Antisense inhibition of gene expression in cells by oligonucleotides incorporating locked nucleic acids: effect of mRNA target sequence and chimera design (original) (raw)
Abstract
Use of antisense oligonucleotides is a versatile strategy for achieving control of gene expression. Unfortunately, the interpretation of antisense-induced phenotypes is sometimes difficult, and chemical modifications that improve the potency and specificity of antisense action would be useful. The introduction of locked nucleic acid (LNA) bases into oligonucleotides confers exceptional improvement in binding affinity, up to 10°C per substitution, making LNAs an exciting option for the optimization of antisense efficacy. Here we examine the rules governing antisense gene inhibition within cells by oligonucleotides that contain LNA bases. LNA- containing oligomers were transfected into cells using cationic lipid and accumulated in the nucleus. We tested antisense gene inhibition by LNAs and LNA–DNA chimeras complementary to the 5′-untranslated region, the region surrounding the start codon and the coding region of mRNA, and identified effective antisense agents targeted to each of these locations. Our data suggest that LNA bases can be used to develop antisense oligonucleotides and that their use is a versatile approach for efficiently inhibiting gene expression inside cells.
INTRODUCTION
Oligonucleotides and their analogs are demonstrating efficacy in clinical applications and are gaining more acceptance as powerful tools for elucidating biological function (1). Many chemically modified bases and backbone linkages are available for optimizing increased oligonucleotide hybridization affinity for intracellular RNA and DNA targets. One such promising modification is locked nucleic acids (LNAs), also known as bridged nucleic acids (BNAs), developed by Wengel and co-workers (2) and Imanishi and co-workers (3).
LNA bases are ribonucleotide analogs containing a methylene linkage between the 2′ oxygen and the 4′ carbon of the ribose ring (Fig. 1). The constraint on the sugar moiety results in a locked 3′-endo conformation that preorganizes the base for hybridization and increases melting temperature (_T_m) values as much as 10°C per base (4,5). LNA bases can be incorporated into oligonucleotides using standard protocols for DNA synthesis. This commonality facilitates the rapid synthesis of chimeric oligonucleotides that contain both DNA and LNA bases and allows chimeric oligomers to be tailored for their binding affinity and ability to activate RNase H. Because oligomers that contain LNA bases have a native phosphate backbone they are readily soluble in water. Introduction of LNA bases also confers resistance to nucleases when incorporated at the 5′ and 3′ ends of oligomers (6). The ability to use LNAs for in vivo applications is also suggested by the finding that LNAs have demonstrated low toxicity when delivered intravenously to animals (7).
Figure 1.

Structure of locked nucleic acid (LNA).
Existing applications for oligomers that contain LNA bases include allele-specific PCR, Taqman probes, single nucleotide polymorphism analysis (8), capture probes and gene arrays, transcription factor decoys (6), triple helix formation (9), alteration of intron splicing (10) and inhibition of HIV-1 Tat-dependent transactivation of gene expression (11). We have shown that LNAs and LNA–DNA chimeras are potent inhibitors of human telomerase and that a relatively short eight base LNA is a 1000-fold more potent agent than an analogous peptide nucleic acid (PNA) oligomer (12). LNAs and LNA–DNA chimeras may also be useful agents for antisense gene inhibition. Wengel and co-workers have used LNAs to inhibit gene expression in mice (7), while Erdmann and colleagues have described the design of LNA-containing oligomers that recruit RNase H and have described the rules governing RNase H activation by LNA–DNA chimeras in cell-free systems (13).
The use of LNA as a tool for improving applications for nucleic acids must be viewed in the context of rapid advances in other areas of nucleic acid recognition. Other oligonucleotide modifications, such as phosphorothioate DNA, 2′-_O_-methyl RNA and 2′-methoxyethyl RNA, are now being tested in clinical trials, morpholino oligomers are proving useful tools for studying embryonic development (14,15), and Formivirsen, a phosphorothioate modified DNA, has been approved by the Federal Drug Administration as a therapeutic (1,16). For studies that use cultured cells, the discovery that small interfering RNAs (siRNAs) can control mammalian gene expression (17,18) has provided a high standard against which novel chemistries must be judged. The high _T_m values that result from introduction of LNA bases, however, lead to the hypothesis that high affinity recognition by LNAs may lead to more potent and reliable antisense oligonucleotides. Such LNA-substituted oligonucleotides might allow more definitive knockdown phenotypes for basic research and be more active for therapeutic applications.
Here we examine whether the high affinity of LNA can be translated into the development of effective antisense agents for blocking gene expression inside cells. We show that LNAs and LNA–DNA chimeras can be potent antisense agents and that they can efficiently inhibit gene expression when targeted to a variety of regions within mRNA.
MATERIALS AND METHODS
Materials
LNAs were obtained as unpurified synthesis products on the 2 µmol scale from Proligo LLC (Boulder, CO) or Cureon A/S (Copenhagen, Denmark). DNA oligonucleotides were obtained from Invitrogen (Rockville, MD) or Proligo LLC. LipofectAMINE was obtained from Invitrogen. RNA oligomers were obtained from Oligos etc. (Wilsonville, OR). Oligomers were solubilized according to the manufacturer’s protocol in sterile RNase- and DNase-free distilled water (Invitrogen). All oligomers were quantitated based on spectrophotometric A260 values and the conversion factor of 30 µg/ml OD260. 9-_cis_-retinoic acid (9-cisRA) was obtained from Sigma (St Louis, MO). Ligand stocks (10 mM) were dissolved in 80% ethanol/20% DMSO (v/v) and stored under nitrogen at –20°C. All manipulations of 9-cisRA were performed under yellow light to minimize the likelihood of isomerization. Plasmids pCMX-hRXR, pCMX-β-gal and pTK-CRBPII-Luc were obtained from Dr David Mangelsdorf (University of Texas Southwestern Medical Center, Dallas, TX) (19).
Annealing of complementary oligonucleotides
Concentrations of LNA, DNA and RNA oligomers were determined as above and 15–40 µl volumes of nucleic acid duplex mixtures (100 µM each) were prepared in thin-walled PCR tubes in 2.5× phosphate-buffered saline (PBS) from a 10× stock (10.0 mM KH2PO4, 1550 mM NaCl, 30 mM Na2HPO4·7H2O, pH 7.4) without calcium or magnesium (Invitrogen). Annealing of oligomers was performed in a thermocycler according to the following temperature profile. Reductions in temperature occurred in 1 min with the hold times indicated: 95°C, 5 min; 85°C, 1 min; 75°C, 1 min; 65°C, 5 min; 55°C, 1 min; 45°C, 1 min; 35°C, 5 min; 25°C, 1 min; 15°C, 1 min; hold at 15°C. After annealing, the oligomer duplexes were maintained at 4°C until evaluation of _T_m was performed.
Melting temperature (_T_m) determination
Melting temperature studies were performed by measuring the change in absorbance at 260 nm using a Cary 100 Bio UV/Vis spectrophotometer (Varian Inc., Walnut Creek, CA) equipped with a 12 position sample holder and a Peltier temperature control accessory. Determinations were performed in a 0.9 ml stoppered semi-micro quartz cuvette (Varian). Sample was prepared by mixing 8 µl of annealed nucleic acid duplex stock solution (100 µM) with 392 µl of 0.1 M sodium phosphate (Na2HPO4, pH 7.4) buffer. Samples were overlaid with 300 µl of mineral oil to prevent evaporation at higher temperatures and to make the upper and lower baselines more consistent. Data were collected with the Cary WinUV Thermal software from 98 to 14°C and from 14 to 98°C in 2°C increments at a rate of 2°C/min, with an equilibration time of 0.2 min at each temperature. An initial 2 min equilibration was included to ensure complete denaturation prior to starting the temperature ramping.
Data were collected in both directions (denaturation and annealing) to ensure that the observed curves were reversible. Data were subjected to non-linear curve fit analysis and the _T_m determined using van’t Hoff parameters included in the software. Independent analyses were performed for the data corresponding to the denaturation and annealing profiles and the average value reported. Unless otherwise noted, _T_m values for LNAs and LNA–DNA chimeras were obtained using DNA complements.
Lipid-mediated transfection LNAs and LNA–DNA chimeras
LNA oligomers were prepared for transfection by equilibrating 6.4 µl of 100 µM LNA in 144 µl of Opti-MEM (Invitrogen). In a separate tube, 1.9 µl of (7 µg/ml) LipofectAMINE (Invitrogen) was activated in 148 µl of Opti-MEM by tapping the tube vigorously for 15 s followed by equilibration for 5–10 min at room temperature. The diluted LNA oligomer and LipofectAMINE aliquots (150 µl each) were mixed together and the contents agitated by tapping the tube vigorously for 15 s. Lipid complexes were allowed to form by incubating the mixture at room temperature for 15–20 min in the dark. The solution containing the LNA–lipid complex (300 µl) was diluted to 3.2 ml with Opti-MEM resultant in a solution containing 200 nM LNA. This solution was then serial diluted to the final working concentrations of 100 nM, and 25 nM for most experiments.
CV-1 cells (CCL-70; American Type Culture Collection, Manassas, VA) were plated at 11 000–13 000 cells/well in 48-well plates using Dulbecco’s minimal essential medium with glutamine supplemented with 10% super-stripped fetal bovine serum (Atlanta Biologicals Norcross, GA), 20 mM HEPES buffer (final concentration, pH 7.4), 500 U/ml penicillin, 0.1 mg/ml streptomycin and 0.06 mg/ml tylosin reagent (Sigma). Super-stripped serum was used to ensure that competing ligands were removed from serum prior to addition of 9-cisRA. Ligand stripping was achieved by twice extracting serum with activated charcoal and cation exchange (CAG 1-X8 resin; Bio-Rad, Hercules, CA). Super-stripped serum was double filtered through a 0.2 µm filter and stored as 50 ml aliquots at –20°C until use. Cells were incubated at 37°C at 5% CO2 for a minimum of 6 h prior to initiating transfection. The cells were then washed once with 250 µl/well Opti-MEM, followed by overnight transfection with either LNA–lipid complex or lipid only. A second transfection of reporter vectors was conducted subsequently as described below.
Lipid-mediated transfection of reporter vector complexes into CV-1 cells
Expression vectors (pCMX-β-Gal, 40 ng/well, pCMX-hRXRα, 20 ng/well, and pTK-CRBPII-Luc, 40 ng/well) were prepared for transfection by equilibrating plasmid DNA in 19.5 µl/well Opti-MEM. Likewise, 0.2 µl of (7 µg/ml) LipofectAMINE was activated in 19.8 µl of Opti-MEM. The two components were mixed and complexed for 15–20 min as described above. The solution of lipid–plasmid complex was diluted with 10 µl/well Opti-MEM and 50 µl of the vector mixture was dispensed into each well. Transfection was carried out for 6 h, after which the composite transfection mixture was removed by aspiration and replaced with the medium containing super-stripped serum. The medium was supplemented with 1 µM 9-cisRA (Sigma) or a solvent control comprised of 80% ethanol/20% DMSO (v/v). Cells were harvested 40 h after addition of ligand and analyzed for luciferase and β-galactosidase activities.
Luciferase assays were conducted with 20 µl of cell lysate, 100 µl of assay buffer and 100 µl of 1 mM luciferin (Biosynth, Naperville, IL) prepared in 0.1 M KH2PO4, pH 7.8, in an opaque, flat bottomed 96-well plate (Costar). Data were collected using enhance flash parameters on a model ML-3000 microplate luminescence system with BioLinx software v.2.22 (Dynex Technologies, Chantilly, VA). β-Galactosidase assays were conducted using 40 µl of cell lysate and 125 µl of phosphate assay buffer containing 2 mg/ml _o_-nitrophenyl β-d-galactopyranoside (ONPG). Color development was conducted at 37°C for 15 min or less, depending on the rate of color development, and the reaction was stopped with the addition of 50 µl of 1 M NaCO3. Data were collected at 410 nm and a 630 nm reference wavelength on a model MR5000 microtiter plate reader with BioLinx software v.2.22 (Dynex). All data points represent the mean of triplicate experiments normalized against β-galactosidase activity and are reported as a percentage of the values of control cells treated with Opti-MEM only.
Fluorescence microscopy
A LNA–DNA chimera was synthesized to include a Cy3 fluorescent label at the 5′-terminus. The LNA was delivered into CV-1 cells as described above adjusted to a Lab-Tek 4-well chambered coverglass (Nalgene Nunc International, Naperville, IL). Transfection of LNA was performed at 100 nM in 500 µl without subsequent vector transfection in order to ascertain the basic intracellular distribution of LNA. After an overnight transfection, cells were washed twice with 500 µl of Opti-MEM at room temperature with a 5 min room temperature incubation between washes. Cells were then incubated for 30 min in 50% (v/v) Opti-MEM and PBS containing 0.05 mg/ml Hoechst 33258 stain (Sigma), then washed five times with 500 µl of Opti-MEM. After the last wash, slides were analyzed using a Zeiss Axiovert 200M inverted transmitted light microscope (Carl Zeiss MicroImaging, Thornwood, NY) equipped with a digital imaging system and Slidebook imaging software (Intelligent Imaging Innovations, Denver, CO).
RESULTS AND DISCUSSION
Luciferase as an antisense target
We chose the mRNA encoding firefly luciferase as a model target to investigate the rules governing antisense inhibition by LNAs and LNA–DNA chimeras inside cells. The major advantage of the use of luciferase is that the activity of luciferase protein can be rapidly detected using a highly sensitive bioluminescence assay. Possession of a convenient assay allowed us to readily test inhibition of gene expression by a large series of LNAs and LNA–DNA chimeras.
In our assay, the plasmid containing the luciferase gene is co-transfected with plasmids containing the β-galactosidase and RXR genes. The β-galactosidase gene is constitutively expressed and serves as a reporter for transfection efficiency and a tool to normalize luciferase measurements. RXR is a nuclear hormone receptor and the promoter for luciferase contains the RXR response element. Addition of 9-cisRA, the ligand for RXR, activates transcription of luciferase, providing a mechanism for tightly controlling expression. LNAs and LNA–DNA chimeras were introduced prior to transfection of the reporter vectors to ensure that they were present before expression of luciferase mRNA was induced.
Choice of mRNA target sequence
The luciferase construct contains a single translation initiation site within the thymidine kinase promoter, resulting in a 90 base 5′-untranslated region (5′-UTR). We had previously shown that PNAs directed to the 5′-terminus of the UTR could inhibit luciferase expression, but that 15mer PNAs directed to the start codon or sequences within the coding region did not (20). Our goal for the current study with LNAs and LNA–DNA chimeras was to test whether their use would afford a wider spectrum of potent antisense agents capable of targeting a variety of regions within mRNA.
One reason for believing that LNAs might be more potent agents than analogous PNAs is that oligomers that are highly substituted with LNA bases possess high _T_m values (4,5). In particular, we speculated that the favorable preorganization of consecutive LNA bases might assist the initiation of mRNA recognition and encourage subsequent binding of the entire oligomer. Another reason is based in Erdmann and colleagues’ report that LNA–DNA chimeras containing a central DNA region of at least seven consecutive DNA bases can recruit RNase H, causing destruction of the mRNA target and providing a mechanism that allows the antisense effects of LNA–DNA chimeras to be amplified (13). Similar PNA–DNA chimeras can be obtained (21), but their synthesis is complicated by the need to combine nucleic acid and peptide synthesis technologies and our earlier study used PNA oligomers that were unable to activate RNase H (20).
To establish the effectiveness of different LNA designs, we divided the transcript into the 5′-UTR, start site and downstream coding regions (Fig. 2). LNAs and LNA–DNA chimeras followed one of four designs: (i) all LNA; (ii) LNA–DNA chimeras with no contiguous DNA stretches of five or more; (iii) LNA–DNA–LNA chimeras with a minimum of five contiguous DNA bases flanked by at least two contiguous sequences of LNA bases; (iv) LNA–DNA chimeras in which the DNA portion contains two phosphorothioate linkages to improve nuclease resistance. Designs (i) and (ii) were intended to bind target mRNA with high affinity but possess little ability to activate RNase H, to test the hypothesis that the high affinity inhibitor would block the progression of the ribosomal complex along the mRNA and inhibit translation of luciferase. Designs (iii) and (iv) were intended to recruit RNase H, following the rules for RNase H recruitment by LNAs developed by Erdmann and co-workers (13).
Figure 2.

Schematic of luciferase mRNA showing the approximate target sites of LNA and LNA–DNA chimeras used in these studies.
Introduction of LNAs into cells
We employed cationic lipid as carrier to assist intracellular delivery of LNAs. To demonstrate delivery into the cytoplasm we obtained a LNA labeled with the fluorophore Cy3, mixed it with lipid and added the complex to cultured CV-1 cells. We and others had previously shown that fluorescently labeled LNAs were evenly distributed throughout cells that had been fixed prior to microscopy (7,12), but recent reports have pointed out that fixing can produce artifacts that confuse conclusions about the localization of oligomers in cells (22,23). Therefore, we repeated the microscopy with live cells (Fig. 3). These experiments revealed that the localization of Cy3 label overlapped with localization of the cell permeable nuclear stain Hoechst 33258, supporting the conclusion that lipid-mediated delivery allows LNAs to enter the cytoplasm and nucleus.
Figure 3.
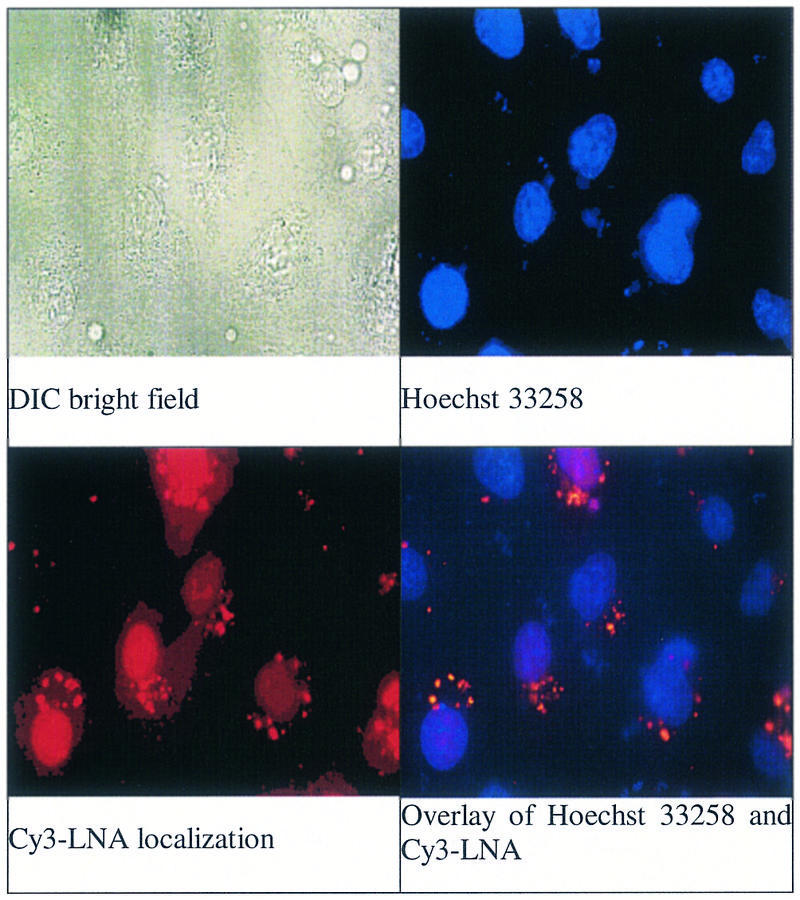
Images of CV-1 cells transfected with a LNA–DNA chimera that was analogous in sequence to XV and labeled with Cy3 fluorophore. All images are magnified 630 times and the field of view is the same. (Left to right) DIC bright field image; staining with the cell permeable nuclear dye Hoechst 32358; localization of Cy3-labeled LNA–DNA chimera; overlay of image of Hoechst 33258 and Cy3-labeled LNA–DNA chimera.
Confocal microscopy of LNA uptake by live cells revealed localization in the center of the cell, also consistent with nuclear uptake (Supplementary Material). We observed punctate localization on the periphery of the nucleus (Fig. 3), which may suggest that LNA-containing oligomers preferentially localize with the large amount of RNA contained in the rRNA component of the rough endoplasmic reticulum. This pattern is consistent with the high affinity of LNA for RNA relative to DNA and with the greater tendency for RNA to offer single-stranded regions that are accessible to LNA binding. The Cy3-labeled LNA possessed no specific intracellular target, therefore, localization reflects the general cellular recognition of the LNA chemistry. It is important to note that cells remain viable, so regardless of the source of the interaction that causes the localization of LNA it does not interfere with essential cellular processes.
As with any oligonucleotide, we observed that LNAs and LNA–DNA chimeras must be carefully purified prior to introduction into cells. Once this purification was achieved, we found that LNA-containing oligomers become toxic only at doses above those used in these experiments (>500 nM), suggesting that the high affinity of LNA does not result in an unacceptable level of non-specific interactions. Cells treated with LNAs and LNA–DNA chimeras remain viable indefinitely (12).
Melting temperature values for hybridization of LNAs and LNA–DNA chimeras
All of the LNAs and LNA–DNA chimeras tested in this study were characterized by determining their _T_m values for hybridization with complementary DNA oligonucleotides (Tables 1–3). DNA oligomers were chosen for routine use as complements because of the high cost of RNA oligomers and the large number of LNA-substituted oligomers that were used in our studies. As expected, we observed that all of the LNAs and LNA–DNA chimeras possessed _T_m values that were much higher than the growth temperature for cultured cells, 37°C. _T_m values increased with the number of LNA substitutions and the length of the oligomer. We did obtain one RNA, complementary to LNA–DNA chimeras XXVI and XXX (Table 3), with _T_m values for hybridization similar to those measured for hybridization to the analogous DNA complement (data not shown).
Table 1. Oligomer sequence, _T_m values and inhibition of luciferase activity data for LNAs and LNA–DNA chimeras that target the 5′-UTR of luciferase mRNA.
| ID | Sequence 5′→3′ | _T_m (°C) DNA:DNA | _T_m (°C) LNA:DNA | Inhibition (%) | ||
|---|---|---|---|---|---|---|
| 25 nM | 100 nM | 200 nM | ||||
| I | AgggTcGcTCGGT | 66 | ND | 50 | 63 | 76 |
| II | AgggTcGcTCAAT | 51 | 65 | 0 | 0 | 21 |
| III | ACACCGAGcGacccT | 71 | ND | 0 | 0 | 0 |
| IV | AgggTcGcTCGGTgT | 71 | ND | 69 | 80 | 95 |
| V | AgggTcGcTCAATgT | 66 | 83 | 20 | 19 | 21 |
| VI | AGGGTCGCTCGGTGT | 66 | ND | 76 | 83 | 93 |
| VII | TAaGcggGTcGcTGC | 66 | 86 | 28 | 36 | 48 |
| VIII | AGGGtcgctcgGTGT | 71 | 78 | 33 | 74 | 81 |
| IX | TCGAgatctgcGGCA | 64 | 68 | 11 | 22 | 25 |
| X | TTACcaacagtACCG | 54 | 71 | 0 | 0 | 5 |
Table 3. Oligomer sequence, _T_m values and inhibition of luciferase activity data for LNA–DNA chimeras that target the downstream coding region of luciferase mRNA.
| ID | Sequence 5′→3′ | _T_m (°C) DNA:DNA | _T_m (°C) LNA:DNA | Inhibition (%) | ||
|---|---|---|---|---|---|---|
| 25 nM | 100 nM | 200 nM | ||||
| XXVI | GtcgTtCgCGGGCgC | 61 | 77 | 23 | 40 | 55 |
| XXVII | TGtAgccATcCaTCC | 47 | 78 | 0 | 0 | 0 |
| XXVIII | CGGTtccatccTCTA | 55 | 69 | 60 | 61 | 78 |
| XXIX | TTGTattcagcCCAT | 52 | 68 | 33 | 39 | 69 |
| XXX | GTCGttcgcggGCGC | 71 | 81 | 82 | 94 | 95 |
| XXXI | GCTGttcgcggCGGC | 69 | 78 | 6 | 7 | 8 |
| XXXII | TGTAgccatccATCC | 47 | 78 | 29 | 39 | 53 |
Effect on luciferase expression of LNA–DNA chimeras targeted to the 5′-UTR
The terminal region of the 5′-UTR is a promising target for antisense oligomers because binding of the oligomer should block association with the ribosome and prevent translation. To test whether LNAs targeted to the 5′-UTR could inhibit translation, we obtained 13 (I and II) and 15 (III–X) base oligomers with partial or full substitution with LNA bases (Table 1). The most potent inhibitors were 15 base oligomers that were either completely substituted with LNA bases (VI) or had at least three consecutive LNA bases (IV and VIII) (Table 1 and Fig. 4A). A 13 base LNA–DNA chimera (I) also inhibited luciferase expression, but not as potently. Inhibition of gene expression by LNA–DNA chimeras I and IV that lack more than three consecutive DNA bases and by LNA (VI) supports the hypothesis that recruitment of RNase H is not necessary for activity.
Figure 4.
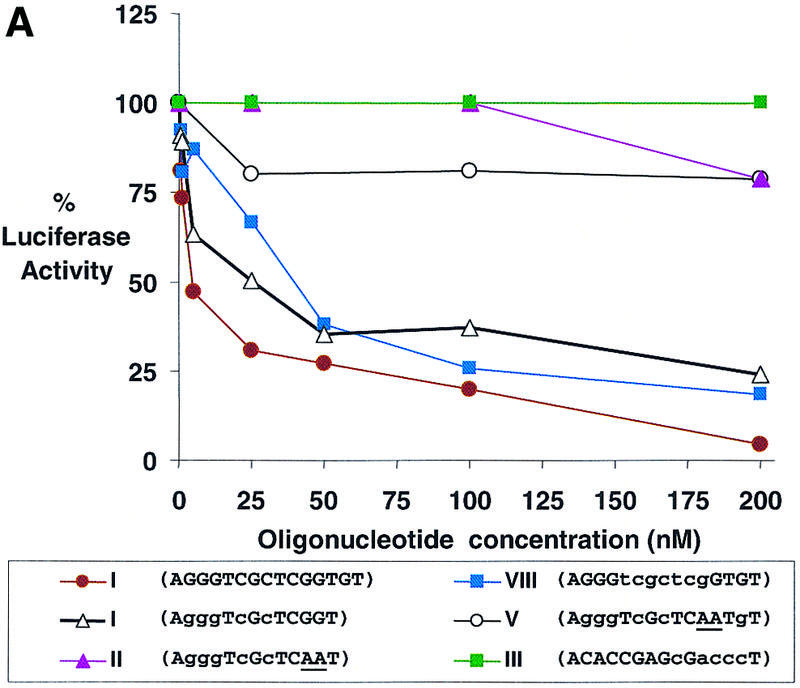
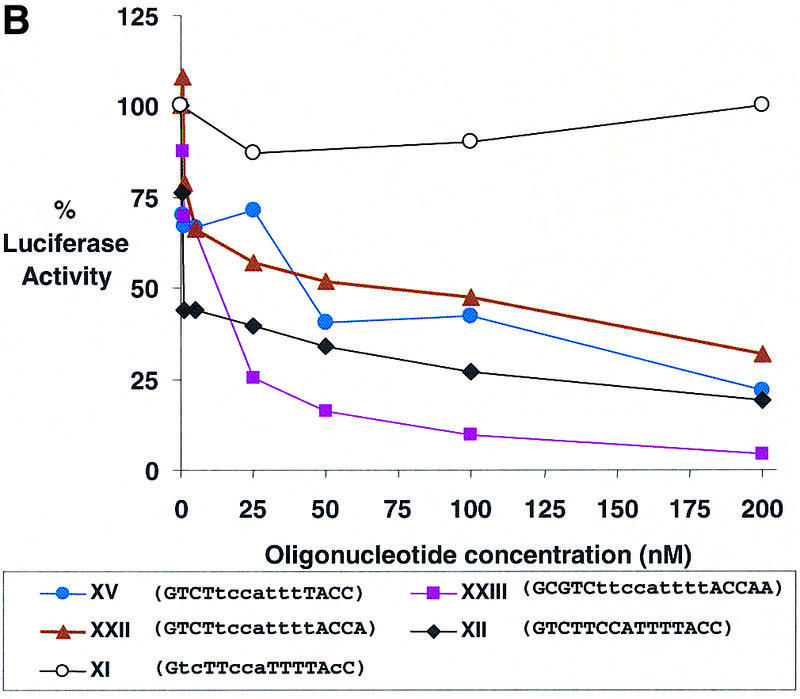
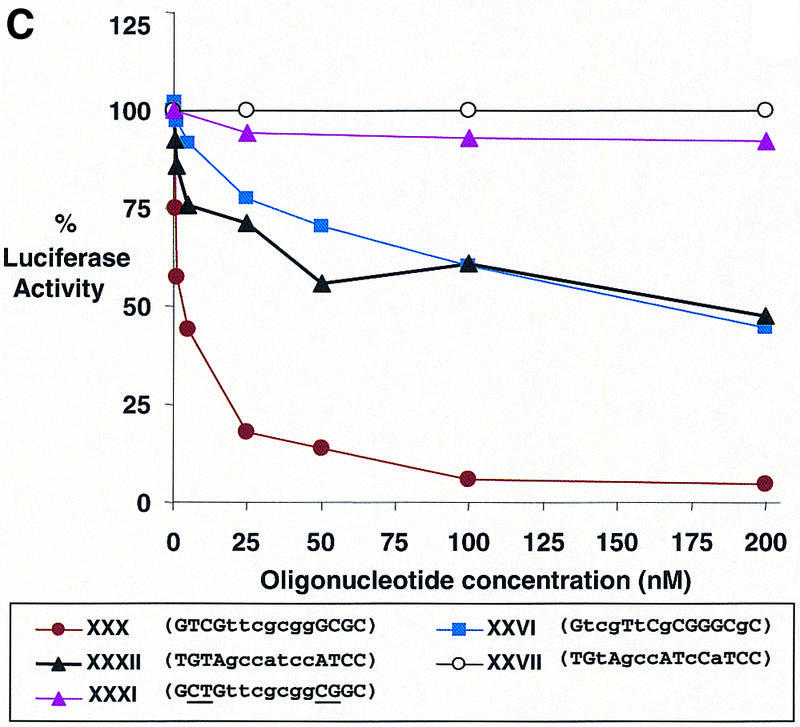
Dose–response curves of antisense gene inhibition by LNAs and LNA–DNA chimeras targeted to (A) the 5′-UTR, (B) the translation start site and (C) the downstream coding region of luciferase mRNA. All points are averages of triplicate determinations and are normalized to an independent measurement of β-galactosidase activity. Capitalized letters within a sequence represent LNA, lower case letters represent DNA bases and underlined bases depict mismatched bases.
Oligomers that contained mismatched bases (II and V) or that were complementary to a control sense strand LNA–DNA chimera (III) did not significantly inhibit luciferase activity. LNA–DNA chimera VII, targeted immediately downstream from the terminal target site, was only a modest inhibitor. LNA–DNA chimeras targeted to sequences further downstream within the 5′-UTR (IX and X) did not significantly inhibit expression of luciferase activity even though they possessed high _T_m values for binding complementary sequences and the same arrangement of LNA bases found in successful inhibitor VIII. These data are similar to those reported earlier for inhibition of luciferase expression by PNA oligomers (20). They support the conclusion that the terminus of the 5′-UTR is a useful site for targeting LNAs and that inhibition decreases dramatically as target sites are moved downstream within the 5′-UTR.
Effect of an LNA and LNA–DNA chimera on luciferase expression when targeted to the translation start site of luciferase mRNA
The second target region that we examined was the translation start site. The start site is an attractive target because its location is predictable and because there have been several reports suggesting that morpholino oligomers that target the start site are reliable tools for controlling gene expression (14,15). However, other reports indicate that the start site is not a generally susceptible site for antisense inhibition (24). To determine the potential for oligomers that contain LNA bases that target the start site to act as effective antisense agents we examined antisense gene inhibition by one fully substituted LNA and 14 LNA–DNA chimeras (XI–XXV) (Table 2 and Fig. 4B). These oligomers can be divided into two groups depending on their predicted ability to recruit RNase H. The LNA and LNA–DNA chimeras XI–XIV have four or fewer consecutive DNA bases and would not be expected to effectively recruit RNase H, while LNA–DNA chimeras XV–XXV have at least six consecutive DNA bases and should be able to activate RNase H upon binding mRNA according to the rules developed by Erdmann (13).
Table 2. Oligomer sequence, _T_m values and inhibition of luciferase activity data by LNAs and LNA–DNA chimeras that target the start site of the luciferase mRNA.
| ID | Sequence 5′→3′ | _T_m (°C) DNA:DNA | _T_m (°C) LNA:DNA | Inhibition (%) | ||
|---|---|---|---|---|---|---|
| 25 nM | 100 nM | 200 nM | ||||
| XI | GtcTTccaTTTTAcC | 50 | 65 | 13 | 10 | 0 |
| XII | GTCTTCCATTTTACC | 50 | 89 | 60 | 73 | 81 |
| XIII | CgtcTtccaTtttaCcaacAgtacC | 70 | 74 | 2 | 0 | 12 |
| XIV | CGtCTtcCATttTacCAaCagTACC | 64 | ND | 38 | 52 | 62 |
| XV | GTCTtccatttTACC | 50 | 64 | 29 | 58 | 78 |
| XVI | TTTTggcgtctTCCA | 48 | 68 | 12 | 42 | 49 |
| XVII | GTCTTCcattttacC | 50 | 60 | 72 | 78 | 86 |
| XVIII | GTCTTCCattttacC | 50 | 66 | 74 | 79 | 87 |
| XIX | GTCTTccattttacC | 50 | 56 | 0 | 0 | 19 |
| XX | GTCTtccattttacC | 50 | 53 | 0 | 0 | 0 |
| XXI | GTCttccattttACC | 50 | 60 | 30 | 52 | 73 |
| XXII | GTCTtccattttACCA | 50 | 66 | 43 | 53 | 68 |
| XXIII | GCGTCttccattttACCAA | 60 | 67 | 75 | 90 | 96 |
| XXIV | GTCTTCCAtttta⁁c⁁c | 50 | 68 | 38 | 51 | 62 |
| XXV | g⁁t⁁cttccATTTTACC | 50 | 67 | 0 | 3 | 48 |
LNA XII, which was completely substituted with LNA bases, was an effective inhibitor of luciferase expression. In contrast, LNA–DNA chimera XI that was complementary to the same target site was not an effective inhibitor at any concentration. This difference in potency between LNA XII and LNA–DNA XI is probably due to the fact that the _T_m value for hybridization by LNA–DNA chimera XI is 24°C lower than the _T_m value for the potent inhibitor LNA XII (Table 2). We also tested 25 base LNA–DNA chimeras XIII and XIV to determine whether increasing length might increase antisense efficacy. LNA–DNA chimera XIII that was sparingly substituted with LNA bases did not significantly inhibit gene expression, while LNA–DNA chimera XIV, which was 60% substituted with LNA bases, was a modest inhibitor. It is likely that XIV is a better inhibitor than XIII because it possesses a high _T_m value for binding (Table 2). These results suggest that LNAs and LNA–DNA chimeras that have a poor ability to activate RNase H are able to block gene expression when they are extensively or completely substituted with LNA bases and have a high affinity for their mRNA target sequences.
All of the other LNA–DNA chimeras were designed to contain at least six consecutive DNA bases and, according to the studies by Kurreck et al. (13), should be able to recruit RNase H. Each oligomer was designed to cover the AUG start codon and possessed a _T_m value between 53 and 68°C. Two LNA–DNA chimeras, XIX and XX, possessed _T_m values below 60°C and did not significantly inhibit luciferase activity. The other chimera with at least six consecutive DNA bases reduced expression of luciferase activity upon delivery into cells (Table 2). Dose–response data indicated that the LNA–DNA chimera containing nine consecutive DNA bases, XXIII, was a better inhibitor than LNA–DNA chimeras containing seven (XV) or eight (XXII) consecutive DNA bases (Fig. 4B).
We also tested LNA–DNA chimeras XXIV and XXV that were a different design. Rather than having LNA bases clustered at both termini to increase affinity and resistance to nuclease digestion, the LNA bases were placed at just one termini, with the other termini being made up of DNA and protected from digestion by two phosphorothioate linkages. This was done to allow a larger cluster of consecutive LNA bases within a 15 base oligomer to provide sufficient affinity, while the remaining DNA bases provided the capacity for efficient activation of RNase H. We found that LNA–DNA XXIV was a moderately effective inhibitor, while LNA–DNA XXV was not. These data suggest that LNA–DNA chimeras containing one domain of consecutive LNA bases and one domain of consecutive DNA bases can act as effective antisense agents but that the orientation of LNA and DNA bases relative to the mRNA target may be important.
Effect of LNA-containing oligomers on luciferase expression when targeted to the downstream coding regions
To test the efficacy of LNA-substituted oligomers directed within the coding region we tested LNA–DNA chimeras XXVI–XXXII (Table 3). Chimeras XXVIII–XXXII were complementary to three different target sequences and were designed to be able to recruit RNase H according to the rules developed by Erdmann and colleagues (13) with seven DNA bases flanked by four LNA bases. Each of these LNA–DNA chimeras was an effective antisense agent (Table 3 and Fig. 4C) with the exception of chimera XXXI that contained mismatches within the LNA regions and was not active. We also tested LNA–DNA chimeras XXVI and XXVII that contained not more than three consecutive DNA bases but were analogous to active LNA–DNA chimeras XXX and XXXII, respectively. Chimera XXVII was not an active inhibitor, but chimera XXVI did block luciferase activity significantly. Repeated efforts to confirm RNase H action by evaluating RNA levels were unsuccessful because of interference from the large amount of transfected plasmid DNA encoding luciferase and instability of the luciferase mRNA.
CONCLUSIONS
Our data show that LNAs and LNA–DNA chimeras can be versatile agents for inhibiting gene expression capable of blocking gene expression when targeted to several different sequences throughout the mRNA. Oligomers that target the terminus of the 5′-UTR are active, presumably because they are able to block binding of the ribosome to the transcript. Oligomers that target the translation start site or the coding region are also active, but potent inhibition requires that consecutive DNA bases capable of recruiting RNase H be included. Significantly, all of the sites targeted within the coding region were susceptible sites for inhibition by LNA–DNA chimeras, suggesting that identification of active LNA-containing antisense oligonucleotides for other mRNA targets may be straightforward. Our ability to potently inhibit gene expression by targeting sequences throughout luciferase mRNA indicates that LNAs and LNA–DNA chimeras are a promising option for further study.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at NAR Online.
[Supplementary Material]
Acknowledgments
ACKNOWLEDGEMENTS
We thank Proligo LLC and Cureon for supply of the LNA oligomers used in these studies and for assistance with their design. This work was supported by grants from the National Institutes of Health (GM 60642) and the Robert A. Welch Foundation (I-1244).
REFERENCES
- 1.Braasch D.A. and Corey,D.R. (2002) Novel antisense strategies for controlling gene expression. Biochemistry, 41, 4503–4510. [DOI] [PubMed] [Google Scholar]
- 2.Koshkin A.A., Singh,S.K., Nielsen,P., Rajwanshi,V.K., Kumar,R., Meldgaard,M., Olsen,C.E. and Wengel,J. (1998) LNA (locked nucleic acid): synthesis of the adenine, cytosine, guanine, 5-methyl cytosine, thymine, and uracil bicyclonucleoside monomers, oligomerisation and unprecedented nucleic acid recognition. Tetrahedron, 54, 3607–3630. [Google Scholar]
- 3.Obika S., Nanbu,D., Hari,Y., Andoh,J., Morio,K., Doi,T. and Imanishi,T. (1998) Stability and structural features of the duplexes containing nucleoside analogues with fixed N-type conformation. Tetrahedron Lett., 39, 5401–5404. [Google Scholar]
- 4.Wengel J. (1999) Synthesis of 3′-C- and 4′-C-branched oligodeoxynucleotides and the development of locked nucleic acid (LNA). Acc. Chem. Res., 32, 301–310. [Google Scholar]
- 5.Braasch D.A. and Corey,D.R. (2001) Locked nucleic acid (LNA): fine-tuning the recognition of DNA and RNA. Chem. Biol., 8, 1–7. [DOI] [PubMed] [Google Scholar]
- 6.Crinelli R., Bianchi,M., Gentilini,L. and Magnani,M. (2002) Design and characterization of decoy oligonucleotides containing locked nucleic acids. Nucleic Acids Res., 30, 2435–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wahlestedt C., Salmi,P., Good,L., Kela,J., Johnsson,T., Hokfelt,T., Broberger,C., Porreca,F., Lai,J., Ren,K. et al. (2000) Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl Acad. Sci. USA, 97, 5633–5638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Orum H., Jakobsen,M.H., Koch,T., Vuust,J. and Borre,M.B. (1999) Detection of the factor V Leiden mutation by direct allele-specific hybridization of PCR amplicons to photoimmobilized locked nucleic acids. Clin. Chem., 45, 1898–1905. [PubMed] [Google Scholar]
- 9.Torigoe H., Hari,Y., Seiguchi,M., Obika,S. and Imanishi,T. (2001) 2′-O,4′-C-methylene bridged nucleic acid modification promotes pyrimidine motif triplex formation at physiologic pH. J. Biol. Chem., 276, 2354–2360. [DOI] [PubMed] [Google Scholar]
- 10.Childs J.L., Disney,M.D. and Turner,D.H. (2002) Oligonucleotide directed misfolding of RNA inhibits Candida albicans group I intron splicing. Proc. Natl Acad. Sci. USA, 99, 11091–11096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arzumanov A., Walsh,A.P., Rajwanshi,V.K., Kumar,R., Wengel,J. and Gait,M.J. (2001) Inhibition of HIV-1 Tat-dependent trans activation by steric block chimeric 2′-O-methyl/LNA oligoribonucleotides. Biochemistry, 40, 14645–14654. [DOI] [PubMed] [Google Scholar]
- 12.Elayadi A.N., Braasch,D.A. and Corey,D.R. (2002) Implications of high-affinity hybridization by locked nucleic acid oligomers for inhibition of human telomerase. Biochemistry, 41, 9973–9981. [DOI] [PubMed] [Google Scholar]
- 13.Kurreck J., Wyszko,E., Gillen,C. and Erdmann,V.A. (2002) Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Res., 30, 1911–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nasevicius A. and Ekker,S.C. (2000) Effective targeted gene “knock down” in zebrafish. Nature Genet., 26, 216–220. [DOI] [PubMed] [Google Scholar]
- 15.Ekker S.C. (2000) Morphants: a new systematic vertebrate functional genomics approach. Yeast, 17, 302–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grillone L.R. and Lanz,R. (2001) Fomivirsen. Drugs Today, 37, 245–255. [DOI] [PubMed] [Google Scholar]
- 17.Elbashir S.M., Harboth,J., Lendeckel,W., Yalcin,A., Weber,K. and Tuschl,T. (2001) Duplexes of 21-nucelotide RNAs mediate RNA interference in cultured mammalian cells. Nature, 411, 494–498. [DOI] [PubMed] [Google Scholar]
- 18.Zamore P.D. (2001) RNA interference: listening to the sound of silence. Nature Struct. Biol., 8, 746–750. [DOI] [PubMed] [Google Scholar]
- 19.Mangelsdorf D.J., Umesono,K., Kliewer,S.A., Borgmeyer,U., Ong,E.S. and Evans,R.M. (1991) A direct repeat in the cellular retinol binding protein type II gene confers differential regulation by RXR and RAR. Cell, 66, 555–661. [DOI] [PubMed] [Google Scholar]
- 20.Doyle D.F., Braasch,D.A, Simmons,C.G., Janowski,B.A. and Corey,D.R. (2001) Inhibition of gene expression inside cells by peptide nucleic acids: effect of mRNA target sequence, mismatched bases and PNA length. Biochemistry, 40, 53–64. [DOI] [PubMed] [Google Scholar]
- 21.Romanelli A., Pedrone,C., Saviano,M., Bianchi,N., Borgatti,M., Mischiati,C. and Gambari,R. (2001) Molecular interactions between nuclear factor kB (NFkB) transcription factors and a PNA-DNA chimera mimicking NF-kB binding sites. Eur. J. Biochem., 268, 6066–6075. [DOI] [PubMed] [Google Scholar]
- 22.Belitsky J.M., Leslie,S.J., Arora,P.S., Beerman,T.A. and Dervan,P.B. (2002) Cellular uptake of N-methylpyrrole/N-methylimidazole polyamide-dye conjugates. Bioorg. Med. Chem., 10, 3313–3318. [DOI] [PubMed] [Google Scholar]
- 23.Koppelhus U., Awasthi,S.K., Zachar,V., Holst,H.U., Ebbesen,P. and Nielsen,P.E. (2002) Cell-dependent differential cellular uptake of PNA, peptides and PNA–peptide conjugates. Antisense Nucleic Acid Drug Dev., 12, 51–63. [DOI] [PubMed] [Google Scholar]
- 24.Flanagan W.M and Wagner,R.W. (1997) Potent and selective gene inhibition using antisense oligonucleotides. Mol. Cell. Biochem., 172, 213–225. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
[Supplementary Material]