A model for the abrogation of the SOS response by an SOS protein: a negatively charged helix in DinI mimics DNA in its interaction with RecA (original) (raw)
Abstract
DinI is a recently described negative regulator of the SOS response in Escherichia coli. Here we show that it physically interacts with RecA and prevents the binding of single-stranded DNA to RecA, which is required for the activation of the latter. DinI also displaces ssDNA from a stable RecA–DNA cofilament, thus eliminating the SOS signal. In addition, DinI inhibits RecA-mediated homologous DNA pairing, but has no effect on actively proceeding strand exchange. Biochemical data, together with the molecular structure, define the C-terminal α-helix in DinI as the active site of the protein. In an unusual example of molecular mimicry, a negatively charged surface on this α-helix, by imitating single-stranded DNA, interacts with the loop L2 homologous pairing region of RecA and interferes with the activation of RecA.
Keywords: DinI, RecA, SOS response, molecular mimicry
When a bacterial cell is subjected to the harmful action of DNA-damaging agents, such as UV (ultraviolet) and ionizing radiation and a variety of chemical compounds, it turns on a cascade of cellular reactions called the SOS response. Several excellent reviews and books have surveyed in detail current models for the regulation of the SOS response (Friedberg et al. 1995; Shinagawa 1996; Walker 1996; Kuzminov 1999). The SOS response results in the induction of over two and one-half dozen genes (Fernandez de Henestrosa et al. 2000) that are under the control of the transcriptional repressor, LexA. Products of many of these genes are known to take part in DNA repair and mutagenesis. Other genes have no function ascribed as yet and are classified as SOS genes solely on the basis of the presence of an SOS box in their promoter region (Friedberg et al. 1995; Koch and Woodgate 1998).
The mechanism of the initiation of the SOS response is fairly well elucidated and consists of the activation of the RecA protein, which acts as a coprotease in the autocatalytic digestion of the LexA repressor. The active form of RecA is a protein filament formed on ssDNA in an ATP-dependent fashion. Thus, the accumulation of ssDNA regions resulting from stalled replication is the intracellular signal for the induction of the SOS system (Walker 1996). Much less is known about the events leading to the termination of the SOS response. Shutdown of the SOS machinery is an important step in the regulation of the SOS response: It ensures the resumption of DNA replication, reduces high mutability, and reverses cell filamentation. It is widely believed that bacteria revert to the steady (uninduced) state in a passive manner, that is, after DNA lesions are healed, the inducing signal disappears and the original pool of LexA protein is restored (Little 1991; Friedberg et al. 1995). No active mechanism for turning down the SOS response has been described so far.
The dinI gene was first discovered as part of a screen for genes induced by DNA-damaging agents (Kenyon and Walker 1980). Subsequently, dinI was independently cloned by Ohmori and coworkers as a multicopy suppressor of the cold-sensitive phenotype of dinD68 mutation, a mutant allele that causes SOS induction without any DNA damaging agent at temperatures <20°C (Yasuda et al. 1996). Later, the same workers showed that when overexpressed from a multicopy plasmid, DinI conferred significant UV sensitivity to cells and prevented induction of an SOS gene (sulA–lacZ fusion) in response to mitomycin C treatment (Yasuda et al. 1998). Overexpression of DinI also impaired cleavage of LexA and UmuD in vivo. In addition, cells bearing a dinI null mutant processed the UmuD protein faster and more extensively than cells having a single chromosomal copy of the wild-type gene. As a result, a three- to fivefold increase in the UmuDC-dependent mutagenesis was observed in the mutant strain (Yasuda et al. 1998). Finally, in the same study Ohmori and coworkers showed that DinI inhibited the ability of the RecA–ssDNA–ATPγS filament to promote the cleavage of UmuD in vitro. In aggregate, these data strongly suggest that DinI is a physiological down-regulator of the SOS response.
Because the RecA protein is involved in the generation of the SOS signal, we have studied the interaction between the purified RecA and DinI proteins and the effect of DinI on the biochemical activities of RecA. Here we show that DinI, interacting with RecA, precludes the formation of the ssDNA–RecA cofilament, thus eliminating the signal for SOS induction.
Furthermore, DinI can dismantle preformed ssDNA–RecA cofilaments.
Results
Evidence for a physical interaction between RecA and DinI in vitro and in whole cell lysates
First, using immunoprecipitation, we show a physical interaction between RecA and DinI in different in vitro conditions and in cell lysates. Figure 1A shows that the RecA and DinI protein interact in vitro. Following the activation of RecA by incubation with ssDNA and ATPγS, the RecA protein could be precipitated by anti-DinI antibody only when both RecA and DinI were present in the reaction (Fig. 1A, lane 7). No coimmunoprecipitation was observed when the preimmune serum was used instead of the anti-DinI serum (Fig. 1A, lanes 2–4). We next examined whether the formation of an active RecA–ssDNA–ATPγS cofilament is required for the formation of a RecA–DinI complex. Figure 1B indicates that DinI and inactive RecA interact, that is, no DNA or nucleotide cofactors are required (Fig. 1B, lanes 3–5,7). These results suggest that activation of RecA is not a prerequisite for its interaction with DinI. However, we would like to point out that the observation of similar RecA intensities on the Western blot (Fig. 1A, top panel) does not exclude the possibility that DinI has different affinities for activated and inactive RecA.
Figure 1.
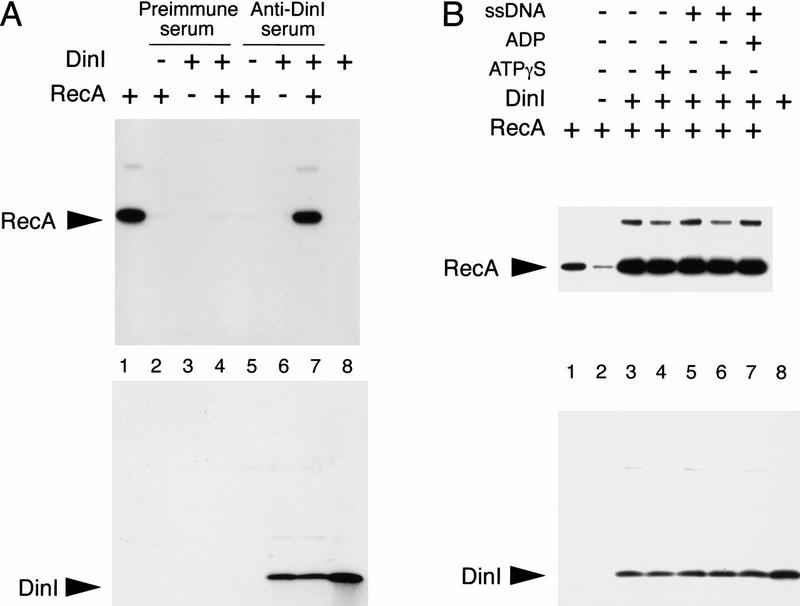
Physical interaction between the RecA and DinI proteins is detected by coimmunoprecipitation. In all cases either monoclonal anti-RecA ARM193 or polyclonal affinity-purified anti-DinI antibodies, in combination with the ELC detection system, were used for immunodetection of RecA and DinI proteins. (A) RecA–DinI complex was formed in the presence of ssDNA and ATPγS. Products of immunoprecipitation performed with either preimmune (lanes 2_–_4) or anti-DinI sera (lanes 5_–_7) were separated on two tricine SDS gels. As controls, RecA alone (lanes 2,5) and DinI alone (lanes 3,6) were subjected to the identical procedure. Lanes 1 and 8 contain purified RecA and DinI. RecA coimmunoprecipitated with DinI is visible in lane 7. (B) Immunoprecipitation of RecA–DinI complex formed in the presence of different combinations of ATPγS, ADP, and ssDNA was performed with antigen-purified anti-DinI antibody. Higher molecular weight bands on the RecA blot correspond to covalently crosslinked RecA dimers formed during prolonged incubation in the absence of reducing agent. The low intensity RecA signal visible in lane 2 is a result of RecA aggregation (see Materials and Methods).
We then took advantage of the coimmunoprecipitation approach to test whether DinI and RecA interact in UV-irradiated and untreated wild-type Escherichia coli K12 cells. As a first step, we examined the levels of DinI and RecA expression in K12 cells after induction of the SOS system with UV. As expected, the levels of both endogenous DinI and RecA increased significantly after UV treatment (Fig. 2A). However, contrary to our expectation, DinI expression reached its highest level at early stages of the SOS response, reaching its maximum (∼60-fold increase) around 20–40 min after irradiation. The maximal expression of RecA was observed 40–60 min after induction. Next, we used anti-DinI antibody to immunoprecipitate DinI from the same cell lysates. The amount of DinI precipitated from UV-treated lysates virtually followed the profile of its expression (Fig. 2, cf. left panels in A and B). Interestingly, the recovery of RecA coimmunoprecipitated with DinI did not exactly mirror the expression of either RecA or DinI (Fig. 2B, left panel). Indeed, when we plotted the amount of coimmunoprecipitated RecA normalized to the amount of recovered DinI, we found that the amount of RecA that coimmunoprecipitated with DinI significantly increases at later stages of the SOS response (Fig. 2B, right panel). This result probably reflects an increase in the affinity of RecA and DinI for each other. Coimmunoprecipitation of RecA and DinI from the cell lysates indicates that they interact in vivo during the SOS response. That the extent of interaction increases at the time expected to down-regulate this response strongly suggests a physiological role for DinI in this regard and that factors, so far unknown, may modulate this interaction (see Discussion).
Figure 2.
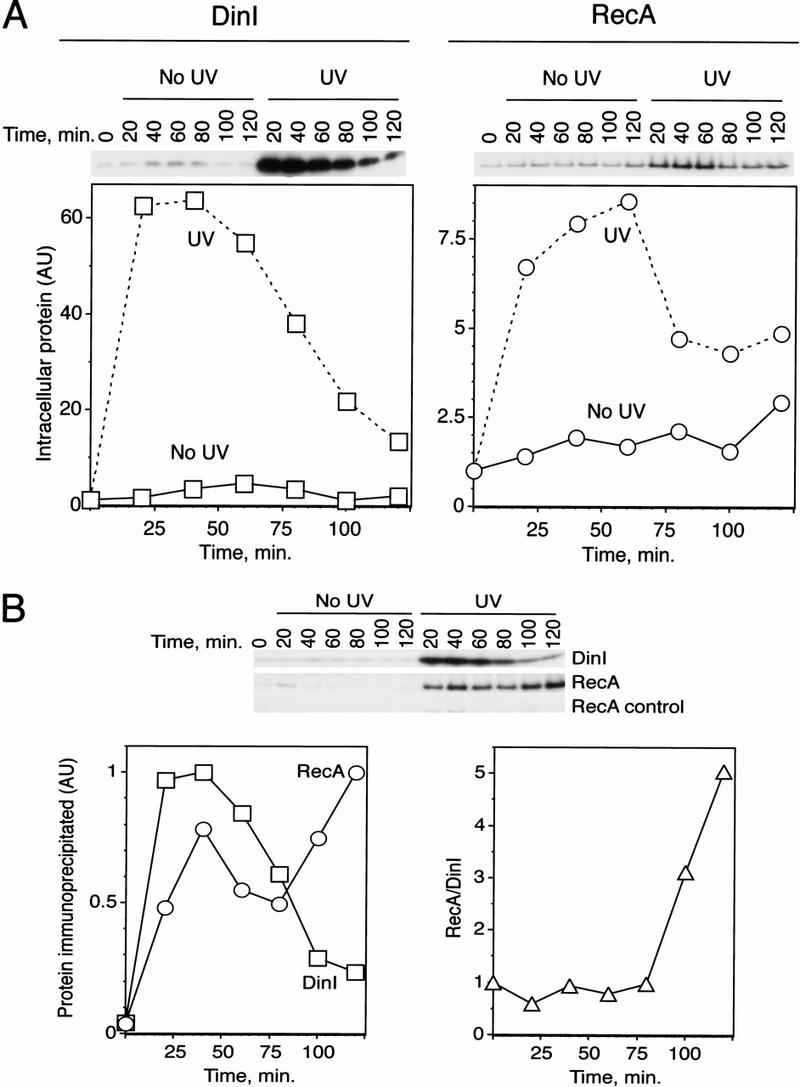
The affinity of RecA and DinI for each other increases late in the SOS response. (A) Kinetics of expression of the DinI and RecA proteins in E. coli cell lysates prepared from UV-irradiated and untreated cultures. Wild-type K12 cells were allowed to recover for different periods of time after irradiation with 25 J/m2 of 254 nm UV light, and cell lysates were prepared. Ten micrograms (for DinI) or 1 μg (for RecA) of total protein were used for Western blot analysis. Polyclonal affinity-purified anti-DinI antibody and anti-RecA serum were used for DinI and RecA detection, respectively. (B) Coimmunoprecipitation of DinI and RecA with anti-DinI antibody. Top panel shows original data obtained with the same lysates and antibodies used in A. RecA controls correspond to mock experiments in which anti-DinI antibody was omitted at the immunoprecitation step and anti-RecA serum was used for detection. Left panel represents relative amounts (in AU or arbitrary units) of DinI and RecA recovered at different time points from irradiated cells. Right panel shows recovery of RecA normalized to the amount of immunoprecipitated DinI. The normalized signal represents the ratio between the RecA and DinI signals in arbitrary units.
DinI displaces ssDNA from DNA–RecA filaments
Next, we tested the ability of DinI to bind DNA. Despite numerous attempts, we failed to detect any DNA–DinI interactions using DNA-agarose columns, filter binding (data not shown), or agarose gel retention assays (Fig. 3B, lane 3). No ssDNA–DinI complexes were detected by electron microscopy (O. Voloshin and A. Stasiak, unpubl.). On the other hand, binding of ssDNA is a basic function of RecA, which is required for the initiation of the SOS response, the coprotease activities, and the recombinational proficiency of the RecA protein (Roca and Cox 1997). Accordingly, we examined the effect of DinI on the DNA binding properties of RecA.
Figure 3.
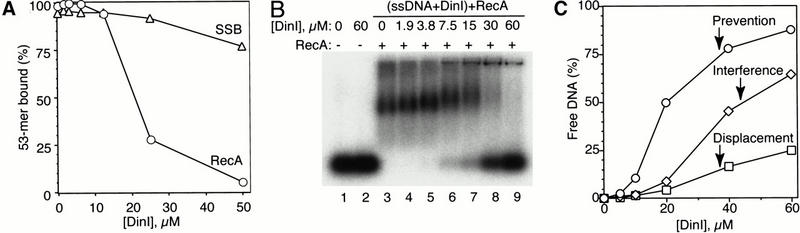
DinI specifically inhibits the ssDNA-binding activity of RecA. (A) Addition of DinI suppresses ssDNA-binding activity of RecA, but not SSB protein, as revealed by filter binding. 0.5 μM RecA or SSB was first preincubated with increasing concentrations of DinI for 10 min at room temperature. After addition of 0.5 μM 32P-labeled 53-mer BS-S1 oligonucleotide, the binding reaction was allowed to proceed at 37°C for 30 min (the buffer conditions are described in Materials and Methods). (B) Displacement of intact ssDNA from ternary ssDNA–RecA–DinI complexes by DinI can be detected by electrophoresis on agarose gels. 2.5 μM 32P-labeled BS-S1 was first preincubated with 0–60 μM DinI at 37°C for 30 min, then 1 μM RecA was added and the reaction was further incubated at 37°C for 30 min. Samples were separated on 1.2% agarose. (C) Time of DinI addition determines extent of inhibition of binding of RecA to ssDNA. The reactions contained the same amounts of ssDNA, RecA, and DinI as in B, but were assembled in three different orders of addition. (1) Prevention: RecA was incubated with DinI first, then ssDNA was added. (2) Interference: DinI and ssDNA were preincubated together and then allowed to compete for RecA. (3) Displacement: DinI was added to preformed stable ssDNA–RecA complexes. In each case, two constituents of the reaction were preincubated at 37°C for 30 min, and after addition of the third component, reactions were allowed to proceed for an additional 30 min. After separation on 1.2% agarose gels, the amount of free DNA was quantitated and plotted as a percentage of the total radioactivity in the lane.
Filter binding revealed that the amount of ssDNA bound to RecA declines if RecA was preincubated for a short period with increasing amounts of DinI. That DinI has only a modest effect on the binding of the same ssDNA to the SSB protein, another single-strand binding protein from E. coli, shows that the prevention of the formation of the ssDNA–RecA complex by RecA is specific (Fig. 3A). To prove that this observation is not the result of ssDNA degradation caused by the presence of intrinsic or contaminating nucleases in the preparation of DinI, or for that matter, other activities that might interfere artifactually with filter binding, we used gel electrophoresis to visualize displaced or unbound DNA. The size of the ssDNA displaced from the RecA–ssDNA complex by DinI corresponds exactly to the size of the original DNA (Fig. 3B, lanes 6–9). Analysis of the products of the reaction in a high-resolution PAGE confirmed that there was no degradation of the released DNA (data not shown). The data in Figure 3B show that ssDNA is released quantitatively from a ternary ssDNA–RecA–DinI complex, which may be an intermediate of the reaction. Formation of such a complex is supported by the DinI-dependent upshift of the major band representing the RecA–DNA complex (Fig. 3B, lanes 3–9).
One can think of at least three possible mechanisms for the inhibitory effect of DinI on the binding of RecA to ssDNA. First, DinI is an ssDNA binding protein competing with RecA for ssDNA. Second, DinI interacts with RecA and blocks its ssDNA binding site. Third, DinI is able to interact with the ssDNA–RecA filament and displace DNA from it. The first possibility seems very unlikely because, as already mentioned above, we could not detect any ssDNA–DinI interactions. To choose from the two remaining possibilities, we performed ssDNA-binding experiments in such a way that DinI was added at different stages of the assembly of the RecA–ssDNA cofilament. The strongest inhibition of ssDNA binding was observed when DinI was allowed to form a complex with RecA before DNA was added (Fig. 3C, prevention). DinI could also effectively compete with DNA for binding to RecA when DNA and DinI were preincubated together before adding RecA (Fig. 3B,C, interference).
Moreover, DinI was capable of displacing of DNA from ssDNA–RecA cofilament (Fig. 3C, displacement), albeit the amount of RecA-free DNA was smaller than in the prevention and interference reactions. Thus, the binding of DinI to RecA significantly alters the ssDNA-binding capability of RecA. Quite surprisingly, DinI is able to displace ssDNA from a stable complex with RecA.
The C-terminal portion of DinI interacts with the L2 homologous pairing region of RecA
To examine the possibility that there is a direct interaction between the ssDNA binding site of RecA and DinI protein, we tested the effect of the DinI protein on the binding of a peptide (WECO) derived from the L2 region of RecA to ssDNA. We have shown previously that this is the ssDNA binding region of RecA, as well as the homologous pairing domain (Gardner et al. 1995; Malkov and Camerini-Otero 1995; Voloshin et al. 1996; Wang et al. 1998). As seen in Figure 4A, DinI significantly reduces the binding of ssDNA–WECO complexes to a nitrocellulose filter. The order in which the components were added is not important for achieving this effect: DinI efficiently depresses ssDNA–WECO complex formation both in interference and displacement reactions. Thus, DinI interacts with at least the L2 region of RecA.
Figure 4.
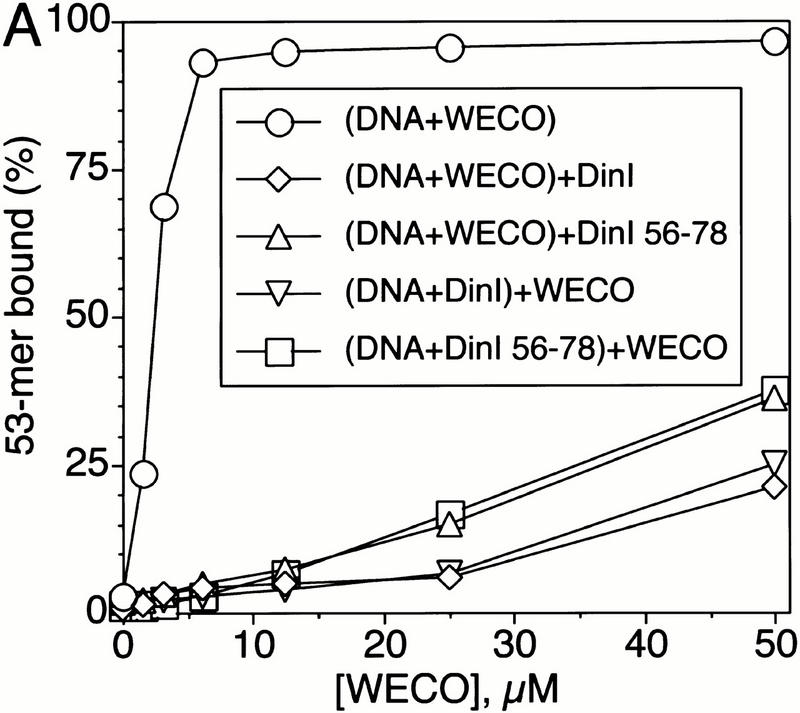
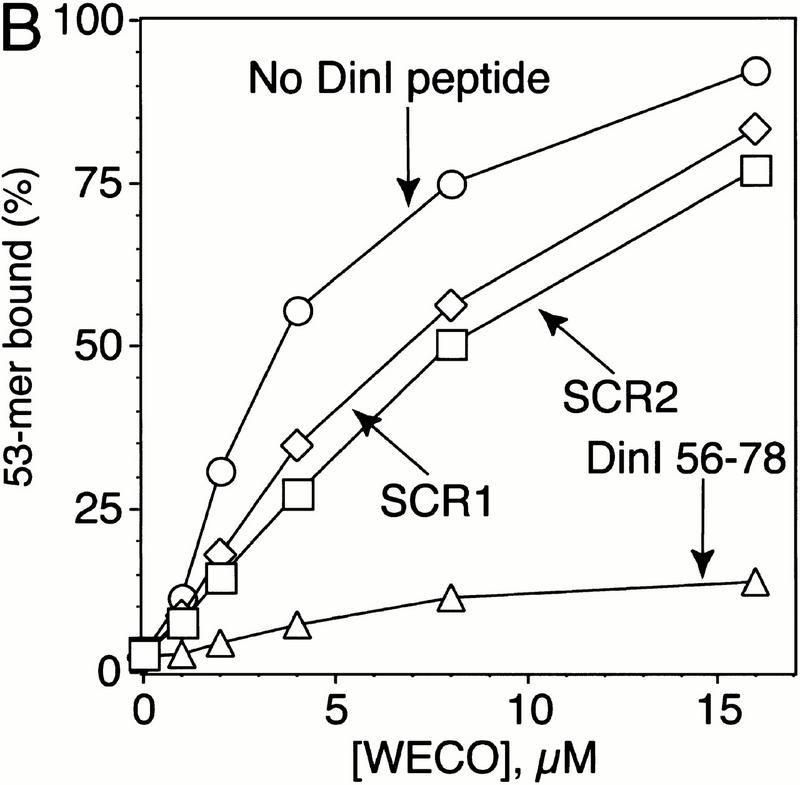
The loop L2-derived peptide from RecA (WECO) interacts with the C-terminal peptide from DinI. DinI and all DinI peptides were present at a concentration of 25 μM in the DNA-binding reaction; the concentration of ssDNA was 0.5 μM. WECO is a loop L2 derived 20-mer peptide comprising the DNA-binding and pairing domain of RecA (Voloshin et al. 1996; Wang et al. 1998). (A) The whole DinI protein and the DinI 56–78 peptide derived from its C terminus prevent binding of WECO to ssDNA and disassemble the ssDNA–WECO complex. (B) Interaction between WECO and DinI 56–78 is specific because peptides with the same amino acid composition but scrambled sequence, SCR1 and SCR2, do not disrupt the WECO–ssDNA complex.
To determine the part of the DinI protein that might interact with L2, we first prepared 40-mer peptides covering the N-terminal (DinI 1–40), middle (DinI 21–60) and C-terminal (DinI 42–81) parts of the whole DinI protein (Table 1). The effect of the DinI-derived peptides on the binding of WECO to ssDNA was examined by filter binding. We could find absolutely no influence of DinI 1–40 and DinI 21–60 peptides on the binding of ssDNA to WECO. However, DinI 42–81 efficiently prevented interactions between ssDNA and the WECO peptide (data not shown).
Table 1.
Amino acid sequences of the DinI protein and the peptides used in this work
| Protein/peptide name | Amino acid sequence | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DinI | M | R | I | E | V | T | I | A | K | T | S | P | L | P | A | G | A | I | D | A | L | A | G | E | L | S | R | R | I | Q | Y | A | F | P | D | N | E | G | H | V | S |
| V | R | Y | A | A | A | N | N | L | S | V | I | G | A | T | K | E | D | K | Q | R | I | S | E | I | L | Q | E | T | W | E | S | A | D | D | W | F | V | S | E | ||
| DinI 1–40 | M | R | I | E | V | T | I | A | K | T | S | P | L | P | A | G | A | I | D | A | L | A | G | E | L | S | R | R | I | Q | Y | A | F | P | D | N | E | G | H | V | |
| DinI 21–60 | L | A | G | E | L | S | R | R | I | Q | Y | A | F | P | D | N | E | G | H | V | S | V | R | Y | A | A | A | N | N | L | S | V | I | G | A | T | K | E | D | K | |
| DinI 42–81 | V | R | Y | A | A | A | N | N | L | S | V | I | G | A | T | K | E | D | K | Q | R | I | S | E | I | L | Q | E | T | W | E | S | A | D | D | W | F | V | S | E | |
| DinI 56–78 | T | K | E | D | K | Q | R | I | S | E | I | L | Q | E | T | W | E | S | A | D | D | W | F | ||||||||||||||||||
| DinI 56–81 | T | K | E | D | K | Q | R | I | S | E | I | L | Q | E | T | W | E | S | A | D | D | W | F | V | S | E | |||||||||||||||
| SCR1 | I | Q | T | T | S | I | Q | K | E | A | D | E | F | D | S | W | E | E | R | L | W | D | K | ||||||||||||||||||
| SCR2 | L | D | A | I | E | S | E | T | E | D | I | T | K | R | Q | W | K | S | F | W | D | E | Q | ||||||||||||||||||
| WECO | N | Q | I | R | M | K | I | G | V | M | W | G | N | P | E | T | T | T | G | G |
Next, the three-dimensional structure of DinI, which we have determined by NMR (nuclear magnetic resonance) spectroscopy (Ramirez et al. 2000), was used to design smaller peptides recapitulating the activity of the whole DinI. This structure reveals an α-β fold consisting of a three-stranded antiparallel β-sheet and two α-helixes (Fig. 5A). Interestingly, the C-terminal α-helix, highly enriched with negatively charged residues (7 of the 13 in whole DinI), forms a negatively charged surface (Fig. 5B,C). Because it is well known that RecA binds to the negatively charged phosphate backbone of DNA (see Discussion), we asked whether the C-terminal portion of DinI might be the RecA binding portion of the molecule. Accordingly, two peptides, 23-mer DinI 56–78 and 26-mer DinI 56–81, were prepared. Whereas the former peptide corresponds to the C-terminal α-helix proper, the latter contains three additional C-terminal amino acids.
Figure 5.
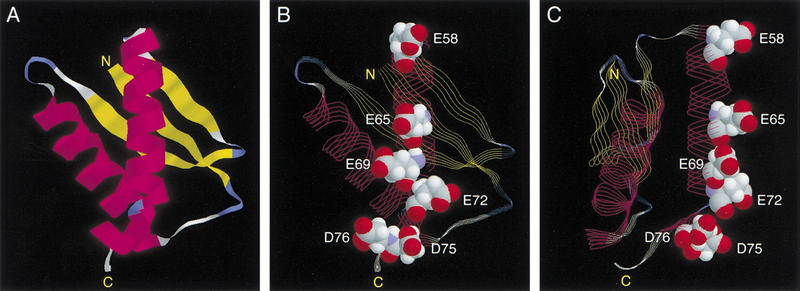
Negatively charged surface on the C-terminal α-helix in DinI mimics DNA. Figure is prepared with RasMol program from the structure determined by Ramirez and coworkers (2000). (A) Overall three-dimensional structure of DinI. (B,C) Space-filling representation of the negatively charged residues in the active site of DinI. Image (C) is rotated by 90° with respect to projection (B) to emphasize that the highlighted acidic residues form a surface.
As expected, both DinI 56–78 and DinI 56–81 were capable of disrupting the ssDNA–WECO complex. We only show the effect of the shortest 23-mer DinI 56–78 peptide (Fig. 4A). Because the C-terminal part of DinI is heavily enriched with negatively charged amino acid residues, and the loop L2-derived peptide contains arginine and lysine residues, one might argue that disruption of the ssDNA–WECO complex is the result of a mere nonspecific electrostatic interaction between these two peptides. To rule out this possibility, we prepared two control peptides, SCR1 and SCR2, in which the DinI 56–78 peptide sequence has been scrambled (Table 1), but the amino acid composition and net negative charge have been conserved. Figure 4B shows that the control peptides have a much smaller effect on the stability of the ssDNA–WECO complex than the DinI 56–78 peptide when added to the reaction at similar concentrations.
We then exploited surface plasmon resonance biosensor technology to follow real-time interactions between wild-type RecA and DinI and their mutant forms. To prove that the positively charged residues from L2 of RecA and the acidic amino acids from the C-terminal α-helix of DinI contribute to the interaction between these proteins, we created mutants carrying point substitutions in these regions: RecAR196M, RecAK198M, and DinIE72A. The overall gross conformation of all three mutant proteins was examined by circular dichroism spectroscopy and proven indistinguishable from that of their wild-type counterparts (data not shown). Whereas the ssDNA binding activity of RecAR196M could not be activated by ATPγS (Voloshin et al. 2000), RecAK198M mutant binds ssDNA at wild-type levels in the presence of ATPγS (data not shown).
Sensorgrams illustrating the binding of the wild-type and mutant RecA proteins to surface-immobilized wild-type DinI are presented in Figure 6A. The interactions between wild-type proteins generated a strong resonance signal, whereas RecA mutants lacking one of two positive charges in loop L2 region produced no signal at all. Figure 6B shows that the binding of wild-type RecA to mutant DinIE72A was affected considerably by single amino acid substitution. Not surprisingly, mutant RecAs did not bind DinIE72A. These biosensor experiments provide additional strong evidence for the direct interaction between RecA and DinI, and that the electrostatic forces between the C-terminal α-helix of DinI and loop L2 of RecA anchor this interaction.
Figure 6.
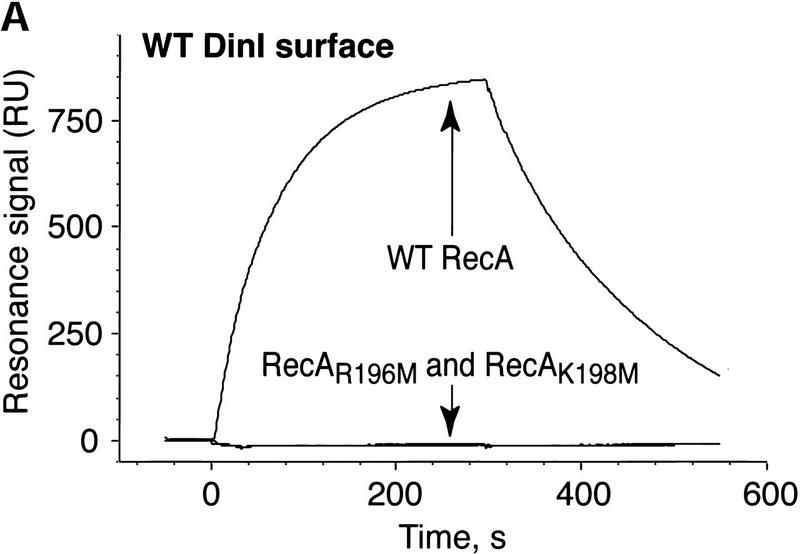
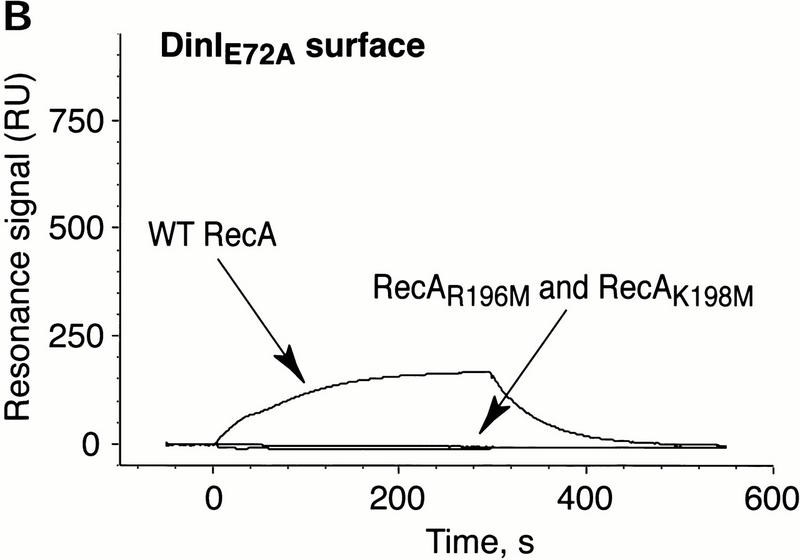
Electrostatic contacts between loop L2 of RecA and the C-terminal α-helix in DinI are critical to the interaction between RecA and DinI. The binding of wild-type and loop L2 mutants of RecA to wild-type DinI (A) and DinIE72A (B) was monitored using surface plasmon resonance on a BIAcore 1000 instrument. The data are presented in the form of sensorgrams that represent the changes in the surface plasmon resonance signal as a function of time.
Finally, we examined the effect of mutations in the C-terminal α-helix on the biological activity of DinI in vivo. E. coli cells overexpressing plasmid-born dinI are not able to mount an SOS response and, consequently, are sensitive to UV irradiation. (Yasuda et al. 1998). In contrast, overexpression of an inactive DinI should not interfere with the induction of the SOS response. Accordingly, UV sensitivity of cells overexpressing DinI can be a sensitive indicator of the protein activity in vivo. Figure 7A shows that single-point substitutions for each of most of the negatively charged residues in the C-terminal of DinI significantly reduce UV sensitivity of host cells.
Figure 7.
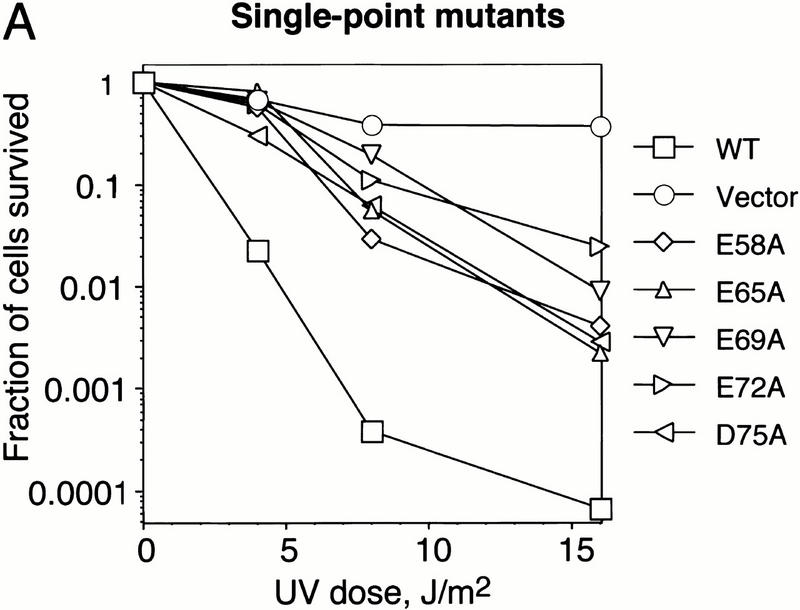
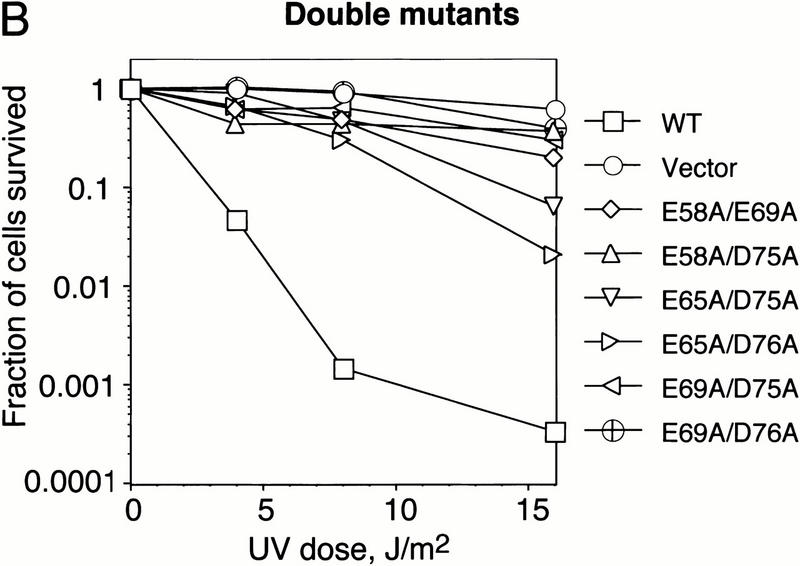
Mutations in the C-terminal α-helix of the DinI protein decrease the UV sensitivity of E. coli SY183 cells (Yasuda et al. 1998) overexpresing plasmid-born DinI. Cells expressing DinI mutant proteins with two acidic amino acids replaced with alanine (B) are less UV sensitive than cells expressing mutant proteins in which a single acidic amino acid has been replaced (A). SY183 carrying pBluescript II SK+ (Vector) or pBluescript II SK+ with the wild-type dinI gene (WT) were used as controls. Details of the experiment are described in Materials and Methods.
Double substitution mutants are even more UV resistant. These findings imply that the acidic residues from the negatively charged surface in the C-terminal part of DinI are important for the biological activity of the protein. These in vivo results are in good agreement with the biochemical experiments performed with synthetic peptides and purified proteins presented in Figures 4 and 6. Taken together, all these experiments prove that the C-terminal α-helix of DinI interacts specifically with loop L2 of RecA, and strongly suggest that this interaction might be entirely responsible for the biological activity of the whole protein.
DinI inhibits homologous DNA pairing but does not affect ongoing strand exchange
RecA promotes two key events in homologous recombination: homologous DNA pairing and strand exchange (West 1992; Camerini-Otero and Hsieh 1995; Bianco et al. 1998). Previous reports have shown that overproduction of the DinI protein reduces the frequency of RecA-mediated homologous recombination at least 100-fold (Yasuda et al. 1998). To examine the effect of DinI on RecA-mediated DNA pairing, we used relatively short DNA substrates (Rosselli and Stasiak 1991). The pairing reaction was launched by the addition of dsDNA to preformed presynaptic filament. No pairing product was observed when two DNA molecules were mixed alone or in the presence of DinI (Fig. 8B, lanes 8,). Addition of RecA (Fig. 8B, lane 4) promoted the reaction and DinI inhibited it in a concentration-dependent manner (Fig. 8B, lanes 5–10). The observed degree of inhibition of DNA pairing was much greater than could be accounted for by a simple ssDNA displacement from ssDNA–RecA complex (cf. Figs. 8C and 3C, displacement). As we have shown previously, in addition to its ssDNA binding activity, 20-mer peptides derived from L2 can promote homologous DNA pairing (Voloshin et al. 1996). Inhibition of the DNA-pairing activity of RecA provides additional evidence for the interaction of DinI with the L2 loop region in RecA and predicts that the pairing promoted by L2 peptides (Voloshin et al. 1996) should also be inhibited by DinI. Indeed, we have found that both DinI and DinI 56–78 inhibited the D-loop formation promoted by WECO (Voloshin et al. 1996), whereas SCR1 and SCR2 had no effect (data not shown).
Figure 8.
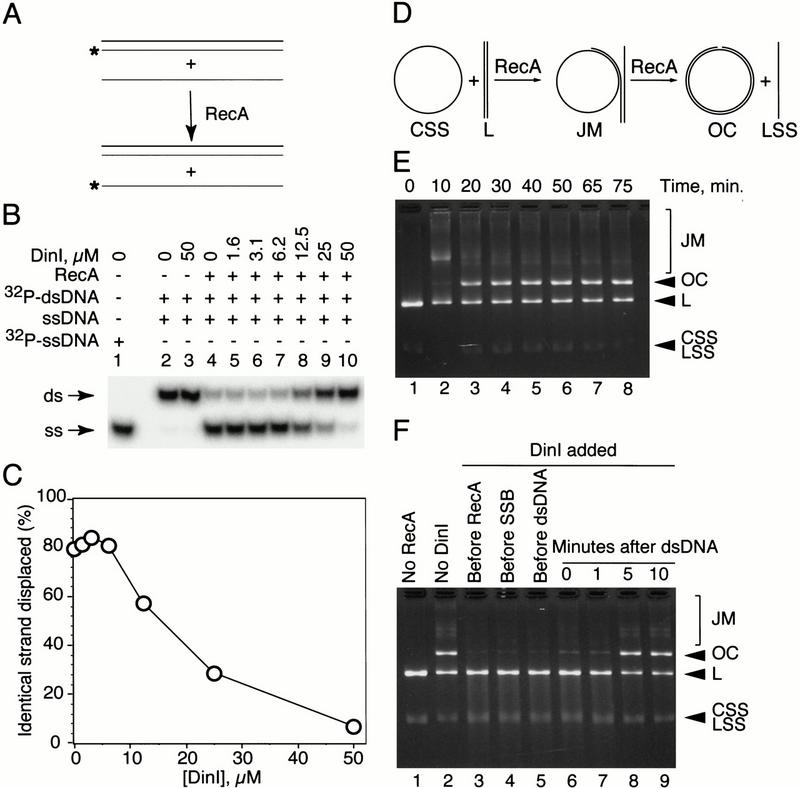
DinI inhibits RecA-mediated homologous DNA pairing but does not prevent completion of the actively proceeding strange exchange reaction. (A) Schematic illustration of the experiment. Presynaptic filament was formed after incubation of unlabeled BS-S1 with a stoichiometric amount of RecA followed by addition of DinI; the pairing reaction was immediately initiated by addition of homologous BS duplex. (B) The amount of displaced 32P-labeled BS-S1 strand, reflecting the extent of the pairing reaction, was estimated after deproteinization and separation of the samples by 12% native PAGE. (C) Quantitation of the gel shown in B. (D) Illustration of the strand exchange reaction. RecA mediates pairing of covalently closed ssDNA (CSS) and homologous linear dsDNA (L) giving rise to joint molecules (JM). RecA-promoted branch migration results in strand exchange and formation of double-stranded open circles (OC) and linear ssDNA (LSS). Substrates and the final product of the reaction (OC) can be easily separated on an agarose gel. (E) Time course of the standard (DinI-free) strand exchange reaction. (F) Addition of DinI to the strand exchange reaction before or right after dsDNA blocks strand transfer. However, the yield of the final product (OC) was not affected when DinI was introduced to the reaction after DNA pairing had occurred.
A partial disruption or modification of the presynaptic filament by DinI could also explain the finding that the inhibition of RecA-mediated pairing is greater than expected from a simple ssDNA displacement mechanism. That the presynaptic filament is altered by DinI is suggested by the upshift of the major ssDNA–RecA band after addition of DinI (Fig. 3B). In addition to the loop L2 region, the interaction of DinI with the presynaptic filament may involve other parts of the RecA molecule.
To study the possible effect of DinI on a strand exchange reaction in progress, we added DinI at different time points to the RecA-promoted reaction between two φX174 DNA molecules (Fig. 8D–F). In the absence of DinI, the yield of nicked molecules (OC) was barely detectable after 10 min of incubation (after the addition of dsDNA) but reached a plateau after 30 min (Fig. 8E). When DinI was added to the assembling strand exchange reaction before, or within 1 min after the addition of dsDNA, very little of the final product was observed even after 30 min of incubation (Fig. 8F, lanes 3–7). If the DNA pairing was allowed to proceed for at least 5 min (a time not sufficient to complete strand exchange) before introduction of DinI, the reaction reached completion (Fig. 8F, cf. lanes 2,8, and 9). Thus, DinI suppresses the initial step of strand exchange, DNA pairing, yet it has no effect on the second stage, RecA-mediated strand transfer. This observation can be rationalized by assuming that the stable incorporation of a second DNA molecule into the homologous DNA pairing site of RecA might interfere with DinI binding. That ongoing strand exchange is not blocked after emergence of an SOS termination signal (DinI) makes biological sense. It allows the cell to complete the repair process.
Discussion
As Ohmori and coworkers showed first, overexpression of the DinI protein prevents initiation of the SOS response in E. coli cells (Yasuda et al. 1998). To elucidate the mechanism of this negative regulation, we studied the effect of the purified DinI protein on the biochemical activity of the RecA protein, a key player in the process of the SOS induction. We have shown that DinI and RecA physically interact (Figs. 1,6). This interaction can be detected with purified proteins, as well as in cell lysates prepared from UV-irradiated cells (Fig. 2). It is believed that formation of the active ssDNA–RecA cofilament is a prerequisite for SOS induction (Friedberg et al. 1995; Walker 1996). We show that DinI can eliminate the SOS induction signal in three different ways: First, DinI is able to interact with the ssDNA binding site of inactive RecA, precluding RecA from being activated by its binding to ssDNA. Second, if ssDNA and DinI are exposed simultaneously to RecA, DinI interferes with DNA binding. Third, DinI can disrupt stable ssDNA–RecA cofilaments (Fig. 3).
Certain findings strongly suggest that DinI may require yet unknown cofactors for modulating its activity. We found that a relatively high concentration of the DinI protein was required for the biochemical activities described here. Similar concentrations were also needed to inhibit the in vitro coprotease activity of RecA in the autocatalytic digestion of the UmuD protein (Yasuda et al. 1998). Thus, the system reconstructed from the purified proteins might be missing one or more components. The fact is that both proteins show very similar kinetics of expression after SOS induction (Fig. 2A) is also consistent with the idea that additional factors could be involved in the regulation of the interaction of DinI and RecA. It would be more reasonable to expect a peak of DinI overexpression at later stages in the SOS response, after the repair of DNA damage and when the cell is ready to return to the uninduced state. Intriguingly, however, the extent of interaction between DinI and RecA increases at these later stages (Fig. 2B), just when one would expect DinI to act as a down-regulator of the SOS response. This enhanced interaction of RecA and DinI at later stage of the SOS response is the most compelling finding arguing for the existence of one or more modulators of the RecA–DinI interaction. These unknown factors could be unidentified proteins or low molecular weight regulators interacting with RecA or DinI. On the other hand, we cannot exclude that a posttranslational modification of RecA or DinI is involved in the regulation of their interaction.
At this point, we do not know whether monomers or multimers of the protein constitute the active form of DinI. The behavior of the protein during purification, NMR results, and preliminary MALDI (matrix-assisted laser desorption ionization) mass-spectroscopic analysis indicate that DinI can form dimers or tetramers (Voloshin et al., unpubl.). If DinI acts as a multimer, the high concentration required for its biochemical activity may reflect the need to shift the monomer-multimer equilibrium to multimer. Finally, we cannot exclude the possibility that additional factors or posttranslational modifications of DinI may stimulate the transition to the multimeric state in vivo.
The recently described chromosomal isfA mutation results in increasing UV sensitivity of E. coli cells and inhibits many SOS-related functions: mutagenesis, cell filamentation, prophage induction, and UmuD processing (Bebenek and Pietrzykowska 1995, 1996; Felczak et al. 1999). The corresponding isfA gene product (still unidentified) is a conceivable candidate for the role of another modulator of RecA activity. It is doubtful that dinI and isfA gene products regulate SOS functions in concert and synergistically enhance each other's activity. More likely, they act in opposite directions (up- and down-regulation of SOS), because attenuation of the SOS response ensues from a mutation of isfA or from overexpression of dinI.
DinI does not inhibit the induction of the SOS response by competing with RecA for ssDNA, but rather competes with DNA for the same binding site on the RecA molecule. Several crucial observations implicate participation of the ssDNA-binding and homologous DNA-pairing domain of RecA, loop L2, in the interaction with the C-terminal part of DinI. First, the DinI 56–78 peptide recapitulates the activity of whole DinI in preventing the binding of ssDNA to a loop L2-derived peptide from RecA (Fig. 3). Second, the C-terminal α-helix in DinI (residues 57–78), because it is highly enriched in glutamic and aspartic residues, forms a negatively charged surface (Fig. 4B,C). These negative charges recall the negative phosphates on DNA that are crucial for its interaction with RecA (Leahy and Radding 1986).
Third, the point substitution E72A in the C-terminal part of DinI significantly impairs interaction with RecA; reciprocally, the replacement of either of the two positive charges in the L2 region of RecA with methionine completely eliminates the interaction of RecA with DinI (Fig. 6). Finally, point substitutions of negatively charged residues with alanines significantly decrease the UV sensitivity of E. coli cells overexpressing DinI from a multicopy plasmid (Fig. 7).
Thus, the active site of DinI mimics the negative charges on a DNA molecule, essentially acting like a competitor for DNA on RecA. Although proteins mimicking the overall shape of a nucleic acid molecule (tRNA) have been described (Selmer et al. 1999), to our knowledge, DinI is the first example of a protein imitating the distribution of negative charges on the phosphate backbone of DNA.
A BLAST sequence similarity search revealed significant homology (31% identity—24 of 77—at the amino acid level) between DinI and the Tum protein, an antirepressor from coliphage 186. Interestingly, Tum has been shown not to compete with the repressor for binding sites on DNA. Instead, it has been proposed to bind to the DNA binding site of the repressor or prevent the repressor's cooperative interactions or oligomerization (Shearwin et al. 1998).
Because this repressor, unlike RecA, binds to a specific cognate DNA sequence, it would be of interest if Tum also acts as a DNA mimic, albeit in a specific manner. Alternatively, DinI might bind not only to the DNA binding site of RecA but also to one or more RecA domains to prevent oligomerization. It is this activity that might be shared with Tum.
In conclusion, our model for the negative role of DinI in the regulation of the SOS response follows this outline: DinI acts as a part of a classic feedback mechanism and does so in the most direct and simple manner. Mainly, as the product of one of the genes turned on during the SOS response, DinI directly disrupts and prevents the formation of the signal that turns on the SOS pathway, the ssDNA–RecA cofilament (Fig. 9). Furthermore, the ability of the DinI protein to control the cornerstone function of RecA—ssDNA binding—implies that its role may not be restricted to the down-regulation of the SOS response. It could regulate other RecA-dependent cellular processes, such as homologous recombination and the reestablishment of collapsed replication forks (Cox et al. 2000).
Figure 9.
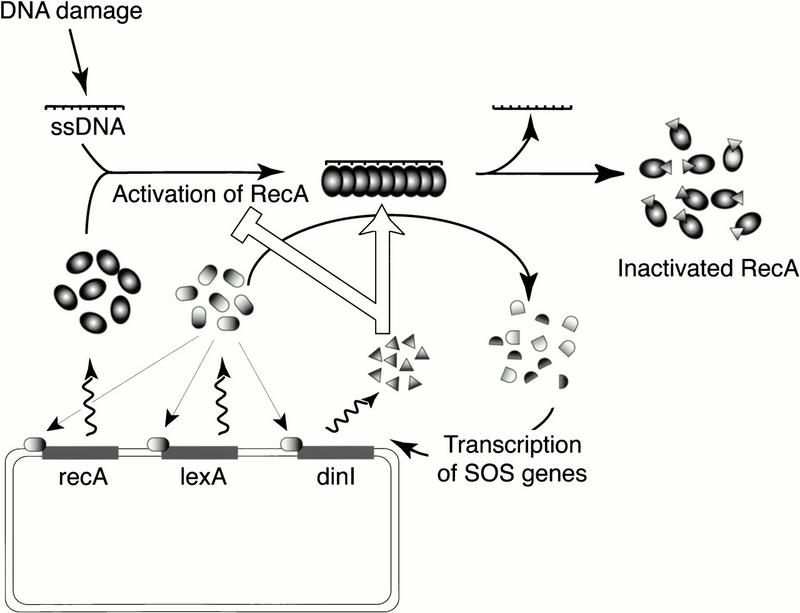
The possible role of DinI in the SOS regulatory network. The SOS response is signaled on by RecA binding to ssDNA that accumulates in the cell as a result of DNA damage. Induction of the SOS response leads to overexpression of at least 31 gene products (Fernandez de Henestrosa et al. 2000). Among them are RecA, LexA, and DinI, the proteins involved in the autoregulation of SOS. The role of DinI consists in turning down the SOS response by abolishing the SOS signal. DinI can eliminate this signal in two ways: by preventing interaction between RecA and ssDNA or by disrupting ssDNA–RecA cofilaments. A detailed description of the SOS response and its induction can be found in several reviews (Friedberg et al. 1995; Walker 1996; Kuzminov 1999).
Materials and methods
All routine procedures and manipulations were performed as described (Sambrook et al. 1989; Ausubel et al. 1998).
DNA substrates
The BS-S1 oligonucleotide, spanning positions 742 to 690 in the polylinker region of pBluescript SK+ plasmid, its complement, BS-S2, and PCR primers were synthesized on Applied Biosystems model 380B synthesizer and purified by denaturing PAGE. BS duplex, formed by annealing of BS-S1 and BS-S2, was purified by native PAGE. Virion (single-stranded) and replicative (double-stranded) forms of φX174 DNA were obtained from New England Biolabs and additionally purified by phenol deproteinization and ethanol precipitation.
DNA concentrations are expressed as base (for ssDNA) or base pair (for dsDNA) concentrations.
Site-directed mutagenesis
Site-directed mutagenesis was performed with the QuikChange Mutagenesis Kit (Stratagene) in accordance with manufacturer's instructions, and introduced mutations were verified by DNA sequencing.
Proteins and peptides
The gene encoding for the DinI protein was amplified from E. coli strain W3110 DNA (purchased from Sigma) using Din-5′-Nde (TTT CAG TTT CAT ATG CGA ATT GAA GTC ACC AT) and Din-3′-Xho (TTT ACG TTA CTC GAG TTA TTC GCT GAC AAA CCA GT) primers and PCR SuperMix (GIBCO BRL). Portions of the oligonucleotides corresponding to the portion of dinI gene are underlined, and the _Nde_I and _Xho_I recognition sites are shown in italics.
The PCR fragment was ligated into pET21a(+) expression vector (Novagen) precut with _Nde_I and Xho_I, allowing for expression of recombinant proteins under the control of the T7_lac promoter. We maintained the stop codon in the dinI gene to avoid a 6-His tag at the C terminus of the protein. The correspondence of the cloned fragment to the published DinI sequence (Yasuda et al. 1996) was confirmed by DNA sequencing. The DinI protein was overexpressed in BLR (DE3) pLysS cells (Novagen) on addition of 1 mM IPTG. In pilot experiments, we did not notice any difference in the intracellular DinI protein level when the IPTG induction was performed over 1 or 3 hr.
One liter of a bacterial culture in LB media supplemented with 100 μg/mL ampicillin and 25 μg/mL chloramphenicol was grown at 37°C to OD600 = 0.5, and the protein expression was induced with 1 mM IPTG over a 1 hr period. We used the same modification of the described procedure (Yasuda et al. 1998) for the purification of both wild-type and mutant E72A DinI. The cells were collected by centrifugation at 5000 rpm for 10 min in a Sorvall SLA-3000 rotor, resuspended in 30 mL of buffer A (20 mM Tris-HCl at pH 7.4, 100 mM NaCl, 1 mM EDTA, 10% glycerol) and sonicated. All purification procedures were performed at 4°C.
The lysate was cleared by centrifugation at 18,000 rpm for 1 hr in an SS-34 Sorvall rotor, and nucleic acids were removed by treatment of the supernatant with 3% (w/v) streptomycin sulfate.
The crude extract was fractionated with ammonium sulfate, and the fraction corresponding to 40%–80% saturation was precipitated. The protein was dissolved in 10 mL of buffer B (20 mM Tris-HCl at pH 7.4, 1 mM EDTA), desalted on a Hi-Trap Desalting column (Pharmacia Biotech), and applied to a SOURCE 30Q column (Pharmacia Biotech). The column was developed with a linear salt gradient (0–1 M NaCl in buffer B). DinI containing fractions were pooled. The protein was precipitated with ammonium sulfate (80% saturation), dissolved in buffer B plus 1 M NaCl, and applied to a HiLoad 16/60 Superdex 75 prep grade column (Pharmacia Biotech) equilibrated with the same buffer. The DinI protein was desalted on a Hi-Trap Desalting column equilibrated with buffer B and passed through a ssDNA-agarose (GIBCO BRL) column to remove contaminating nuclease activities. The protein from the flowthrough of this column was concentrated by ammonium sulfate precipitation and stored at −80°C after dialysis against 20 mM Tris-HCl (pH 7.4), 1 mM EDTA, and 50% glycerol.
13C- or 15N-labeled DinI proteins used in NMR studies were purified from BL21(DE3) cells grown in minimal media supplemented by 13C-glucose and 15N-NH4Cl as sole sources of carbon and nitrogen, respectively.
All the peptides were prepared on an ABI model 430A synthesizer and purified on a C18 reverse phase column (218TP1022, Vydac). WECO peptide, derived from the loop L2 region of RecA, has been previously described (Voloshin et al. 1996; Wang et al. 1998). The numbers in the names of the DinI-derived peptides correspond to the amino acid positions in the whole protein.
The amino acid sequences of all the peptides used in this study are shown in Table 1.
Rabbit polyclonal antibodies were prepared by Covance. Anti-DinI antibodies were raised against the DinI 42–81 peptide, whereas for anti-RecA antibody preparation, the whole protein was used. Anti-DinI antibodies were affinity-purified on a DinI 42–81 peptide column using an AminoLink Immobilization Kit and Immunopure Gentle Ag/Ab Buffers from Pierce in accordance with the manufacturer's recommendations. Anti-RecA serum was used without prior purification. Anti-RecA ARM193 monoclonal antibody was purchased from PanVera. Restriction endonucleases and DNA modifying enzymes were from New England Biolabs. E. coli SSB protein was obtained from USB Biochemicals.
The wild-type and mutant RecA proteins were purified as described (Voloshin et al. 2000).
Preparation of cell lysates for Western blot analysis and immunoprecipitation
We used the M9 medium supplemented with 0.4% glucose and 0.5% casein hydrosylate (peptone #140, Difco) for these experiments. An overnight culture of the wild-type E. coli K12 cells was diluted 1:100 in 35 mL of the media and grown at 37°C to OD600 = 0.4. The culture was transferred to 100 mm petri dish and irradiated with 25 J/m2 at room temperature in Stratalinker 1800 (Stratagen) equipped with germicidal lamps. A control culture was kept at room temperature for the same period but was not subjected to UV treatment. Both cultures were allowed to grow at 37°C, and aliquots were taken at 20 min intervals. Cells were collected, resuspended in 500 μL of TE buffer at pH 7.4, and disrupted by sonication. The lysates were cleared by centrifugation and the concentration of the total protein was determined with a Bio-Rad Protein Assay Kit.
Immunoprecipitation assay
In general, “insoluble or highly polymerized antigens may not be able to be studied using immunoprecipitations, because they pellet in the centrifugation step regardless of whether or not specific antibodies are present” (Harlow and Lane 1999). Such is the case for RecA, which, depending on the protein concentration and ionic conditions, forms a variety of quaternary structures, including highly aggregated filaments and bundles of rods (Brenner et al. 1988). Accordingly, we modified the experimental procedure so that multiple centrifugations of the immunoprecipitation reactions were replaced by a very intensive (at least 2000 volumes) wash in disposable columns (see below). This approach significantly reduced the background attributable to RecA aggregation. Nevertheless, in some cases we observed signals from trace amounts of RecA even in the absence of specific antibody.
For in vitro assays, 10 μM RecA and 25 μM DinI were incubated in 20 mM Tris-HCl (pH 7.5), 10 mM MgCl2, and 20 mM NaCl in a total volume of 20 μL at 37°C for 30 min. Where indicated, RecA was preincubated with 30 μM BS-S1 oligonucleotide (Voloshin et al. 1996), 200 μM ATPγS or 200 μM ADP at 37°C for 30 min, and combined with DinI. Reactions were diluted 10-fold with buffer IP (20 mM Tris-HCl at pH 7.5, 50 mM NaCl, 0.2% Tween 20, 0.02% NP-40, 0.2% SDS) and 5 μL of preimmune or immune serum, or 4 μg of antigen-purified antibodies, were added. The immunoprecipitation reactions were performed at 4°C for 14 hr. After addition of 50 μL of UltraLink Immobilized Protein G gel (Pierce), reactions were left for 2 hr at room temperature on a rotary shaker. The slurry was transferred to empty disposable columns, washed with 100 mL of buffer PI, and transferred to microcentrifuge tubes. After brief centrifugation, the supernatant was discarded and the gel was resuspended in 50 μL of SDS-PAGE sample buffer. Five microliters of each reaction were separated in two identical 10%–20% tricine SDS gels (Novex), and the proteins were transferred to the PVDF membranes. Each membrane was probed with monoclonal anti-RecA ARM193 or polyclonal antigen-purified anti-DinI antibody. We used the ECL Western blotting detection system from Amersham Pharmacia Biotech.
Immunoprecipitation of proteins in lysates prepared from UV-irradiated or untreated E. coli cells was performed similarly except for these changes: (1) 200 μg of the total protein were used; (2) 100 μL of UltraLink Immobilized Protein A gel (Pierce) were incubated with 200 μL of the lysate for 4 h in the cold room; (3) polyclonal antiserum was used for RecA detection.
DNA binding assays
DNA–RecA binding reaction contained 0.5–2.5 μM BS-S1, 0–1 μM RecA, 0–60 μM DinI, 300 μM ATPγS, 10 mM MgCl2, 20 mM NaCl, and 20 mM Tris-HCl at pH 7.5. After incubation at 37°C, products were filtered using a double filter system (Wong and Lohman 1993) or separated on 1.2% agarose in TAE buffer supplemented with 3 mM MgCl2.
Peptide–DNA complexes were formed as described (Voloshin et al. 1996). Filters and dried gels were quantitated with a Fuji 2000 imaging system.
NMR structure determination
The NMR structure of DinI was determined using heteronuclear triple resonance NMR on a 0.6 mM sample in H2O, 100 mM NaCl (pH 6.6). Experiments were performed at 500, 600, and 800 MHz 1H frequency, using standard two- and three-dimensional NMR (Bax and Grzesiek 1993). In addition to the conventional NOE-derived interproton distances (an average of 14 restraints per residue) and J-coupling derived torsion angle restraints, ∼500 global orientation restraints were derived from heteronuclear dipolar couplings, measured in dilute liquid crystalline media of planar micelles (bicelles) (Tjandra and Bax 1997) and filamentous phage Pf1 (Hansen et al. 1998). Details regarding the NMR structure determination have been published elsewhere (Ramirez et al. 2000).
In vivo assay for DinI activity
The coding sequence and the promoter region of the dinI gene were amplified from E. coli strain W3110 DNA, using Din-5′-Xba (TTT CAG TTT TCT AGA GGA AAC GGT ACG CTG GTC) and Din-3′-Xho (TTT ACG TTA CTC GAG TTA TTC GCT GAC AAA CCA GT) primers and PCR SuperMix (GIBCO BRL). The _Xba_I and _Xho_I recognition sites are shown in italics. The dinI gene, alone with its promoter, was inserted between _Xba_I and _Xho_I recognition sites of the plasmid pBluesript II SK+. For the UV sensitivity assay, we used SY183 strain kindly provided by Haruo Ohmori ([araD139], Δ(araA-leu)7697, Δ(codB-lacI)3, _galK16, galE15, λ−, mcrA_−, relA1, rpsL150(strR), _spoT1, mcrB_−, hsdR2, ΔdinI2::kan) (Yasuda et al. 1998). Levels of plasmid-driven expression of mutant dinI genes in SY183 cells were verified by Western blotting procedure and found to be comparable with expression of the wild-type gene.
Overnight cultures of SY183 cells carrying plasmid-born wild-type or mutant dinI genes were prepared in LB media supplemented with 100 μg/mL ampicillin. After a 50-fold dilution, cells were grown in the same media at 37°C to OD600 = 0.5. One hundred microliters of serial dilutions were plated on LB plates with 100 μg/mL ampicillin, and plates were irradiated with different doses of short-wave UV light in a Stratalinker 1800 (Strategene). Colonies produced by surviving cells were counted after an overnight incubation in the dark at 37°C.
Surface plasmon resonance measurements
All surface plasmon resonance measurements were performed on a BIAcore 1000 instrument (BIAcore) at 25°C and at a flow rate of 10 μL/min. DinI proteins were immobilized on a CM 5 sensor chip using standard amino coupling chemistry (Johnsson et al. 1991). The wild-type and mutant E72A proteins immobilized on the same sensor chip produced response signals of 550 RU (resonance units) and 750 RU, correspondingly. A control empty surface was created in the same batch run. The flow buffer was 20 mM Tris-HCl (pH 7.4), 50 mM NaCl, 10 mM MgCl2, 0.4 mM DTT, and 0.005% surfactant P20. RecA proteins were injected at a concentration of 1 μM in 50 μL of the flow buffer. The signal generated by an empty surface was always subtracted from the experimental sensorgrams. Surfaces were regenerated by injection of 3 M NaCl in flow buffer.
Homologous DNA pairing and strand exchange reactions
Homologous DNA pairing was studied using 53-mer BS-S1 oligonucleotide and BS duplex as substrates. Unlabeled BS-S1 was incubated with RecA at 37°C for 20 min. DinI and BS duplex containing 32P-label in BS-S1 strand were added sequentially and the reaction was incubated for an additional 20 min at 37°C. Twenty microliters of the final reaction mixture contained 6 μM BS-S1, 2 μM RecA, 0–50 μM DinI, 0.6 μM BS duplex, 50 mM Tris-HCl (pH 7.4), 12.5 MgCl2, 0.4 mM DTT, and 0.3 mM ATPγS. The reaction was terminated by addition of EDTA and SDS to final concentrations of 12.5 mM and 2.5%, respectively, and the products were separated on 12% nondenaturing PAGE gels.
Strand exchange reactions contained 60 ng of single-stranded φX174 DNA, 60 ng of the replicative form linearized with _Xho_I, 3 μM RecA, 2.5 μM SSB, 2 mM ATP, 6 mM phosphocreatine, 10 units/mL creatine phosphokinase, 100 μg/mL BSA, 50 mM Tris-HCl (pH 7.4), 12.5 mM MgCl2, and 0.4 mM DTT in a total volume of 20 μL. Where indicated, 50 μM DinI was added.
The reaction was assembled in the following order: ssDNA was preincubated with RecA at 37°C for 2 min, followed by addition of SSB and additional incubation at 37°C for 2 min. The strand exchange reaction was initiated by addition of dsDNA. The DinI protein was added at different stages in the assembling of the strand exchange reaction: before RecA, before SSB, before dsDNA, or 0–10 min after dsDNA was added. After incubation at 37°C for 10–75 min, reactions were deproteinized by the addition of SDS to 2.5% and separated on 1% agarose TAE gel followed by staining with ethidium bromide. The reactions shown in the Figure 8F were performed for 30 min after the addition of dsDNA.
Acknowledgments
We thank Oksana Gavrilova, Peggy Hsieh, Howard Nash, Susan Lovett, Katsumi Morimatsu, Haruo Ohmori, Charles Radding, Andrzej Stasiak, Steve West, and members of the Camerini-Otero laboratory for many valuable suggestions and discussions. James Elmer participated in the early stages of this work. We thank Haruo Ohmori for providing us with the dinI deletion strain SY183, George Poy for oligonucleotide and peptide synthesis, and Linda Robinson for secretarial assistance.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL camerini@ncisun1.ncifcrf.gov; FAX (301) 496-9878.
Article and publication are at www.genesdev.org/cgi/doi/10.1101/gad.862901.
References
- Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struhl K, Albright LM, Coen DM, Varki A. Current protocols in molecular biology. New York: John Wiley; 1998. [Google Scholar]
- Bax A, Grzesiek S. Methodological advances in protein NMR. Accounts Chem Res. 1993;26:131–138. [Google Scholar]
- Bebenek A, Pietrzykowska I. A new mutation in Escherichia coli K12, isfA, which is responsible for inhibition of SOS functions. Mol Gen Genet. 1995;248:103–113. doi: 10.1007/BF02456619. [DOI] [PubMed] [Google Scholar]
- ————— The isfA mutation inhibits mutator activity and processing of UmuD protein in Escherichia coli recA730 strains. Mol Gen Genet. 1996;250:674–680. doi: 10.1007/BF02172978. [DOI] [PubMed] [Google Scholar]
- Bianco PR, Tracy RB, Kowalczykowski SC. DNA strand exchange proteins: A biochemical and physical comparison. Front Biosci. 1998;3:D570–D603. doi: 10.2741/a304. [DOI] [PubMed] [Google Scholar]
- Brenner SL, Zlotnick A, Griffith JD. RecA protein self-assembly. Multiple discrete aggregation states. J Mol Biol. 1988;204:959–972. doi: 10.1016/0022-2836(88)90055-1. [DOI] [PubMed] [Google Scholar]
- Camerini-Otero RD, Hsieh P. Homologous recombination proteins in prokaryotes and eukaryotes. Annu Rev Genet. 1995;29:509–552. doi: 10.1146/annurev.ge.29.120195.002453. [DOI] [PubMed] [Google Scholar]
- Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- Felczak M, Bebenek A, Pietrzykowska I. The isfA mutation specifically inhibits the SOS-dependent mutagenic pathway and does not selectively affect any particular base substitution. Mutagenesis. 1999;14:295–300. doi: 10.1093/mutage/14.3.295. [DOI] [PubMed] [Google Scholar]
- Fernandez de Henestrosa AR, Ogi T, Aoyagi S, Chafin D, Hayes JJ, Ohmori H, Woodgate R. Identification of additional genes belonging to the LexA regulon in Escherichia coli. Mol Microbiol. 2000;35:1560–1572. doi: 10.1046/j.1365-2958.2000.01826.x. [DOI] [PubMed] [Google Scholar]
- Friedberg EC, Walker GC, Siede W. DNA repair and mutagenesis. Washington, D.C.: ASM Press; 1995. [Google Scholar]
- Gardner RV, Voloshin ON, Camerini-Otero RD. The identification of the single-stranded DNA-binding domain of the Escherichia coli RecA protein. Eur J Biochem. 1995;233:419–425. doi: 10.1111/j.1432-1033.1995.419_2.x. [DOI] [PubMed] [Google Scholar]
- Hansen MR, Mueller L, Pardi A. Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat Struct Biol. 1998;5:1065–1074. doi: 10.1038/4176. [DOI] [PubMed] [Google Scholar]
- Harlow E, Lane D. Using antibodies: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1999. [Google Scholar]
- Johnsson B, Lofas S, Lindquist G. Immobilization of proteins to a carboxymethyldextran-modified gold surface for biospecific interaction analysis in surface plasmon resonance sensors. Anal Biochem. 1991;198:268–277. doi: 10.1016/0003-2697(91)90424-r. [DOI] [PubMed] [Google Scholar]
- Kenyon CJ, Walker GC. DNA-damaging agents stimulate gene expression at specific loci in Escherichia coli. Proc Natl Acad Sci. 1980;77:2819–2823. doi: 10.1073/pnas.77.5.2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch WH, Woodgate R. The SOS response. In: Nockoloff JA, Hoekstra MF, editors. DNA damage and repair. Totowa, NJ: Humana Press; 1998. pp. 107–134. [Google Scholar]
- Kuzminov A. Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda. Microbiol Mol Biol Rev. 1999;63:751–813. doi: 10.1128/mmbr.63.4.751-813.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leahy M, Radding CM. Topography of the interaction of recA protein with single-stranded deoxyoligonucleotides. J Biol Chem. 1986;261:6954–6960. [PubMed] [Google Scholar]
- Little JW. Mechanism of specific LexA cleavage: Autodigestion and the role of RecA coprotease. Biochimie. 1991;73:411–421. doi: 10.1016/0300-9084(91)90108-d. [DOI] [PubMed] [Google Scholar]
- Malkov VA, Camerini-Otero RD. Photocross-links between single-stranded DNA and Escherichia coli RecA protein map to loops L1 (amino acid residues 157–164) and L2 (amino acid residues 195–209) J Biol Chem. 1995;270:30230–30233. doi: 10.1074/jbc.270.50.30230. [DOI] [PubMed] [Google Scholar]
- Ramirez BE, Voloshin ON, Camerini-Otero RD, Bax A. Solution structure of DinI provides insight into its mode of RecA inactivation. Protein Sci. 2000;9:2161–2169. doi: 10.1110/ps.9.11.2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roca AI, Cox MM. RecA protein: Structure, function, and role in recombinational DNA repair. Prog Nucleic Acid Res Mol Biol. 1997;56:129–223. doi: 10.1016/s0079-6603(08)61005-3. [DOI] [PubMed] [Google Scholar]
- Rosselli W, Stasiak A. The ATPase activity of recA is needed to push the DNA strand exchange through heterologous regions. EMBO J. 1991;10:4391–4396. doi: 10.1002/j.1460-2075.1991.tb05017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Selmer M, Al-Karadaghi S, Hirokawa G, Kaji A, Liljas A. Crystal structure of Thermotoga maritima ribosome recycling factor: A tRNA mimic. Science. 1999;286:2349–2352. doi: 10.1126/science.286.5448.2349. [DOI] [PubMed] [Google Scholar]
- Shearwin KE, Brumby AM, Egan JB. The Tum protein of coliphage 186 is an antirepressor. J Biol Chem. 1998;273:5708–5715. doi: 10.1074/jbc.273.10.5708. [DOI] [PubMed] [Google Scholar]
- Shinagawa H. SOS response as an adaptive response to DNA damage in prokaryotes. In: Feige U, Morimoto RI, Yahara I, Polla B, editors. Stress-inducible cellular responses. Basel: Burkhäuser Verlag; 1996. pp. 221–235. [DOI] [PubMed] [Google Scholar]
- Tjandra N, Bax A. Direct measurement of distances and angles in biomolecules by NMR in a dilute liquid crystalline medium. Science. 1997;278:1111–1114. doi: 10.1126/science.278.5340.1111. [DOI] [PubMed] [Google Scholar]
- Voloshin ON, Wang L, Camerini-Otero RD. Homologous DNA pairing promoted by a 20-amino acid peptide derived from RecA. Science. 1996;272:868–872. doi: 10.1126/science.272.5263.868. [DOI] [PubMed] [Google Scholar]
- Voloshin ON, Wang L, Camerini-Otero RD. The homologous pairing domain of RecA also mediates the allosteric regulation of DNA binding and ATP hydrolysis: A remarkable concentration of functional residues. J Mol Biol. 2000;303:709–720. doi: 10.1006/jmbi.2000.4163. [DOI] [PubMed] [Google Scholar]
- Walker GC. The SOS Response of Escherichia coli. In: Neidhardt FC, Curtiss III R, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE, editors. Escherichia coli and salmonella: Cellular and molecular biology. Washington, D.C.: ASM Press; 1996. pp. 1400–1416. [Google Scholar]
- Wang L, Voloshin ON, Stasiak A, Stasiak A, Camerini-Otero RD. Homologous DNA pairing domain peptides of RecA protein: Intrinsic propensity to form β-structure and filaments. J Mol Biol. 1998;277:1–11. doi: 10.1006/jmbi.1997.1591. [DOI] [PubMed] [Google Scholar]
- West SC. Enzymes and molecular mechanisms of genetic recombination. Annu Rev Biochem. 1992;61:603–640. doi: 10.1146/annurev.bi.61.070192.003131. [DOI] [PubMed] [Google Scholar]
- Wong I, Lohman TM. A double-filter method for nitrocellulose-filter binding: Application to protein-nucleic acid interactions. Proc Natl Acad Sci. 1993;90:5428–5432. doi: 10.1073/pnas.90.12.5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Nagata T, Ohmori H. Multicopy suppressors of the cold-sensitive phenotype of the pcsA68 (dinD68) mutation in Escherichia coli. J Bacteriol. 1996;178:3854–3859. doi: 10.1128/jb.178.13.3854-3859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda T, Morimatsu K, Horii T, Nagata T, Ohmori H. Inhibition of Escherichia coli RecA coprotease activities by DinI. EMBO J. 1998;17:3207–3216. doi: 10.1093/emboj/17.11.3207. [DOI] [PMC free article] [PubMed] [Google Scholar]