Archaeal RNA polymerase subunits F and P are bona fide homologs of eukaryotic RPB4 and RPB12 (original) (raw)
Abstract
The archaeal and eukaryotic evolutionary domains diverged from each other ~2 billion years ago, but many of the core components of their transcriptional and translational machineries still display a readily recognizable degree of similarity in their primary structures. The F and P subunits present in archaeal RNA polymerases were only recently identified in a purified archaeal RNA polymerase preparation and, on the basis of localized sequence homologies, tentatively identified as archaeal versions of the eukaryotic RPB4 and RPB12 RNA polymerase subunits, respectively. We prepared recombinant versions of the F and P subunits from Methanococcus jannaschii and used them in in vitro and in vivo protein interaction assays to demonstrate that they interact with other archaeal subunits in a manner predicted from their eukaryotic counterparts. The overall structural conservation of the M.jannaschii F subunit, although not readily recognizable on the primary amino acid sequence level, is sufficiently high to allow the formation of an archaeal–human F-RPB7 hybrid complex.
INTRODUCTION
Archaea are prokaryotic organisms that are generally thought to have retained a substantial degree of similarity to the universal ancestor of all organisms living ~1.8 billion years ago (1). Nowadays they can be found in many different environments, ranging from thermal vents on ocean floors and hot volcanic springs to arctic oceans (2). Despite their morphological similarity to bacteria they contain a transcriptional machinery that is very similar to the ones found, in a more elaborate manner, in eukaryotic cells (3). A series of studies have for instance identified clearly recognizable archaeal homologs of the eukaryotic basal transcription factors TATA-binding protein (TBP) and TFIIB (4,5) that are known from studies on minimal promoters to constitute components of the core RNA polymerase II (RNAPII) transcriptional machinery (6). This exceptional degree of similarity between the archaeal and eukaryotic transcriptional core machineries also extends to the polymerases. Archaeal RNAPs contain, depending on the species, between 10 and 12 different subunits (7,8). The majority of these subunits display a considerable amount of primary sequence homology to the subunits present in eukaryotic RNAPIIs, suggesting that many of the fundamental structural and functional aspects of contemporary eukaryotic RNAPIIs have remained remarkably unchanged throughout evolution.
Until recently it was thought that three subunits (RPB4, RPB8 and RPB12) found in all eukaryotic RNAPIIs studied so far (9) lacked archaeal counterparts and thus may have only evolved specifically in eukaryotic organisms. A recent systematic study of the polypeptides present in highly purified RNAP from the archaeon Methanobacterium thermoautotrophicum by MALDI-TOF analysis revealed, however, the presence of two polypeptides, subunits F and P, that display partial homology to the eukaryotic RPB4 and RPB12 subunits, respectively (8). A closer inspection of the data shows why F and P previously failed to be classified as RNAP subunits. The overall sequence identity of subunit F to representatives of the eukaryotic RPB4 family is very limited (∼15% sequence identity between Methanococcus jannaschi F and human RPB4; Fig. 1A) and had therefore not been previously detected. On the other hand, the sequence similarity between P and eukaryotic RPB12 homologs is much more distinct (Fig. 1B), but is restricted to a small region because P is an extremely short polypeptide (46 amino acids) lacking the N-terminal sequences present in the eukaryotic RPB12 subunits.
Figure 1.

Sequence alignment of archaeal F and P subunits with eukaryotic RPB4 and RPB12 proteins, respectively. (A) Alignment of two representative archaeal F subunit sequences with two eukaryotic RPB4 subunits (mj, M.jannaschii; mt, M.thermoautotrophicum; sp, Schizosaccharomyces pombe; hs, Homo sapiens). Note the limited degree of overall homology and the rather disperse location of similar or identical present in all four polypeptide sequences shown. (B) Alignment of two representative archaeal P subunit sequences with two eukaryotic RPB12 subunits (species abbreviations as above). Dark shaded residues are identical and light shaded residues are similar.
In order to subject the hypothesis, that F and P are genuine homologs of RPB4 and RPB12, respectively, to a more stringent test we have studied the protein interaction properties of these archaeal proteins in relation to other RNAP subunits in more detail. In eukaryotic systems, RPB4 and RPB12 have been implicated in specific interaction with two other RNAP subunits and we would therefore expect similar functional properties from their archaeal counterparts. Extensive genetic and biochemical studies in yeast, human and plant model systems have shown that RPB4 forms a distinct heterodimeric complex with the RNAP subunit RPB7 (10–12). In yeast cells the yRPB4–yRPB7 heterodimer occurs in substoichiometric quantities within the RNAPII of exponentially growing cells (13), but becomes preferentially integrated into RNAPII during stressful growth conditions (e.g. stationary phase; 13,14) to induce a ‘closed’ RNAPII conformation. Electron microscopy studies have shown that the two subunits are located near the DNA-binding cleft of RNAPII, suggesting that they couple the entry of DNA into the active center cleft with the closure of the cleft (15,16). All archaeal genomes sequenced up to now contain a homolog of RPB7, subunit E, that is clearly identifiable on the basis of sequence similarity to its eukaryotic counterpart (17). In order to test whether subunit F represents a genuine homolog of RPB4 we tested if E and F can interact with each other in a manner that would mimic the interaction between RPB4 and RPB7. A similar test can be applied to subunit P: in human RNAPII a distinct and highly specific interaction has been observed to occur between hsRPB12 and hsRPB3 (18). The recent X-ray crystallographic study of yeast RNAPII has indeed confirmed the existence of direct contacts between yRPB3 and yRPB12 (19). We have previously described the assembly of a heterotrimeric RNAP subcomplex consisting of subunits D (RPB3), L (RPB11) and N (RPB10) (20). In this report we show that the P subunit interacts with subunit D and thus allows the assembly of a tetrameric D–L–N–P complex. This D–L–N–P complex is a key assembly intermediate because it provides the structural framework for the recruitment of the large catalytic subunits (21), and thus represents an important stepping stone towards the production of archaeal and eukaryotic RNAPs from recombinant subunits.
MATERIALS AND METHODS
Plasmid constructions
The cloning and expression of the RNAP subunit D from M.jannaschii have been previously described (20). For the study described here we amplified the complete open reading frames encoding RNAP subunits E, F and P by PCR from 1 µg M.jannaschii genomic DNA using the oligonucleotides listed in Table 1. The coding regions of the human subunits hsRPB4 and hsRPB7 were amplified from full-length cDNAs in plasmids generously provided by E. Golemis (Fox Chase Cancer Center, Philadelphia, PA). The resulting PCR fragments were cloned into pGEM-T (Promega) and completely sequenced before they were transferred into either the _Bam_HI, _Xma_I or _Eco_RI sites of the pGEX-2TK for bacterial expression (Amersham-Pharmacia Biotech). All cloning procedures were performed according to standard molecular biological protocols.
Table 1. Oligonucleotide primers used for cloning full-length archaeal and human RNAP subunits.

Bicistronic expression strategy
Bacterial coexpression of recombinant proteins was achieved by cloning the open reading frames encoding the RNAP subunits into the _Bam_HI, _Xma_I and _Eco_RI sites, respectively, of the same pGEX-2TK plasmid. The first open reading frame cloned into the _Bam_HI site was thus fused in frame to the GST coding region, whereas translational initiation of the downstream coding region was initiated by a Shine–Dalgarno ribosome recruitment sequence (AGGAGG) located upstream of the internal ATG start codon (see Table 1 and Figs 2A, 3A and 5A).
Figure 2.

Coexpression of GST–hsRPB4 and hsRPB7 subunits in E.coli. (A) Schematic representation of the expression constructs used. (B) Comparison of the protein expression profiles of bacterial cells harboring the constructs illustrated above. The insoluble and soluble fractions, together with the glutathione column eluates (GCE), are shown for the GST–hsRPB4 (lanes 1, 4 and 7), GST–hsRPB7 (lanes 2, 5 and 8) and GST–hsRPB4/hsRPB7 (lanes 3, 6 and 9) expression strains. The GST–hsRPB4 and GST–hsRPB7 fusion proteins are highly insoluble when expressed by themselves and are thus exclusively present in the pellet fractions (lanes 1 and 2). The bicistronic construct expressing GST–hRPB4 and hRPB7 leads, however, to the formation of a heterodimeric complex with increased solubility that can be purified by glutathione affinity chromatography (lane 9).
Figure 3.

Coexpression of GST–mjF and mjE subunits in E.coli. (A) Schematic representation of the expression constructs used. (B) Comparison of the protein expression profiles of bacterial cells harboring the constructs illustrated above. The insoluble and soluble fractions, together with GCE, are shown for the GST–mjF (lanes 1, 4 and 7), GST–mjE (lanes 2, 5 and 8) and GST–mjF/mjE (lanes 3, 6 and 9) expression strains. The GST–mjE fusion protein is highly insoluble when expressed on its own and is thus exclusively present in the pellet fractions (lane 2). The bicistronic construct expressing GST–mjF and mjE leads, however, to the formation of a heterodimeric complex with increased solubility that can be purified by glutathione affinity chromatography (lane 9).
Figure 5.

Assembly of an archaeal–human hybrid complex. (A) Schematic representation of the expression constructs used. (B) Analysis of the protein expression profiles of bacterial cells harboring the constructs illustrated above. The insoluble and soluble fractions, together with GCE, are shown for the GST–mjF/mjE (lanes 1, 3 and 5) and GST–mjF/hsRPB7 (lanes 2, 4 and 6) expression strains. The cross-species bicistronic construct expressing GSTmjF and hsRPB7 expresses an archaeal–human hybrid complex (lane 6). Partial GST–mjF products present in this lane are indicated by an asterisk.
Recombinant protein production and purification
Escherichia coli BL21 cells containing the various expression plasmids were grown in Terrific Broth medium (EZ Mix, Sigma) to an approximate optical density of A600 = 0.8 before inducing them with 1 mM IPTG for 4 h. The preparation of extracts and purification of recombinant RNAP subunits was essentially performed as described previously (20,22).
Yeast two-hybrid assays
The assays were performed according to the Clontech ‘Yeast Protocols Handbook’. All β-galactosidase liquid assays were done at least in triplicate.
RESULTS
In vivo assembly of recombinant hRPB4–hRPB7 and mjE–mjF binary complexes
The high degree of sequence conservation of the recently identified F subunits (8) across the archaeal domain allowed an unambiguous identification of the corresponding homolog in the genome of M.jannaschii. We therefore retrieved the corresponding open reading frames encoding the M.jannaschii F subunit (henceforth referred to as mjF) by PCR in order to test its protein interaction properties. A specific binary interaction between the RPB4 and RPB7 subunits has been demonstrated in several eukaryotic model systems (10–12). If mjF represents an archaeal version of RPB4 we would expect to observe a specific interaction with the mjE archaeal RNAP subunit, which (based on primary sequence similarity) is a clearly identifiable homolog of the eukaryotic RPB7 subunit. Recombinant mjF can be expressed in soluble form in bacterial expression systems, but its potential interaction partner, mjE, forms insoluble inclusion bodies under a variety of induction conditions (Fig. 3B; J.J.Eloranta and R.O.J.Weinzierl, data not shown). Similarly, the human subunits hsRPB4 and hsRPB7 also fail to be expressed in soluble form in bacterial expression systems (Fig. 2B). Since the lack of solubility of these subunits could at least partially be due to a lack of a suitable protein interaction partner we investigated a coexpression strategy based on bicistronic expression operons. Briefly, we produced one of the subunits as a GST-fusion protein and inserted the coding region for the second subunit downstream of the GST-fusion protein-encoding region, but upstream of the transcription terminator sequence of the expression cassette. Such constructs are expected to give rise to a bicistronic mRNA from which two recombinant proteins are translated in a coordinated manner. We initially tested the validity of this approach by coexpressing the hsRPB4 and hsRPB7 proteins to see whether a soluble hsRPB4–hsRPB7 heterodimeric complex could be produced in such a way. As shown in Figure 2B, the coexpression of GST–hsRPB4 with hsRPB7 does indeed result in a substantial expression of a soluble heterodimeric complex containing the two subunits in an equimolar amount. Thrombin cleavage and further chromatographic purification steps show that the interaction between hRPB4 and hRPB7 is highly specific and stable (data not shown).
To test whether it is possible to obtain a comparable archaeal complex we applied this strategy to the mjE and mjF subunits. Coexpression of GST–mjF with mjE yields a heterodimeric complex that can be specifically affinity-purified on glutathione columns and contains both proteins in stoichiometric quantities. Following the removal of the GST tag, the heterodimeric mjE–mjF complex copurifies in a variety of chromatography steps (Fig. 4). We therefore conclude that mjF and mjE interact with each other in a manner that is comparable to the interaction between hsRPB4 and hsRPB7, and hence mjF does indeed behave like a genuine homolog of RPB4.
Figure 4.
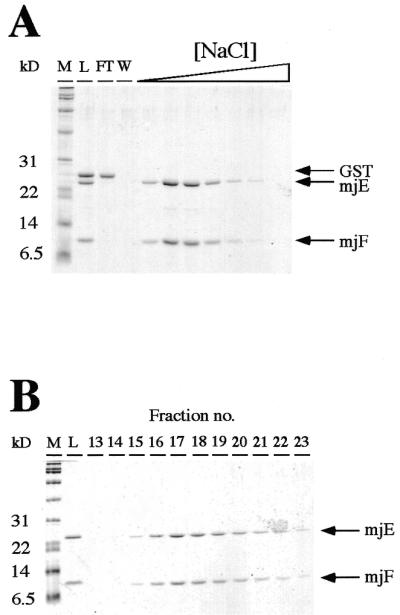
Copurification of mjF–mjE. (A). Thrombin-cleavage of the affinity purified GST–mjF/mjE complex releases the GST moiety from mjF, resulting in a mixture of three polypeptides (lane L). GST does not bind to DEAE–Sepharose (lane FT, flowthrough; lane W, wash fractions), whereas mjE and mjF coelute with each other as the ionic strength in the elution buffer is increased. (B) Native size exclusion chromatography of the mjF–mjE complex. Eluates from the high-salt fractions from the DEAE–Sepharose purification step were chromatographed on an S-100HR size exclusion column. mjE and mjF, despite their different sizes, coelute as a symmetrical peak migrating in a position consistent with their combined molecular weights.
In their original description of the M.jannaschii genome Bult et al. (17) proposed, on the basis of weak sequence homology, the existence of a second RPB7 homolog, E″. Distinct genes encoding homologs of E″ also exist in other euryarchaeal genomes, whereas in the crenarchaeote Sulfolobus acidocaldarius the E″ reading frame appears to be directly fused to the E′ gene (23,24). In order to address the question as to whether the mjE″ displays protein interaction properties similar to mjE (and thus could serve as an alternative/additional subunit in archaeal RNAPs) we coexpressed mjE″ with the mjF subunit from a bicistronic expression construct, and also with both mjF and mjE subunits from a tricistronic construct. mjE″ was not found to be associated with either the mjF, mjE or the mjF–mjE complex (data not shown). This result is compatible with the findings by Darcy et al. (8), showing no evidence for the presence of E″ in purified Methanobacterium RNAP.
A specific interaction of mjF with human RPB7 indicates a high degree of structural conservation
The interactions between the Methanococcus mjE–mjF and human hsRPB4–hsRPB7 subunits observed in the bacterial coexpression systems suggests a basic degree of structural similarity between the two complexes. To investigate further whether the subunit interactions between the archaeal and human subunits is sufficiently highly conserved to allow the formation of an archaeal–human hybrid complex we set up a bacterial GST–mjF and hsRPB7 coexpression system. This bicistronic construct gives rise to a stable and soluble heterodimeric complex that can be purified to near-homogeneity by glutathione affinity chromatography (Fig. 5). This proves conclusively that the archaeal F subunits resemble the eukaryotic RPB4 subunits (at least in the region involved in the heterodimerization) so that a specific complex can be formed between proteins that have been separated by ~1.8 billion years of evolutionary divergence (1). This observation is reminiscent of the interaction that we previously documented to occur between the archaeal D subunit and the yeast RPB11 or AC19 subunits (20).
Interactions between mjD and mjP mirror the ones observed between the eukaryotic RPB3 and RPB12 subunits
All the characterized members of the eukaryotic RPB12 family of RNAP subunits are small polypeptides (∼70–80 amino acids) that are mostly distinguished by the presence of a zinc-binding motif and a highly positively charged C-terminus (25). The archaeal homolog of RPB12 in M.jannaschii, subunit P, contains these features but is even shorter (∼46 amino acids) and its gene was therefore originally not annotated (17). The discovery of a homolog of P in purified Methanobacterium RNAP (8) provided, however, the final piece of evidence that P is actually expressed in vivo and constitutes an integral component of archaeal RNAPs.
A series of interaction assays based on the pairwise coexpression of human RNAPII subunits in a baculovirus expression system have shown that hsRPB12 selectively and exclusively binds to hsRPB3 (18). In order to show that the archaeal homolog, subunit P, behaves similarly we would expect to see a specific interaction between P and subunit D, which is the archaeal homolog of hsRPB3. Due to the small size of P, which might cause technical problems with the expression and identification of the recombinant protein in E.coli extracts, we initially decided to investigate the proposed interaction in the yeast two-hybrid system (26). The open reading frames encoding mjD and mjP where cloned into the appropriate yeast two-hybrid vectors (pGBT9 and pGAD424) and tested for β-galactosidase production after cotransfection in various combinations into the yeast strain AH109. A positive signal observed between the fusion protein of mjD to the GAL4 activation domain and a fusion of mjP to the GAL4 DNA binding domain suggests that the two proteins interact with each other as expected from the properties of their eukaryotic counterparts (Fig. 6). The strength of the signal caused by the interaction is comparable to that observed in the positive control (SV40 large T antigen/p53).
Figure 6.

The yeast two-hybrid system demonstrates a specific in vivo interaction between mjP and mjD. β-Galactosidase assay of yeast strains cotransformed with the indicated GAL4 activation (AAD) and GAL4 DNA-binding (DBD) domain fusion expression plasmids. The β-galactosidase activity produced in the cells containing the various plasmid combinations shown are as a percentage relative to the p53/large T positive control.
Encouraged by the success of using a bacterial coexpression system to produce soluble and stoichiometric hsRPB4–hsRPB7, mjE–mjF and mjF–hsRPB7 complexes we next proceeded to construct a mjP–mjD coexpression plasmid based on these principles. mjP is highly expressed and soluble as a GST-fusion protein. The solubility of GST–mjP is, however, substantially affected when expressed in the presence of mjD. Coexpression of GST–mjP with mjD results in the successful production of both recombinant proteins within the same cell, but in a mostly (>95%) insoluble form (data not shown). This observation is compatible with the hypothesis that specific heterodimeric complexes are formed in vivo between mjD and mjP, causing the normally highly soluble mjP to accumulate as inclusion bodies with its interaction partner mjD.
Assembly of an archaeal D–N–L–P (RPB3–10–11–12) subcomplex
The recent determination of the yeast RNAPII structure at 3.2 Å (19) highlights the key function of a subcomplex consisting of RPB3, RPB10, RPB11 and RPB12. The four subunits are clustered on one end of the enzyme and represent a small complex that almost certainly functions as an assembly platform for the two large subunits. RPB3 and RPB11 display a good structural resemblance to the α subunits of bacterial RNAPs which are known to homodimerize during the first step in the assembly pathway of bacterial RNAPs (27). Experimental evidence in yeast has shown that the association between RPB3 and RPB11 is followed by the recruitment of the second largest subunit, RPB2 (21), suggesting that the assembly patterns of archaeal and eukaryotic enzymes closely mimic the one observed in bacterial systems.
We have previously reported the in vitro assembly of the M.jannaschii homologs of RPB3, RPB10 and RPB11 (mjD–mjN–mjL; 20). Having shown that mjP associates specifically with mjD (see above) we next tested whether it would be possible to incorporate it into the mjD–mjL–mjN complex. mjP was expressed as a GST-fusion protein and purified away from its fusion partner, after thrombin cleavage, by size exclusion chromatography. We prepared the mjD–mjN–mjL complex as described previously (20), added purified recombinant mjP and subjected this combination of proteins to size exclusion chromatography. Figure 7 demonstrates that mjP coelutes with the much larger mjD–mjN–mjL complex, indicating that the subunit becomes incorporated to form the expected tetrameric mjD–mjL–mjN–mjP subcomplex.
Figure 7.
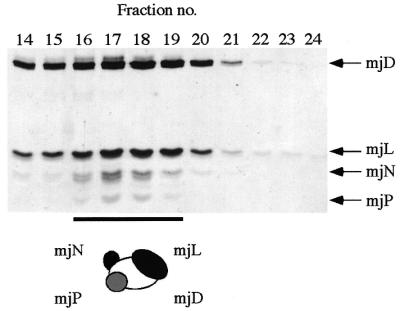
Formation of a heterotetrameric mjD–mjL–mjN–mjP complex. Tricine SDS–PAGE gel of size exclusion chromatography fractions of the mjD–mjL–mjN–mjP complex run on an S-100HR size exclusion chromatography column (Amersham-Pharmacia Biotech).
DISCUSSION
The relative simplicity of the archaeal transcriptional machinery, combined with its close structural similarity to the core components of the eukaryotic RNAPII system, offers an opportunity for studying the most fundamental aspects of gene regulation in a biochemically accessible and highly defined system. Many key aspects of the eukaryotic RNAPII basal machinery are faithfully reproduced in archaea, such as the presence of TBP–TFIIB complexes to initiate the assembly of preinitiation complexes in the vicinity of transcription start sites, and the use of RNAPs with complex subunit organizations to execute the transcriptional programme (3). The majority of archaeal RNAP subunits display a readily recognizable degree of sequence similarity to their eukaryotic counterparts (7,8), indicating that the basic structural framework of archaeal and eukaryotic RNAPs has almost certainly evolved before the divergence of the two evolutionary domains, which is estimated to have occurred ~1.8 billion years ago (1). The sequence similarities in the largest subunits are perhaps least surprising because numerous biochemical and genetic studies have indicated that these contain the domains required for nucleotide binding and RNA synthesis. More unexpected was the discovery, made ~10 years ago, that many of the small archaeal RNAP subunits are very similar in their primary sequence organization to the small subunits present in eukaryotic RNAPs, especially in RNAPII. Certain subunits, such as the archaeal N subunits and their eukaryotic RPB10 homologs, are not only similar in length, but also in their amino acid sequences, and their three-dimensional shapes are essentially identical (19,28). Other eukaryotic subunits, such as RPB5, are longer than their archaeal homologs because of the presence of more recently evolved eukaryote-specific domains, but the C-terminal domains of these proteins nevertheless display a high degree of structural similarity (29,30). The majority of the archaeal RNAP subunits are thus readily recognizable due to their homology to the subunits previously characterized in a number of eukaryotic model systems. Until recently the existence of archaeal versions of RPB4, RPB8 and RPB12 was unclear. A systematic analysis of several archaeal genomes failed to identify potential candidates on the basis of sequence homology, leading to the conclusion that these subunits were either altogether absent in archaea, or that they diverged so substantially in their primary sequence that their detection was not possible by homology searches.
At this stage there is still no evidence for the presence of an archaeal RPB8 homolog, but a recent systematic study of the polypeptides present in purified RNAP from the archaeon M.thermoautothrophicum helped to clarify the situation by revealing the presence of two previously uncharacterized RNAP subunits, F and P (8). Darcy et al. tentatively identified F as a putative homolog of the eukaryotic RPB4 family of RNAPII subunits, but the overall degree of primary sequence identity between these proteins is at best marginal (Fig. 1A). Here we present new evidence which shows that recombinant subunit F from M.jannaschii does indeed reveal protein interaction properties that are entirely consistent with it being a true RPB4 homolog. mjF forms a highly specific complex with another archaeal subunit, mjE, which mirrors the formation of a specific complex between the human homologs of these proteins, i.e. hsRPB4 and hsRPB7. Despite the low degree of primary sequence similarity between mjF and hsRPB4 the archaeal protein can form a specific complex with hsRPB7, thus proving that important structural motifs responsible for this specific heterodimeric interaction have been highly conserved during more than 1.8 billion years of evolutionary divergence (1). It is therefore very likely that the archaeal F and eukaryotic RPB4 subunits fulfill a similar role when present in their respective RNAPs. The formation of specific archaeal–eukaryotic hybrid subunit complexes has also been shown to occur between an archaeal D subunit and eukaryotic RPB11 or AC19 (20). Taken together these observations strongly emphasize the substantial degree of structural similarity between the archaeal enzymes and eukaryotic RNAPIIs.
The recently identified archaeal P subunit (8) is more clearly identifiable on the primary amino acid sequence level as a homolog of the eukaryotic RPB12 protein, but its small size (46 amino acids in M.jannaschii) initially cast some doubt on whether it could carry out its interactions with other RNAP subunits with the same degree of specificity as its eukaryotic counterparts. Our yeast two-hybrid study indicates that mjP does indeed bind very specifically to mjD, giving rise to a signal that is comparable in strength and specificity to the p53–large T antigen interaction, widely used as a positive control in this type of assay. Additional supporting evidence for a specific interaction between these two proteins is derived from the fact that coexpression of GST–mjP with mjD in a bacterial coexpression system results in the insolubilization of GST–mjP, hinting at the formation of a specific (but mostly insoluble) complex between these proteins. Interestingly, while the mjD–mjP binary complex by itself is not soluble, the addition of the two other interaction partners of subunit D, namely subunits L and N (20), results in a tetrameric complex which is stable in solution. Although the recombinant archaeal RNAP subunits used in our work are derived from a hyperthermophilic organism, the observed protein–protein interactions readily occur at experimental temperatures (4–37°C) that are well below the temperatures under which these proteins would normally assemble in their in vivo environment (48–94°C; 31).
In summary, our data concerning the protein interaction properties of two recently discovered RNAP subunits confirms that they are bona fide homologs of the eukaryotic subunits RPB4 and RPB12. The successful assembly of the mjD–mjN–mjL–mjP and mjE–mjF complexes described here brings the number of archaeal RNAP subunits assembled into soluble complexes to six. Our current efforts focus on developing strategies for utilizing these complexes as assembly platforms for the incorporation of the large subunits that harbor the catalytic activity.
Acknowledgments
ACKNOWLEDGEMENTS
We would like to thank Dr Erica Golemis for providing us with cDNA clones containing the full-length human RPB4 and RPB7 open reading frames. R.O.J.W. acknowledges financial support from the Wellcome Trust.
REFERENCES
- 1.Doolittle R.F., Feng,D.F., Tsang,S., Cho,G. and Little,E. (1996) Science, 271, 470–471. [DOI] [PubMed] [Google Scholar]
- 2.Aravalli R.N., She,Q. and Garrett,R.A. (1998) Trends Ecol. Evol., 13, 190–194. [DOI] [PubMed] [Google Scholar]
- 3.Bell S.D. and Jackson,S.P. (1998) Trends Microbiol., 6, 222–228. [DOI] [PubMed] [Google Scholar]
- 4.Qureshi S.A., Bell,S.D. and Jackson,S.P. (1997) EMBO J., 16, 2927–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bell S.D. and Jackson,S.P. (2000) J. Biol. Chem., 275, 12934–12940. [DOI] [PubMed] [Google Scholar]
- 6.Parvin J.D. and Sharp,P.A. (1993) Cell, 73, 533–540. [DOI] [PubMed] [Google Scholar]
- 7.Langer D., Hain,J., Thuriaux,P. and Zillig,W. (1995) Proc. Natl Acad. Sci. USA, 92, 5768–5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Darcy T.J., Hausner,W., Awery,D.E., Edwards,A.M., Thomm,M. and Reeve,J.N. (1999) J. Bacteriol., 181, 4424–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Woychik N.A., Liao,S.M., Kolodziej,P.A. and Young,R.A. (1990) Genes Dev., 4, 313–323. [DOI] [PubMed] [Google Scholar]
- 10.Edwards A.M., Kane,C.M., Young,R.A. and Kornberg,R.D. (1991) J. Biol. Chem., 266, 71–75. [Google Scholar]
- 11.Khazak V., Estojak,J., Cho,H., Majors,J., Sonoda,G., Testa,J.R. and Golemis,E.A. (1998) Mol. Cell. Biol., 18, 1935–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Larkin R.M. and Guilfoyle,T.J. (1998) J. Biol. Chem., 273, 5631–5637. [DOI] [PubMed] [Google Scholar]
- 13.Choder M. and Young,R.A. (1993) Mol. Cell. Biol., 13, 6984–6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maillet I., Buhler,J.M., Sentenac,A. and Labarre,J. (1999) J. Biol. Chem., 274, 22586–22590. [DOI] [PubMed] [Google Scholar]
- 15.Asturias F.J., Meredith,G.D., Poglitsch,C.L. and Kornberg,R.D. (1997) J. Mol. Biol., 272, 536–540. [DOI] [PubMed] [Google Scholar]
- 16.Jensen G.J., Meredith,G., Bushnell,D.A. and Kornberg,R.D. (1998) EMBO J., 17, 2353–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bult C.J., White,O., Olsen,G.J., Zhou,L., Fleischmann,R.D., Sutton,G.G., Blake,J.A., Fitzgerald,L.M., Clayton,R.A., Gocayne,J.D., Kerlavage,A.R., Dougherty,B.A., Tomb,J.F., Adams,M.D., Reich,C.I., Overbeek,R., Kirkness,E.F., Weinstock,K.G., Merrick,J.M., Glodek,A., Scott,J.L., Geoghagen,N.S.M. and Venter,J.C. (1996) Science, 273, 1058–1073. [DOI] [PubMed] [Google Scholar]
- 18.Acker J., de Graaff,M., Cheynel,I., Khazak,V., Kedinger,C. and Vigneron,M. (1997) J. Biol. Chem., 272, 16815–16821. [DOI] [PubMed] [Google Scholar]
- 19.Cramer P., Bushnell,D.A., Fu,J., Gnatt,A.L., Maier-Davis,B., Thompson,N.E., Burgess,R.R., Edwards,A.M., David,P.R. and Kornberg,R.D. (2000) Science, 288, 640–649. [DOI] [PubMed] [Google Scholar]
- 20.Eloranta J.J., Kato,A., Teng,M.S. and Weinzierl,R.O. (1998) Nucleic Acids Res., 26, 5562–5567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kimura M., Ishiguro,A. and Ishihama,A. (1997) J. Biol. Chem., 272, 25851–25855. [DOI] [PubMed] [Google Scholar]
- 22.Hodach M., Todone,F., Eloranta,J.J., Onesti,S. and Weinzierl,R.O.J. (1999) Acta Crystallogr., D55, 1373–1374. [DOI] [PubMed] [Google Scholar]
- 23.Langer D., Lottspeich,F. and Zillig,W. (1994) Nucleic Acids Res., 22, 694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Makarova K.S., Aravind,L., Galperin,M.Y., Grishin,N.V., Tatusov,R.L., Wolf,Y.I. and Koonin,E.V. (1999) Genome Res., 9, 608–628. [PubMed] [Google Scholar]
- 25.Rubbi L., Labarre-Mariotte,S., Chedin,S. and Thuriaux,P. (1999) J. Biol. Chem., 274, 31485–31492. [DOI] [PubMed] [Google Scholar]
- 26.Fields S. and Song,O. (1989) Nature, 340, 245–246. [DOI] [PubMed] [Google Scholar]
- 27.Ishihama A. (1981) Adv. Biophys., 14, 1–35. [PubMed] [Google Scholar]
- 28.Mackereth C.D., Arrowsmith,C.H., Edwards,A.M. and McIntosh,L.P. (2000) Proc. Natl Acad. Sci. USA, 97, 6316–6321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yee A., Booth,V., Dharamasi,A., Engel,A., Edwards,A. and Arrowsmith,C.H. (2000) Proc. Natl Acad. Sci. USA, 97, 6311–6315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Todone F., Weinzierl,R.O.J., Brick,P. and Onesti,S. (2000) Proc. Natl Acad. Sci. USA, 97, 6306–6310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones W.J., Leigh,J.A., Mayer,F., Woese,C.R. and Wolfe,R.S. (1983) Arch. Microbiol., 136, 254–261. [Google Scholar]