Prevalence of small inversions in yeast gene order evolution (original) (raw)
Abstract
Gene order evolution in two eukaryotes was studied by comparing the Saccharomyces cerevisiae genome sequence to extensive new data from whole-genome shotgun and cosmid sequencing of Candida albicans. Gene order is substantially different between these two yeasts, with only 9% of gene pairs that are adjacent in one species being conserved as adjacent in the other. Inversion of small segments of DNA, less than 10 genes long, has been a major cause of rearrangement, which means that even where a pair of genes has been conserved as adjacent, the transcriptional orientations of the two genes relative to one another are often different. We estimate that about 1,100 single-gene inversions have occurred since the divergence between these species. Other genes that are adjacent in one species are in the same neighborhood in the other, but their precise arrangement has been disrupted, probably by multiple successive multigene inversions. We estimate that gene adjacencies have been broken as frequently by local rearrangements as by chromosomal translocations or long-distance transpositions. A bias toward small inversions has been suggested by other studies on animals and plants and may be general among eukaryotes.
The order and transcriptional orientation of genes along a chromosome can change during evolution by DNA inversions and transpositions or by chromosomal translocations. In bacteria, long strings of genes show conserved order and orientation between closely related species or strains, so that most rearrangements involve large pieces of DNA (1–4). Comparative genetic mapping in vertebrates and plants has shown that large syntenic regions are conserved, with breakpoints corresponding to interchromosomal translocations (5, 6). More detailed mapping of some of these syntenic regions, however, has revealed several examples where the local gene order is not conserved (7–11).
The extent of gene order conservation among ascomycete fungi previously has been estimated by comparing the S. cerevisiae genome sequence (12) to DNA sequences from other species, using either random “genome survey” sequences from both ends of small clones (13–15) or existing European Molecular Biology Laboratory database sequences (16). Between Saccharomyces cerevisiae and_Candida albicans_, two species separated by 140–330 million years (17, 18), only one example of conserved gene order and orientation has been reported so far (STE6_–_UBA1; ref. 19), whereas there are three cases of gene pairs that are adjacent in both species but where one gene has been inverted (RAD16–LYS2, NFS1–LEU2 and_RPS31–SEC10_; refs. 15, 16, and 20–22). To investigate whether the apparently high frequency of inversions is general throughout the C. albicans genome we analyzed the genome sequence data currently available.
Methods
The 1,680 contig DNA sequences in the May 2000 data release (7× coverage) from the C. albicans whole genome shotgun sequencing project at Stanford University were downloaded fromhttp://www-sequence.stanford.edu/group/candida. The contigs range from 2 to 151 kb and total 16.2 Mb, which is approximately equal to the estimated genome size. The S. cerevisiae proteome was searched against these contigs by using gappedtblastn (23) with the seg filter (24) and a cutoff E value of 10-10. Reanalysis of the data using other cutoffs (10−6, 10−20) did not change the results significantly. Gene locations in the Stanford contigs were estimated solely on the basis oftblastn hits to S. cerevisiae proteins, and we did not annotate them further. The sequences of six cosmids completely sequenced at the Sanger Centre were obtained from GenBank (accession numbers AL033391, AL033396, AL033497, AL033501,AL033502, and AL033503). Gene identifications for the Sanger Centre cosmids shown in Fig. 4 were made manually, and some of these involve matches weaker than E = 10−10.C. albicans genes without orthologs in S. cerevisiae were ignored in the analysis of Stanford contigs, but are shown for the cosmids in Fig. 4.
Figure 4.

Gene order relationships of four C. albicans cosmids, sequenced at the Sanger Centre, to parts of the S. cerevisiae genome. Vertical lines connect orthologous genes. Curved arrows indicate genes with inverted orientations. C. albicans genes are named after their S. cerevisiae orthologs; unnamed genes have no close relative in_S. cerevisiae_. Numbers in parentheses indicate numbers of intervening genes in S. cerevisiae that are not shown. S. cerevisiae regions in or near duplicated chromosomal blocks (26) are labeled. The scale at the top refers to_C. albicans_ only.
Results
Changes in Gene Order and Orientation.
We used tblastn searches (23) with all S. cerevisiae proteins to estimate the locations of genes in the_C. albicans_ contigs assembled at Stanford, thereby ignoring any C. albicans genes that do not have S. cerevisiae counterparts. The contigs contain 3,188 pairs of genes that appear to be adjacent in C. albicans (i.e., either they are adjacent, or any intervening genes do not have S. cerevisiae orthologs). For 298 pairs (9%), the S. cerevisiae orthologs are also adjacent. Despite remaining as neighbors, 103 of these pairs (35%) have different gene orientation or order in the two species. Ninety-one pairs can be explained by inversions of one gene, and 12 pairs require two inversions each (Fig.1). From this observation (115 single-gene inversions among 298 intergenic links), we estimate that the total number of single-gene inversions that have occurred in their genomes following the divergence of these species is about 1,100 (= 5,800 genes in the genome × 115/298 ÷ 2 links broken per inversion).
Figure 1.

Order and orientation relationships between 298 gene pairs that are adjacent in both S. cerevisiae and C. albicans. All 10 possible relationships between two adjacent genes are shown, with the number of inversions needed to convert any combination into any other. The names of gene pairs in each category are listed at www.gen.tcd.ie/khwolfe/candida. The categories labeled as “2 inversions” also could be explained by one gene leapfrogging over the other, but we consider this unlikely.
The set of 298 adjacent pairs includes 21 runs of three genes that have conserved gene order in the two species. Among these, 16 examples of apparent single-gene inversions are seen (Fig.2). The most dramatic example is the cluster SLU7–RRP1–SSS1, where the order is conserved but all three genes have reversed orientations. This example could be explained either by three independent single-gene inversions or by two short-distance transpositions, both of which seem quite improbable.
Figure 2.

Examples of single-gene inversions. The three genes in each set are adjacent (ignoring any C. albicans genes without homologs) and in the same order in the two species. Directions of transcription in S. cerevisiae and C. albicans are shown above and below gene names, respectively. Genes named in bold italics have different orientations in the two species. S. cerevisiae gene names are used.
Other pairs of adjacent C. albicans genes have S. cerevisiae orthologs that are physically close to each other but are not immediate neighbors (Fig. 3). The Stanford contig data include 97 pairs of adjacent C. albicans genes whose S. cerevisiae orthologs are separated by 1–5 intervening genes. Gene orientation and relative order are conserved in 28 of these pairs, which is only slightly more than the 24.25 expected by chance. These findings suggest that multigene inversions may have occurred, moving genes over short distances.
Figure 3.

Histogram showing the distance apart in S. cerevisiae of the orthologs of gene pairs that are adjacent in C. albicans. The distance between two genes is expressed in terms of the number of other genes between them on the chromosome.
To further examine local gene order we studied six C. albicans cosmids (25) that were completely sequenced at the Sanger Centre. These sequence comparisons point to numerous rearrangements, both interchromosomal (translocations) and intrachromosomal (small inversions). Most of the long C. albicans sequences contain small clusters of genes whose S. cerevisiae orthologs also are physically clustered (Fig. 4). These clusters are generally shorter than 10 genes in C. albicans and often are interspersed with genes from other S. cerevisiae chromosomes. The ends of the clusters probably correspond to sites of chromosomal translocations (16, 26, 27). In some cases a cluster of genes in C. albicans is related to two_S. cerevisiae_ genomic regions (blocks) that are paired by whole-genome duplication in the S. cerevisiae lineage (26), as predicted by our model (16, 27). The relationships shown in Fig. 4 comprise 32 orthologous genes and at least 11 independent inversions. It is not possible to estimate the exact sizes of these inversions (i.e., the numbers of genes involved) because, in all cases, the genes immediately upstream and downstream of the inverted ones are different in the two species. For example, the inversion of YLR423C in cosmid Ca49C10 might have included some of the four genes downstream of it in S. cerevisiae. However, the inversions must be relatively small because gene order is conserved at a coarser level (e.g., YLR423C is in-between YLR418C and_YLR424W_ in both species). Similar scrambling of local gene order recently was reported by Mallet et al. (28) for the region around the CHS6 gene compared between S. cerevisiae and C. albicans.
The conservation of small neighborhoods of genes, without absolute conservation of order or orientation, suggests that small DNA inversions have contributed significantly to the evolution of ascomycete genomes. A further example is seen in cosmid Ca49C4 (Fig.4), which contains a pseudogene related to the C. albicans oligopeptide transporter gene OPT1 (29) and its S. cerevisiae homolog YJL212C. The pseudogene has 98% DNA sequence identity over 2 kb to part of OPT1, but a 0.3-kb internal segment has been inverted relative to OPT1 and other members of this gene family. There is also evidence for small inversions within the S. cerevisiae genome itself, where 11 of 655 duplicated genes associated with whole-genome duplication now show inverted orientation with respect to the flanking chromosomal regions (30).
Relative Rates of Intrachromosomal Versus Interchromosomal Rearrangements.
Small rearrangements keep genes within a local neighborhood, so we can use the C. albicans/S. cerevisiae comparisons from the Stanford dataset (Figs. 1 and 3) to estimate the rate of small rearrangements (which we suggest are mostly inversions) relative to large rearrangements (translocations, larger inversions, and long-distance transpositions). Even if there had been no other chromosomal rearrangements, we would expect about half of the links between immediate neighbors in S. cerevisiae and C. albicans to have been broken by the process of random gene loss due to differential silencing after genome duplication in the S. cerevisiae lineage (26, 31). The remaining breaks are the combined result of inversions, translocations, and transpositions. The fraction of links that has been conserved is under 10%, but this fraction has been reduced by a factor of 2 by genome duplication in S. cerevisiae. Consequently, chromosomal rearrangements are responsible for breaking over 80% of the links between neighbors. Assuming that breakpoints are made randomly, and using a Poisson distribution to correct for multiple hits, this implies that there have been an average of 1.6 breaks per link, or approximately 9,000 breakpoints in total since speciation. This argument assumes that the_S. cerevisiae_ genome duplication occurred recently, but an identical conclusion is reached if the genome duplication is assumed to have occurred shortly after speciation. It also assumes that no other genome duplications have occurred in either lineage.
Statistical methods have been developed previously to estimate relative numbers of intrachromosomal and interchromosomal rearrangements between species (32), but these methods are not adaptable to the current problem because the kind of data being considered is local (the Stanford contigs are short relative to chromosomes) and because the number of rearrangements is close to saturation. It is problematic to model the small inversions directly because not enough is known about their size distribution. Instead, to model the combined processes of large and small chromosomal rearrangements, adjacent genes in C. albicans having orthologs on the same chromosome in S. cerevisiae were divided into two categories: gene pairs that are also adjacent in_S. cerevisiae_ (state A), and gene pairs that are “near-neighbors” (syntenic but separated by a small number of genes) in S. cerevisiae (state B).
The number of gene pairs in the sequenced sample that are in state A is_P_A. In a time interval Δ_t_ the change in population of state A is
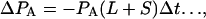 |
1 |
|---|
where L and S are the rates at which single intergenic links are broken by large and small rearrangements, respectively. Let I be the mean number of intervening genes for gene pairs that are near-neighbors in S. cerevisiae, so that I + 1 is the mean number of intervening links. If we make the assumption that the average separation of this category of gene pair has been similar throughout the evolutionary history then
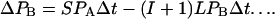 |
2 |
|---|
This assumption is justified because gene pairs in state B are unlikely to drift too far apart before their linkage is broken by a translocation. Large rearrangements (translocations) are taken to be the only way in which gene pairs leave state B because the number of gene pairs that are syntenic but not near-neighbors is small (Fig. 3).
Eq. 1 and 2 above can be treated as differential equations and solved, giving
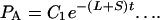 |
3 |
|---|
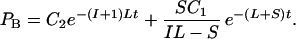 |
4 |
|---|
At time 0 _P_A = J/2, where J is the number of gene pairs in the sample that are adjacent in C. albicans and have orthologs in S. cerevisiae, because S. cerevisiae has undergone genome duplication followed by differential silencing. Therefore_C_1 = J/2. At time 0_P_B = 0, therefore
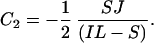
Eq. 3 and 4 provide an estimate of the proportion of all rearrangements that are small (S/S+L), given values for the number of conserved adjacent gene pairs (_P_A, which is 298; Fig. 3), the number of pairs that are adjacent in C. albicans but near-neighbors in S. cerevisiae (_P_B) and the average spacing between near-neighbors (I). The values of_P_B and I can be calculated from the data in Fig. 3 but depend on the maximum number of intervening genes that is permitted in the definition of near-neighbors (_I_max). In Fig. 3 there appears to be an excess of conserved linkages over short distances, up to a limit of at least five intervening genes and possibly as many as 20. The relationship between the estimated proportion of small rearrangements and _I_max is shown in Fig.5. Allowing a maximum of five genes between near-neighbors, 38% of broken links are attributed to small rearrangements. This increases to 67% for_I_max = 20 genes. These results suggest that approximately equal numbers of linkages have been broken by small and large rearrangements.
Figure 5.

Relationship between the maximum permitted number of intervening genes (_I_max) between near-neighbors (gene pairs in state B), and the estimate of the proportion of rearrangements that are small (S/S+L). Calculated numerically from Eqs. 3 and 4 using data for PB at different values of_I_max from Fig. 3.
A limit of I_max = 5 also was suggested by an experiment where we compared the number of adjacent pairs in_C. albicans whose homologs are syntenic in S. cerevisiae to those whose homologs are located on specific pairs of different chromosomes, as a way of estimating the “background” level of random gene associations in Fig. 3 (data not shown).
Discussion
This study compares gene order between two eukaryotes based on whole genome sequence data. Our results suggest that successive random small inversions frequently cause a gene's chromosomal position and orientation to drift during its evolution. This process would alter gene order and orientation without moving any genes very far from their starting points. It also would tend to blur the endpoints of interchromosomal translocations. The mechanism by which small inversions occur is unknown, and our data are uninformative in this regard because intergenic sequences are highly diverged between_C. albicans_ and S. cerevisiae. Our results also suggest that gene order in yeasts is relatively unconstrained by natural selection. The orientations of some pairs of adjacent genes, particularly those that are transcribed divergently from a shared regulatory region (such as the histone pair HTA1–HTB1) may be under selection, but the high frequency of rearrangement indicates that this type of constraint is the exception rather than the rule (cf. ref. 33). It is notable that divergently transcribed adjacent gene pairs are broken up at approximately the same frequency as pairs transcribed convergently or in parallel (Fig. 1).
In our analysis we made an arbitrary distinction between small and large rearrangements, using a limit of five or 20 intervening genes based on inspection of Fig. 3. The size distribution of inversions during evolution is unknown but it seems likely that there is a skewed distribution with a bias toward smaller sizes, either because of mechanistic reasons or natural selection against disruption of meiosis. A more accurate description of the size distribution is clearly needed but will require comparisons between more closely related yeast species. One evolutionary inversion in the S. cerevisiae genome whose size can be estimated by comparing to Kluyveromyces marxianus comprises eight genes and approximately 13 kb (34).
Small inversions also may be frequent in eukaryotes other than fungi. For animals, Gilley and Fried (10) proposed that small gene order differences between Fugu rubripes and human may have been caused by inversions, and local rearrangements including gene inversions are seen in comparison of the DiGeorge syndrome region between human and mouse (8). There also have been several reports of conserved synteny, but not gene order, between_Caenorhabditis elegans_ and Drosophila melanogaster or mammals (35–38). Small inversions have been observed directly in comparisons of C. elegans vs. C. briggsae (39) and of D. melanogaster vs. D. buzzatii (40) and have been suggested by a genomewide analysis of the organization of tandem gene arrays in C. elegans (41). For plants, analysis of duplicated regions in the Arabidopsis thaliana genome has revealed several inversions with sizes ranging from megabases (hundreds of genes) (42–44) down to single genes (44). Quantifying the size distributions of local rearrangements in different eukaryotic kingdoms will require more extensive comparative sequence data.
Acknowledgments
C. albicans sequencing is supported at the Sanger Centre by the Wellcome Trust/Beowulf Genomics initiative and at Stanford by the National Institute of Dental Research and the Burroughs Wellcome Fund.
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.240462997.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.240462997
References
- 1.Himmelreich R, Plagens H, Hilbert H, Reiner B, Herrmann R. Nucleic Acids Res. 1997;25:701–712. doi: 10.1093/nar/25.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alm R A, Ling L S, Moir D T, King B L, Brown E D, Doig P C, Smith D R, Noonan B, Guild B C, deJonge B L, et al. Nature (London) 1999;397:176–180. doi: 10.1038/16495. [DOI] [PubMed] [Google Scholar]
- 3.Kalman S, Mitchell W, Marathe R, Lammel C, Fan J, Hyman R W, Olinger L, Grimwood J, Davis R W, Stephens R S. Nat Genet. 1999;21:385–389. doi: 10.1038/7716. [DOI] [PubMed] [Google Scholar]
- 4.Read T D, Brunham R C, Shen C, Gill S R, Heidelberg J F, White O, Hickey E K, Peterson J, Utterback T, Berry K, et al. Nucleic Acids Res. 2000;28:1397–1406. doi: 10.1093/nar/28.6.1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carver E A, Stubbs L. Genome Res. 1997;7:1123–1137. doi: 10.1101/gr.7.12.1123. [DOI] [PubMed] [Google Scholar]
- 6.Gale M D, Devos K M. Science. 1998;282:656–659. doi: 10.1126/science.282.5389.656. [DOI] [PubMed] [Google Scholar]
- 7.Johansson M, Ellegren H, Andersson L. Genomics. 1995;25:682–690. doi: 10.1016/0888-7543(95)80011-a. [DOI] [PubMed] [Google Scholar]
- 8.Lindsay E A, Botta A, Jurecic V, Carattini-Rivera S, Cheah Y C, Rosenblatt H M, Bradley A, Baldini A. Nature (London) 1999;401:379–383. doi: 10.1038/43900. [DOI] [PubMed] [Google Scholar]
- 9.Yang Y P, Womack J E. Genome Res. 1998;8:731–736. doi: 10.1101/gr.8.7.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gilley J, Fried M. Hum Mol Genet. 1999;8:1313–1320. doi: 10.1093/hmg/8.7.1313. [DOI] [PubMed] [Google Scholar]
- 11.Cavell A C, Lydiate D J, Parkin I A, Dean C, Trick M. Genome. 1998;41:62–69. [PubMed] [Google Scholar]
- 12.Goffeau A, Aert R, Agostini-Carbone M L, Ahmed A, Aigle M, Alberghina L, Allen E, Alt-Mörbe J, André B, Andrews S, et al. Nature (London) 1997;387,Suppl.:5–105. [Google Scholar]
- 13.Altmann-Jöhl R, Philippsen P. Mol Gen Genet. 1996;250:69–80. doi: 10.1007/BF02191826. [DOI] [PubMed] [Google Scholar]
- 14.Ozier-Kalogeropoulos O, Malpertuy A, Boyer J, Tekaia F, Dujon B. Nucleic Acids Res. 1998;26:5511–5524. doi: 10.1093/nar/26.23.5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartung K, Frishman D, Hinnen A, Wolfl S. Yeast. 1998;14:1327–1332. doi: 10.1002/(SICI)1097-0061(1998100)14:14<1327::AID-YEA321>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 16.Keogh R S, Seoighe C, Wolfe K H. Yeast. 1998;14:443–457. doi: 10.1002/(SICI)1097-0061(19980330)14:5<443::AID-YEA243>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 17.Berbee M L, Taylor J W. In: The Fungal Holomorph: Mitotic, Meiotic and Pleomorphic Speciation in Fungal Systematics. Reynolds D R, Taylor J W, editors. Wallingford, U.K.: CAB International; 1993. pp. 67–78. [Google Scholar]
- 18.Pesole G, Lotti M, Alberghina L, Saccone C. Genetics. 1995;141:903–907. doi: 10.1093/genetics/141.3.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raymond M, Dignard D, Alarco A M, Mainville N, Magee B B, Thomas D Y. Mol Microbiol. 1998;27:587–598. doi: 10.1046/j.1365-2958.1998.00704.x. [DOI] [PubMed] [Google Scholar]
- 20.Suvarna K, Seah L, Bhattacherjee V, Bhattacharjee J K. Curr Genet. 1998;33:268–275. doi: 10.1007/s002940050336. [DOI] [PubMed] [Google Scholar]
- 21.Plant E P, Becher D, Poulter R T. Yeast. 1998;14:287–295. doi: 10.1002/(SICI)1097-0061(199802)14:3<287::AID-YEA213>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 22.Roig P, Martinez J P, Gil M L, Gozalbo D. Yeast. 2000;16:1413–1419. doi: 10.1002/1097-0061(200011)16:15<1413::AID-YEA632>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 23.Altschul S F, Madden T L, Schaffer A A, Zhang J, Zhang Z, Miller W, Lipman D J. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wootton J C, Federhen S. Methods Enzymol. 1996;266:554–571. doi: 10.1016/s0076-6879(96)66035-2. [DOI] [PubMed] [Google Scholar]
- 25.Tait E, Simon M C, King S, Brown A J, Gow N A, Shaw D J. Fungal Genet Biol. 1997;21:308–314. doi: 10.1006/fgbi.1997.0983. [DOI] [PubMed] [Google Scholar]
- 26.Wolfe K H, Shields D C. Nature (London) 1997;387:708–713. doi: 10.1038/42711. [DOI] [PubMed] [Google Scholar]
- 27.Seoighe C, Wolfe K H. Proc Natl Acad Sci USA. 1998;95:4447–4452. doi: 10.1073/pnas.95.8.4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mallet L, Renault G, Jacquet M. Yeast. 2000;16:959–966. doi: 10.1002/1097-0061(200007)16:10<959::AID-YEA592>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 29.Lubkowitz M A, Hauser L, Breslav M, Naider F, Becker J M. Microbiology. 1997;143:387–396. doi: 10.1099/00221287-143-2-387. [DOI] [PubMed] [Google Scholar]
- 30.Seoighe C, Wolfe K H. Gene. 1999;238:253–261. doi: 10.1016/s0378-1119(99)00319-4. [DOI] [PubMed] [Google Scholar]
- 31.Seoighe C, Wolfe K H. Curr Opin Microbiol. 1999;2:548–554. doi: 10.1016/s1369-5274(99)00015-6. [DOI] [PubMed] [Google Scholar]
- 32.Ehrlich J, Sankoff D, Nadeau J H. Genetics. 1997;147:289–296. doi: 10.1093/genetics/147.1.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kruglyak S, Tang H. Trends Genet. 2000;16:109–111. doi: 10.1016/s0168-9525(99)01941-1. [DOI] [PubMed] [Google Scholar]
- 34.Ladrière J M, Georis I, Guerineau M, Vandenhaute J. Gene. 2000;255:83–91. doi: 10.1016/s0378-1119(00)00310-3. [DOI] [PubMed] [Google Scholar]
- 35.Ruddle F H, Bentley K L, Murtha M T, Risch N. Development (Cambridge, U.K.) 1994. Suppl., 155–161. [PubMed] [Google Scholar]
- 36.Trachtulec Z, Hamvas R M, Forejt J, Lehrach H R, Vincek V, Klein J. Genomics. 1997;44:1–7. doi: 10.1006/geno.1997.4839. [DOI] [PubMed] [Google Scholar]
- 37.Pébusque M-J, Coulier F, Birnbaum D, Pontarotti P. Mol Biol Evol. 1998;15:1145–1159. doi: 10.1093/oxfordjournals.molbev.a026022. [DOI] [PubMed] [Google Scholar]
- 38.Ruvkun G, Hobert O. Science. 1998;282:2033–2041. doi: 10.1126/science.282.5396.2033. [DOI] [PubMed] [Google Scholar]
- 39.Hutter H, Vogel B E, Plenefisch J D, Norris C R, Proenca R B, Spieth J, Guo C, Mastwal S, Zhu X, Scheel J, et al. Science. 2000;287:989–994. doi: 10.1126/science.287.5455.989. [DOI] [PubMed] [Google Scholar]
- 40.Robin G C d Q, Claudianos C, Russell R J, Oakeshott J G. J Mol Evol. 2000;51:149–160. doi: 10.1007/s002390010075. [DOI] [PubMed] [Google Scholar]
- 41.Semple C, Wolfe K H. J Mol Evol. 1999;48:555–564. doi: 10.1007/pl00006498. [DOI] [PubMed] [Google Scholar]
- 42.Lin X, Kaul S, Rounsley S, Shea T P, Benito M I, Town C D, Fujii C Y, Mason T, Bowman C L, Barnstead M, et al. Nature (London) 1999;402:761–768. doi: 10.1038/45471. [DOI] [PubMed] [Google Scholar]
- 43.Mayer K, Schuller C, Wambutt R, Murphy G, Volckaert G, Pohl T, Dusterhoft A, Stiekema W, Entian K D, Terryn N, et al. Nature (London) 1999;402:769–777. [Google Scholar]
- 44.Blanc G, Barakat A, Guyot R, Cooke R, Delseny M. Plant Cell. 2000;12:1093–1102. doi: 10.1105/tpc.12.7.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]