Human DNA ligase I efficiently seals nicks in nucleosomes (original) (raw)
Abstract
The access to DNA within nucleosomes is greatly restricted for most enzymes and _trans_-acting factors that bind DNA. We report here that human DNA ligase I, which carries out the final step of Okazaki fragment processing and of many DNA repair pathways, can access DNA that is wrapped about the surface of a nucleosome in vitro and carry out its enzymatic function with high efficiency. In addition, we find that ligase activity is not affected by the binding of linker histone (H1) but is greatly influenced by the disposition of the core histone tail domains. These results suggest that the window of opportunity for human DNA ligase I may extend well beyond the first stages of chromatin reassembly after DNA replication or repair.
Keywords: chromatin/gene regulation/human DNA ligase/nucleosome
Introduction
Instability of DNA within the human genome is the basis for a large number of genetic disorders including myotonic dystrophy, Huntington’s disease, several ataxias, fragile X syndrome (Reddy and Housman, 1997) and several forms of cancer (Gordenin et al., 1997). The enzymes that carry out replication, repair and recombination are fundamental for maintaining an intact genome. These three essential processes involve similar reaction pathways and use many of the same enzymes. Two examples are the flap endonuclease 1 (FEN1) and DNA ligase I. Aberrant function of FEN1 is thought to be a major contributing factor for several of the genetic diseases listed above, and mutations in ligase I can contribute to aberrant DNA replication and repair (Prigent et al., 1994; Bambara et al., 1997).
An extensive body of research has elucidated many of the enzymes and pathways involved in eukaryotic DNA replication and repair (Waga and Stillman, 1998). The activities of these enzymes have been studied in great detail using naked DNA as a substrate (Bambara et al., 1997; Waga and Stillman, 1998). However, in vivo the substrate for these processes is not naked DNA but chromatin. Indeed, assembly of chromatin is likely to be directly coupled to DNA replication and repair in vivo (Almouzni and Mechali, 1988; Smith and Stillman, 1989; Gaillard et al., 1997; for review see Wolffe, 1998). Chromatin is comprised of nuclear DNA and histone proteins, and virtually all genomic DNA exists within this complex. At first consideration, chromatin appears to be a poor substrate for the molecular machines that carry out these activities. Nucleosomes, the basic repeating subunit of chromatin, are remarkably stable to physical perturbation. Under physiological conditions nucleosomal arrays fold into stable higher order structures that self-associate within the nucleus to concentrations >50–75 mg/ml (Widom, 1986; van Holde, 1989). In spite of this apparent stability and compaction, complex metabolic processes utilizing chromatin occur very efficiently in vivo. Unfortunately, our current understanding of how various enzymes and activities are able to utilize chromatin as a substrate is inadequate.
The cell uses several strategies to allow transcription factors, polymerases and other proteins that carry out DNA metabolism to overcome the steric and structural impediments of chromatin. Post-translational modifications such as acetylation are thought to destabilize the highly compacted chromatin fiber, causing greater exposure of individual nucleosomes (Garcia-Ramirez et al., 1995; Tse et al., 1998). Access to nucleosomal DNA may then occur by any of several mechanisms. First, factors may be accommodated by a transient dissociation or uncoiling of a portion of the nucleosomal DNA from the histone proteins (Polach and Widom, 1995). However, if site exposure occurs only by this mechanism the competing histone–DNA interactions are thought to reduce the association constant for DNA binding proteins by a factor of 102–105, depending upon the location of their binding sites within the nucleosome (Polach and Widom, 1995; Anderson and Widom, 2000). Additionally, nucleosomes may exhibit translational mobility, ‘sliding’ along the DNA to allow transcription factors to gain access to previously occluded binding sites (Sera and Wolffe, 1998). Sliding occurs when the histone octamer equilibrates with alternate binding positions along the DNA molecule. These alternate positions may not be highly populated at any particular moment but could allow transient exposure of sequences sequestered within the main translational position occupied by the nucleosome. Interestingly, the association of linker histones is thought to eliminate this translational mobility and thus depletion of these proteins from chromatin may be a prerequisite to transcriptional activation (Pennings et al., 1994; Ura et al., 1997; Sera and Wolffe, 1998). However, the degree to which spontaneous nucleosome sliding contributes to site exposure and DNA-dependent processes such as replication or repair has not been accurately determined. Finally, a small number of transcription factors, including some steroid hormone receptors, can bind DNA on the surface of the nucleosome with high affinity and selectivity (Perlmann and Wrange, 1988; Cirillo et al., 1998; Wolffe, 1998). Presumably, these factors can accommodate the distorted, highly bent conformation of DNA within the nucleosome and bind without significant disruption of histone–DNA interactions (Polach and Widom, 1995).
Access to nucleosomal DNA via the above mechanisms is further mediated by ATP-dependent nucleosome remodeling activities and post-translational modifications of the core histone proteins. Several multi-subunit complexes have been identified that utilize energy from ATP hydrolysis to facilitate greater access of nucleosomal DNA to DNA binding proteins via a poorly understood mechanism (for recent reviews see Kingston and Narlikar, 1999; Vignali et al., 2000). In some cases, the efficiency of these complexes is dependent upon the presence of the core histone tail domains (Georgel et al., 1997; Logie et al., 1999). In addition, acetylation of the tails, especially those of histones H3 and H4, may subtly enhance DNA site exposure within individual nucleosomes in vivo (Lee et al., 1993; Vettese-Dadey et al., 1994; Howe et al., 1998; Polach et al., 2000; Vitolo et al., 2000). Clearly, the conformations and interactions of the core histone tails can mediate DNA accessibility within individual nucleosomes.
Human DNA ligase I carries out the final enzymatic step completing Okazaki fragment joining during DNA replication and several major DNA repair pathways (Prigent et al., 1994). After synthesis of DNA on both leading and lagging DNA strands or after the excision of damaged DNA bases and the re-synthesis of DNA, this enzyme catalyzes the formation of a phosphodiester bond from a 5′ phosphomonoester terminus and a 3′ hydroxyl to create an uninterrupted DNA strand. Failure to ligate nascent DNA strands together results in a potentially deleterious nick in the DNA backbone, making the position vulnerable to exonuclease degradation, increased frequency of recombination and formation of a double-strand break. Ligation is especially prevalent on the lagging strand during DNA replication, occurring every 100–200 bp in eukaryotic cells (Anderson and DePamphilis, 1979).
Immediately after passage of the replication fork, the new DNA is assembled into ‘immature’ nucleosome structures, which are then processed into mature nucleosomes (Seale, 1978; Annunziato and Seale, 1982; Cusick et al., 1983). In fact, during replication of the SV40 genome, nucleosome assembly was found to occur as soon as there was sufficient room to accommodate a nucleosome after passage of the replication fork (Sogo et al., 1986). In human cells, this assembly is thought to occur in stages, with the (H3/H4)2-tetramer being assembled rapidly with the DNA immediately after passage of the replication fork, a short lag period then occurs before the H2A/H2B dimers are incorporated, followed by the linker histones to produce the ‘mature’ nucleosomes (Almouzni et al., 1990; Smith and Stillman, 1991). Okazaki fragment maturation, which includes RNA primer removal, synthesis to create a nick and then ligation, must occur in this environment. Early work suggests that at least some Okazaki fragments are assembled into nucleosomes before ligation (Herman et al., 1981). Moreover, Okazaki fragments in yeast cells containing a temperature-sensitive mutant of ligase I remain unligated at the non-permissive temperature but these cells otherwise appear to complete DNA replication and chromatin assembly (Bielinsky and Gerbi, 1999). Transfer of these cells to the permissive temperature results in efficient ligation of Okazaki fragments throughout the genome (Bielinsky and Gerbi, 1999). In addition, nucleotide excision repair, which must occur at all phases of the cell cycle, can be carried out on nucleosomal DNA in vivo (Smerdon and Thoma, 1990).
In order to determine how such enzymes may access DNA within chromatin, we have characterized the activity of human DNA ligase I on model nucleosome substrates. Surprisingly, we found that the activity of human DNA ligase I was only moderately inhibited at sealing nicks placed at the center of a nucleosome, suggesting that the trans-esterification catalyzed by ligase can be carried out without significant disruption of histone–DNA interactions. Furthermore, ligase I was unaffected by the binding of linker histone to the nucleosomal substrate or by other changes in conditions that would be expected to dampen nucleosome sliding. However, our results suggest that in some cases appropriate positioning of the core histone tail domains can lead to a drastic inhibition of ligase activity.
Results
Construction of ligation substrates
In order to investigate the activity of human DNA ligase I in nucleosomal DNA we designed ligation substrates to place nicks at selected sites within model nucleosome complexes. A key feature of these substrates is that they contain a precise nucleosome-positioning element (NPE) based on a DNA fragment containing a Xenopus borealis somatic-type 5S rRNA gene (Hayes and Wolffe, 1992; Lee and Hayes, 1998) (see Figure 1). Thus, after reconstitution with purified core histone proteins, the nucleosome will adopt a precise translational and rotational setting along the DNA fragment. Substrate DNAs were constructed by annealing full-length ‘bottom’ strands for the 154 or 218 bp substrates with complementary oligonucleotides, producing ligatable nicks at selected locations (see Figure 1 and Materials and methods). Substrates were constructed so that ligase activity could be assessed independently for individual nicks by including only one 5′-32P-phos phorylated oligonucleotide per template in separate annealing reactions with the remaining unlabeled and unphosphorylated oligonucleotides (see Materials and methods). Thus, only the nick just upstream of the single labeled oligomer in each template can be ligated, resulting in a single longer product, avoiding multiple ligation products and greatly simplifying the quantitation of ligation at individual sites. We found that the fully assembled nicked ligation substrates migrated identically to the corresponding double-stranded DNAs on either 6% polyacrylamide or 1.8% agarose gels, and that >95% of the labeled nicks were competent as sites of ligation, indicating proper annealing of the substrates (data not shown).

Fig. 1. Nucleosomal DNA substrates for human DNA ligase I. Three double-stranded substrates for human DNA ligase I were constructed as described in Materials and methods. Each contains the same NPE with the expected position of the nucleosome indicated by the oval. The 154 bp substrate contains a single nick near the predicted location of the nucleosome dyad. The 218 bp substrates consist of the 154 bp sequence with 64 bp appended to the right-hand end and either contains a single nick near the nucleosome dyad or three nicks at the indicated locations. The length of oligonucleotides used to assemble the substrates is indicated. The positions of radioactive phosphate labels are indicated (circles). Identical DNAs lacking nicks were also constructed (not shown; see Figure 2).
Nucleosomes were reconstituted on the ligation substrates and checked for proper positioning and DNA organization by hydroxyl radical footprinting. The hydroxyl radical footprinting technique can precisely detect histone–DNA interactions along the nucleosome (Hayes and Wolffe, 1992). Nucleosomes formed on nicked and intact DNAs were subjected to hydroxyl radicals and the cleavage patterns analyzed on 6% sequencing gels (Figure 2). Hydroxyl radical footprinting of naked DNA templates yields an approximately even distribution of single-stranded cuts at every nucleotide position (Figure 2, lanes 2 and 5). Nucleosomes reconstituted with intact (un-nicked) DNA generated a series of more biased 10 bp protections, indicative of the DNA backbone being transiently exposed and protected from the bulk solvent because of its helical structure and association with the surface of the histone octamer (Figure 2, lane 3). The pattern observed corresponds to the expected nucleosome position on this fragment (Hayes and Wolffe, 1992). The nicked templates generated the same pattern of 10 bp protections, indicating that these nucleosomes adopt the same translational and rotational positions as nucleosomes reconstituted with the original 5S DNA fragments (Figure 2, lane 4).

Fig. 2. Hydroxyl radical characterization of nucleosomal ligase substrates. Nucleosomes (Nuc) and free DNA (FD) were reacted with hydroxyl radicals (⋅OH), isolated, and then cleavage patterns analyzed on 6% sequencing gels as described in Materials and methods. Shown are reactions for the 218 (left) or 154 bp (right) intact double-stranded fragments (lanes 1–3) or nicked substrates (lanes 4–6). Lane G contains products of the Maxam–Gilbert G-specific sequencing reaction for each DNA. Lanes 1 and 6 in each gel contain free DNA not reacted with hydroxyl radicals while products from cleavage of naked DNA or nucleosomes are shown in lanes 2 and 5 or lanes 3 and 4, respectively. A schematic depicting the 5S DNA fragment and the position of the nucleosome (oval) based on the footprinting results is shown. The locations of nicks when present on the opposite strand are indicated (circles).
Human DNA ligase I can seal nicks within nucleosomal DNA
We first tested the ability of human DNA ligase I to seal each of the three nicks within the 218 bp ligation substrate when this DNA was assembled into a nucleosome. This substrate was designed such that after nucleosome reconstitution the nicks would be located near the nucleosome dyad, at the edge of the nucleosome and ∼30 bp away from the edge of the nucleosome, within the histone-free ‘linker DNA’ region (Figures 1 and 2). Nucleosomes assembled with the 218 bp substrate were subjected to human DNA ligase I for increasing times before separation of the nucleosome complexes and naked DNA on 0.7% nucleoprotein gels. The labeled DNA was recovered from each band and analyzed on 6% sequencing gels (Figure 3). No ligation is evident in the free DNA or nucleosome samples in the absence of human DNA ligase I (Figure 3, lanes 1 and 6). As expected, approximately equal amounts of ligation occurred at the linker position (a protein-free region) in both substrates (Figure 3, compare lanes 2–5 with 7–10, linker band). Unlike the linker region, the edge and dyad positions in the nucleosome ligate with slightly slower kinetics compared with their respective sites in the free DNA (Figure 3, lanes 2–5 and 7–10, Edge, Dyad). Quantitation of the ligation products shows a decrease in ligase activity of ∼10-fold at the dyad and 4- to 6-fold at the edge position (see Table I). Experiments with nucleosomes reconstituted with the analogous 218 bp substrate containing only a single nick near the dyad gave identical results for this site (Table I; see below).
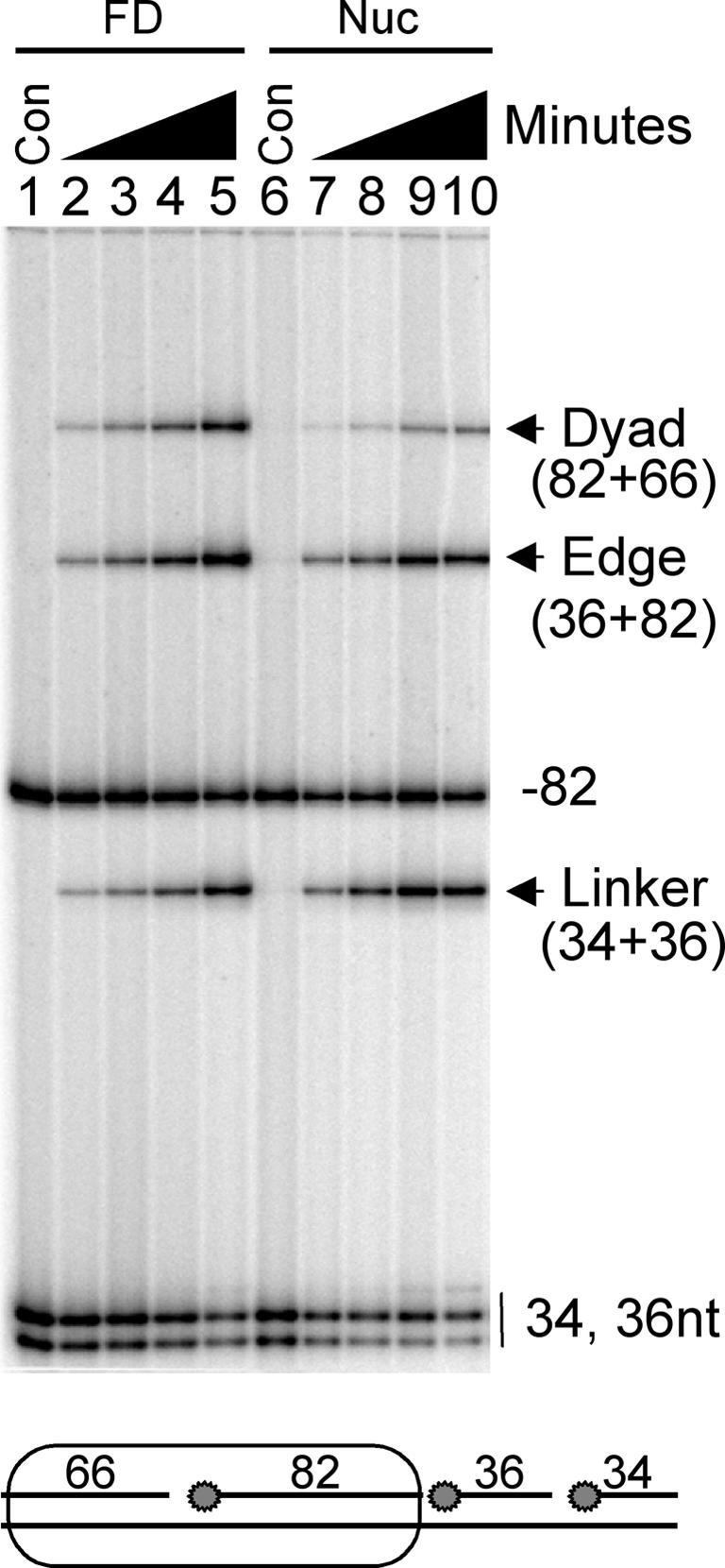
Fig. 3. Efficient ligation of a nick in a nucleosome. Nucleosomes were reconstituted with the 218 bp substrate containing three nicks and treated with human DNA ligase I as described in Materials and methods. Labeled reactants and products from the nucleosome (Nuc) or the free DNA (FD) were isolated from a 0.7% agarose gel and analyzed on a 6% sequencing gel. Lanes 1–5 and 6–10 contain ligation products from free DNA or nucleosomes generated after 0, 5, 10, 30 and 60 min, respectively. Oligonucleotides in the original substrate are shown in the schematic below the gel and the products of ligation at each of the three labeled sites (circles, schematic) are indicated alongside the gel.
Table I. Nucleosome-dependent fold inhibition of human DNA ligase I and _Sac_I endonuclease.
| Nucleosome: Site | 218 | 154 | 154-try | 218+H1 | 218–4° | 218* | 154* | 154-try* |
|---|---|---|---|---|---|---|---|---|
| Dyad | 10 | 1 × 105 | 5 | 7.5 | 6 | 5.5 × 104* | 6.5 × 104* | 3.4 × 104* |
| Edge | 4–6 | – | – | 2 | 2 | – | – | – |
| Linker | 1 | – | – | 1 | 1 | – | – | – |
Ligase accessibility to nucleosomal DNA is not due to nucleosome sliding or alternate translational positions
The relatively efficient ligation at the site near the nucleosome dyad suggests that human DNA ligase I can catalyze trans-esterification without significant disruption of histone–DNA interactions. For example, if ligase required complete release of the DNA from the histone surface, the measured rate of ligation of nucleosomal DNA would be ∼10 000-fold less than that of naked DNA based on the expected probability of site exposure for a site near the nucleosome dyad (Polach and Widom, 1995; Anderson and Widom, 2000; see below). Alternatively, it is possible that unanticipated processes such as nucleosome sliding, i.e. equilibration between related translational positions along the DNA, or populations of unexpected alternative translational positions within the ensemble of reconstituted nucleosomes contribute to site exposure.
The hydroxyl radical footprinting studies argue against large populations of unexpected translational positions that would result in exposure of the dyad nick (Figure 2). To further exclude this possibility, we performed ligation assays after resolution of nucleosome ‘translational isomers’ on 5% acrylamide gels (Figure 4). Such gel systems have been used to separate nucleosome translational isomers produced by in vitro reconstitution (Meersseman et al., 1992; Pennings et al., 1994). We found two apparent major translational isomers for nucleosomes reconstituted with the 218 bp substrate used in these studies. Hydroxyl radical footprinting showed that both isomers occupied related positions near the left edge of the DNA fragment, as expected (results not shown; see Figures 2 and 4A). Importantly, both isomers exhibited the same kinetics with regard to ligation (Figure 4B). Note that these experiments were carried out with the 218 bp substrate containing a single nick near the nucleosome dyad (see Materials and methods). We also determined that the bulk of the substrate within each species ligated with the same kinetic parameters, indicating that unresolved heterogeneity does not exist within each translational isomer band (data not shown). Taken together, these results indicate that translational heterogeneity within the ensemble of nucleosomal ligase substrates does not lead to unexpected exposure of the ‘dyad’ nick in a fraction of reconstituted nucleosomes.

Fig. 4. Rate of ligation within the 218 bp nucleosome substrate is not affected by nucleosome translational heterogeneity or binding of linker histone H1. (A) Isolation of nucleosome translational isomers. Nucleosomes were assembled with the 218 bp template containing a single nick (Figure 1), subjected to human DNA ligase I, and labeled ligation products were isolated on a 5% polyacrylamide ‘translational’ gel. Lanes 1–8 contain nucleosomes treated with 15 U of human DNA ligase I for 0.25, 0.5, 1, 2, 4, 8, 16 and 32 min. Schematics of nucleosome positions for each translational isomer as determined by hydroxyl radical footprinting are shown (right). (B) Characterization of ligation within individual nucleosome translational isomers. Labeled DNA was isolated from nucleosome and naked DNA bands as shown in (A) and ligation products analyzed on 6% sequencing gels as in Figure 3. Free DNA (lanes 1–8) and nucleosome samples (lanes 9–24) were obtained from reactions containing 1 or 15 U of ligase, respectively. Lanes 9–16 or 17–24 show results for the ‘upper’ or ‘lower’ nucleosomes from (A). (C) Binding of linker histone H1 to nucleosome substrates does not affect the efficiency of ligase. The 218 bp nucleosome substrate containing three nicks was prepared and incubated in the presence or absence of H1 before the addition of 1 U of ligase for 10 min. Reactions were loaded onto nucleoprotein gels to isolate linker histone-bound and/or unbound nucleosomes before sequencing gel analysis of ligase products. Lane 1, no ligase; lanes 2, 3 and 4, ligation within free DNA, nucleosomes or nucleosomes bound by linker histone, respectively. Bands are as indicated in Figure 3.
To determine whether facile equilibration between alternate translational positions, i.e. sliding, is a contributing factor to ligase activity, we performed ligation experiments at a reduced temperature and in the presence of linker histone. It has been shown previously that nucleosome mobility is greatly reduced at temperatures <37°C and as a result of H1 binding (Meersseman et al., 1992; Pennings et al., 1994). If nucleosome mobility increases the accessibility of ligase to the nucleosomal DNA, then reducing the temperature should result in greater inhibition of ligation within the nucleosome relative to naked DNA. Nucleosomes reconstituted with the 218 bp singly nicked template were treated with human DNA ligase for increasing times at 4°C. We still observed only a modest ∼10-fold inhibition of ligase activity relative to naked DNA, as seen previously at higher temperature (Table I). In addition, we find that H1 preferentially binds nucleosomes containing three ligatable nicks in a fashion identical to that of the un-nicked nucleosome (results not shown). Ligation assays carried out in the presence of the linker histone H1 or with H1 and at low temperature also exhibited only a ∼10-fold inhibition of ligase activity at the site near the nucleosome dyad (Figure 4C; Table I). These results together indicate that rapid nucleosome repositioning (sliding) does not contribute to the accessibility of the site near the nucleosomal dyad to human DNA ligase I.
The core histone tail domains can inhibit ligation of nucleosome core DNA
In addition to assessing ligase function with a nucleosome containing extra-nucleosomal ‘linker’ DNA, we also tested the ability of ligase to seal a nick near the center of a nucleosome assembled on a shorter 154 bp DNA substrate. This substrate contains only the sequence occupied by the nucleosome on the 218 bp substrate and retains the single nick at the same position near the dyad (see Figure 1). The DNA is just long enough to wrap about 1¾ turns around the core histone octamer and lacks any ‘linker’ DNA. Thus, reconstitution with this DNA yields a nucleosome core particle (Lee and Hayes, 1998). The 154 bp nucleosome core substrate was subjected to human DNA ligase I for various periods of time, and the rate of ligation within naked and nucleosomal DNA analyzed as described above for the 218 bp nucleosome (Figure 5). As expected, no ligation is evident in the absence of added human DNA ligase I (Figure 5, lane 1). Within the free DNA in the sample, substantial ligation of the original labeled 88 nucleotide primer is observed, resulting in a 154 bp ligation product after 1 min with 1 U of ligase (Figure 5, lane 2). However, in nucleosomes from the same reaction tube no ligation was evident, suggesting that the 154 bp nucleosome structure was highly inhibitory to human DNA ligase I. Indeed, 50 U of ligase were required for 30 min to observe a measurable amount of ligation product in the 154 bp nucleosome substrate (Figure 5, lane 4). Quantitation of such results revealed a decrease in ligation of ∼10 000-fold at the dyad site in the 154 bp nucleosome compared with the identical site in the free DNA (Table I).
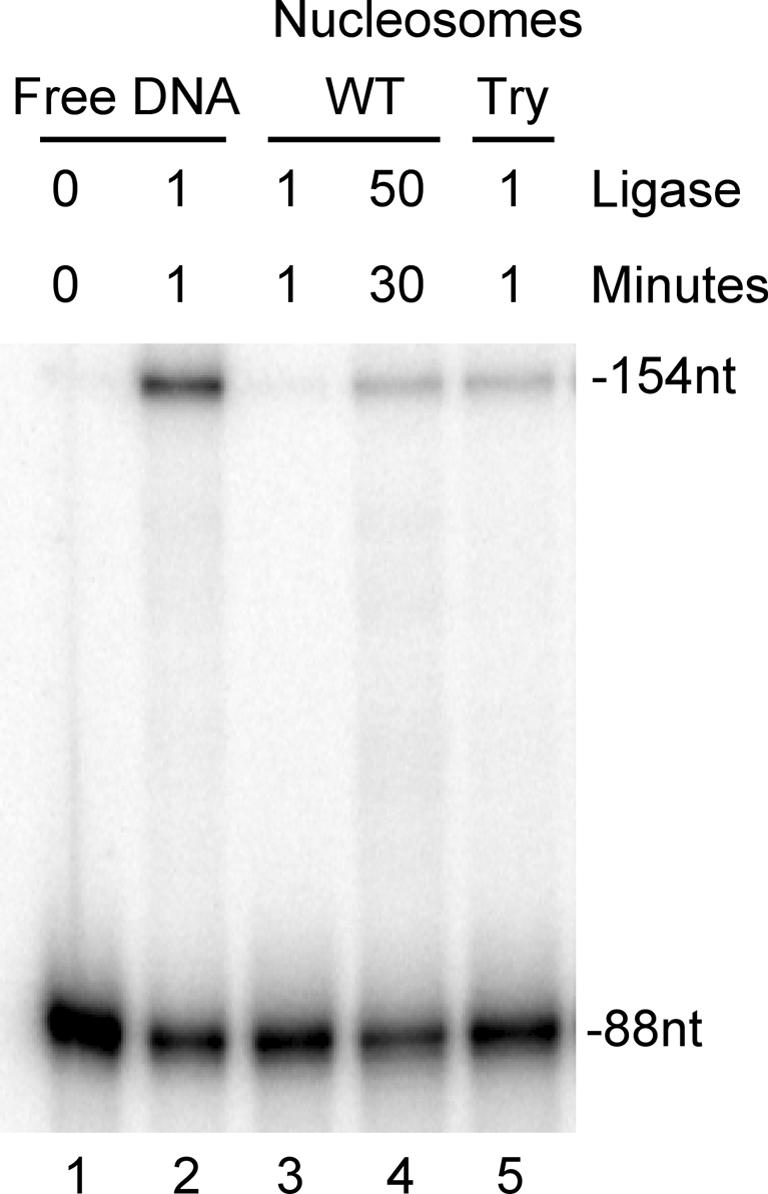
Fig. 5. Ligation of 154 bp nucleosome core substrates. Nucleosome cores were assembled with the 154 bp substrate (Figure 1), then subjected to the indicated levels of human DNA ligase for the indicated times before separation of free DNA and nucleosomes on preparative nucleoprotein gels and analysis of ligation on 6% sequencing gels. Lane 1, no ligase added; lane 2, ligation within free DNA; lanes 3 and 4, ligation of nucleosomal DNA containing full-length histones (WT) with 1 U of ligase for 1 min or with 50 U of ligase for 30 min, respectively; lane 5, ligation within trypsinized 154 bp nucleosomes lacking core histone tail domains (Try). Positions of 88 nucleotide labeled unligated oligonucleotide and 154 bp ligation product are indicated to the right of the gel.
Ligation within the 154 bp nucleosome is ∼1000× slower than at the same site in the 218 bp nucleosome. We then asked what structural feature of the 154 bp substrate modulates the accessibility of human DNA ligase I. It has been shown that some of the highly basic core histone tails bind to different regions of nucleosomal DNA, depending upon the presence or absence of the linker DNA (Usachenko et al., 1994; Lee and Hayes, 1998). It is possible that the lack of linker DNA in the 154 bp substrate constrains the core histone tails to bind DNA within the nucleosome core, thus sterically inhibiting ligase from accessing the nick near the dyad. If this were the case, then removing the core histone tails in the shorter 154 bp substrate would allow ligase to interact with the nucleosomal DNA and result in a relatively rapid rate of ligation. Thus, we reconstituted nucleosomes on the 154 bp substrate with core histone proteins in which the tail domains had been removed by light trypsin digestion (Bohm and Crane-Robinson, 1984; van Holde, 1989). In contrast to the large inhibition of ligase observed when nucleosomes were formed using wild-type (wt) histone proteins, nucleosomes reconstituted with ‘tailless’ histones exhibited significantly increased rates of ligation, similar to that observed for the 218 bp nucleosome (Figure 5, lane 5; Table I).
Removal of the core histone tails does not result in substantially increased exposure of DNA near the nucleosome dyad
The effect of removing the core histone tail domains on ligase activity suggests that one or more tails sterically block access of ligase to DNA in the 154 bp nucleosomal substrate. However, it is possible that removal of the tail domains results in significantly greater site exposure by some mechanism. Site exposure before and after tail removal can be accurately measured based on the ability of a DNA restriction enzyme to digest nucleosome DNA, as described by Polach and Widom (1995). By comparing the rate of restriction endonuclease digestion of nucleosomes and free DNA, the equilibrium distribution of fully site-exposed to unexposed nucleosomes can be quantified. Typically, the activity of proteins requiring full exposure of a nucleosomal DNA site is reduced ∼10 000-fold for sites located near the nucleosome dyad compared with the same DNA free of histones (Polach and Widom, 1995; Anderson and Widom, 2000). In order to determine whether removal of the core histone tails caused a large increase in the extent of DNA site exposure within the tailless nucleosome substrate we performed _Sac_I restriction enzyme digestion experiments with 154 bp nucleosomes containing either intact or trypsinized core histones. _Sac_I digestion cuts these 5S DNA fragments at the –5 position, very near the location of the nick in the ligation substrate (–12) (Figure 6A; see Materials and methods). Digestion of the nucleosomal DNA was compared with the digestion of a different length (but otherwise identical) naked DNA fragment included in the same reaction (corrected for the amount of enzyme). When 154 bp nucleosome substrates containing intact core histones were subjected to _Sac_I for increasing times, digestion occurred at a rate ∼5 orders of magnitude slower than that for the naked DNA control (Figure 6; Table I). Surprisingly, and in contrast to the ligase results, removal of the core histone tails did not significantly increase the accessibility of the nucleosomal DNA to _Sac_I restriction endonuclease (Figure 6B and C; Table I). Moreover, _Sac_I digested a 218 bp nucleosome substrate with nearly identical kinetics (Table I). These results indicate that neither removal of the core histone tails nor the presence of linker DNA results in a large increase in accessibility of nucleosomal DNA near the dyad for a restriction endonuclease.

Fig. 6. Removal of histone tail domains does not substantially increase site exposure within the nucleosome. (A) Schematic of nucleosomes. Positions of nucleosome (oval) and _Sac_I sites (block) are indicated. Except for the introduction of the _Sac_I site (see text), templates are identical to those shown in Figure 1. (B) _Sac_I digestion of nucleosomes and naked DNA. Nucleosomes reconstituted with the 154 bp substrate were mixed with the 218 bp free DNA and subjected to either 4 U/ml (not shown) or 10 000 U/ml (shown) of _Sac_I restriction endonuclease for various periods of time, then digestion products analyzed on non-denaturing acrylamide gels. Digestion products from wt or trypsinized nucleosomes are shown. Samples in lanes 1–9 were digested for 0, 0.5, 5, 15, 30, 45, 60, 90 and 120 min, respectively. Note that _Sac_I cleavage of both the nucleosomal template and the naked DNA control yields the same size DNA product (Frags). (C) Kinetics of _Sac_I digestion of nucleosomes and naked DNA. Data such as those shown in (B) were quantitated and plotted as the log fraction of uncut DNA remaining versus time of digestion. Shown are plots from digestions of 154 bp nucleosomes containing wt or trypsinized (tailless) nucleosomes with 10 000 U/ml _Sac_I and naked DNA digested with 4 U/ml _Sac_I, as indicated in the inset. Lines represent the results of fits of a single exponential first-order decay to the data.
Discussion
The major conclusion from this work is that human DNA ligase I can seal nicks in the midst of histone–DNA interactions. Ligase activity is reduced only ∼10-fold for a site within the nucleosome, much less than expected if this enzyme required full release of the DNA from the surface of the histone octamer. In addition, we find that the binding of linker histone H1 does not affect the ability of human DNA ligase I to ligate nicks within the nucleosome, but that ligase is sensitive to the disposition of the core histone tail domains. These results provide a simple explanation for the observed accessibility of ligase I to nicks dispersed throughout the yeast genome in vivo (Bielinsky and Gerbi, 1999) and imply that human DNA ligase I will have a much greater range of potential substrates than previously expected.
Nucleosomal DNA is accessible to DNA ligase
We demonstrated that human DNA ligase I retains high activity towards nicks in nucleosomal DNA. Nucleosomes can transiently expose internal DNA, most likely via an unwrapping mechanism, thereby allowing DNA-binding factors to gain access to their cognate sites (Polach and Widom, 1995). This unwrapping occurs with a probability of ∼1:10 000 (or greater) for a site near the nucleosomal dyad (Polach and Widom, 1995; Anderson and Widom, 2000). Thus, the affinity of a DNA-binding factor that requires histone-free DNA for a site near the center of the nucleosome would be reduced ∼10 000-fold relative to its affinity for naked DNA, due to the energetic cost of breaking ∼75 bp worth of histone–DNA interactions. Likewise, if complete release of the DNA site surrounding the nick were required for human DNA ligase activity, we would expect a corresponding ∼10 000-fold reduction in the rate of ligation upon nucleosome formation. Surprisingly, at the most inaccessible portion of the nucleosome (dyad), we see only a modest 10-fold reduction in ligase activity, suggesting that human DNA ligase I can readily access nucleosomal DNA. These results indicate that human DNA ligase I does not require significant disruption of histone–DNA interactions to carry out its enzymatic function on the surface of the nucleosome.
The basis for this higher than expected accessibility is not clear but several possibilities exist. First, human DNA ligase I molecules most likely have a smaller recognition sequence than restriction endonucleases (see below) or most other DNA-binding proteins. Thus, ligase would require interactions with only a few base pairs-worth of DNA, which are available without disruption of histone–DNA interactions. Secondly, all of the nicks we have examined were designed in regions of the nucleosomal DNA that orient the nick toward the bulk solvent and away from the surface of the histone octamer. At this point it is unclear exactly what tangential orientations on the nucleosome surface are available to the ligase or if this is a general property of all DNA ligases or just eukaryotic DNA ligases that have evolved to accommodate chromatin structure.
It is unlikely that translational heterogeneity within the ensemble of nucleosomes or nucleosome sliding contributes to the observed rapid rate of ligase activity on nucleosome DNA. Considerable information supports this conclusion. First, nucleosome positions detectable by hydroxyl radical footprinting are located as expected, directly over the NPE of the 5S fragment (Figure 2). Secondly, a nick placed within the ‘linker DNA’, outside of the nucleosome binding region, at the 3′ end of the template ligates with kinetics identical to free DNA, suggesting that this region is free of histone–DNA interactions. Thirdly, ligation assays carried out at reduced temperature did not result in a further reduction in the activity with nucleosome substrates compared with naked DNA. Fourthly, reconstituted 5S nucleosome substrates analyzed on polyacrylamide ‘translational’ gels revealed two major translational isomers, both of which contained nucleosomes positioned near the 5′ end of the DNA template separated in translational positions by only 10–20 bp (results not shown; see Figure 4A). Moreover, both of these nucleosome translational isomers ligated with identical kinetics and the binding of linker histone H1 to nucleosome substrates did not further inhibit activity of the ligase (Figure 4C). Finally, analysis of site exposure by restriction enzyme digestion indicated that >95% of nucleosomes exhibited substantially reduced accessibility of DNA near the predicted dyad (Figure 6C; Table I).
Repositioning of the core histone tail domains
We have shown that the relatively rapid rate of ligation within the 218 bp nucleosome substrate was drastically reduced when a nearly identical 154 bp substrate was prepared lacking ‘linker’ DNA. However, the ∼10 000-fold inhibition we observed with the 154 bp nucleosome core substrate was nearly completely lost upon removal of the core histone tail domains (see Table I). Importantly, we find that removal of the tail domains or the addition of linker DNA (as in the 218 bp nucleosome) does not result in a significant change in exposure of DNA sites near the nucleosome dyad, as measured by digestion with a restriction endonuclease (Figure 6; Table I). Assembly of a nucleosome on the 154 bp DNA reduces the rate of _Sac_I digestion by ∼5 orders of magnitude, consistent with previous results (Polach and Widom, 1995; Anderson and Widom, 2000). In contrast to the large effect observed with DNA ligase, removal of the core histone tail domains results in a small, ∼2× increase in the rate of digestion by _Sac_I restriction endonuclease, again consistent with a recent report (Polach et al., 2000). Thus, the large effect of tail removal on ligase activity must be due to a mechanism other than increased release of the DNA from the histone octamer surface. Moreover, and again in contrast to the ligase results, we find an equivalent degree of inhibition of _Sac_I digestion for both the 154 and 218 bp nucleosomes. Thus, the existence of extra-nucleosomal ‘linker’ DNA does not cause a significant increase in exposure of intra-nucleosomal DNA sites that would account for the differences in ligase activity between these two substrates.
Our data support a model in which the core histone tails physically block the interaction of human DNA ligase I within the 154 bp nucleosome core substrate but not in the 218 bp substrate containing linker DNA or the 154 bp substrate containing trypsinized core histones lacking their tail domains. The core histone tail domains have been implicated in a variety of regulatory functions in vivo due to the interactions of these highly basic moieties with the negatively charged nucleosomal DNA (Hansen, 1997; Wolffe, 1998). The exact DNA sites bound by the tails are likely to be dependent upon the configuration and the condensation state of the chromatin fiber (Hansen, 1997). For example, the C-terminal tail of H2A contacts DNA near the dyad in the nucleosome core particle but ‘moves’ to contact DNA near the edge of the core region when linker DNA is present (Usachenko et al., 1994; Lee and Hayes, 1998). Moreover, UV-laser-directed cross-linking reveals that, in general, most core histone tails prefer to bind linker DNA outside of the core DNA region (S.Dimitrov, personal communication). Thus, we conclude that in the 154 bp nucleosome, the tails are constrained to bind intra-core DNA, occluding access of DNA ligase. Upon the addition of the extra-nucleosomal DNA to the nucleosome core (218 bp nucleosome), the tails ‘reposition’ to bind extra-core linker DNA sites, thereby allowing DNA ligase free access to the DNA on the surface of the nucleosome. This model also explains why ligase accessibility is greatly increased by direct removal of the tails by limited trypsin digestion. Thus, the nucleosome core may be considered a somewhat non-physiological substrate in this regard (Zlatanova and van Holde, 1999). However, it is possible that in some configurations of the chromatin fiber in vivo the tails would be appropriately placed to occlude ligase. Thus, the tails can directly mediate access of enzymes like human DNA ligase I to nucleosomal DNA.
Biological relevance
Thousands of sites on the genome of mammalian cells sustain damage every day. This damage is in the form of modified bases caused by UV light [T–T pairs, cyclobutane pyrimidine dimers (CPDs), etc.], missing bases (abasic sites), double- and single-strand breaks. Studies indicate that many types of damage can be accommodated by the structure of the nucleosome. For instance, Mann et al. (1997) reported that lesions produced from the addition of (+/–)-anti-benzo[_a_]pyrene diol epoxide (BPDE) enhanced nucleosome formation, whereas lesions caused by UV light (CPDs) disrupted nucleosome formation. Even so, CPDs are still incorporated into nucleosome structures but in some instances are accommodated by orienting the damage away from the histone surface (Gale et al., 1987; Suquet and Smerdon, 1993; Liu et al., 2000). Not surprisingly, we found that placement of 1–3 nicks in the 5S DNA backbone does not interfere with nucleosome formation or the translational or rotational phasing of the histone octamer (Figure 2) and it is likely that the presence of nicks within DNA in vivo will not hinder nucleosome formation. Moreover, DNA damage events occur directly within nucleosomes and partial repair of such damage will result in a nicked nucleosomal substrate (Liu et al., 2000).
The ability of human DNA ligase I to use nucleosomal DNA as a substrate represents a significant advantage to the cell. This ability potentially circumvents the need for utilization of ATP by activities that ‘remodel’ nucleosomes to allow access to nucleosomal DNA. This saving is quite substantial considering the frequent occurrence of nicks (about one in every 100 bp) during lagging strand DNA synthesis. Furthermore, our results suggest that the ‘window of opportunity’ for ligase activity following replication is much longer than expected. After passage of the replication fork, nucleosome deposition occurs in a stepwise manner where the H3/H4 histones initially wrap the DNA loosely into tetramer–DNA complexes (Figure 7). Subsequently, two copies each of the H2A/H2B histone dimers join the tetramer complexes and form nucleosomes. Finally, linker histone H1 binding completes assembly of the nucleosome. Most transcription factors access DNA relatively freely until the assembly of the H2A/H2B dimers onto the DNA (Wolffe, 1998). However, our data suggest that the assembly of nucleosomes and even the binding of H1 within nascent chromatin will present only a minor barrier for human DNA ligase I (Figure 7). Not until the nucleosomal array compacts into the chromatin fiber is ligase activity expected to be significantly reduced, and even then regions on the exterior of the folded fiber would be available to the enzyme. Indeed, reactivation of a temperature-sensitive ligase mutant in yeast suggests that the bulk of nicks generated during DNA replication can be sealed long after chromatin assembly is expected to be completed (Bielinsky and Gerbi, 1999). These observations are consistent with a model in which at least the last step in the processing of Okazaki fragments is not strictly constrained by the kinetics of nucleosome deposition. It will be interesting to determine whether other Okazaki fragment processing enzymes exhibit similar levels of accessibility on physiological nucleosome substrates.

Fig. 7. Accessibility of human DNA ligase I during chromatin assembly after passage of the replication fork. Structures predicted to be relatively accessible or inaccessible to ligase I are indicated.
Materials and methods
Construction of substrates for nucleosome assembly and DNA ligase
The three different ligase substrates used in this study were based on the X.borealis somatic-type 5S rDNA nucleosome-positioning sequence (Hayes and Wolffe, 1992). Reconstitution of DNAs containing this sequence results in nucleosomes having their dyad axis (center) located near the transcriptional start site of the 5S gene (position +1; see Figure 1). First, asymmetric PCR was performed with _Eco_RI-digested plasmid pXP-10 and either primer CTCGAATGGCAAAAGTGCAAAAGCC or ACTAACCAGGCCCGACCCTGCTTGGCT, and products were purified on 1.8% agarose gels. This generated single-stranded DNAs extending from positions –78 to +140 or –78 to +76 in the 5S sequence to form the bottom strands for the 218 or 154 bp templates, respectively (see Figure 1). Double-stranded substrates containing nicks for DNA ligase were then prepared by annealing oligonucleotides complementary to the single-stranded bottom strands. Three separate annealing reactions were used to prepare the 218 bp substrate containing three nicks. Each reaction contained 1 µg of either the 34, 36 or 82mer oligonucleotide radiolabeled at its 5′ end along with 1 µg each of the remaining three cold oligonucleotides (see Figure 1) and 50 ng of the bottom strand DNA from the asymmetric PCR. All labeling reactions were carried out with T4 polynucleotide kinase and [γ-32P]ATP. DNAs were mixed in annealing buffer (10 mM Tris–HCl pH 8.0, 50 mM NaCl) and heated to 90°C for 10 min. The reactions were cooled slowly to room temperature and the double-stranded products isolated by agarose gel electrophoresis. The purified double-stranded DNAs from the three annealing reactions were then mixed together for nucleosome reconstitution. For the 218 bp substrate containing a single nick, the 34 and 36mer oligonucleotides were first exhaustively 5′-phosphorylated with ATP and T4 polynucleotide kinase. These oligonucleotides were then annealed to the bottom strand DNA with the 82mer and the annealed product exhaustively treated with T4 DNA ligase. The continuous 152 nucleotide product was purified by denaturing gel electrophoresis and radiolabeled at the 5′ end with T4 kinase and [γ-32P]ATP. Approximately 0.5 µg of this product were annealed with 1 µg of the 66 nucleotide oligo and 50 ng of the 218 nucleotide single-stranded bottom strand DNA from the asymmetric PCR (Figure 1). The 154 bp template was prepared by direct annealing of the complementary 66mer and a 5′ radiolabeled 88mer oligonucleotide with the 154 nucleotide single-stranded bottom strand (Figure 1).
Nucleosome reconstitution
Nucleosomes were reconstituted onto nicked DNA substrates by a standard salt step dialysis procedure (Hayes and Lee, 1997). Briefly, equi molar ratios of purified chicken erythrocyte core histone proteins were mixed with 4.5 µg of unlabeled calf thymus DNA, 160 µl of 5 M NaCl, 750 000 c.p.m. of labeled substrates from the annealing reactions (∼50 ng) and TE buffer in 400 µl total volume. Samples were dialyzed against TE buffer, 1 mM dithiothreitol (DTT) containing 1.2, 1.0, 0.8 and 0.6 M NaCl for 2 h each at 4°C, then finally against TE buffer containing no salt overnight. Nucleosome preparations were analyzed on analytical 0.7%/0.5× TBE agarose gels to ensure proper formation.
Hydroxyl radical footprinting of nucleosomes
Approximately 10 000 c.p.m. (∼90 ng of total DNA) of nucleosomes were diluted to 28 µl with TE buffer. Four microliters of 1 mM Fe-EDTA and 4 µl of 20 mM sodium ascorbate were pipetted to the side of the reaction vessel. The reaction was started by pipetting 4 µl of a 1:250 dilution of a 30% solution of H2O2 into the footprinting reagents and then quickly mixing the reagents with the nucleosome solution by pipetting. After 2 min, 4 µl of 50% glycerol and 10 mM EDTA were added to stop the reaction. The reactions were then loaded onto preparative 0.7% agarose/0.5× TBE gels to isolate nucleosomes and naked DNA.
Ligation reactions
Typical 40 µl reactions contained 4 µl of 10× ligation buffer (200 mM Tris–HCl pH 8.0, 0.1 mM DTT, 500 µg/ml bovine serum albumin, 2 mM ATP, 20 mM MgCl2), 36 µl of reconstituted nucleosomes (∼320 ng of total DNA) and the indicated units of human DNA ligase I for the indicated times. For the experiment in Figure 5, reconstituted nucleosomes were incubated with 20–40 ng of linker histone H1 as described (Hayes and Wolffe, 1992). Reactions were stopped by adding 4 µl of 50% glycerol and 100 mM EDTA. Ligation reactions were loaded directly onto either 0.7% preparative agarose gels run in 1× TBE, or 5% polyacrylamide ‘translational’ gels run in 20 mM HEPES pH 7.5 to separate nucleosome complexes from naked DNA. Translational gels were performed with the 218 bp template containing a single nick because the 218 bp template containing multiple nicks did not yield sharp bands in our hands on the 5% acrylamide ‘translational’ gels. After separation, labeled DNA was purified from nucleosome and naked DNA bands. The DNAs were denatured and separated on 6% sequencing gels, the gels were dried and ligation products quantitated using a PhosphorImager. Fold inhibition for nucleosome samples was calculated at each time point as the amount of ligation product (band density) observed in the nucleosome with respect to that obtained with the naked DNA sample, normalized for enzyme concentration, then all time points averaged. Alternatively, slopes of plots of the ln (band density) versus time for nucleosome and naked DNA samples were compared, again corrected for the amount of enzyme used in the particular reaction. Both methods yielded similar fold inhibitions.
Conformational equilibrium studies
Conformational equilibrium experiments were carried out as described with modifications (Polach and Widom, 1995). A DNA fragment identical to the 154 bp ligation substrate was prepared (see Figure 1) except for the introduction of a single _Sac_I site located near the nucleosomal dyad. This was achieved by mutating two base pairs within the 5S sequence by standard PCR at positions –71 A to G (to destroy a _Sac_I site) and a T to A at position –5 to introduce a _Sac_I cleavage site at position –5. This 154 bp fragment was radiolabeled at the 5′ _Eco_RI site with [γ-32P]ATP and T4 polynucleotide kinase by standard methods and reconstituted into nucleosomes containing either wt or tailless core histones prepared by limited trypsin digestion (Vitolo et al., 2000). Nucleosomes were purified by 5–20% sucrose gradients as previously described (Hayes and Lee, 1997). In some experiments, a 218 bp _Eco_RI–_Dde_I fragment containing the 154 bp sequence was radiolabeled and added to the preparations of purified nucleosomes to serve as an internal naked DNA control. Nucleosomes and naked DNA were then digested with _Sac_I. Typical digestion reactions contained ∼100 ng of total DNA in 1× New England Biolabs buffer 2 and _Sac_I (10 000 U/ml for nucleosomal DNA or 4 U/ml for naked DNA). Reactions were incubated at 37°C and 10 µl aliquots were removed at the indicated times, and the digestion quenched by the addition of EDTA and SDS to 40 mM and 0.2% final concentration, respectively. DNA products were separated by electrophoresis on 5% polyacrylamide gels containing 0.2% SDS. Gels were dried and quantitated by PhosphorImager analysis and the fraction of uncut DNA determined. Alternatively, in some experiments the naked DNA control was omitted and the relative rates of nucleosome digestions determined by following the appearance of the cleaved fragment. Data were fit to a first-order exponential decay and conformational equilibrium constants were calculated as previously described (Polach and Widom, 1995) or fold inhibition was calculated as described above.
Acknowledgments
Acknowledgements
We thank Woong Kim for preparation of histones and Drs Jonathan Widom and Jeffrey Hansen for helpful discussions. This work was supported by NIH grant GM52426 and an American Cancer Society grant RPG-00-080-01-GMC. J.M.V. was supported by NIH grant T32 DE07202.
References
- Almouzni G. and Mechali,M. (1988) Assembly of spaced chromatin promoted by DNA synthesis in extracts from Xenopus eggs. EMBO J., 7, 665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almouzni G., Clark,D.J., Mechali,M. and Wolffe,A.P. (1990) Chromatin assembly on replicating DNA in vitro. Nucleic Acids Res., 18, 5767–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J.D. and Widom,J. (2000) Sequence and position-dependence of the equilibrium accessibility of nucleosomal DNA target sites. J. Mol. Biol., 296, 979–987. [DOI] [PubMed] [Google Scholar]
- Anderson S. and DePamphilis,M.L. (1979) Metabolism of Okazaki fragments during simian virus 40 DNA replication. J. Biol. Chem., 254, 11495–11504. [PubMed] [Google Scholar]
- Annunziato A.T. and Seale,R.L. (1982) Maturation of nucleosomal and non-nucleosomal components of nascent chromatin: differential requirements for concurrent protein synthesis. Biochemistry, 21, 5431–5438. [DOI] [PubMed] [Google Scholar]
- Bambara R.A., Murante,R.S. and Henricksen,L.A. (1997) Enzymes and reactions at the eukaryotic replication fork. J. Biol. Chem., 272, 4647–4650. [DOI] [PubMed] [Google Scholar]
- Bielinsky A.-K. and Gerbi,S.A. (1999) Chromosomal ARS1 has a single leading strand start site. Mol. Cell, 3, 477–486. [DOI] [PubMed] [Google Scholar]
- Bohm L. and Crane-Robinson,C. (1984) Proteases as structural probes for chromatin: the domain structure of histones. Biosci. Rep., 4, 365–386. [DOI] [PubMed] [Google Scholar]
- Cirillo L.A., McPherson,C.E., Bossard,P., Stevens,K., Cherian,S., Shim,E.Y., Clark,K.L., Burley,S.K. and Zaret,K.S. (1998) Binding of the winded-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J., 17, 244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusick M.E., Lee,K.S., DePamphilis,M.L. and Wassarman,P.M. (1983) Structure of chromatin at replication forks: nuclease hypersensitivity results from both prenucleosomal deoxyribonucleic acid and an immature chromatin structure. Biochemistry, 22, 3873–3884. [DOI] [PubMed] [Google Scholar]
- Gaillard P.H., Moggs,J.G., Roche,D.M., Quivy,J.P., Becker,P.B., Wood,R.D. and Almouzni,G. (1997) Initiation and bidirectional propagation of chromatin assembly from a target site for nucleotide excision repair. EMBO J., 16, 6281–6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale J.M., Nissen,K.A. and Smerdon,M.J. (1987) UV-induced formation of pyrimidine dimers in nucleosome core DNA is strongly modulated with a period of 10.3 bases. Proc. Natl Acad. Sci. USA, 84, 6644–6648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Ramirez M., Rocchini,C. and Ausio,J. (1995) Modulation of chromatin folding by histone acetylation. J. Biol. Chem., 270, 17923–17928. [DOI] [PubMed] [Google Scholar]
- Georgel P.T., Tsukiyama,T. and Wu,C. (1997) Role of the histone tails in nucleosome remodeling by Drosophila NURF. EMBO J., 16, 4717–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordenin D.A., Kunkel,T.A. and Resnick,M.A. (1997) Repeat expansion—all in a flap? Nature Genet., 16, 116–118. [DOI] [PubMed] [Google Scholar]
- Hansen J. (1997) The core histone amino termini: combinatorial interaction domains that link chromatin structure with function. Chemtracts: Biochem. Mol. Biol., 10, 56–69. [Google Scholar]
- Hayes J.J. and Lee,K.-M. (1997) In vitro reconstitution and analysis of mononucleosomes containing defined DNAs and proteins. Methods, 12, 2–9. [DOI] [PubMed] [Google Scholar]
- Hayes J.J. and Wolffe,A.P. (1992) Transcription factor interaction with nucleosomal DNA. BioEssays, 14, 597–603. [DOI] [PubMed] [Google Scholar]
- Herman T.M., DePamphilis,M.L. and Wassarman,P.M. (1981) Structure of chromatin at deoxyribonucleic acid replication forks: location of the first nucleosomes on newly synthesized simian virus 40 deoxyribonucleic acid. Biochemistry, 20, 621–630. [DOI] [PubMed] [Google Scholar]
- Howe L., Ranalli,T.A., Allis,C.D. and Ausio,J. (1998) Transcriptionally active Xenopus laevis somatic 5S ribosomal RNA genes are packaged with hyperacetylated histone H4, whereas transcriptionally silent oocyte genes are not. J. Biol. Chem., 273, 20693–20696. [DOI] [PubMed] [Google Scholar]
- Kingston R.E. and Narlikar,G.J. (1999) ATP-dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev., 13, 2339–2352. [DOI] [PubMed] [Google Scholar]
- Lee D., Hayes,J.J., Pruss,D. and Wolffe,A.P. (1993) A positive role for histone acetylation in transcription factor binding to nucleosomal DNA. Cell, 72, 73–84. [DOI] [PubMed] [Google Scholar]
- Lee K.-M. and Hayes,J.J. (1998) Linker DNA and H1-dependent reorganization of histone tail–DNA interactions within the nucleosome. Biochemistry, 37, 8622–8628. [DOI] [PubMed] [Google Scholar]
- Liu X., Mann,D.B., Suquet,C., Springer,D.L. and Smerdon,M.J. (2000) Ultraviolet damage and nucleosome folding of the 5S ribosomal RNA gene. Biochemistry, 39, 557–566. [DOI] [PubMed] [Google Scholar]
- Logie C., Tse,C., Hansen,J.C. and Peterson,C.L. (1999) The core histone N-terminal domains are required for multiple rounds of catalytic chromatin remodeling by the SWI/SNF and RSC complexes. Biochemistry, 38, 2514–2522. [DOI] [PubMed] [Google Scholar]
- Mann D.B., Springer,D. and Smerdon,M.J. (1997) DNA damage can alter the stability of nucleosomes: effects are dependent on damage type. Proc. Natl Acad. Sci. USA, 94, 2215–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meersseman G., Pennings,S. and Bradbury,E.M. (1992) Mobile nucleosomes—a general behavior. EMBO J., 11, 2951–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennings S., Meersseman,G. and Bradbury,E.M. (1994) Linker histones H1 and H5 prevent the mobility of positioned nucleosomes. Proc. Natl Acad. Sci. USA, 91, 10275–10279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlmann T. and Wrange,O. (1988) Specific glucocorticoid receptor binding to DNA reconstituted in a nucleosome. EMBO J., 7, 3073–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polach K.J. and Widom,J. (1995) Mechanism of protein access to specific DNA sequences in chromatin: a dynamic equilibrium model for gene regulation. J. Mol. Biol., 254, 130–149. [DOI] [PubMed] [Google Scholar]
- Polach K.J., Lowary,P.T. and Widom,J. (2000) Effects of core histone tail domains on the equilibrium constants for dynamic DNA site accessibility in nucleosomes. J. Mol. Biol., 298, 211–223. [DOI] [PubMed] [Google Scholar]
- Prigent C., Satoh,M.S., Daly,G., Barnes,D.E. and Lindahl,T. (1994) Aberrant DNA repair and DNA replication due to an inherited enzymatic defect in human DNA ligase I. Mol. Cell. Biol., 14, 310–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy P.S. and Housman,D.E. (1997) The complex pathology of trinucleotide repeats. Curr. Opin. Cell Biol., 9, 364–372. [DOI] [PubMed] [Google Scholar]
- Seale R.L. (1978) Nucleosomes associated with newly replicated DNA have an altered conformation. Proc. Natl Acad. Sci. USA, 75, 2717–2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sera T. and Wolffe,A.P. (1998) Role of histone H1 as an architectural determinant of chromatin structure and as a specific repressor of transcription on Xenopus oocyte 5S rRNA genes. Mol. Cell. Biol., 18, 3668–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smerdon M.J. and Thoma,F. (1990) Site-specific DNA repair at the nucleosome level in a yeast minichromosome. Cell, 61, 675–684. [DOI] [PubMed] [Google Scholar]
- Smith S. and Stillman,B. (1989) Purification and characterization of CAF-I, a human cell factor required for chromatin assembly during DNA replication in vitro. Cell, 58, 15–25. [DOI] [PubMed] [Google Scholar]
- Smith S. and Stillman,B. (1991) Stepwise assembly of chromatin during DNA replication in vitro. EMBO J., 10, 971–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sogo J.M., Stahl,H., Koller,T. and Knippers,R. (1986) Structure of replicating simian virus 40 minichromosomes. The replication fork, core histone segregation and terminal structures. J. Mol. Biol., 189, 189–204. [DOI] [PubMed] [Google Scholar]
- Suquet C. and Smerdon,M.J. (1993) UV damage to DNA strongly influences its rotational setting on the histone surface of reconstituted nucleosomes. J. Biol. Chem., 268, 23755–23757. [PubMed] [Google Scholar]
- Tse C., Sera,T., Wolffe,A.P. and Hansen,J.C. (1998) Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol. Cell. Biol., 18, 4629–4638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ura K., Kurumizaka,H., Dimitrov,S., Almouzni,G. and Wolffe,A.P. (1997) Histone acetylation: influence on transcription, nucleosome mobility and positioning and linker histone-dependent transcriptional repression. EMBO J., 16, 2096–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usachenko S.I., Bavykin,S.G., Gavin,I.M. and Bradbury,E.M. (1994) Rearrangement of the histone H2A C-terminal domain in the nucleosome. Proc. Natl Acad. Sci. USA, 91, 6845–6849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Holde K.E. (1989) Chromatin. Springer-Verlag, New York, NY. [Google Scholar]
- Vettese-Dadey M., Walter,P., Chen,H., Juan,L.-J. and Workman,J.L. (1994) Role of the histone amino termini in facilitated binding of a transcription factor, GAL4-AH to nucleosome cores. Mol. Cell. Biol., 14, 970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignali M., Hassan,A.H., Neely,K.E. and Workman,J.L. (2000) ATP-dependent chromatin-remodeling complexes. Mol. Cell. Biol., 20, 1899–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitolo J.M., Thiriet,C. and Hayes,J.J. (2000) The N-terminal tail domains of H3 and H4 are the primary mediators of transcription factor IIIA access to DNA within a nucleosome. Mol. Cell. Biol., 20, 2167–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waga S. and Stillman,B. (1998) The DNA replication fork in eukaryotic cells. Annu. Rev. Biochem., 67, 721–751. [DOI] [PubMed] [Google Scholar]
- Widom J. (1986) Physicochemical studies of the folding of the 100Å nucleosome filament into the 300Å filament: cation dependence. J. Mol. Biol., 190, 411–424. [DOI] [PubMed] [Google Scholar]
- Wolffe A.P. (1998) Chromatin Structure and Function, 3rd edn. Academic Press, San Diego, CA. [Google Scholar]
- Zlatanova J. and van Holde,K.E. (1999) The nucleosome core particle: does it have structural and physiologic relevance? BioEssays, 21, 776–780. [DOI] [PubMed] [Google Scholar]