Identification of the enzymatic mechanism of nitroglycerin bioactivation (original) (raw)
Abstract
Nitroglycerin (glyceryl trinitrate, GTN), originally manufactured by Alfred Nobel, has been used to treat angina and heart failure for over 130 years. However, the molecular mechanism of GTN biotransformation has remained a mystery and it is not well understood why “tolerance” (i.e., loss of clinical efficacy) manifests over time. Here we purify a nitrate reductase that specifically catalyzes the formation of 1,2-glyceryl dinitrate and nitrite from GTN, leading to production of cGMP and relaxation of vascular smooth muscle both in vitro an_d in vivo_, and we identify it as mitochondrial aldehyde dehydrogenase (mtALDH). We also show that mtALDH is inhibited in blood vessels made tolerant by GTN. These results demonstrate that the biotransformation of GTN occurs predominantly in mitochondria through a novel reductase action of mtALDH and suggest that nitrite is an obligate intermediate in generation of NO bioactivity. The data also indicate that attenuated biotransformation of GTN by mtALDH underlies the induction of nitrate tolerance. More generally, our studies provide new insights into subcellular processing of NO metabolites and suggest new approaches to generating NO bioactivity and overcoming nitrate tolerance.
It is generally assumed that glyceryl trinitrate (GTN) is converted in vascular smooth muscle cells to NO or an NO congener (_S_-nitrosothiol, SNO), which activates guanylate cyclase and thus relaxes vascular smooth muscle (1, 2). However, the molecular mechanism involved is not known, and there is no unifying explanation for the pharmacological profile of the drug (e.g., the preferential effects on capacitance vessels and sulfhydryl requirement for relaxation) or the phenomenon of nitrate tolerance (i.e., the loss of clinical efficacy over time), which leaves patients vulnerable to ischemia and may contribute to other deleterious consequences of long-term nitrate use (3, 4).
GTN biotransformation is tissue and cell specific and dose dependent, and it yields 1,2-glyceryl dinitrate (1,2-GDN), 1,3-GDN, inorganic nitrite, and NO (or SNO) in differing amounts and ratios. In vascular smooth muscle cells, 1,2-GDN is the predominant dinitrate metabolite (2, 5, 6) and the vasorelaxation-related formation of 1,2-GDN is diminished in GTN-tolerant blood vessels (7, 8). These data suggest that an enzyme with product specificity for 1,2-GDN (over 1,3-GDN) generates NO bioactivity from GTN and that loss of this activity at least partly accounts for tolerance. Several enzymatic mechanisms of vascular smooth muscle relaxation by GTN have been proposed (9–15), and candidate enzymes include glutathione _S_-transferases, cytochrome P450 reductase, cytochrome P450, xanthine oxidoreductase, and an unidentified microsomal protein. But none of these enzymes catalyzes the selective formation of 1,2-GDN (12, 15, 16) or is inhibited in tolerant vessels, and the roles of these enzymes in generating NO bioactivity remain controversial. Here we report on the purification of a GTN reductase that specifically generates 1,2-GDN, identify it as mitochondrial aldehyde dehydrogenase (mtALDH), and establish its central role in GTN bioactivation, vasorelaxation, and tolerance.
Materials Methods
Chemicals and Drugs.
GTN, 1,2-GDN, and 1,3-GDN standards were bought from Radian International (Austin, TX). GTN was synthesized according to ref. 17 and purified by TLC. 1,2-GDN and 1,3-GDN was prepared by hydrolysis of GTN and purified by TLC (18). [2-14C] GTN (55 mCi/mmol) was obtained from American Radiolabeled Chemicals (St. Louis). Disulfiram, chloral hydrate, cyanamide, acetaldehyde, phenylephrine, sodium nitroprusside (SNP), Q-Sepharose, DEAE-cellulose, and butyl-Sepharose were obtained from Sigma. Hydroxyapatite and protease inhibitor (mixture set III) were from CalBiochem. The cGMP assay (125I) kit was purchased from Amersham Pharmacia.
Enzyme Purification.
Mouse RAW264.7 cells (50-g pellet; ≈1010 cells) were disrupted by sonication in 30 mM phosphate buffer (KPi), pH 7.5 containing 1 mM DTT, 0.5 mM EDTA, and protease inhibitors (1:200 dilution). The 100,000-g supernatant was loaded onto a DEAE-cellulose column, and the flow-through fractions, which contained the enzyme activity, were pooled and concentrated by ultrafiltration (Amicon, 10-kDa cut-off membrane). After 3-fold dilution with cold water, the fractions were loaded onto a Q-Sepharose column, and the activity was eluted by a phosphate gradient (10 mM to 150 mM, pH 7.5); the active fractions were pooled and solid potassium chloride (KCl) was added to 1.25 M. The sample was then loaded onto a butyl-Sepharose column, and the enzyme activity was eluted by decreasing the salt concentration. The concentrated active fractions were then diluted and loaded onto a hydroxyapatite column. After washing the column with 10 mM KPi/0.4 M KCl, the enzyme was eluted by a phosphate gradient from 10 mM KPi to 150 mM KPi, pH 7.5. All steps were performed at 4°C, and the elution buffers contained 1 mM DTT and 0.5 mM EDTA unless otherwise specified.
GTN Biotransformation.
1,2-GDN and 1,3-GDN formation were measured by the TLC and liquid scintillation spectrometry method as described by Brien et al. (19) with a few modifications. During protein purification, the assay mixture (1 ml) contained: 100 mM KPi (pH 7.5), 0.5 mM EDTA, 1 mM NADH, 1 mM NADPH, 0.1 or 1 μM GTN, and protein (with DTT) from column fractions. After incubation at 37°C for 10–30 min, the reaction was stopped (on dry ice or 4°C) and GTN and its metabolites were extracted with 3 × 4 ml ether and pooled, and the solvent was evaporated by a stream of nitrogen. The final volume was kept to less than 100 μl in ethanol for subsequent TLC separation and scintillation counting. For the activation of dialyzed enzyme samples and assay of the activity of purified enzyme, the assay mixture contained: 100 mM KPi (pH 7.5), 0.5 mM EDTA, 0–1 mM DTT or other reductants (0.5 mM DTT for the bovine enzyme), and 1 μM GTN. For assays in tissues such as rabbit aorta, rings were blotted and weighed after sitting for 1 h in Krebs solution (control) (composition described below). When used, inhibitors were added for 20 min before the addition of 1 μM GTN; the mixture (1 ml) was then kept at 37°C for 5 min. The extraction and TLC-liquid scintillation spectrometry analysis were as described above. Buffer control (Krebs buffer plus GTN) and nonspecific biotransformation (heat-inactivated rings plus GTN) activities were also measured, and the results were corrected accordingly.
Aortic Ring Bioassays.
New Zealand White rabbits (2.5–3 kg) were killed by carbon dioxide inhalation. Thoracic aorta were removed, cleaned of fat and connecting tissue, and cut into 3-mm rings. The rings were mounted under 2 g of resting tension in tissue baths (25 ml) filled with Krebs solution (37°C) containing: 118 mM NaCl, 4.8 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 2.5 mM CaCl2, 25 mM NaHCO3, and 11 mM glucose, pH 7.4. The solution was bubbled with 20% O2, 5% CO2, and balance N2. Changes in isometric tension were recorded with Statham (Hato Rey, PR) transducers and a Grass Instruments (Quincy, MA) polygraph, and contractions were initiated with phenylephrine. Tolerance to GTN was induced by incubating rings with 0.3 mM GTN for 30 min. Rings were then washed several times with Krebs solution and left for 1 h before being further tested.
Nitrite and Nitrate.
Nitrite and nitrate concentrations (in the aqueous phase after ether extraction, see GTN biotransformation) were determined in an NO analyzer (model 280, Sievers, Boulder, CO) according to the manufacturer's instructions.
cGMP Assay.
Aortic rings were blotted and weighed before being incubated in aerated bioassay chambers for 1 h. Then, after treatment with different inhibitors, the rings were exposed to 1 μM GTN for 1 min and immediately frozen in liquid nitrogen and stored at −80°C until the time of analysis. cGMP extraction and measurements were performed according to the manufacturer's protocol.
ALDH Assay.
Rabbit aortic rings were homogenized with a Kontes tissue grinder in 30 mM KPi buffer (deoxygenated with nitrogen gas), pH 7.5, and the homogenate was then sonicated and centrifuged at 10,000 g for 10 min. ALDH activity in the supernatant was monitored at room temperature by following NADH formation at 340 nm. The assay mixture (1 ml) contained: 100 mM Tris⋅HCl (pH 8.5), 1 mM NAD+, 1 mM propionaldehyde, and 1 mM 4-methylpyrazole.
GTN Infusions in Vivo.
New Zealand White rabbits (2.5–3 kg) were anesthetized with isoflurane (1.5%). The carotid artery and jugular vein were isolated via cut down and cannulated. The blood pressure was monitored continuously with a Viggo Spectramed pressure transducer (AD Instruments, Grand Junction, CO) attached to a Gould (Cleveland) recorder. Drugs were infused via the jugular vein. mtALDH inhibitors (≈5 mM) were infused 20 min before initiating studies with GTN. Cyanamide had no effect on mean arterial blood pressure, whereas chloral hydrate had a modest hypotensive effect. Similar studies were performed in Sprague–Dawley rats.
Statistical Analysis.
Results were statistically analyzed by the Student's t test, and two sets of data were considered statistically different when P < 0.05.
Results and Discussion
We reasoned that the difficulty in isolating the enzyme responsible for GTN biotransformation from blood vessels may reflect a lack of starting material and/or exposure to excessive concentrations of GTN (under which conditions GTN metabolism may not represent biotransformation), and we thus screened alternative animal tissues and cell lines by monitoring 1,2-GDN formation from physiological amounts of GTN (0.1 μM). We found that mouse macrophage RAW264.7 cells, which can be grown in large numbers, resemble vascular smooth muscle cells in generating mainly 1,2-GDN, and that after cellular disruption by sonication, the enzyme activity was present predominantly in the 100,000-g supernatant. We then developed a four-step purification procedure to isolate the enzyme from RAW264.7 cells (Table 1). The enzyme activity passed freely through a DEAE-cellulose column and was preserved as a single peak after each of the following chromatographic steps: ion exchange (Q-Sepharose), hydrophobicity (butyl-Sepharose), and hydroxyapatite column chromatography. At the end of the procedure the protein had been purified to near homogeneity (90-fold), with 14.4% overall yield (Fig. 1a); its estimated subunit mass on SDS gel is about 53 kDa. N-terminal sequence of 30 aa residues (SAAATSAVPAPNHQPEVFXNQIFINNEWHD) was obtained by Edman degradation and matched exactly with mouse mtALDH (the unidentified residue 19 is a cysteine in the database). To confirm that this enzyme is located in mitochondria and responsible for the production of 1,2-GDN, we assayed purified preparations of mitochondria (20) that we had isolated from macrophage cells and found that they generated 1,2-GDN from GTN, whereas the mitochondria-free supernatant had no residual activity (data not shown). We then pretreated the macrophage cells with known ALDH inhibitors that either modify the active site residue (disulfiram or cyanamide) or compete for substrate binding (chloral hydrate) and found that all of the inhibitors, tested at concentrations from 50 μM to 1 mM, blocked formation of 1,2-GDN (see below).
Table 1.
Purification of GTN reductase from mouse macrophage RAW264.7 cells
| Procedures | Total units, nmol/h | Total protein, mg | Specific activity, nmol/h per mg | Yield, % | Fold purification |
|---|---|---|---|---|---|
| 100,000 g supernatant | 837.4 | 2,265 | 0.37 | 100 | 1.0 |
| DEAE-cellulose | 756.0 | 345 | 2.19 | 90.3 | 5.9 |
| Q-Sepharose | 528.5 | 123 | 4.29 | 63.1 | 11.6 |
| Butyl-Sepharose | 205.4 | 10.9 | 18.81 | 24.5 | 50.8 |
| Hydroxyapatite | 120.9 | 3.6 | 33.36 | 14.4 | 90.2 |
Figure 1.

(a) Purification of mtALDH from mouse macrophage RAW264.7 cells. The gradual enrichment of the 53-kDa band is shown on a 7% SDS polyacrylamide gel. Lane A, protein molecular mass marker (kDa); lane B, 100,000-g supernatant; lane C, after DEAE-cellulose; lane D, after Q-Sepharose; lane E, after butyl-Sepharose; lane F, after hydroxyapatite. (b) Kinetic analysis of purified GTN reductase in the absence of NAD+ (_K_M = 12 μM; _V_max = 3 nmol/mg per min)._V_max in the presence of NAD+ = ≈30 nmol/mg per min.
In addition to the classical NAD+-dependent dehydrogenation activity, ALDH possesses an esterase activity (21), which is initiated by nucleophilic attack of the active site cysteine on substrate esters, such as _p_-nitrophenyl acetate, and is followed by hydrolysis to generate the acetic acid product. The analogous reaction with GTN would be expected to yield an E-S-NO2 active site intermediate that would then hydrolyze to yield nitrate (NO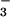 ). However, we could not detect any nitrate formation. Rather, the purified enzyme catalyzed the stoichiometric formation of 1,2-GDN and nitrite (NO
). However, we could not detect any nitrate formation. Rather, the purified enzyme catalyzed the stoichiometric formation of 1,2-GDN and nitrite (NO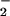 ) from GTN. Specifically, the rates of 1,2-GDN formation were linear with physiological GTN concentrations in the range of 0.1 to 10 μM (data not shown), whereas 1,3-GDN was below detection limits, and nitrite was generated in stoichiometric amounts. The product ratio of 1,2-GDN/nitrite (0.83 ± 0.14, n = 3) was similar to that of 1,2-GDN/NOx (nitrite plus nitrate) (0.73 ± 0.05, n = 3). Thus, the overall reaction can be expressed as:
) from GTN. Specifically, the rates of 1,2-GDN formation were linear with physiological GTN concentrations in the range of 0.1 to 10 μM (data not shown), whereas 1,3-GDN was below detection limits, and nitrite was generated in stoichiometric amounts. The product ratio of 1,2-GDN/nitrite (0.83 ± 0.14, n = 3) was similar to that of 1,2-GDN/NOx (nitrite plus nitrate) (0.73 ± 0.05, n = 3). Thus, the overall reaction can be expressed as:
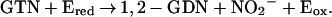
To show that the GTN metabolizing activity and enantiomeric selectivity of mtALDH is not a function of cell type or species, we purified the enzyme from bovine liver (22). Bovine mtALDH also specifically catalyzes 1,2-GDN formation (1,2-GDN/1,3-GDN = ≈8:1) from GTN (0.1 to 10 μM), with classical Michaelis–Menten kinetics (_K_m = 11.98 ± 3.04 μM and _V_max = 3.03 ± 0.20 nmol/min per mg) (Fig. 1b) [and with the same pH dependence as the dehydrogenase activity (optimum pH of 9.0)]. Addition of NAD+ (20–1,000 μM) to the reaction mixture increases the rate of 1,2-GDN formation by about 10-fold (i.e., _V_max ≈ 30 nmol/min per mg), easily adequate for NO bioactivity such as vasodilation. By comparison, the purified bovine enzyme has a specific ALDH activity of 0.54 μmol/min per mg. It is important to appreciate that whereas the GTN reductase activity is ≈15- to 20-fold lower than the classical dehydrogenase activity of mtALDH, bioactive NO concentrations are as much as 10,000-fold lower than circulating or tissue acetaldehyde concentrations (23), and the mitochondrial GTN reductase activity is large from the standpoint of NO biology, being significantly greater than that of mitochondrial NO synthase activity (24, 25).
We reasoned that acetaldehyde (the natural enzyme substrate) should competitively inhibit GTN turnover by mtALDH, and we also tested the effects of the classical ALDH inhibitors on GTN-metabolizing activity of the purified bovine enzyme. Both chloral hydrate and acetaldehyde inhibited 1,2-GDN formation, with IC50 = 99.5 and 815.5 μM, respectively. The relatively high IC50s in the reductase assay [as compared with the _K_m (acetaldehyde) and _K_i (chloral hydrate) for dehydrogenase activity] reflect the absence of NAD+, which increases substrate binding (26). As expected, cyanamide did not have inhibitory effects on the purified enzyme because it first requires bioconversion in vivo.
The classical sulfhydryl requirement for relaxation of vascular smooth muscle by GTN (2, 27) has never been satisfactorily explained. It has been suggested that thiols potentiate GTN activity through formation of bioactive SNOs and/or protect against nitrate tolerance that results from depletion of intracellular thiols. However, the formation of SNO and depletion of intracellular thiol have not been confirmed (28), and alternative hypotheses, such as oxygen radical scavenging by thiol (29), have more recently found favor. We found that the purified mtALDH could catalyze 1,2-GDN formation only when DTT or 2-mercaptoethanol (2ME) were present (although these concentrations of thiol had no effect on their own). Further, catalytic activity that was lost by dialysis could be fully restored by addition of DTT or 2ME (to a lesser extent) but not by glutathione.
Human mtALDH has two cysteines immediately adjacent to the active site thiol. The formation of an intramolecular disulfide bond has been demonstrated by MS (30, 31) and accounts for the “inactive form” of ALDH that is produced by ALDH inhibitors. Notably, active site participation in a disulfide bond has been previously implicated in the mechanism-based inhibition of mtALDH by NO (32), SNO (33), and GTN (21, 34), accounting for disulfram-like reactions in GTN-treated patients ingesting alcohol. But whereas the generation of disulfide has been viewed as indicative of enzyme inactivation (i.e., loss of dehydrogenase and esterase activities), the new data suggest that it can represent a reaction intermediate in a novel reductase reaction requiring thiol (and perhaps other reductant) cofactor (Scheme S1). That is, GTN is an inhibitor of mtALDH activity because it serves as a substrate for its reductase activity. Collectively, our data indicate that mtALDH is in fact a GTN reductase.
Scheme 1.

The specific conversion of GTN to 1,2-GDN and dependence on thiol cofactor (as reductant) make mtALDH a compelling candidate for the elusive enzymatic system that is responsible for catalyzing the bioactivation of GTN in the vasculature. We tested this hypothesis by using an aortic ring bioassay. Preincubation of blood vessels with mtALDH inhibitors chloral hydrate and cyanamide or with the mtALDH substrate acetaldehyde (a competitive inhibitor of GTN metabolizing activity) produced a rightward shift in the GTN relaxation curves (Fig. 2 a1_–_c1) that depended on inhibitor concentration (Fig. 2a1 and data not shown). The mtALDH inhibitor disulfiram also blocked GTN relaxations, but interpretation of these data was confounded by a direct affect on vessel tone. By contrast, preincubation with the mtALDH inhibitors did not attenuate relaxation induced by the nitrosovasodilator SNP (Fig. 2 a2_–_c2), by NO solutions or verapamil, which works through a NO-independent mechanism (not shown). These results suggest a specific inhibition of the organic nitrate ester reductase activity in the arterial vasculature. Indeed, we confirmed that 1,2-GDN generation in the aortic rings was diminished by chloral hydrate, cyanamide, and acetaldehyde (1 mM of each) to 27.6%, 56.9%, and 52.5% of the control activity, respectively (Fig. 3a). The GTN reductase inhibitors and competitive substrate also significantly decreased cGMP accumulation in aortic rings (Fig. 4). For example, chloral hydrate and acetaldehyde completely blocked the increase in cGMP produced by GTN (1 μM for 1 min) and cyanamide abrogated the cGMP increase by 46%. Thus, we conclude that the reductase activity of mtALDH is largely responsible for the increase in cGMP that at least partly mediates vasorelaxation by GTN.
Figure 2.

Effects of inhibitor treatment and GTN tolerance on GTN-induced (Left) and SNP-induced (Right) relaxation in rabbit aorta. The dose–response curves of control rings are shown as ●, and inhibitor-treated rings (1 mM) are shown as ○. (a) Chloral hydrate dose–response (○, 1 mM; ▴, 5 mM). (b) Cyanamide. (c) Acetaldehyde. (d) GTN tolerant. All inhibitor curves (a_–_c) are significantly different from GTN control (P < 0.01), but none are different from SNP control (P = not significant). The GTN tolerance curve (d) is significantly different from control to P < 0.05. Data are expressed as the means ± SD of 4–6 aortic rings.
Figure 3.

Inhibition of GTN (1 μM) biotransformation by ALDH inhibitors and tolerance induction in rabbit aorta. (a) Concentration-dependent inhibition of 1,2-GDN formation by chloral hydrate (●), cyanamide (■), and acetaldehyde (▴). Activities are presented as the percentage of control rings (without inhibitor treatment). Data are the average of two experiments. (b) 1,2-GDN formation is reduced in tolerant vessels relative to control (P < 0.01). Data are the means ± SD of three experiments. (c) mtALDH activity is inhibited in tolerant vessels (P < 0.01). Data are the means ± SD of three experiments.
Figure 4.

Inhibition of cGMP accumulation in rabbit aorta by ALDH inhibitors (1 mM × 20 min, followed by washout) and tolerance (Tol)-inducing amounts of GTN (0.3 mM × 30 min, followed by washout). Rings were (+) or were not (−) exposed to 1 μM GTN for 1 min. All bars are significantly less than control (Con) + GTN to P < 0.01, except cyanamide (Cya) + GTN, where P < 0.05. Data are presented as the means ± SD of three aortic rings. Chl, chloral hydrate; Ace, acetaldehyde.
It has been reported that vascular rings also generate some 1,3-GDN from GTN, and we verified that a small amount of 1,3-GDN was produced under our assay conditions (1,2-GDN/1,3-GDN = 4.98 ± 0.64, n = 6). However, 1,3-GDN formation was not mechanism based, because heat-inactivated rings generated the same amounts.
Chronic therapy with organic nitrates produces “tolerance,” a desensitization of blood vessels that compromises antianginal efficacy. This poorly understood phenomenon is at least partly attributable to impaired bioactivation of GTN in both small animals and humans and is readily recapitulated in vitro by lengthy high-dose exposures. We induced tolerance with a regimen of 0.3 mM GTN for 30 min, followed by washout. Whereas these conditions had no effect on SNP-induced relations (Fig. 2d2), the EC50 value for GTN increased significantly (Fig. 2d1). Furthermore, in aorta made tolerant to GTN, both GTN reductase activity (Fig. 3b), as measured by 1,2-GDN-formation, and classical mtALDH activity (Fig. 3c) decreased significantly (and comparably), and cGMP accumulation was abolished (Fig. 4). Reports that ALDH (dehydrogenase activity) is also potently inhibited in patients undergoing chronic treatment with GTN and other nitrate esters (34) provide strong support for the clinical relevance of these findings.
As a further test of the physiological role of mtALDH in bioactivation of GTN, we assessed the effects of the mtALDH inhibitors cyanamide and chloral hydrate on the systemic effects of GTN infusions in anesthetized rabbits. Both inhibitors significantly attenuated the hypotensive effects of GTN (Fig. 5a), whereas they had little or no effect on SNP-mediated (i.e., NO dependent) or adenosine-mediated (i.e., NO independent) (not shown) hypotension (Fig. 5b). Similar results were obtained in rats, suggesting a generalized role for mtALDH in mammalian GTN metabolism (data not shown).
Figure 5.

mtALDH inhibitors block the hypotensive effects of GTN in vivo. (a) i.v. infusions of GTN in rabbits produce dose-dependent decrements in blood pressure (before treatment, ●) that are attenuated significantly (P < 0.05) by both cyanamide (17 mg/kg, ○) and chloral hydrate (66 mg/kg, ▵) (n = 5–7). (b) Cyanamide and chloral hydrate have no effect on SNP-mediated decreases in blood pressure (n = 7) (data for cyanamide treatment are shown). ●, before treatment; ○, after treatment.
The classical studies by Needleman et al. (27) and Ignarro et al. (2, 35), suggesting a sulfhydryl requirement for relaxation by GTN, have been interpreted in terms of a “thiol receptor model” for organic nitrates in vascular smooth muscle, which provided for the possibility of an enzyme that generated inorganic nitrite as an intermediate and rationalized nitrate tolerance in terms of a depletion of intracellular thiols (27). Almost every feature of this model has been challenged in recent years by studies that have: (i) implicated alternative metalloenzymes in the mechanism of GTN bioactivation (11, 12); (ii) failed to detect a depletion of intracellular thiol in tolerant vessels (28); (iii) rejected the role of inorganic nitrite as an intermediate (36, 37); (iv) offered alternative interpretations for the effects of thiols on GTN bioactivity, and (v) suggested new mechanisms of tolerance (29). There is even recent evidence to suggest that GTN activation of guanylate cyclase may be independent of NO (38). None of these explanations, however, can reconcile the entire set of data. In contrast, the identification of the sulfhydryl-dependent mtALDH with GTN metabolic activity provides a viewpoint from which the models of Needleman et al. and Ignarro et al. are revived and shown to be in line with the original ideas of Murad et al. (1) as well as the newer data on enzymatic biotransformation of GTN and impairment of this mechanism in tolerant vessels. In particular, our findings are strongly supported by recent studies in GTN-treated patients, which have shown a profound decrease in mitochondrial-type (low _K_m) ALDH activity in the blood (34) and lower amounts of 1,2-GDN in tolerant vessels (8).
Several lines of evidence have been offered against the original proposal (2, 39) that GTN acts via liberation of inorganic nitrite, including studies showing that GTN is readily transformed into NO bioactivity within cellular homogenates and subcellular fractions whereas nitrite is not (36, 37). However, these previous studies did not consider the possibility of biotransformation within the mitochondria, which is highly competent for nitrite bioactivation to NO. It has been shown that members of mitochondria electron transport chain, cytochrome bc1 complex (complex III) and cytochrome c oxidase (complex IV) can reduce nitrite to NO (40, 41), and nitrite might also be transported to the intermembrane space where the higher proton concentration may facilitate its conversion to NO or SNO. Mitochondria can also reduce NO to HNO, which has been shown to produce both reversible [e.g., intramolecular disulfide (32)] and irreversible [e.g., sulfinamide (33)] modifications of the active site cysteine in mtALDH and thus may explain why complete reversal of nitrate tolerance requires new protein synthesis (42). Interestingly, the organic nitrate nicorandil [_N_-(2-hydroxyethyl)nicotinamide nitrate ester] was recently shown to generate NO in rat myocardial mitochondria (43). It is therefore conceivable that other nitrates and endogenous nitrogen oxides may be ALDH substrates. More generally, our studies provide new insights into subcellular processing of NO metabolites and suggest new approaches for generating NO bioactivity and overcoming nitrate tolerance.
Residual, albeit attenuated, relaxations by GTN were observed in the presence of biotransformation inhibitors that completely blocked the GTN-mediated increase in cGMP. Likewise, the relaxations in tolerant blood vessels appeared to be partly cGMP independent. These data are consistent with a number of studies showing that the mechanism of smooth muscle relaxation by NO and GTN is partly mediated by a direct effect on ion channels (44–46).
Our discovery that GTN is metabolized within mitochondria by the actions of ALDH has implications for patient care. It has long been a mystery why nitrates do not provide survival benefits, unlike agents that increase NO synthase activity, and the possibility that they may impair mitochondrial metabolism would provide an explanation. Indeed, this notion is supported by the findings of mitochondrial damage, impaired respiration, and increased production of proatherogenic superoxide in GTN-exposed tissues (29, 47). In addition, our studies suggest that certain classes of drugs such as sulfonylurea hypoglycemics, chloral hydrate, and acetaminophen (48), which inhibit mtALDH activity, may be contraindicated in patients taking organic nitrates esters for the treatment of acute ischemic syndromes and congestive heart failure. Finally, a polymorphism in the mtALDH gene (ALDH2) has been linked to impairments in ethanol metabolism and risk of developing cancer and dementia (48), and it follows that the ALDH genotype may predict GTN responsiveness and even therapeutic efficacy. A variety of new approaches to providing NO bioactivity and overcoming tolerance are suggested by our results.
Acknowledgments
We thank Drs. D. Hess, T. McMahon, A. Hausladen, J. Eu, and L. Liu for their support and many valuable discussions and M. Dewhirst for assistance.
Abbreviations
GTN
glyceryl trinitrate
GDN
glyceryl dinitrate
mtALDH
mitochondrial aldehyde dehydrogenase
SNO
_S_-nitrosothiol
SNP
sodium nitroprusside
Footnotes
See commentary on page 7816.
References
- 1.Murad F, Mittal C K, Arnold W P, Katsuki S, Kimura H. Adv Cyclic Nucleotide Res. 1978;9:145–158. [PubMed] [Google Scholar]
- 2.Ignarro L J, Lippton H, Edwards J C, Baricos W H, Hyman A L, Kadowitz P J, Gruetter C A. J Pharmacol Exp Ther. 1981;218:739–749. [PubMed] [Google Scholar]
- 3.Munzel T. J Am Coll Cardiol. 2001;38:1102–1105. doi: 10.1016/s0735-1097(01)01507-8. [DOI] [PubMed] [Google Scholar]
- 4.Nakamura Y, Moss A J, Brown M W, Kinoshita M, Kawai C. Am Heart J. 1999;138:577–585. doi: 10.1016/s0002-8703(99)70163-8. [DOI] [PubMed] [Google Scholar]
- 5.Brien J F, McLaughlin B E, Kobus S M, Kawamoto J H, Nakatsu K, Marks G S. J Pharmacol Exp Ther. 1988;244:322–327. [PubMed] [Google Scholar]
- 6.Kawamoto J H, McLaughlin B E, Brien J F, Marks G S, Nakatsu K. J Cardiovasc Pharmacol. 1990;15:714–719. doi: 10.1097/00005344-199005000-00005. [DOI] [PubMed] [Google Scholar]
- 7.Slack C J, McLaughlin B E, Brien J F, Marks G S, Nakatsu K. Can J Physiol Pharmacol. 1989;67:1381–1385. doi: 10.1139/y89-221. [DOI] [PubMed] [Google Scholar]
- 8.Sage P R, de la Lande I S, Stafford I, Bennett C L, Phillipov G, Stubberfield J, Horowitz J D. Circulation. 2000;102:2810–2815. doi: 10.1161/01.cir.102.23.2810. [DOI] [PubMed] [Google Scholar]
- 9.Tsuchida S, Maki T, Sato K. J Biol Chem. 1990;265:7150–7157. [PubMed] [Google Scholar]
- 10.Yeates R A, Schmid M, Leitold M. Biochem Pharmacol. 1989;38:1749–1753. doi: 10.1016/0006-2952(89)90408-5. [DOI] [PubMed] [Google Scholar]
- 11.Millar T M, Stevens C R, Benjamin N, Eisenthal R, Harrison R, Blake D R. FEBS Lett. 1998;427:225–228. doi: 10.1016/s0014-5793(98)00430-x. [DOI] [PubMed] [Google Scholar]
- 12.McDonald B J, Bennett B M. Can J Physiol Pharmacol. 1990;68:1552–1557. doi: 10.1139/y90-236. [DOI] [PubMed] [Google Scholar]
- 13.McDonald B J, Bennett B M. Biochem Pharmacol. 1993;45:268–270. doi: 10.1016/0006-2952(93)90403-j. [DOI] [PubMed] [Google Scholar]
- 14.Seth P, Fung H L. Biochem Pharmacol. 1993;46:1481–1486. doi: 10.1016/0006-2952(93)90115-d. [DOI] [PubMed] [Google Scholar]
- 15.McGuire J J, Anderson D J, McDonald B J, Narayanasami R, Bennett B M. Biochem Pharmacol. 1998;56:881–893. doi: 10.1016/s0006-2952(98)00216-0. [DOI] [PubMed] [Google Scholar]
- 16.Nigam R, Anderson D J, Lee S F, Bennett B M. J Pharmacol Exp Ther. 1996;279:1527–1534. [PubMed] [Google Scholar]
- 17.Dean T W, Baun D C. Am J Hosp Pharm. 1975;32:1036–1038. [PubMed] [Google Scholar]
- 18.Crew M, Dicarlo F. J Chromatogr. 1968;35:506–512. [Google Scholar]
- 19.Brien J F, McLaughlin B E, Breedon T H, Bennett B M, Nakatsu K, Marks G S. J Pharmacol Exp Ther. 1986;237:608–614. [PubMed] [Google Scholar]
- 20.Madden E A, Storrie B. Anal Biochem. 1987;163:350–357. doi: 10.1016/0003-2697(87)90235-1. [DOI] [PubMed] [Google Scholar]
- 21.Mukerjee N, Pietruszko R. J Biol Chem. 1994;269:21664–21669. [PubMed] [Google Scholar]
- 22.Ikawa M, Impraim C C, Wang G, Yoshida A. J Biol Chem. 1983;258:6282–6287. [PubMed] [Google Scholar]
- 23.Weiner H. In: Biochemistry and Pharmacology of Ethanol. Majchrowicz E, Noble E P, editors. Vol. 1. New York: Plenum; 1979. pp. 125–144. [Google Scholar]
- 24.Kobzik L, Stringer B, Balligand J L, Reid M B, Stamler J S. Biochem Biophys Res Commun. 1995;211:375–381. doi: 10.1006/bbrc.1995.1824. [DOI] [PubMed] [Google Scholar]
- 25.Lacza Z, Puskar M, Figueroa J P, Zhang J, Rajapakse N, Busija D W. Free Radical Biol Med. 2001;31:1609–1615. doi: 10.1016/s0891-5849(01)00754-7. [DOI] [PubMed] [Google Scholar]
- 26.Feldman R I, Weiner H. J Biol Chem. 1972;247:267–272. [PubMed] [Google Scholar]
- 27.Needleman P, Jakschik B, Johnson E M., Jr J Pharmacol Exp Ther. 1973;187:324–331. [PubMed] [Google Scholar]
- 28.Boesgaard S, Aldershvile J, Poulsen H E, Loft S, Anderson M E, Meister A. Circ Res. 1994;74:115–120. doi: 10.1161/01.res.74.1.115. [DOI] [PubMed] [Google Scholar]
- 29.Munzel T, Sayegh H, Freeman B A, Tarpey M M, Harrison D G. J Clin Invest. 1995;95:187–194. doi: 10.1172/JCI117637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vallari R C, Pietruszko R. Science. 1982;216:637–639. doi: 10.1126/science.7071604. [DOI] [PubMed] [Google Scholar]
- 31.Shen M L, Lipsky J J, Naylor S. Biochem Pharmacol. 2000;60:947–953. doi: 10.1016/s0006-2952(00)00435-4. [DOI] [PubMed] [Google Scholar]
- 32.DeMaster E G, Redfern B, Quast B J, Dahlseid T, Nagasawa H T. Alcohol. 1997;14:181–189. doi: 10.1016/s0741-8329(96)00142-5. [DOI] [PubMed] [Google Scholar]
- 33.DeMaster E G, Redfern B, Nagasawa H T. Biochem Pharmacol. 1998;55:2007–2015. doi: 10.1016/s0006-2952(98)00080-x. [DOI] [PubMed] [Google Scholar]
- 34.Towell J, Garthwaite T, Wang R. Alcohol Clin Exp Res. 1985;9:438–442. doi: 10.1111/j.1530-0277.1985.tb05579.x. [DOI] [PubMed] [Google Scholar]
- 35.Ignarro L J, Gruetter C A. Biochim Biophys Acta. 1980;631:221–231. doi: 10.1016/0304-4165(80)90297-4. [DOI] [PubMed] [Google Scholar]
- 36.Kowaluk E A, Chung S J, Fung H L. Drug Metab Dispos. 1993;21:967–969. [PubMed] [Google Scholar]
- 37.Romanin C, Kukovetz W R. J Mol Cell Cardiol. 1988;20:389–396. doi: 10.1016/s0022-2828(88)80130-5. [DOI] [PubMed] [Google Scholar]
- 38.Artz J D, Toader V, Zavorin S I, Bennett B M, Thatcher G R. Biochemistry. 2001;40:9256–9264. doi: 10.1021/bi002885x. [DOI] [PubMed] [Google Scholar]
- 39.Hay M. Practitioner. 1883;30:422–433. [Google Scholar]
- 40.Kozlov A V, Staniek K, Nohl H. FEBS Lett. 1999;454:127–130. doi: 10.1016/s0014-5793(99)00788-7. [DOI] [PubMed] [Google Scholar]
- 41.Brudvig G W, Stevens T H, Chan S I. Biochemistry. 1980;19:5275–5285. doi: 10.1021/bi00564a020. [DOI] [PubMed] [Google Scholar]
- 42.Hinz B, Schroder H. Biochem Biophys Res Commun. 1998;252:232–235. doi: 10.1006/bbrc.1998.9630. [DOI] [PubMed] [Google Scholar]
- 43.Sakai K, Akima M, Saito K, Saitoh M, Matsubara S. J Cardiovasc Pharmacol. 2000;35:723–728. doi: 10.1097/00005344-200005000-00007. [DOI] [PubMed] [Google Scholar]
- 44.Brunner F, Schmidt K, Nielsen E B, Mayer B. J Pharmacol Exp Ther. 1996;277:48–53. [PubMed] [Google Scholar]
- 45.Cohen R A, Weisbrod R M, Gericke M, Yaghoubi M, Bierl C, Bolotina V M. Circ Res. 1999;84:210–219. doi: 10.1161/01.res.84.2.210. [DOI] [PubMed] [Google Scholar]
- 46.Bolotina V M, Najibi S, Palacino J J, Pagano P J, Cohen R A. Nature (London) 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- 47.Needleman P, Hunter F E., Jr Mol Pharmacol. 1966;2:134–143. [PubMed] [Google Scholar]
- 48.Vasiliou V, Pappa A, Petersen D R. Chem Biol Interact. 2000;129:1–19. doi: 10.1016/s0009-2797(00)00211-8. [DOI] [PubMed] [Google Scholar]