from the base of the arrow to the arrowhead. C) Quantification of the percentage of cells containing ASC specks in WT BMDMs treated with LPS + ATP for 10 min. D) Compositional analysis of ASC specks (n ≥ 95) induced by LPS + ATP in (C). Mean ± standard error is shown. E) Expansion microscopy views of a RIPK3/CASP8-containing ASC speck induced by IAV. Images are representative of at least three independent experiments.
Supp Figure 3. Supplemental Figure 3. RIPK3 plays no observable functional role in LPS + ATP-mediated inflammasome activation.
Bone marrow-derived macrophages (BMDMs) from indicated genotypes were primed with 100 ng/μl LPS for 4 h, then stimulated with the indicated concentration of ATP for 30 min. A) Western blots of RIPK3 phosphorylation (Thr231/Ser232; pRIPK3) and total RIPK3 (tRIPK3) in whole cell lysates from indicated genotypes. B) Cell death measured by Sytox Green staining in the indicated genotypes. Microscopy images (right) showing dead cells denoted by red masking. Sytox green detects lytic types of cell death, where the cellular membranes are compromised. C) Caspases-1, −3, −7, −8, GSDMD, and GSDME cleavage, and total and phosphorylated MLKL in whole cell lysates from the indicated genotypes in mock-treated or LPS + ATP-treated cells. Asterisk denotes nonspecific band. D) Quantification of IL-1β and IL-18 release in the supernatant of cells of the indicated genotypes after LPS + ATP treatment. Lysate and supernatant were prepared at 30 min after ATP stimulation. Mean ± standard error is shown. Results are representative of three independent experiments.
Acknowledgements
We thank all members of the Kanneganti lab for discussions. We also thank R. Tweedell, PhD, and J. Gullett, PhD, for scientific editing and writing support.
Funding
Research in the Kanneganti lab is supported by grants from the US National Institutes of Health (AI101935, AI124346, AI160179, AR056296, and CA253095) and the American Lebanese Syrian Associated Charities to T.D.-K. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Competing interests
T.-D.K. is a consultant for Pfizer.
Ethics approval
Studies were conducted under protocols approved by the St. Jude Children’s Research Hospital committee on the Use and Care of Animals.
Data availability
All datasets generated during this study are contained within the figures and supplement of this manuscript.
REFERENCES
- 1.Gullett JM, Tweedell RE, Kanneganti TD. It’s All in the PAN: Crosstalk, Plasticity, Redundancies, Switches, and Interconnectedness Encompassed by PANoptosis Underlying the Totality of Cell Death-Associated Biological Effects. Cells. 2022;11(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuriakose T, Man SM, Malireddi RK, Karki R, Kesavardhana S, Place DE, et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol. 2016;1(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malireddi RKS, Karki R, Sundaram B, Kancharana B, Lee S, Samir P, et al. Inflammatory Cell Death, PANoptosis, Mediated by Cytokines in Diverse Cancer Lineages Inhibits Tumor Growth. Immunohorizons. 2021;5(7):568–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kesavardhana S, Malireddi RKS, Burton AR, Porter SN, Vogel P, Pruett-Miller SM, et al. The Zα2 domain of ZBP1 is a molecular switch regulating influenza-induced PANoptosis and perinatal lethality during development. Journal of Biological Chemistry. 2020;295(24):8325–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Banoth B, Tuladhar S, Karki R, Sharma BR, Briard B, Kesavardhana S, et al. ZBP1 promotes fungi-induced inflammasome activation and pyroptosis, apoptosis, and necroptosis (PANoptosis). Journal of Biological Chemistry. 2020:jbc.RA120.01592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Christgen S, Zheng M, Kesavardhana S, Karki R, Malireddi RKS, Banoth B, Place DE, Briard B, Sharma BR, Tuladhar S, Samir P, Burton A, Kanneganti T-D Identification of the PANoptosome: A molecular platform triggering pyroptosis, apoptosis, and necroptosis (PANoptosis). Frontiers in cellular and infection microbiology. 2020;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karki R, Sharma BR, Lee E, Banoth B, Malireddi RKS, Samir P, et al. Interferon regulatory factor 1 regulates PANoptosis to prevent colorectal cancer. JCI insight. 2020;5(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng M, Williams EP, Malireddi RKS, Karki R, Banoth B, Burton A, et al. Impaired NLRP3 inflammasome activation/pyroptosis leads to robust inflammatory cell death via caspase-8/RIPK3 during coronavirus infection. Journal of Biological Chemistry. 2020:jbc.RA120.01503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malireddi RK, Ippagunta S, Lamkanfi M, Kanneganti TD. Cutting edge: proteolytic inactivation of poly(ADP-ribose) polymerase 1 by the Nlrp3 and Nlrc4 inflammasomes. J Immunol. 2010;185(6):3127–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malireddi RKS, Gurung P, Kesavardhana S, Samir P, Burton A, Mummareddy H, et al. Innate immune priming in the absence of TAK1 drives RIPK1 kinase activity–independent pyroptosis, apoptosis, necroptosis, and inflammatory disease. Journal of Experimental Medicine. 2020;217(3). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Malireddi RKS, Kesavardhana S, Karki R, Kancharana B, Burton AR, Kanneganti TD. RIPK1 Distinctly Regulates Yersinia-Induced Inflammatory Cell Death, PANoptosis. Immunohorizons. 2020;4(12):789–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zheng M, Karki R, Vogel P, Kanneganti TD. Caspase-6 Is a Key Regulator of Innate Immunity, Inflammasome Activation, and Host Defense. Cell. 2020;181(3):674–87.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Karki R, Sharma BR, Tuladhar S, Williams EP, Zalduondo L, Samir P, et al. Synergism of TNF-α and IFN-γ Triggers Inflammatory Cell Death, Tissue Damage, and Mortality in SARS-CoV-2 Infection and Cytokine Shock Syndromes. Cell. 2021;184(1):149–68.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee S, Karki R, Wang Y, Nguyen LN, Kalathur RC, Kanneganti TD. AIM2 forms a complex with pyrin and ZBP1 to drive PANoptosis and host defence. Nature. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Karki R, Sundaram B, Sharma BR, Lee S, Malireddi RKS, Nguyen LN, et al. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep. 2021;37(3):109858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karki R, Lee S, Mall R, Pandian N, Wang Y, Sharma BR, et al. ZBP1-dependent inflammatory cell death, PANoptosis, and cytokine storm disrupt IFN therapeutic efficacy during coronavirus infection. Sci Immunol. 2022:eabo6294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kesavardhana S, Kuriakose T, Guy CS, Samir P, Malireddi RKS, Mishra A, et al. ZBP1/DAI ubiquitination and sensing of influenza vRNPs activate programmed cell death. J Exp Med. 2017;214(8):2217–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cookson BT, Brennan MA. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9(3):113–4. [DOI] [PubMed] [Google Scholar]
- 19.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–26. [DOI] [PubMed] [Google Scholar]
- 20.Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. British journal of cancer. 1972;26(4):239–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90(3):405–13. [DOI] [PubMed] [Google Scholar]
- 22.Kim HE, Du F, Fang M, Wang X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc Natl Acad Sci U S A. 2005;102(49):17545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, et al. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91(4):479–89. [DOI] [PubMed] [Google Scholar]
- 24.Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1- and TNF receptor-induced cell death. Cell. 1996;85(6):803–15. [DOI] [PubMed] [Google Scholar]
- 25.Muzio M, Chinnaiyan AM, Kischkel FC, O’Rourke K, Shevchenko A, Ni J, et al. FLICE, a novel FADD-homologous ICE/CED-3-like protease, is recruited to the CD95 (Fas/APO-1) death--inducing signaling complex. Cell. 1996;85(6):817–27. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94(4):491–501. [DOI] [PubMed] [Google Scholar]
- 27.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94(4):481–90. [DOI] [PubMed] [Google Scholar]
- 28.Gross A, Yin XM, Wang K, Wei MC, Jockel J, Milliman C, et al. Caspase cleaved BID targets mitochondria and is required for cytochrome c release, while BCL-XL prevents this release but not tumor necrosis factor-R1/Fas death. J Biol Chem. 1999;274(2):1156–63. [DOI] [PubMed] [Google Scholar]
- 29.Dhuriya YK, Sharma D. Necroptosis: a regulated inflammatory mode of cell death. J Neuroinflammation. 2018;15(1):199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murphy JM, Czabotar PE, Hildebrand JM, Lucet IS, Zhang JG, Alvarez-Diaz S, et al. The pseudokinase MLKL mediates necroptosis via a molecular switch mechanism. Immunity. 2013;39(3):443–53. [DOI] [PubMed] [Google Scholar]
- 31.Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan K, et al. The role of necroptosis in cancer biology and therapy. Mol Cancer. 2019;18(1):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nailwal H, Chan FK. Necroptosis in anti-viral inflammation. Cell Death Differ. 2019;26(1):4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao J, Jitkaew S, Cai Z, Choksi S, Li Q, Luo J, et al. Mixed lineage kinase domain-like is a key receptor interacting protein 3 downstream component of TNF-induced necrosis. Proc Natl Acad Sci U S A. 2012;109(14):5322–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sun L, Wang H, Wang Z, He S, Chen S, Liao D, et al. Mixed lineage kinase domain-like protein mediates necrosis signaling downstream of RIP3 kinase. Cell. 2012;148(1–2):213–27. [DOI] [PubMed] [Google Scholar]
- 35.Newton K, Wickliffe KE, Dugger DL, Maltzman A, Roose-Girma M, Dohse M, et al. Cleavage of RIPK1 by caspase-8 is crucial for limiting apoptosis and necroptosis. Nature. 2019;574(7778):428–31. [DOI] [PubMed] [Google Scholar]
- 36.Man SM, Hopkins LJ, Nugent E, Cox S, Glück IM, Tourlomousis P, et al. Inflammasome activation causes dual recruitment of NLRC4 and NLRP3 to the same macromolecular complex. Proc Natl Acad Sci U S A. 2014;111(20):7403–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Zhu R, Zhong J, Ying Y, Wang W, Cao Y, et al. Mosaic composition of RIP1-RIP3 signalling hub and its role in regulating cell death. Nat Cell Biol. 2022;24(4):471–82. [DOI] [PubMed] [Google Scholar]
- 38.Man SM, Tourlomousis P, Hopkins L, Monie TP, Fitzgerald KA, Bryant CE. Salmonella infection induces recruitment of Caspase-8 to the inflammasome to modulate IL-1beta production. J Immunol. 2013;191(10):5239–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sagulenko V, Thygesen SJ, Sester DP, Idris A, Cridland JA, Vajjhala PR, et al. AIM2 and NLRP3 inflammasomes activate both apoptotic and pyroptotic death pathways via ASC. Cell Death Differ. 2013;20(9):1149–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pierini R, Juruj C, Perret M, Jones CL, Mangeot P, Weiss DS, et al. AIM2/ASC triggers caspase-8-dependent apoptosis in Francisella-infected caspase-1-deficient macrophages. Cell Death Differ. 2012;19(10):1709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Y, Kanneganti TD. From pyroptosis, apoptosis and necroptosis to PANoptosis: A mechanistic compendium of programmed cell death pathways. Comput Struct Biotechnol J. 2021;19:4641–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oberst A, Dillon CP, Weinlich R, McCormick LL, Fitzgerald P, Pop C, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471(7338):363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24(4):1464–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ozören N, Masumoto J, Franchi L, Kanneganti TD, Body-Malapel M, Ertürk I, et al. Distinct roles of TLR2 and the adaptor ASC in IL-1beta/IL-18 secretion in response to Listeria monocytogenes. J Immunol. 2006;176(7):4337–42. [DOI] [PubMed] [Google Scholar]
- 45.Ishii KJ, Kawagoe T, Koyama S, Matsui K, Kumar H, Kawai T, et al. TANK-binding kinase-1 delineates innate and adaptive immune responses to DNA vaccines. Nature. 2008;451(7179):725–9. [DOI] [PubMed] [Google Scholar]
- 46.Jones JW, Kayagaki N, Broz P, Henry T, Newton K, O’Rourke K, et al. Absent in melanoma 2 is required for innate immune recognition of Francisella tularensis. Proc Natl Acad Sci U S A. 2010;107(21):9771–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kanneganti TD, Ozoren N, Body-Malapel M, Amer A, Park JH, Franchi L, et al. Bacterial RNA and small antiviral compounds activate caspase-1 through cryopyrin/Nalp3. Nature. 2006;440(7081):233–6. [DOI] [PubMed] [Google Scholar]
- 48.Hoffmann E, Neumann G, Kawaoka Y, Hobom G, Webster RG. A DNA transfection system for generation of influenza A virus from eight plasmids. Proc Natl Acad Sci U S A. 2000;97(11):6108–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang Y, Karki R, Zheng M, Kancharana B, Lee S, Kesavardhana S, et al. Cutting Edge: Caspase-8 Is a Linchpin in Caspase-3 and Gasdermin D Activation to Control Cell Death, Cytokine Release, and Host Defense during Influenza A Virus Infection. J Immunol. 2021;207(10):2411–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang C, Kang JS, Asano SM, Gao R, Boyden ES. Expansion Microscopy for Beginners: Visualizing Microtubules in Expanded Cultured HeLa Cells. Curr Protoc Neurosci. 2020;92(1):e96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Malireddi RKS, Gurung P, Mavuluri J, Dasari TK, Klco JM, Chi H, et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med. 2018;215(4):1023–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Messaoud-Nacer Y, Culerier E, Rose S, Maillet I, Rouxel N, Briault S, et al. STING agonist diABZI induces PANoptosis and DNA mediated acute respiratory distress syndrome (ARDS). Cell Death Dis. 2022;13(3):269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cui Y, Wang X, Lin F, Li W, Zhao Y, Zhu F, et al. MiR-29a-3p Improves Acute Lung Injury by Reducing Alveolar Epithelial Cell PANoptosis. Aging Dis. 2022;13(3):899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin JF, Hu PS, Wang YY, Tan YT, Yu K, Liao K, et al. Phosphorylated NFS1 weakens oxaliplatin-based chemosensitivity of colorectal cancer by preventing PANoptosis. Signal Transduct Target Ther. 2022;7(1):54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu X, Lan X, Fu S, Zhang Q, Gui F, Jin Q, et al. Dickkopf-1 exerts protective effects by inhibiting PANoptosis and retinal neovascularization in diabetic retinopathy. Biochem Biophys Res Commun. 2022;617(Pt 2):69–76. [DOI] [PubMed] [Google Scholar]
- 56.Chi D, Lin X, Meng Q, Tan J, Gong Q, Tong Z. Real-Time Induction of Macrophage Apoptosis, Pyroptosis, and Necroptosis by Enterococcus faecalis OG1RF and Two Root Canal Isolated Strains. Frontiers in cellular and infection microbiology. 2021;11:720147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Song M, Xia W, Tao Z, Zhu B, Zhang W, Liu C, et al. Self-assembled polymeric nanocarrier-mediated co-delivery of metformin and doxorubicin for melanoma therapy. Drug Deliv. 2021;28(1):594–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Masumoto J, Taniguchi S, Ayukawa K, Sarvotham H, Kishino T, Niikawa N, et al. ASC, a novel 22-kDa protein, aggregates during apoptosis of human promyelocytic leukemia HL-60 cells. J Biol Chem. 1999;274(48):33835–8. [DOI] [PubMed] [Google Scholar]
- 59.Chen F, Tillberg PW, Boyden ES. Optical imaging. Expansion microscopy. Science. 2015;347(6221):543–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Glück IM, Mathias GP, Strauss S, Ebert TS, Stafford C, Agam G, et al. Nanoscale organization of the endogenous ASC speck. bioRxiv. 2022:2021.09.17.460822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Samson AL, Fitzgibbon C, Patel KM, Hildebrand JM, Whitehead LW, Rimes JS, et al. A toolbox for imaging RIPK1, RIPK3, and MLKL in mouse and human cells. Cell Death Differ. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bao G, Tang M, Zhao J, Zhu X. Nanobody: a promising toolkit for molecular imaging and disease therapy. EJNMMI Res. 2021;11(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supp Figure 1. Supplemental Figure 1. ASC, RIPK3, and CASP8 form a complex under PANoptotic conditions.
A) Quantification of the percentage of cells containing ASC specks in IAV-infected wild type (WT) bone marrow-derived macrophages (BMDMs) at 9 h and 12 h post-infection (h.p.i.). B) BMDMs from the indicated genotypes were mock treated, infected with HSV-1, or treated with a KPT-330 + IFN-β and stained for ASC, RIPK3, and CASP8. Representative images of ASC, RIPK3, CASP8-containing specks are shown in WT cells. C) Quantification of the percentage of cells containing ASC specks in WT BMDMs treated with KPT-330 + IFN-β for 24 h. D) Compositional analysis of ASC specks in (C). Mean ± standard error is shown. Images are representative of at least three independent experiments.
Supp Figure 2. Supplemental Figure 2. “Firework”-like RIPK3 structure forms during LPS + ATP stimulation, and ASC and CASP8 form a core surrounded by RIPK3 during IAV infection.
A) Wild type (WT) bone marrow-derived macrophages (BMDMs) were treated with LPS + ATP and stained for ASC, RIPK3, and CASP8. The cells were expanded as described in the methods section. ASC, RIPK3, and CASP8-containing specks with “firework”-like RIPK3 structure are shown. B) Fluorescence intensity of ASC, RIPK3, and CASP8 across the “firework” section along the white arrow. X axis indicates the distance along the white arrow traveling from the base of the arrow to the arrowhead. C) Quantification of the percentage of cells containing ASC specks in WT BMDMs treated with LPS + ATP for 10 min. D) Compositional analysis of ASC specks (n ≥ 95) induced by LPS + ATP in (C). Mean ± standard error is shown. E) Expansion microscopy views of a RIPK3/CASP8-containing ASC speck induced by IAV. Images are representative of at least three independent experiments.
Supp Figure 3. Supplemental Figure 3. RIPK3 plays no observable functional role in LPS + ATP-mediated inflammasome activation.
Bone marrow-derived macrophages (BMDMs) from indicated genotypes were primed with 100 ng/μl LPS for 4 h, then stimulated with the indicated concentration of ATP for 30 min. A) Western blots of RIPK3 phosphorylation (Thr231/Ser232; pRIPK3) and total RIPK3 (tRIPK3) in whole cell lysates from indicated genotypes. B) Cell death measured by Sytox Green staining in the indicated genotypes. Microscopy images (right) showing dead cells denoted by red masking. Sytox green detects lytic types of cell death, where the cellular membranes are compromised. C) Caspases-1, −3, −7, −8, GSDMD, and GSDME cleavage, and total and phosphorylated MLKL in whole cell lysates from the indicated genotypes in mock-treated or LPS + ATP-treated cells. Asterisk denotes nonspecific band. D) Quantification of IL-1β and IL-18 release in the supernatant of cells of the indicated genotypes after LPS + ATP treatment. Lysate and supernatant were prepared at 30 min after ATP stimulation. Mean ± standard error is shown. Results are representative of three independent experiments.
Data Availability Statement
All datasets generated during this study are contained within the figures and supplement of this manuscript.