Regulated secretion of neurotrophins by metabotropic glutamate group I (mGluRI) and Trk receptor activation is mediated via phospholipase C signalling pathways (original) (raw)
Abstract
Neurotrophins (NTs) play an essential role in modulating activity-dependent neuronal plasticity. In this context, the site and extent of NT secretion are of crucial importance. Here, we demonstrate that the activation of phospolipase C (PLC) and the subsequent mobilization of Ca2+ from intracellular stores are essential for NT secretion initiated by both Trk and glutamate receptor activation. Mutational analysis of tyrosine residues, highly conserved in the cytoplasmic domain of all Trk receptors, revealed that the activation of PLC-γ in cultured hippocampal neurons and nnr5 cells is necessary to mobilize Ca2+ from intracellular stores, the key mechanism for regulated NT secretion. A similar signalling mechanism has been identified for glutamate-mediated NT secretion—which in part depends on the activation of PLC via metabotropic receptors—leading to the mobilization of Ca2+ from internal stores by inositol trisphosphate. Thus, PLC-mediated signal transduction pathways are the common mechanisms for both Trk- and mGluRI-mediated NT secretion.
Keywords: adenovirus/BDNF/Ca2+ stores/hippocampal neurons/NGF
Introduction
Neurotrophins (NTs), a gene family of neurotrophic molecules comprising nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3 (NT3) and neurotrophin 4/5 (NT4/5), bind with virtually identical affinity to a common receptor (p75NTR) and with high selectivity to the tyrosine kinase receptors (Trks). NGF preferentially binds to TrkA, BDNF and NT4/5 to TrkB, and NT3 to TrkC. NTs regulate the survival and differentiation of specific populations of neurons during embryonic development and the maintenance of specific neuronal functions in adulthood (see Bothwell, 1995; Lewin and Barde, 1996). However, there is increasing evidence that NTs also play an essential role in modulating activity-dependent neuronal plasticity (Thoenen, 1995; Bonhoeffer, 1996; Cellerino and Maffei, 1996; McAllister et al., 1999). The modulatory actions of NTs on synaptic transmission are mediated by both pre- and post-synaptic mechanisms. Pre-synaptically, NTs enhance activity-mediated neurotransmitter release (Lohof et al., 1993; Knipper et al., 1994a,b; Lessmann et al., 1994; Blöchl and Sirrenberg, 1996; Gottschalk et al., 1998; Y.X.Li et al., 1998). Post-synaptically, BDNF enhances transmission via _N_-methyl-d-aspartate (NMDA) receptors (Levine et al., 1995, 1998; Suen et al., 1997) and attenuates transmission via γ-aminobutyric acid (GABA)A receptors (Tanaka et al., 1997). Recently, Kafitz et al. (1999) demonstrated that NTs also activate a tetrodotoxin-resistant sodium channel within a time frame of milliseconds, resulting in the initiation of repetitive action potentials. In a relatively simply organized organotypic in vitro system, namely hippocampal slices, it has been shown that BDNF is essential for the formation of long-term potentiation (LTP) (Korte et al., 1995; Patterson et al., 1996). The fact that LTP is impaired in both homozygous and heterozygous BDNF-defective mice suggests that a critical quantity of BDNF is required for LTP formation in the CA3/CA1 hippocampal system. Either exogenous administration (Patterson et al., 1996) or local re-expression of BDNF (Korte et al., 1996) could restore LTP. How these highly selective effects are elicited in an integrated physiological system in vivo is dependent on the quantity of NTs locally available to the corresponding Trk receptors. In addition to the understanding of the mechanisms of activity-dependent NT synthesis (see Lindholm et al., 1994; Shieh et al., 1998; Tao et al., 1998), the understanding of the mechanism(s) and site(s) of NT secretion is of crucial importance. In previous experiments, it has been demonstrated that the secretion of NTs from hippocampal neurons is regulated by neuronal activity and mediated via the excitatory neurotransmitters glutamate and acetylcholine (Blöchl and Thoenen, 1995, 1996; Canossa et al., 1997; Griesbeck et al., 1999). More recently, it became apparent that NTs also regulate their own secretion (Canossa et al., 1997; Krüttgen et al., 1998). Both neurotransmitters (Blöchl and Thoenen, 1995; Griesbeck et al., 1999) and NTs (Canossa et al., 1997) initiate NT secretion with a similar time course via activation of the corresponding receptors. On the basis of the use of specific receptor antagonists, glutamate is thought to induce NT secretion via the ionotropic α-amino-3-hydroxyl-5- methyl-4-isoxazolpropionic acid (AMPA) receptors and the metabotropic glutamate receptors (mGluRs), but not NMDA receptors (Blöchl and Thoenen, 1995, 1996). The NT-mediated NT secretion can be triggered by all Trk receptors: in hippocampal neurons via TrkB and TrkC receptors (Canossa et al., 1997) and in the rat phaeochromocytoma PC12 cells via TrkA receptors (Krüttgen et al., 1998). Virtually nothing is known about the signal transduction cascade leading to NT secretion, although mobilization of Ca2+ from endogenous stores seems to be the common denominator of all pathways that lead to regulated NT secretion (Blöchl and Thoenen, 1995, 1996; Griesbeck et al., 1999).
The goal of the present investigation was to elucidate, for both NTs and glutamate, the signal transduction pathways resulting in NT secretion. To this end, we first used TrkA receptor constructs mutated on the tyrosine residues of the intracellular domain that are highly conserved in all Trk receptors (Inagaki et al., 1995). The activation of Trk receptors results in the phosphorylation of specific tyrosine residues. These tyrosine residues initiate the binding and phosphorylation of adaptor molecules such as SHC and SNT, and the activation of enzymes such as phospholipase C-γ (PLC-γ) and phosphatidylinositol 3-kinase (PI3-K) (see Kaplan and Miller, 2000). We demonstrate that Trk-mediated NT secretion depends on the phosphorylation of PLC-γ leading to Ca2+ release from intracellular stores. Moreover, we demonstrate that the glutamate-induced NT secretion—mediated by mGluRI—also results from the activation of PLC and the subsequent release of Ca2+ from intracellular stores.
Results
Evidence that PLC-γ mediates NT secretion in nnr5 cells
We analysed the TrkA-mediated signalling pathways of NT secretion by exploring the functional importance of individual tyrosine residues in the cytoplasmic domain. A set of TrkA receptor mutants had been produced previously by systematically replacing the tyrosine residues (Y499, Y594, Y643, Y704, Y726, Y732, Y760 or Y794) by phenylalanines (Inagaki et al., 1995). In preliminary experiments, we transiently transfected nnr5 cells, which are variants of PC12 cells (Green et al., 1986) that express p75NTR receptors but no Trk receptors (Loeb et al., 1991), with different TrkA receptor mutants. We obtained evidence that the replacement of Y794 by phenylalanine abolished NGF-mediated BDNF secretion. All the other mutants in which individual tyrosines were replaced by phenylalanines did not interfere with NGF-mediated BDNF secretion. In order to substantiate further the role played by Y794 in NT-mediated NT secretion, we produced nnr5 cells stably expressing wild-type or mutated TrkA constructs (Figure 1A). We used either a construct in which Y794 of the wild-type is replaced (Y794F) or one in which only the Y794 (Re794Y) is preserved together with residues Y679, Y683 and Y684, which are putative autophosphorylation sites and are required for the receptor tyrosine kinase activity (Stephens et al., 1994). We selected stable clones (nnr5-TrkA, nnr5-Y794F and nnr5-Re794Y) that expressed about the same levels of receptor protein as evaluated by western blotting with an anti-pan-Trk antibody (Figure 1B). Cells were exposed for 0, 5 or 10 min to 100 ng/ml NGF and the level of receptor phosphorylation was determined by western blot using an anti-phosphotyrosine antibody. Wild-type TrkA shows a weak tyrosine kinase activity (Figure 2A) in the absence of NGF (0 min). However, the signal is strongly increased after 5 and 10 min exposure to NGF. The Y794F mutant showed a similar pattern of tyrosine phosphorylation. The ‘rescue mutant’ Re794Y showed a clear tyrosine phosphorylation signal after 5 and 10 min of NGF exposure, although it was distinctly weaker than that in wild type and Y794F mutants. Tyrosine residue Y794 has been identified by Obermeier et al. (1993) as the binding site for PLC-γ. After exposure of the different stably transfected nnr5 clones to 100 ng/ml NGF, the cells were lysed and immunoprecipitated with an anti-phosphotyrosine antibody. The precipitates were subjected to western blotting and evaluated by a specific anti-PLC-γ antibody. In the nnr5-Re794Y clone, the PLC-γ phosphorylation after NGF exposure was as strong as in the wild-type clones. Conversely, NGF could not induce any PLC-γ phosphorylation in the nnr5-Y794F clone (Figure 2B).
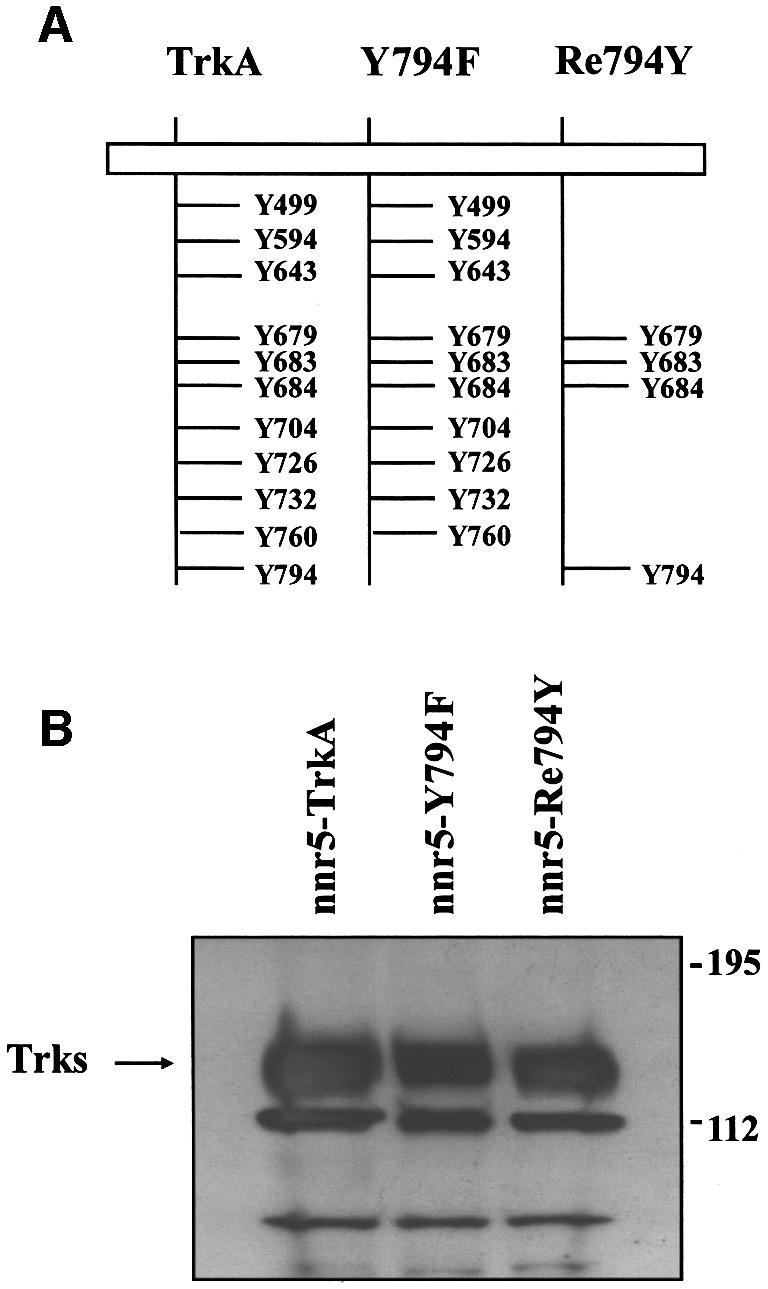
Fig. 1. Stable expression of the wild-type TrkA and receptor mutants in nnr5 cells. (A) Schematic representation of the cytosolic domain of the wild-type TrkA receptor and selected receptor mutants. Y794F represents a TrkA receptor in which the PLC-γ-binding site was inactivated by substituting the tyrosine residue Y794 with a phenyl alanine, while in Re794Y all tyrosine residues have been replaced by phenylalanines, with the exception of Y794. Tyrosine residues Y679, Y683 and Y684 are essential for receptor autophosphorylation and were maintained in all mutants. (B) Western blot of wild-type (nnr5-TrkA) and mutated TrkA receptors (nnr5-Y794F and nnr5-Re794Y) stably expressed in nnr5 cells. Wild-type receptor and mutants were assayed for their levels of expression. We selected clones that, by probing with an anti-pan-Trk antibody, showed comparable levels of receptor protein.

Fig. 2. Comparison between receptor tyrosine and PLC-γ phosphoryl ation in wild-type and mutated TrkA receptors in stably transfected nnr5 cells and adenovirally transduced hippocampal neurons. (A) NGF-mediated tyrosine phosphorylation in nnr5-TrkA, nnr5-Y794F and nnr5-Re794Y clones. Each clone was stimulated for 0, 5 or 10 min with 100 ng/ml NGF and assayed for receptor tyrosine phosphorylation. Wild-type and mutated receptors were immunoprecipitated from cell lysates by an anti-pan-Trk antibody and the samples separated on an SDS–polyacrylamide gel. Western blots were probed with an anti-phosphotyrosine antibody. NGF administered for 5 or 10 min produced a marked tyrosine phosphorylation in wild-type and receptor mutants. (B) NGF-mediated PLC-γ tyrosine phosphorylation in nnr5 clones. The individual clones were stimulated with 100 ng/ml NGF for 0 or 5 min. The cell lysates were immunoprecipitated by agarose-linked anti-phosphotyrosine antibodies and subjected to western blot analysis with a specific anti-PLC-γ antibody. NGF treatment for 5 min was sufficient to induce PLC-γ phosphorylation in nnr5-TrkA and nnr5-Re794Y clones, whereas nnr5-Y794F clones did not show a PLC-γ phosphorylation signal in either the presence or absence of NGF. (C) NGF-mediated tyrosine phosphorylation in cultured hippocampal neurons transduced with TrkA and Re794Y receptors. Transduced neurons were stimulated for 0 and 5 min with 100 ng/ml NGF and assayed for receptor tyrosine phosphorylation. The western blot was performed as in (A). NGF produced a marked tyrosine phosphorylation in the wild-type receptor and mutant. (D) NGF- and BDNF-mediated PLC-γ tyrosine phosphorylation in cultured hippocampal neurons transduced with Re794Y. Cultured hippocampal neurons infected with the adenovirus coding for the Re794Y were stimulated for 0 or 5 min with 100 ng/ml either NGF or BDNF. Lysates of hippocampal neurons were immunoprecipitated by an agarose-conjugated anti-phosphotyrosine antibody. PLC-γ phosphorylation was identified by western blotting as described in (A). Both NGF and BDNF induced PLC-γ tyrosine phosphorylation to a similar extent.
In order to evaluate whether there is a causal relationship between the selective PLC-γ phosphorylation and the NT-mediated NT secretion, we investigated whether and to what extent in the different clones NGF could promote BDNF secretion in the absence or presence of PLC-γ phosphorylation. Owing to the lack of detectable BDNF expression in nnr5 cells, we expressed BDNF using an adenoviral vector (AdCMV-BDNF). After transduction, the cells were placed in a perfusion chamber and the perfusate was collected at 5 min intervals. BDNF was quantified in each fraction by a two-site enzyme-linked immunosorbent assay (ELISA). The NGF-induced BDNF secretion was strictly correlated with the ability of NGF to phosphorylate PLC-γ. NGF induced BDNF secretion in nnr5-TrkA and nnr5-Re794Y, but not in nnr5-Y794F cells (Figure 3). However, all the clones evaluated secreted BDNF in response to high K+ (50 mM).

Fig. 3. High potassium- and NGF-mediated BDNF secretion from nnr5-TrkA, nnr5-Y794F and nnr5-Re794Y. Cells were perfused at a flow rate of 0.1 ml/min. Fractions were collected every 5 min. After collecting two fractions, a first stimulation by 50 mM KCl was initiated and four additional fractions collected. After a recovery of 30 min, cells were stimulated by 100 ng/ml NGF. BDNF concentrations were measured by two-site ELISA. Increased BDNF concentrations (expressed in pg/ml) occurred immediately after stimulation. The peak concentration occurred in the following fraction. The 5 min delay between the onset of stimulation and the peak of BDNF secretion is due to the dead volume of the perfusion system. NGF induced BDNF secretion in nnr5-TrkA and nnr5-Re794Y cells, but not in nnr5 or nnr5-Y794F cells. The values given represent the mean ± SE (n = 6).
The PLC-γ signal transduction pathway is also responsible for NT-mediated NT secretion in hippocampal neurons
In hippocampal neurons, NGF does not initiate NT secretion (Canossa et al., 1997), owing to the absence of functioning TrkA receptors (Aibel et al., 1998). This experimental situation provided the opportunity to analyse the TrkA-mediated signalling pathways. We constructed adenoviral vectors that carry the cDNA for either wild-type TrkA (AdCMV-TrkA) or the mutant receptors Y794F (AdCMV-Y794F) and Re794Y (AdCMV-Re794Y). The use of these viruses, together with AdCMV-BDNF, necessitated double infection procedures. Since we had no reliable anti-TrkA-specific antibody for immunohistochemical analysis, we estimated the degree of double transduction in nnr5 cells that do not express any Trk receptors, and hence an anti-pan-Trk antibody could be used specifically to identify wild-type TrkA and the different mutants. Nnr5 cells transduced with the Re794Y construct showed a predominant signal at the plasma membrane (Figure 4B, middle panel). In contrast, BDNF shows the characteristic discontinuous scattered pattern (Figure 4B, left panel), reflecting its localization in the endoplasmic reticulum (ER) and Golgi (Gärtner et al., 2000). Similar patterns of intracellular distribution were obtained for doubly transduced AdCMV-BDNF/AdCMV-TrkA or AdCMV-BDNF/AdCMV-Y794F nnr5 cells (data not shown). The intensity of expression of both BDNF and wild-type TrkA, or Y794F and Re794Y mutants varies from one cell to another, but the quantitative evaluation of clearly double-stained cells showed a level of double infection of ∼90%. Importantly, the double-infected nnr5 cells showed the same BDNF secretion characteristics (Figure 4A) as stably transfected nnr5 cells (Figure 3). In adenovirally transduced hippocampal neurons, NGF applied for 5 min mediated tyrosine phosphorylation of TrkA wild-type receptors and its mutant Re794Y with a ratio that was similar to that obtained in nnr5 cells (Figure 2C). Furthermore, Re794Y proved to be sufficient to elicit NGF-mediated PLC-γ phosphorylation to a level comparable to that mediated by BDNF via endogenous TrkB receptors (Figure 2D). PLC-γ phosphorylation was in accordance with the observation that NGF resulted in BDNF secretion to a similar extent to that demonstrated by NT4/5 via the activation of endogenous TrkB receptors (Figure 5B). Cultured hippocampal neurons expressing wild-type TrkA or the Re794Y mutant have shown enhanced BDNF secretion in response to NGF, whereas neurons that were transduced with AdCMV-Y794F have not. These experiments were conducted under ‘static’ conditions (see Materials and methods) with the intention of excluding the possibility of a difference between ‘static’ and ‘perfusion’ conditions, an aspect that has been analysed and discussed extensively by Griesbeck et al. (1999).

Fig. 4. Adenovirally transduced nnr5 cells. (A) Time course of BDNF secretion from nnr5 cells double transduced with AdCMV-Re794Y and AdCMV-BDNF. Perfusion was performed as described in Figure 3. Two subsequent stimulations by 50 mM KCl and 100 ng/ml NGF were separated by a recovery period of 30 min. Fractions were collected every 5 min and the BDNF concentrations were determined by ELISA. The values presented are the mean ± SE (n = 4). (B) Expression of BDNF and Re794Y mutant in nnr5 cells co-transduced with AdCMV-BDNF and AdCMV-Re794Y. The expression of both BDNF and Re794Y was detected by probing with the hybridoma supernatant (9E10), which recognizes the myc epitope added to the C-terminal domain of BDNF, and with the anti-pan-Trk antibody, which recognizes the C-terminus of Re794Y. Cells were analysed by confocal microscopy. The pictures represent a single confocal scan along a given x–y plane. The BDNFmyc-infected nnr5 cells showed a discontinuous staining pattern characteristic for the localization of BDNF in ER and Golgi compartments (green). The staining obtained with an anti-pan-Trk antibody was localized predominantly at the plasma membrane (red).

Fig. 5. BDNF secretion from cultured hippocampal neurons expressing TrkA, Y794F and Re794Y receptors. (A) Hippocampal neurons transduced with AdCMV-TrkA, AdCMV-Y794F and AdCMV-Re794Y were stimulated with 100 ng/ml NGF for 10 min under ‘static’ conditions. NGF-induced BDNF secretion occurred only in neurons expressing wild-type TrkA and the Re794Y receptors, but not Y794F or the green fluorescence protein (GFP) taken as a control of virus infection. The values given are the mean ± SE (n = 10). (B) Time course of BDNF secretion induced by NT4/5 via endogenous TrkB or by NGF via transuced Re794Y. Cells were perfused and fractions were collected before, during and after stimulation with 100 ng/ml NT4/5. A second stimulation was initiated by the administration of 100 ng/ml NGF. NT4/5 activated endogenous TrkB receptors, and NGF the TrkA receptor mutant Re794Y. The values given are the mean ± SE (n = 6).
PLC-γ tyrosine phosphorylation correlates with IP3 accumulation and Ca2+ release from intracellular stores
In previous experiments, it has been shown that NT secretion is dependent on intact intracellular Ca2+ stores and the release of Ca2+ from them (Blöchl and Thoenen, 1995, 1996; Canossa et al., 1997; Griesbeck et al., 1999). Hence, the most likely relationship between PLC-γ activation and NT secretion is the formation of d-myo-inositol 1,4,5-trisphosphate (IP3) and subsequent mobilization of Ca2+ from the ER via activation of IP3 receptors (Obermeier et al., 1996; Tinhofer et al., 1996). In nnr5-Re794Y cells, NGF (100 ng/ml) elicited a 3-fold IP3 increase (18.8 ± 0.1 pg/106 cells versus 5.5 ± 0.05 pg/106 cells). In contrast, in control nnr5 cells, NGF did not induce any IP3 formation above the basal level. NGF-mediated Ca2+ mobilization from the ER was assessed by Ca2+ imaging procedures, using the Ca2+ fluorophor Fura-2. In hippocampal neurons infected with an adenovirus carrying the Re794Y mutant, application of either BDNF or NGF elicited intracellular Ca2+ signals in Ca2+-free medium supplemented with 10 µM of the high-affinity Ca2+ chelator BAPTA (Figure 6A). In accordance with previous experiments (Canossa et al., 1997), no Ca2+ signal was obtained after administration of NGF to native, non-transduced cultivated hippocampal neurons (Figure 6A). That the observed increase of cytosolic Ca2+ resulted from the release of Ca2+ from intracellular stores was supported further by the observation that depletion of these stores by pre-treatment with a combination of caffeine and thapsigargine blocked both NGF- and BDNF-mediated Ca2+ signalling in AdCMV-Re794Y-infected hippocampal neurons (Figure 6B).

Fig. 6. Changes of intracellular Ca2+ concentrations in hippocampal neurons expressing the Re794Y receptors. (A) Non-transduced hippocampal neurons (controls) and those transduced by Re794Y were loaded with Fura-2/AM. Changes in intracellular Ca2+ concentrations were determined by the ratio of the fluorescence at excitation wavelengths of 340 and 380 nm. In the absence of extracellular Ca2+ (BAPTA), hippocampal neurons expressing the TrkA receptor mutant showed an NGF-mediated increase in [Ca2+]i that could not be distinguished from that obtained by BDNF acting via endogenous TrkB receptors. This is a representative example of eight independent experiments. (B) Effect of the depletion of the intracellular Ca2+ stores by thapsigargine and caffeine on the subsequent Ca2+ signalling by BDNF or NGF. In the absence of extracellular Ca2+ (BAPTA), caffeine/thapsigargine initiated a strong, prolonged Ca2+ signal resulting from the depletion of the Ca2+ stores. Subsequent administration of BDNF and NGF did not elicit a detectable Ca2+ signal, in distinct contrast to the hippocampal neurons not treated with caffeine and thapsigargine. This is a representative example of three independent experiments.
PLC-γ-mediated BDNF secretion is dependent on Ca2+ release from intact intracellular Ca2+ stores
After showing that NGF induces an increase in cytosolic Ca2+ in hippocampal neurons expressing the Re794Y construct, we next evaluated the role of PLC-γ-mediated Ca2+ release in initiating NT secretion. In previous experiments, we have demonstrated that BDNF, acting via endogenous TrkB receptors, could mediate NGF secretion from hippocampal neurons in the absence of extracellular Ca2+ in a manner similar to that of neurotransmitter-mediated NT secretion (Canossa et al., 1997). A membrane-permeable form of the high-affinity Ca2+ chelator BAPTA-AM abolished the secretion of NGF by sequestering cytosolic Ca2+, indicating that Ca2+ release from intracellular stores is critical in NT-mediated NT secretion. This interpretation is now supported further by the observation that the selective activation of PLC-γ initiates BDNF secretion under Ca2+-free conditions. We compared the response of cells infected with AdCMV-Re794Y to NT4/5 or NGF (Figure 7A). Removal of extracellular Ca2+ from the perfusion medium supplied with 10 µM of the high-affinity Ca2+ chelator BAPTA did not prevent NGF-mediated BDNF secretion (Figure 7A). However, pre-treatment with the membrane-permeable Ca2+ chelator BAPTA-AM abolished the secretion of BDNF in a manner similar to that of emptying the stores by pre-treatment with caffeine and thapsigargine (Figure 7A). Similar results were obtained with nnr5-TrkA and nnr5-Re794Y clones (Figure 7B).

Fig. 7. Dependence on intact Ca2+ stores of NT-mediated NT secretion in hippocampal neurons. (A) BDNF secretion from hippocampal neurons in the absence of extracellular Ca2+ (BAPTA), chelation of intracellular calcium by BAPTA-AM and after depletion of the intracellular Ca2+ stores by caffeine and thapsigargine. Cultured hippocampal neurons co-infected with AdCMV-BDNF and AdCMV-Re794Y were perfused as described in Figure 3. After stimulation with 100 ng/ml NT4/5 for 5 min in normal Ca2+, a second stimulation with 100 ng/ml NGF was performed in Ca2+-free medium or medium containing the Ca2+ chelator BAPTA-AM (10 µM). Removal of extracellular Ca2+ (BAPTA) did not affect the ability of NGF to induce BDNF secretion. However, the effect of NGF on BDNF secretion was abolished by pre-treating hippocampal neurons for 30 min with the cell-permeable BAPTA-AM. Similarly, the depletion of intracellular Ca2+ stores with a combination of caffeine (3 mM) and thapsigargine (10 µM) abolished the NGF-mediated BDNF secretion. The values given represent the mean ± SE (n = 6). (B) Role of Ca2+ in BDNF secretion from nnr5-TrkA or nnr5-Re794Y clones. Similarly to the effects shown in hippocampal neurons, NGF induced BDNF secretion from nnr5 clones in the absence of extracellular Ca2+ (BAPTA). In contrast, chelating the intracellular Ca2+ with BAPTA-AM or depleting intracellular Ca2+ stores with caffeine/thapsigargine prevented NGF-mediated secretion of BDNF. The values given represent the mean ± SE (n = 7).
Evidence that mGluRI mediates NT secretion
We now approached the question of whether the activation of the PLC signal transduction pathway might also be responsible for glutamate-mediated NT secretion. It is known that mGluRI is coupled specifically to PLC- mediated IP3 production (Frenguelli et al., 1993). Hence, this signal transduction pathway lends itself to a more detailed analysis. We first analysed the secretion of endogenous BDNF from hippocampal slices of adult rats in a perfusion chamber. The perfusate was collected in 5 min fractions and the BDNF concentrations measured by a two-site ELISA. Administration of 50 µM 1_S_,3_R_-1- aminocyclopentane-1,3-dicarboxylic acid (t-ACPD), an agonist of mGluRI and II receptors, resulted in an increase of endogenous BDNF secretion after 5 min stimulation (Figure 8). We obtained a similar pattern of secretion in dissociated hippocampal cultures infected with AdCMV-BDNF (Figure 9A). In order to demonstrate that the specific activation of mGluRI is responsible for BDNF secretion, we pre-treated hippocampal neurons with the specific mGluRI inhibitor 1-aminoindan-1,5-dicarboxylacid (AIDA). It has been shown that this inhibitor lowers the production of IP3 elicited by t-ACPD (100 µM) in hippocampal slices without affecting the activation of group II receptors (Moroni et al., 1997). In agreement with the reduction of IP3 accumulation, 20 min pre-treatment with 500 µM AIDA resulted in a strong reduction of BDNF secretion mediated by t-ACPD (Figure 9B). Accordingly, the effects of t-ACPD proved to be strictly dependent on intact Ca2+ stores. Indeed, the addition of thapsigargine and caffeine (Figure 9C), which themselves initiate BDNF secretion (Griesbeck et al., 1999), abolished the BDNF secretion induced by t-ACPD in the presence of extracellular Ca2+. Thus, mGluRI-mediated BDNF secretion in hippocampal slices and transduced hippocampal neurons is initiated by activation of mGluRI receptors, resulting in IP3-mediated Ca2+ mobilization from intracellular stores.

Fig. 8. Time course of BDNF secretion in native, non-transduced hippocampal slices. Acute hippocampal slices were perfused and fractions were collected over consecutive 5 min periods. BDNF concentrations were determined by ELISA. After a recovery period of 30 min, an initial stimulation by 50 µM glutamate was followed by a second stimulation with 100 µM t-ACPD, AMPA or NMDA. The values given are the mean ± SE (n = 4).

Fig. 9. Analysis of the signal transduction pathway(s) leading to BDNF secretion by glutamate. (A) BDNF secretion initiated by glutamate receptor agonists in cultured hippocampal neurons. Primary cultures of hippocampal neurons were infected with AdCMV-BDNF for 36–48 h. After an equilibration time of 60 min, basal levels of secreted BDNF were determined in the medium collected over a 10 min period under ‘static’ conditions. Stimulation of neurons for 10 min with glutamate (50 µM), AMPA (100 µM) or t-ACPD (100 µM) resulted in increased concentrations of BDNF in the incubation medium. NMDA had no effect. (B) Hippocampal neurons, pre-treated with the specific antagonists of AMPA and mGluRI receptors, CNQX (50 µM) and AIDA (500 µM), respectively, were tested for the effects of AMPA and t-ACPD. (C) Influence of Ca2+ stores on AMPA- and t-ACPD-mediated BDNF secretion. In hippocampal neurons, treatment with thapsigargine (10 µM) and caffeine (3 mM) initiated BDNF secretion but abolished the subsequent secretion of BDNF induced by AMPA and t-ACPD. The values given represent the mean ± SE (n = 6).
BDNF secretion is mediated by AMPA but not NMDA glutamate receptors
In previous experiments (Blöchl and Thoenen, 1996), it has been demonstrated that the glutamate-mediated NT secretion could be impaired by antagonists of AMPA receptors, but not those of NMDA receptors. We now used specific agonists for AMPA and NMDA receptors and demonstrated that the selective activation of AMPA, but not NMDA receptors also mediated a 3- to 4-fold increase of BDNF secretion from hippocampal slices (Figure 8) and cultured neurons (Figure 9A). The stimulatory effect of AMPA resulted from the selective activation of AMPA receptors and not from an indirect _trans_-activation of mGluRI. Indeed, the selective AMPA receptor antagonist 6-cyano-7-nitro-quinoxaline-2,3-dione (CNQX) could only prevent BDNF secretion induced by AMPA, but not that by t-ACPD (Figure 9B). Conversely, AIDA could only prevent BDNF secretion induced by t-ACPD without affecting the stimulatory effect of AMPA (Figure 9B). Similarly to mGluRI, AMPA receptor-mediated BDNF secretion was prevented by depletion of intracellular Ca2+ stores (Figure 9C).
Discussion
Use of nnr5 cells and cultured hippocampal neurons as analytical systems for studying the signal transduction pathway of TrkA-mediated NT secretion
TrkA receptors that were mutated in their intracellular tyrosine residues (Inagaki et al., 1995) were used to identify the signal transduction pathways leading to BDNF secretion from both nnr5 cells and cultured hippocampal neurons. Wild-type TrkA and selected mutants were stably expressed in nnr5 cells (defective PC12 cells expressing p75NTR but no Trk receptors) (Loeb et al., 1991). These cells were tested for their ability to mediate regulated BDNF secretion after administration of NGF. The presence of p75NTR alone was not sufficient to induce BDNF secretion by NGF (Figure 3). These data are in agreement with our previous observations (Canossa et al., 1997), but are partially in disagreement with those of Krüttgen et al. (1998) in PC12 cells, in which NT secretion was obtained by not only TrkA receptor stimulation, but also activation of p75NTR, i.e. after blockade of TrkA receptors. These discordant results can most probably be explained by the differing properties of the different PC12 cells used.
In order to validate the results obtained in nnr5 cells, we took advantage of the fact that hippocampal neurons do not express detectable levels of functional TrkA receptors (Aibel et al., 1998) and, accordingly, they do not show any response to NGF. However, after transfection with TrkA, NGF promoted hippocampal neuron differentiation, as reflected by stimulation of fibre outgrowth (Aibel et al., 1998). Hence, cultivated hippocampal neurons represent a valid cellular system with appropriate contextual properties for investigating signal transduction via (transduced) TrkA receptors and their mutants. Adenoviral gene transfer (Figure 5) accomplished expression of wild-type TrkA and corresponding mutants in hippocampal neurons. The quantity of BDNF secreted by activation of TrkA receptors with NGF was comparable to that mediated by NT4/5 through the activation of endogenous TrkB receptors (Figure 5). In accordance with the results obtained in nnr5 cells, the signal transduction via the PLC-γ-activating pathway of the TrkA receptor and the corresponding mutants also proved to be crucial for the NGF-mediated BDNF secretion in hippocampal cultures.
Ca2+ signalling and regulated NT secretion
In previous studies, it has been demonstrated that regulated NT secretion initiated by glutamate (Blöchl and Thoenen, 1995, 1996; Griesbeck et al., 1999) and the activation of Trk receptors by NTs (Canossa et al., 1997) are mediated by the release of Ca2+ from intracellular stores rather than Ca2+ influx. This represents a mechanism that is distinctly different from the activity- mediated secretion of conventional neurotransmitters and the majority of neuropeptides (Hökfelt et al., 1980; Thureson-Klein and Klein, 1990; Matteoli and DeCamilli, 1991; Südhof, 1995; Berridge, 1998), raising pertinent questions concerning the storage/release compartments of NTs and the mechanism(s) of their secretion. In the present investigation, we have focused on the signal transduction pathway leading from the activation of Trk and glutamate receptors to the intracellular release of Ca2+ and subsequent NT secretion. In both primary cultures of hippocampal neurons and nnr5 cells, the signal transduction mediated by Y794, the binding site for PLC-γ, proved to be crucial for TrkA receptor-mediated NT secretion (Figures 3 and 4A). The binding of PLC-γ to Y794 results in PLC-γ activation by phosphorylation, which then cleaves phosphatidylinositol-4,5-bisphosphate to generate IP3. IP3 mobilizes Ca2+ from intracellular ER stores by activating IP3 receptors, as demonstrated by Obermeier et al. (1996) in NIH 3T3 cells. Here we demonstrate that in nnr5 cells stably transfected with the ‘rescue mutant’ Re794Y (nnr5-Re794Y), NGF produced an increase in IP3 comparable to that of wild-type TrkA receptors. NGF-mediated IP3 production via Re794Y was correlated with the Ca2+ release from intracellular ER stores and the Ca2+ mobilization was correlated with the NGF-mediated BDNF secretion, supporting the concept of a causal relationship between IP3 production, Ca2+ mobilization from intracellular stores and NT secretion. In order to provide the most direct evidence for the involvement of IP3 formation in NT secretion, we attempted to demonstrate NT secretion from hippocampal neurons by UV activation of a cell membrane-permeable form of caged IP3 (W.H.Li et al., 1998). However, the intensity of the UV flash necessary to uncage intraneuronal IP3, and to produce a detectable Ca2+ signal, itself resulted in a massive BDNF secretion from hippocampal neurons that did not contain caged IP3 (O.Griesbeck and M.Canossa, unpublished results). Hence, the relationship between Ca2+ mobilization and NT secretion had to rely on pharmacological evidence, namely that regulated NT secretion was blocked by pharmacologically depleting Ca2+ stores with caffeine/thapsigargine or by loading the cells with the intracellular high-affinity Ca2+ chelator BAPTA-AM (Figure 7). Stimulated BDNF secretion was reduced under all these experimental conditions. In contrast, the absence of extracellular Ca2+ did not interfere with the regulated secretion of NTs. Since PLC-γ activation leads to the formation of not only IP3, but also diacylglycerol, leading in turn to the activation of protein kinase C (PKC) (Kikkawa et al., 1989; Nishizuka, 1992), we evaluated the possible involvement of this signal transduction pathway by pre-treating hippocampal neurons with specific PKC inhibitors. This, however, did not interfere with the NGF-mediated BDNF secretion (data not shown).
Of particular interest is the glutamate-mediated NT secretion, since glutamate is the most prominent excitatory neurotransmitter in the central nervous system. We have demonstrated that activation of mGluRI resulted in an NT secretion from hippocampal slices and cultured hippocampal neurons comparable to that mediated by 50 mM potassium (Figures 8 and 9A). Conversely, specific inhibitors of mGluRI markedly reduced, although did not completely abolish, the glutamate-mediated NT secretion. mGluRI is a G-protein-coupled receptor that activates PLC, resulting in IP3 formation (Frenguelli et al., 1993) and subsequent Ca2+ release from intracellular stores by the activation of IP3 receptors. Although the glutamate-mediated NT secretion is mediated by mGluRI, previous experiments conducted by Blöchl and Thoenen (1996) demonstrated that AMPA receptor inhibitors also reduced the glutamate-mediated NGF secretion. In the present experiments, we have demonstrated that selective activators of AMPA receptors also evoked NT secretion, which did not result from cross-activation of mGluRI. This AMPA receptor-mediated NT secretion must be considered in the context of previous investigations from which it was thought to be mediated by sodium influx, given that sodium replacement by _N_-methyl-glucamine abolished the glutamate-mediated NGF secretion (Blöchl and Thoenen, 1995). This interpretation was wrong, since a more thorough analysis demonstrated that _N_-methyl-glucamine exhibited a blocking effect independent of sodium replacement (Höner, 2000). This excludes the fact that AMPA receptors initiate NT secretion via Na+ influx, leading to a mechanism of secretion that has yet to be characterized. The mobilization of Ca2+ from endogenous stores seems to be the only common denominator of all pathways that lead to regulated NT secretion. As previously demonstrated for glutamate (Blöchl and Thoenen, 1995), AMPA-mediated NT secretion depends on intact Ca2+ stores, as demonstrated by the observation that pre-treatment with thapsigargine and caffeine prevented BDNF secretion (Figure 9C). This suggests that, as with mGluRI, AMPA triggers intracellular signalling pathway(s) that lead to Ca2+ mobilization from intracellular stores. However, the elucidation of the signal transduction mechanism(s) that lead to NT secretion by AMPA receptor activation requires more detailed future analysis.
Materials and methods
Cell lines
Rat nnr5 cells (Green et al., 1986) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 5% fetal calf serum (FCS; Gibco) and 10% horse serum (Boehringer Mannheim), and were incubated at 37°C in 10% CO2. Nnr5 clones, nnr5-TrkA, nnr5-Y794F or nnr5-Re794Y were kept under G418 (100 mg/l) selection. Nnr5 cells and clones were plated at a density of 100 000 per collagen-coated glass coverslip (10 mm). At a confluence of 80%, cells were infected with adenoviral vectors in a reduced volume of 300 µl for 8–12 h before the release experiments.
Acute hippocampal slices
Slices of 350 µm were prepared from hippocampi of adult Wistar rats and placed in a perfusion chamber as reported by Blöchl and Thoenen (1995). Release experiments were initiated after a recovery phase of 10 min with a flow rate of 0.1 ml/min.
Primary culture of hippocampal neurons
Hippocampal neurons were prepared from E17 Wistar rats according to Zafra et al. (1990). Dissociated hippocampal neurons obtained from embryonic E17 rats were grown for 5–7 days in complete medium. Under our culture conditions, only 2% of the total cell population are of astroglial origin, as demonstrated by immunocytochemistry probing with glial fibrillary acidic protein (GFAP), an astroglial-specific marker (data not shown). The remaining cells were shown to express the microtubule-associated protein 2 (MAP2), an established neuronal marker. Neurons were plated at a density of 200 000 per 10 mm glass coverslip, coated with poly-dl-ornithine (0.5 mg/ml) and infected with adenoviral vectors in a reduced volume of 300 µl for 36–48 h before the release experiments.
Transfection and selection of nnr5 clones
The cDNA coding for wild-type TrkA and the mutants cloned into a mammalian expression vector (pEF-BOS), described by Inagaki et al. (1995), were transiently transfected in nnr5 cells using a conventional calcium phosphate method. For the generation of stably expressing clones, nnr5 cells were co-transfected with constructs expressing TrkA, Y794F or Re794Y, and a vector expressing the resistance gene for gentamycin. After selection in the presence of G418 (500 mg/l), the resistant colonies were expanded in a medium with 100 mg/l G418.
Recombinant adenovirus construction
The cDNA coding for wild-type TrkA, or the mutants Y794F and Re794Y, were subcloned into the _Xba_I site of the pXCJL1-CMV-BGH vector (provided by C.Gravel, Quebec, Canada). Recombinant replication-deficient virus was obtained by homologous recombination in 293 cells (McGrory et al., 1988; Graham and Prevec, 1992). AdCMV-TrkA, AdCMV-Y794F and AdCMV-Re794Y contained the cDNA sequence coding for TrkA, Y794F and Re794Y receptors, respectively. AdCMV-BDNF, previously described in Canossa et al. (1997), contained the cDNA sequence coding for mouse pre-proBDNF tagged with the myc epitope at the C-terminus. AdVGFP contains the _Hin_dIII–_Not_I fragment of the N1-EGFP vector (Clontech). Virus titres estimated by a plaque assay were in the range of 1010–1011 plaque-forming units/ml.
Characteristics of the perfusion set up and release experiments
Acute slices, cultured hippocampal neurons or nnr5 cells on glass coverslips were placed in a perfusion chamber and perfused as described by Canossa et al. (1997). Both slides and culture cells were stimulated by either replacing 50 mM NaCl in the perfusion medium with 50 mM KCl or adding glutamate (50 µM), AMPA (Biotrend, 100 µM), NMDA (Biotrend, 50 µM), t-ACPD (Biotrend, 100 µM), NGF or NT4/5 (100 ng/ml). Pre-treatment was initiated during the recovery phase with several inhibitors: CNQX (Biotrend, 50 µM), AIDA (Biotrend, 500 µM), caffeine (Sigma, 3 mM), BAPTA-AM (Molecular Probes, 10 µM), BAPTA (Sigma, 10 µM) and thapsigargine (Alexis, 10 µM). BDNF secretion was also quantified under ‘static’ conditions. In these experiments, infected hippocampal neurons were equilibrated for 60 min in the tissue culture plate in Hanks buffer. After four basal collection values of 10 min, neurons were stimulated for 10 min with several stimuli. In each sample, the amount of BDNF was determined by ELISA.
Enzyme immunoassays (ELISAs)
BDNF concentrations were determined by a two-site ELISA according to Canossa et al. (1997) and Kolbeck et al. (1999). The ELISAs showed a sensitivity of 0.5–1.0 pg/ml of BDNF.
Western blot
Confluent 10 cm dishes of nnr5-TrkA, nnr5-Re794Y and nnr5-Y794F cells and cultured hippocampal neurons (5 × 106) infected with AdCMV-TrkA, AdCMV-Re794Y or AdCMV-Y794F were stimulated with 100 ng/ml NGF for 0, 5 or 10 min and lysed. Overnight immunoprecipitation was performed by using either anti-phosphotyrosine antibody-conjugated agarose (Upstate Biotechnology) or wheat germ agglutinin (WGA)-conjugated Sepharose. Samples were then separated by 8% SDS–PAGE and transferred to Immobilon-P membranes (0.45 µm; Millipore) using standard procedures. After blocking unspecific sites, the membranes were incubated overnight at 4°C with the primary antibody: anti-PLC-γ (4 µg/ml; Upstate Biotechnology) or pan-Trk antiserum (1:1000 provided by Mariano Barbacid). Detection was performed after 1 h incubation with the appropriate secondary antibody conjugated to horseradish peroxidase (Dianova) with subsequent conversion of a chemiluminescent substrate (Pierce).
IP3 determination
IP3 was measured using the Biotrak [3H]IP3 assay system (Amersham).
Ca2+ imaging
Free intracellular Ca2+ concentrations were determined by Ca2+ imaging procedures according to Canossa et al. (1997).
Immunohistochemistry and confocal microscopy
For intracellular detection of Re794Y and BDNFmyc, nnr5 cells were fixed for 20 min with 4% paraformaldehyde in phosphate-buffered saline, permeabilized, and unspecific sites blocked. The first antibody was added overnight at 4°C; the other steps were performed at room temperature. A hybridoma (9E10) supernatant (1:10) recognizing the myc epitope was used for detection of BDNFmyc. For Re794Y, rabbit pan-Trk antiserum was used (1:500). Secondary antibodies were fluorescein isothiocyanate-conjugated anti-mouse in combination with an anti-rabbit lissamine– rhodamine antibody (both Dianova; 1:150). Immunofluorescence was analysed by confocal microscopy (Leica).
Acknowledgments
Acknowledgements
We thank Jens Richter and Claudia Huber for excellent technical assistance and Michael Ashdown for linguistic revision of the manuscript. This research was supported by the Deutsche Forschungsgemeinschaft (to H.T.), RFTF-96l00203 and AIRC, MURST ex 60% (to M.C.) and Regeneron Pharmaceuticals, Tarrytown, USA.
References
- Aibel L., Martinzanca,D., Perez,P. and Chao,M.V. (1998) Functional expression of TrkA receptors in hippocampal neurons. J. Neurosci. Res., 54, 424–431. [DOI] [PubMed] [Google Scholar]
- Berridge M.J. (1998) Neuronal calcium signaling. Neuron, 21, 13–26. [DOI] [PubMed] [Google Scholar]
- Blöchl A. and Sirrenberg,C. (1996) Neurotrophins stimulate the release of dopamine from rat mesencephalic neurons via Trk and p75Lntr receptors. J. Biol. Chem., 271, 21100–21107. [DOI] [PubMed] [Google Scholar]
- Blöchl A. and Thoenen,H. (1995) Characterization of nerve growth factor (NGF) release from hippocampal neurons: evidence for a constitutive and an unconventional sodium-dependent regulated pathway. Eur. J. Neurosci., 7, 1220–1228. [DOI] [PubMed] [Google Scholar]
- Blöchl A. and Thoenen,H. (1996) Localization of cellular storage compartments and sites of constitutive and activity-dependent release of nerve growth factor (NGF) in primary cultures of hippocampal neurons. Mol. Cell. Neurosci., 7, 173–190. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer T. (1996) Neurotrophins and activity-dependent develop ment in the neocortex. Curr. Opin. Neurobiol., 6, 119–126. [DOI] [PubMed] [Google Scholar]
- Bothwell M. (1995) Functional interactions of neurotrophins and neurotrophin receptors. Annu. Rev. Neurosci., 18, 223–253. [DOI] [PubMed] [Google Scholar]
- Canossa M., Griesbeck,O., Berninger,B., Campana,G., Kolbeck,R. and Thoenen,H. (1997) Neurotrophin release by neurotrophins: implic ations for activity-dependent neuronal plasticity. Proc. Natl Acad. Sci. USA, 94, 13279–13286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellerino A. and Maffei,L. (1996) The action of neurotrophins in the development and plasticity of the visual cortex. Prog. Neurobiol., 49, 53–63. [DOI] [PubMed] [Google Scholar]
- Frenguelli B.G., Potier,B., Slater,N.T., Alford,S. and Collingridge,G.L. (1993) Metabotropic glutamate receptors and calcium signalling in dendrites of hippocampal CA1 neurones. Neuropharmacology, 32, 1229–1237. [DOI] [PubMed] [Google Scholar]
- Gärtner A., Shostak,Y., Hackel,N., Ethell,I.M. and Thoenen,H. (2000) Ultrastructural identification of storage compartments and localization of activity-dependent secretion of neurotrophin 6 in hippocampal neurons. Mol. Cell. Neurosci., 15, 215–234. [DOI] [PubMed] [Google Scholar]
- Gottschalk W., Pozzo-Miller,L.D., Figurov,A. and Lu,B. (1998) Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J. Neurosci., 18, 6830–6839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham F.L. and Prevec,L. (1992) Adenovirus-based expression vectors and recombinant vaccines. Biotechnology, 20, 363–390. [DOI] [PubMed] [Google Scholar]
- Green S.H., Rydel,R.E., Connolly,J.L. and Greene,L.A. (1986) PC12 cell mutants that possess low- but not high-affinity nerve growth factor receptors neither respond to nor internalize nerve growth factor. J. Cell Biol., 102, 830–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbeck O., Canossa,M., Campana,G., Gärtner,A., Hoener,M.C., Nawa,H., Kolbeck,R. and Thoenen,H. (1999) Are there differences between the secretion characteristics of NGF and BDNF? Implications for the modulatory role of neurotrophins in activity-dependent neuronal plasticity. Microsc. Res. Tech., 45, 262–275. [DOI] [PubMed] [Google Scholar]
- Hökfelt T., Johansson,O., Ljungdahl,A., Lundberg,J.N. and Schultzberg,M. (1980) Peptidergic neurones. Nature, 284, 515–520. [DOI] [PubMed] [Google Scholar]
- Höner M.C. (2000) Role played by sodium in activity-dependent secretion of neurotrophins—revisited. Eur. J. Neurosci., 12, 1–11. [DOI] [PubMed] [Google Scholar]
- Inagaki N., Thoenen,H. and Lindholm,D. (1995) TrkA tyrosine residues involved in NGF-induced neurite outgrowth of PC12 cells. Eur. J. Neurosci., 7, 1125–1133. [DOI] [PubMed] [Google Scholar]
- Kafitz K.W., Rose,C.R., Thoenen,H. and Konnerth,A. (1999) Neurotrophin-evoked rapid excitation through TrkB receptors. Nature, 401, 918–921. [DOI] [PubMed] [Google Scholar]
- Kaplan D.R. and Miller,F.D. (2000) Neurotrophin signal transduction in the nervous system. Curr. Opin. Neurobiol., 10, 381–391. [DOI] [PubMed] [Google Scholar]
- Kikkawa U., Kishimoto,A. and Nishizuka,Y. (1989) The protein kinase-C family: heterogeneity and its implications. Annu. Rev. Biochem., 58, 31–44. [DOI] [PubMed] [Google Scholar]
- Knipper M., Da Penha Berzaghi,M., Blöchl,A., Breer,H., Thoenen,H. and Lindholm,D. (1994a) Positive feedback between acetylcholine and the neurotrophins nerve growth factor and brain-derived neurotrophic factor in the rat hippocampus. Eur. J. Neurosci., 6, 668–671. [DOI] [PubMed] [Google Scholar]
- Knipper M., Leung,L.S., Zhao,D. and Rylett,R.J. (1994b) Short-term modulation of glutamatergic synapses in adult rat hippocampus by NGF. Neuroreport, 5, 2433–2436. [DOI] [PubMed] [Google Scholar]
- Kolbeck R., Bartke,I., Eberle,W. and Barde,Y.A. (1999) Brain-derived neurotrophic factor levels in the nervous system of wild-type and neurotrophin gene mutant mice. J. Neurochem., 72, 1930–1938. [DOI] [PubMed] [Google Scholar]
- Korte M., Carroll,P., Wolf,E., Brem,G., Thoenen,H. and Bonhoeffer,T. (1995) Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc. Natl Acad. Sci. USA, 92, 8856–8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte M., Griesbeck,O., Gravel,C., Carroll,P., Staiger,V., Thoenen,H. and Bonhoeffer,T. (1996) Virus-mediated gene transfer into hippocampal CA1 region restores long-term potentiation in brain-derived neurotrophic factor mutant mice. Proc. Natl Acad. Sci. USA, 93, 12547–12552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüttgen A., Möller,J.C., Heymach,J.V. and Shooter,E.M. (1998) Neurotrophins induce release of neurotrophins by the regulated secretory pathway. Proc. Natl Acad. Sci. USA, 95, 9614–9619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann V., Gottmann,K. and Heumann,R. (1994) BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. Neuroreport, 6, 21–25. [DOI] [PubMed] [Google Scholar]
- Levine E.S., Dreyfus,C.F., Black,I.B. and Plummer,M.R. (1995) Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc. Natl Acad. Sci. USA, 92, 8074–8077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine E.S., Crozier,R.A., Black,I.B. and Plummer,M.R. (1998) Brain-derived neurotrophic factor modulates hippocampal synaptic transmission by increasing _N_-methyl-d-aspartic acid receptor activity. Proc. Natl Acad. Sci. USA, 95, 10235–10239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin G.R. and Barde,Y.A. (1996) Physiology of the neurotrophins. Annu. Rev. Neurosci., 19, 289–317. [DOI] [PubMed] [Google Scholar]
- Li W.H., Llopis,J., Whitney,M., Zlokarnik,G. and Tsien,R.Y. (1998) Cell-permeant caged INSP3 ester shows that Ca2+ spike frequency can optimize gene expression. Nature, 392, 936–941. [DOI] [PubMed] [Google Scholar]
- Li Y.X., Zhang,Y.O., Lester,H.A., Schuman,E.M. and Davidson,N. (1998) Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J. Neurosci., 18, 10231–10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D., Berzaghi,M.D., Cooper,J., Thoenen,H. and Castren,E. (1994) Brain-derived neurotrophic factor and neurotrophin-4 increase neurotrophin-3 expression in the rat hippocampus. Int. J. Dev. Neurosci., 12, 745–751. [DOI] [PubMed] [Google Scholar]
- Loeb D.M., Maragos,J., Martin-Zanca,D., Chao,M.V., Parada,L.F. and Greene,L.A. (1991) The trk proto-oncogene rescues NGF responsiveness in mutant NGF-nonresponsive PC12 cell lines. Cell, 66, 961–966. [DOI] [PubMed] [Google Scholar]
- Lohof A.M., Ip,N.Y. and Poo,M.M. (1993) Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature, 363, 350–353. [DOI] [PubMed] [Google Scholar]
- Matteoli M. and DeCamilli,P. (1991) Molecular mechanisms in neurotransmitter release. Curr. Opin. Neurobiol., 1, 91–97. [DOI] [PubMed] [Google Scholar]
- McAllister A.K., Katz,L.C. and Lo,D.C. (1999) Neurotrophins and synaptic plasticity. Annu. Rev. Neurosci., 22, 295–318. [DOI] [PubMed] [Google Scholar]
- McGrory W.J., Bautista,D.S. and Graham,F.L. (1988) A simple technique for the rescue of early region I mutations into infectious human adenovirus type 5. Virology, 163, 614–617. [DOI] [PubMed] [Google Scholar]
- Moroni F. et al. (1997) Pharmacological characterization of 1-aminoindan-1,5-dicarboxylic acid, a potent mGluR1 antagonist. J. Pharmacol. Exp. Ther., 281, 721–729. [PubMed] [Google Scholar]
- Nishizuka Y. (1992) Intracellular signaling by hydrolysis of phospholipids and activation of protein kinase C. Science, 258, 607–614. [DOI] [PubMed] [Google Scholar]
- Obermeier A., Halfter,H., Wiesmuller,K.H., Jung,G., Schlessinger,J. and Ullrich,A. (1993) Tyrosine 785 is a major determinant of Trk–substrate interaction. EMBO J., 12, 933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermeier A., Tinhofer,I., Grunicke,H.H. and Ullrich,A. (1996) Transforming potentials of epidermal growth factor and nerve growth factor receptors inversely correlate with their phospholipase Cγ affinity and signal activation. EMBO J., 15, 73–82. [PMC free article] [PubMed] [Google Scholar]
- Patterson S.L., Abel,T., Deuel,T.A.S., Martin,K.C., Rose,J.C. and Kandel,E.R. (1996) Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron, 16, 1137–1145. [DOI] [PubMed] [Google Scholar]
- Shieh P.B., Hu,S.C., Bobb,K., Timmusk,T. and Ghosh,A. (1998) Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron, 20, 727–740. [DOI] [PubMed] [Google Scholar]
- Stephens R.M., Loeb,D.M., Copeland,T.D., Pawson,T., Greene,L.A. and Kaplan,D.R. (1994) Trk receptors use redundant signal transduction pathways involving SHC and PLC-γ1 to mediate NGF responses. Neuron, 12, 691–705. [DOI] [PubMed] [Google Scholar]
- Südhof T.C. (1995) The synaptic vesicle cycle: a cascade of protein–protein interactions. Nature, 375, 645–653. [DOI] [PubMed] [Google Scholar]
- Suen P.C., Wu,K., Levine,E.S., Mount,H.T., Xu,J.L., Lin,S.Y. and Black,I.B. (1997) Brain-derived neurotrophic factor rapidly enhances phosphorylation of the postsynaptic _N_-methyl-d-aspartate receptor subunit 1. Proc. Natl Acad. Sci. USA, 94, 8191–8195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T., Saito,H. and Matsuki,N. (1997) Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J. Neurosci., 17, 2959–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao X., Finkbeiner,S., Arnold,D.B., Shaywitz,A.J. and Greenberg,M.E. (1998) Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron, 20, 709–726. [DOI] [PubMed] [Google Scholar]
- Thoenen H. (1995) Neurotrophins and neuronal plasticity. Science, 270, 593–598. [DOI] [PubMed] [Google Scholar]
- Thureson-Klein A. and Klein,R.L. (1990) Exocytosis from neuronal large dense-cored vesicles. Int. Rev. Cytol., 121, 67–126. [DOI] [PubMed] [Google Scholar]
- Tinhofer I., Maly,K., Dietl,P., Hochholdinger,F., Mayr,S., Obermeier,A. and Grunicke,H.H. (1996) Differential Ca2+ signalling induced by activation of the epidermal growth factor and nerve growth factor receptors. J. Biol. Chem., 271, 30505–30509. [DOI] [PubMed] [Google Scholar]
- Zafra F., Hengerer,B., Leibrock,J., Thoenen,H. and Lindholm,D. (1990) Activity dependent regulation of BDNF and NGF mRNAs in the rat hippocampus is mediated by non-NMDA glutamate receptors. EMBO J., 9, 3545–3550. [DOI] [PMC free article] [PubMed] [Google Scholar]