Sphingosine 1-Phosphate, a Diffusible Calcium Influx Factor Mediating Store-operated Calcium Entry (original) (raw)
. Author manuscript; available in PMC: 2011 Nov 2.
Published in final edited form as: J Biol Chem. 2003 May 13;278(30):27540–27547. doi: 10.1074/jbc.M301763200
Abstract
Store-operated calcium entry (SOCE) is a fundamental mechanism of calcium signaling. The mechanisms linking store depletion to SOCE remain controversial, hypothetically involving both diffusible messengers and conformational coupling of stores to channels. Sphingosine 1-phosphate (S1P) is a bioactive sphingolipid that can signal via cell surface G-protein-coupled receptors, but S1P can also act as a second messenger, mobilizing calcium directly via unknown mechanisms. We show here that S1P opens calcium entry channels in human neutrophils (PMNs) and HL60 cells without prior store depletion, independent of G-proteins and of phospholipase C. S1P-mediated entry has the typical divalent cation permeability profile and inhibitor profile of SOCE in PMNs, is fully inhibited by 1 _μ_m Gd3+, and is independent of [Ca2+]i. Depletion of PMN calcium stores by thapsigargin induces S1P synthesis. Inhibition of S1P synthesis by dimethylsphingosine blocks thapsigargin-, ionomycin-, and platelet-activating factor-mediated SOCE despite normal store depletion. We propose that S1P is a “calcium influx factor,” linking calcium store depletion to downstream SOCE.
Store-operated calcium entry (SOCE)1 is the primary mechanism of regulated calcium entry into “non-excitable” cells like immunocytes. The mechanisms governing SOCE are controversial but have centered on two main hypotheses (1). In one model, emptying of endoplasmic reticulum (ER) calcium stores leads to the release of small diffusible messenger molecules that act on channels to increase Ca2+ entry (2). Randriamampita and Tsien (3) found strong suggestions of such a messenger molecule in extracts from Jurkat T-cells. They termed this molecule “calcium influx factor” (CIF).
Other hypotheses suggest that store depletion leads to calcium channel activation via direct physical interactions between cell membranes and intracellular structures. The “exocytosis model” (4, 5) of physical interactions suggests that vesicles bearing calcium channels fuse with the cell membrane after store depletion. The “conformational coupling” model suggests that ER calcium store depletion can lead to direct physical interactions between ER proteins such as the IP3 receptor and cell membrane calcium channels such as TRPC3 (6, 7). No specific molecular mechanisms have been proposed however, that might initiate such physical interactions. Moreover, data has continued to accumulate supporting the existence of an unidentified, low molecular weight CIF (8, 9).
Sphingosine 1-phosphate (S1P) is a universally bioactive, small (_M_r 380) sphingolipid metabolite. S1P is derived from the metabolism of sphingolipids and, most notably, from the actions of sphingosine kinase (10, 11) on sphingosine. In 1991, Zhang et al. (12) showed that S1P induces proliferation in Swiss 3T3 fibroblasts. Since that time, the actions of S1P have been studied in detail, and it has been found to be an important extracellular messenger molecule affecting cell growth, differentiation, adhesion, motility, and survival in many organisms and cell types (13, 14). The calcium-mobilizing effects of S1P are often mediated through G-protein coupled receptors (GPCR), and S1P is the natural ligand for at least five cell-surface GPCRs now known as S1PR1–5 (previously _Edg_1, -5, -3, -6, and -8) (15–18). S1P is also known to enter cells directly, however (19, 20), and in some systems S1P can regulate calcium homeostasis by acting as a second messenger (17, 21). The mechanisms by which S1P acts in this intracellular role have not yet been identified, although direct effects on ER calcium stores have been suggested (22). In yeast, however, intracellular S1P accumulation clearly mediates its biologic effects via calcium entry into the cell (23).
Polymorphonuclear leukocytes (PMN) are central effectors of the innate immune response to inflammation in humans. Calcium entry into the PMN typically occurs through store-operated mechanisms, and we have noted dysfunctional PMNs store-operated calcium entry (SOCE) after injury (24). Human PMN have been found to mobilize Ca2+ in response to extracellular stimuli via the activation of SphK (14), but yet it has also been shown that neutrophil-like HL-60 cells show a complete absence of specific S1P binding (21), suggesting an absence of functional S1PR. We therefore hypothesized that S1P might mobilize PMN calcium by acting as a second messenger to initiate SOCE.
Experimental Procedures
Sphingosine 1-Phosphate
S1P was purchased from Sigma (St. Louis, MO). This preparation of S1P is formulated to be soluble in methanol, which is the optimal vehicle for S1P (25). A 5 mm stock solution was prepared according to the manufacturer's recommendations. The concentration (1–4 _μ_l/ml) of methanol vehicle used has no effect calcium release or entry. Binding of S1P to plasma proteins has been reported to attenuate its biologic effects (26). We confirmed this in preliminary studies of S1P calcium mobilization in PMN. Therefore, we felt that BSA-free environments were optimal for evaluating the biologic effects of S1P, and avoided the use of BSA in S1P preparation.
Other Materials
Sphingosine, pertussis toxin, and other general chemicals were also purchased from Sigma. d-_erythro-N,N_-Dimethylsphingosine (DMS) was purchased from Biomol Research Laboratories Inc. (Plymouth Meeting, PA). d-_erythro_-[3H]Sphingosine (1.85 MBq) was purchased from PerkinElmer Life Sciences (Boston, MA). Thapsigargin and Fura-2AM were purchased from Molecular Probes (Eugene, OR). PAF, fMLP, ionomycin, and U73122 were purchased from Calbiochem (La Jolla, CA).
Neutrophil Preparations
Studies were performed in compliance with the Institutional Review Board of the University of Medicine and Dentistry of New Jersey (UMDNJ)-New Jersey Medical School. Informed consent was obtained for blood withdrawn from healthy human volunteers. A detailed description of our techniques for PMN isolation is available elsewhere (25). Briefly, neutrophils were isolated from heparinized whole blood using a one-step centrifugation procedure. The neutrophil layer was collected, osmolarity was restored, and cells were washed and suspended in HEPES buffer. The number and purity of PMN were checked by flow cytometry. Cells were loaded with 1 _μ_m Fura-2AM and kept on ice until for study. Aliquots of 2 × 106 cells were re-warmed just prior to each experiment.
HL-60 Cell Preparations
HL60-G cells, a subclone of the human promyeloblastic leukemia HL60 cell line, were the kind gift of Dr. George Studzinski (UMDNJ, Newark, NJ). Cells were cultured at 37 °C in RPMI 1640 with l-glutamine (Invitrogen) supplemented with 20% heat-inactivated, fetal bovine serum (Invitrogen). 1.3% Me2SO was added for 2–5 days to differentiate them into PMN-like cells. Differentiation was assured by assessment of CD11b using flow cytometry (27, 28).
Divalent Cation Measurements by Spectrofluorometry
Intracellular calcium ([Ca2+]i) was monitored by Fura fluorescence at 505 nm, using 340/380-nm dual wavelength excitation at 37 °C with constant stirring. Calibration was performed after each experiment using digitonin (_R_max) and EGTA (_R_min). [Ca2+]i was calculated from the 340/380-nm fluorescence ratio as per the methods of Grynkiewicz et al. (29). More detailed descriptions of our techniques are available elsewhere (30).
Measurement of Total PMN S1P Production by Thin Layer Chromatography (TLC)
PMN isolated as above (2 × 106 in 100 _μ_l for each reaction) were incubated in Hanks' balanced salt solution buffer with 1 mm calcium. Aliquots were incubated with DMS or vehicle as indicated for 10 min prior to reactions. At T = 0, 100 _μ_l of a mixture containing [3H]sphingosine (PerkinElmer Life Sciences, 70,000 cpm/reaction) with or without 1 _μ_m TG (final concentration, 500 nm) was added to warmed PMN to begin the reaction. Aliquots were allowed to incubate at 37 °C for 0, 2, 5, and 10 min. The reactions were stopped by the addition of 1 ml of ice-cold methanol:chloroform (2:1). The samples were then vortexed for 10 s and spun for 10 min at 2000 × g. Supernatants were collected, and a second 100-_μ_l mixture of methanol:chloroform (2:1) was applied, vortexed, and spun as before. The combined supernatants were dried, resuspended in 60 _μ_l of methanol, and applied to Silica Gel 60 TLC plates. Unlabeled sphingosine and sphingosine 1-phosphate were applied to control lanes to identify [3H]sphingosine and [3H]sphingosine 1-phosphate, respectively. Lipids were separated by TLC using a 1-butanol/acetic acid/water (3:1:1) mixture. Standard control lipids were visualized using ninhydrin (for S1P and SP) and molybdenum blue (for S1P). The radioactivity of the corresponding areas of the experimental lanes was measured by liquid scintillation counting. The mean ± S.E. of three observations at each time point is shown. More detailed methods can be found elsewhere (31).
Results
S1P Induces Calcium Influx Directly
S1P was applied to human PMN in the absence of external calcium (0.3 mm EGTA added) in order to study calcium store release and influx (Fig. 1). After equilibration, cells were treated with S1P or vehicle (methanol). PMN show no store release transient at all in response to S1P. In contrast, dose-dependent calcium influx is seen on re-addition of external 1 mm Ca2+. Calcium influx is immediate under these conditions, rises to a typical plateau, and subsequently decays. In other experiments no store release transient was seen in Ca2+-free media out to 900 s, although brisk store release responses to the GPCR agonists formyl-Met-Leu-Phe (fMLP) and platelet-activating factor (PAF) continued to be present during this time (data not shown).
Fig. 1. Sphingosine 1-phosphate induces Ca2+ influx in human PMN.
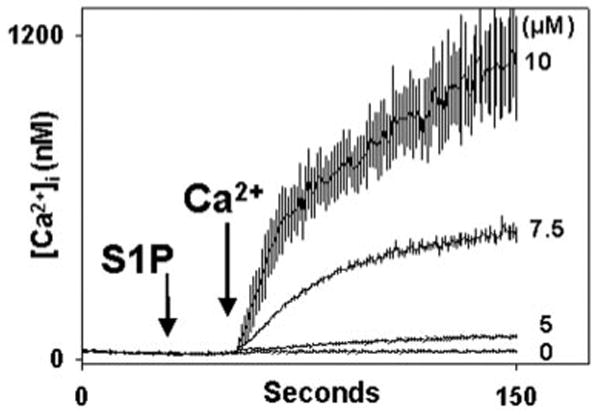
PMN were Fura-loaded in HEPES buffer with 1 mm Ca2+ and 0.1% BSA. Immediately prior to study, PMNs were spun for 5 s at 4500 rpm, washed once with BSA-free/Ca2+ free buffer, spun again, and resuspended in BSA-free, nominally Ca2+-free (0.3 mm EGTA) buffer for study. S1P was added at t = 30 s in the concentrations shown. For the zero S1P control, 3 _μ_l of vehicle (methanol) was added. 1 mm Ca2+ was added to the medium at t = 50 s. Dose-dependent calcium entry is immediately noted. The morphology of calcium entry is typical of SOCE in the PMN. Traces represent the mean ± S.E. of 3–4 experiments at each S1P dose.
S1P Induction of Calcium Influx Is Independent of ER Store Depletion
Although no store release transient was seen in response to S1P in the experiments presented in Fig. 1, we evaluated the possibility that undetected store release might have occurred. Fura-loaded PMN samples in calcium-free media were treated with 10_μ_m S1P or methanol vehicle. The cells were stimulated 30 s later with fMLP to assess the status of IP3-releasable ER calcium stores (Fig. 2). fMLP-induced store release was morphologically identical with and without prior S1P, and no statistically significant evidence of S1P-initiated store depletion could be found at this time point. Moreover, the degree of SOCE seen (on Ca2+ re-addition at t = 300, Fig. 2) was clearly a reflection of the presence of S1P rather than prior store depletion by fMLP. In longer experiments (data not shown), a slow but significant depletion of Ca2+ stores was noted after S1P exposure, but this was limited to a 50% fall in fMLP-releasable stores in comparison to vehicle-treated cells after 15 min. These findings therefore confirm that calcium entry in response to S1P occurs in the absence of significant ER calcium store depletion.
Fig. 2. S1P does not induce emptying of PMN calcium stores.
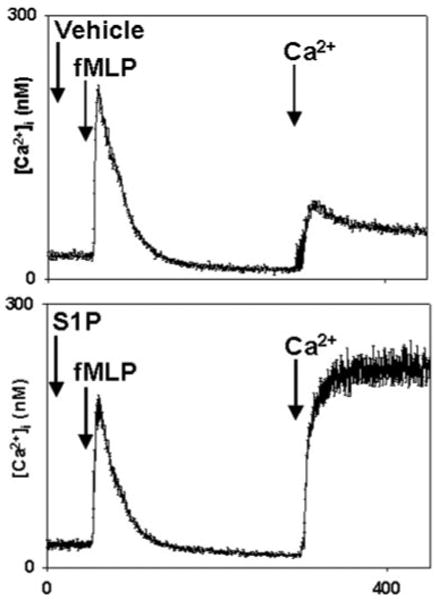
PMN were studied in BSA-free, nominally calcium-free conditions. Cells were treated with 10 _μ_m S1P or methanol at 30 s. The cells were then stimulated with 10 nm fMLP (EC90) at 60 s. 1 mm Ca2+ was added to the medium at t = 300 s. The store release transients seen on fMLP treatment were indistinguishable. After S1P treatment, however, SOCE was magnified severalfold. The traces shown are the mean [Ca2+]i ± S.E. from four separate experiments.
S1P Induction of Calcium Influx Is Independent of G-proteins
Prior studies have shown that PMN-like HL60 cells show no specific binding of S1P (21) and that endogenous S1P production in PMN activates the cells at S1P concentrations far lower than those required for signaling through any of the known S1PR (22). These findings, plus the lack of GPC store depletion by S1P noted in Fig. 2, make it very unlikely that S1P acts via GPC, cell-surface S1PR in human PMN. Nonetheless, we studied the possibility that S1P-mediated Ca2+ influx might depend on prior store release by cell surface G-protein-coupled S1PR.
S1PR1–5 can all signal through the pertussis toxin (PTX)-sensitive G_α_i (15, 18). Activation of G_α_i in the PMN activates phospholipase C_β_ (PLC) and causes IP3-dependent release of cell calcium stores within seconds. To further exclude the possibility that PMN responses to S1P were initiated via G_α_i-coupled S1PR, we treated PMN with pertussis toxin (PTX, 1 _μ_g/ml, for 3 h at 37 °C) prior to stimulation with S1P. G_α_i-coupled calcium store release in the neutrophil is fully inhibited at this PTX dose (32–34). We found that, in the neutrophil, PTX had no effect upon S1P-induced calcium entry (Fig. 3A), whereas [Ca2+]i responses to fMLP were uniformly abolished (Fig. 3B). Thus PMN calcium influx in response to S1P was independent of PTX-sensitive G_α_i-linked S1PR.
Fig. 3. Calcium mobilization by S1P is pertussis toxin (PTX)-insensitive.
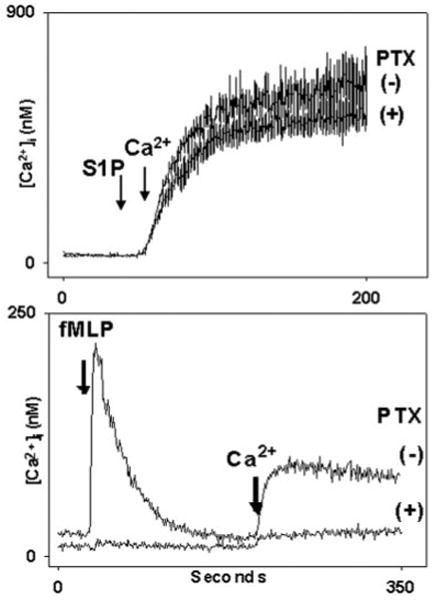
PMN were incubated with PTX (1 _μ_g/ml) or vehicle for 3 h at 37 °C. In the upper panel, PMN were suspended in nominal Ca2+, BSA-free buffer. At 30 s cells were treated with 5 _μ_m S1P. 1 mm Ca2+ was added to the medium at t = 50 s. Calcium entry in response to S1P was indistinguishable in the presence and absence of PTX. Note, again, that S1P elicits no measurable store emptying in this time frame (Fig. 2). In the lower panel, cells incubated identically with PTX were exposed to 10 nm fMLP at 30 s. PTX completely blocked the Ca2+ store release response to fMLP. The subsequent calcium entry response was limited to the level of the “leak.” (i.e. that Ca2+ entry always seen on re-calcification of the medium irrespective of prior stimulation or store depletion. PMN [Ca2+]i changes due to leak can vary from 20 to 40 nm in PMN using our methods.) PMN incubated 3 h without PTX responded normally to fMLP (lower panel, upper trace).
In addition, however, the S1P2 and SIP3 receptors are also known to bind to Gq/11 (18). The Gq/11 pathway is not PTX-inhibited, but like G_α_i, Gq/11 acts through phospholipase C_β_ (PLC) and IP3 to release cell calcium stores (15, 18). Thus S1P acting on S1P2 or SIP3 could potentially deplete PMN calcium stores in a PTX-insensitive manner but in a PLC-dependent fashion. We therefore incubated PMN with or without the PLC inhibitor U73122 (20 _μ_m, 5 min at 37 °C) prior to stimulation with S1P. This agent inhibits calcium store depletion by both G_α_i and Gq. U73122 had no effect whatsoever on calcium entry in response to S1P (Fig. 4), whereas, again, [Ca2+]i mobilization in response to fMLP was fully blocked. These experiments demonstrate that PMN calcium entry after S1P is independent of G_α_i, Gq, the PLC/IP3 pathway, and the depletion of cell Ca2+ stores.
Fig. 4. Calcium influx caused by S1P is insensitive to PLC inhibition.
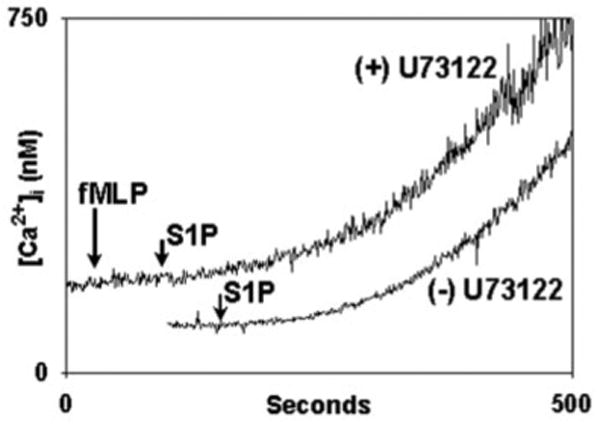
PMN were treated with 20 _μ_m U73122 (upper trace) for 15 min in the cuvette in the presence of 1 mm calcium and 0.1% fatty-acid free BSA. Recordings were begun at t = 0. At 30 s 10 nm fMLP was applied and no [Ca2+]i flux was detected. At 100 s 20 _μ_m S1P was applied. S1P caused calcium influx. In the lower trace (displaced for clarity) PMN were exposed to S1P at the time marked without prior exposure to U73122. [Ca2+]i flux is indistinguishable in the presence and absence of U73122. Representative traces are shown, three to four experiments were done per condition. No S1P-mediated [Ca2+]i flux is seen in the absence of external Ca2+. S1P is bound strongly by BSA, and this slow pattern of calcium influx is typical of PMN responses to S1P in BSA-containing medium. The BSA was required to prevent cell clumping after adding the U73122.
S1P-stimulated Ca2+ Entry Is Independent of G-protein Expression in HL60 Cells
To further exclude the possibility that G-protein-coupled, cell-surface S1PR might play a role in PMN Ca2+ entry response to S1P, we evaluated the PMN-like, HL60-G cell line. We found that in their basal (undifferentiated) state, these HL60 cells showed no Ca2+ store release at all in response to fMLP (Fig. 5A, lower trace) or any other typical PMN GPCR agonist (complement fragment 5a, interleukin-8, GRO-α, and leukotriene B4, data not shown). After treatment with 1.3% Me2SO, however, the cells differentiate into a PMN-like (CD11b+, data not shown) phenotype. We found that such “HL60-PMN” cells showed brisk Ca2+ store release responses to fMLP (Fig. 5A, upper trace) as well as all the other PMN agonists noted above. S1P-induced Ca2+ store release remained totally absent despite this generalized increase in the expression of functional GPCR (Fig. 5B). Conversely, S1P induced very similar amounts of calcium entry in HL60-PMN and in HL60 cells (Fig. 5B). These findings support the lack of involvement of GPCR in S1P-mediated PMN calcium mobilization.
Fig. 5. Responses of HL-60 cells to S1P.
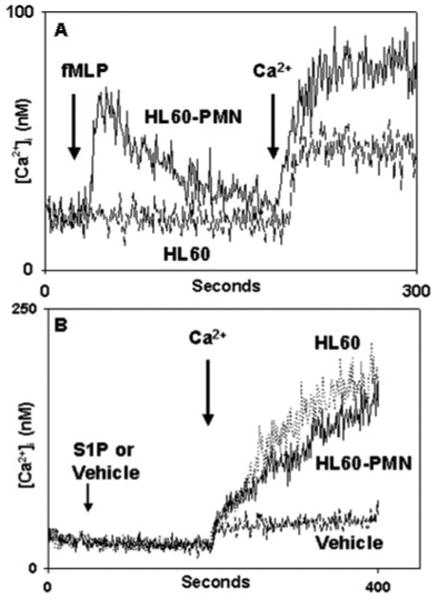
Undifferentiated HL60 or differentiated HL60-PMN cells (see “Experimental Procedures”) were exposed to 10 nm fMLP (A) or to S1P (B). Undifferentiated cells showed no response to fMLP and a typical small Ca2+ “leak” upon re-addition of external Ca2+. HL60-PMNs show a morphologically normal Ca2+ store release transient after fMLP stimulation, followed by typical SOCE. Similar induction of Ca2+ store release was seen in response to multiple GPCR agonists (see text). In B, neither HL60 nor HL60-PMN showed any Ca2+ store release response to S1P, but brisk Ca2+ entry was seen upon re-addition of external Ca2+. Only the Ca2+ “leak” was seen in the absence of S1P, which was very similar (note the change in scale) in HL60 (A) and HL60-PMN (B). These findings demonstrate that S1P-mediated Ca2+ entry is independent of the overall degree of GPCR expression in PMN-like cells.
Ionic Selectivity of SIP-induced Ca2+ Entry
Having determined that PMN calcium entry responses to S1P were independent of store depletion and of GPCR, we compared the known attributes of PMN SOCE with S1P-mediated Ca2+ entry. We have previously reported that total SOCE in human PMN reflects the combined contributions of Ca2+-specific and Sr2+-permeable pathways (30). We therefore exposed PMN to 10 _μ_m S1P and serially added 1 mm Sr2+ and 1 mm Ca2+ to the medium (Fig. 6). We detected both Sr2+ influx and Ca2+ influx on serial cation addition. As with TG, ionomycin, fMLP, and PAF stimulation in our prior studies, we found that, after S1P stimulation, additional calcium entry through Ca2+-specific pathways always persisted after Sr2+ entry had reached equilibrium. Thus S1P activates influx through the specific and nonspecific PMN Ca2+ entry pathways and acts upon them in a fashion consistent with that seen previously with traditional agonists eliciting SOCE in the PMN.
Fig. 6. S1P elicits both Sr2+ and Ca2+ entry in PMN.
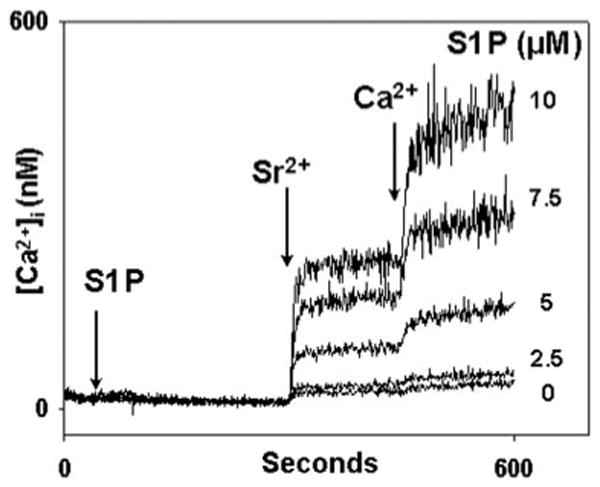
After Fura-2AM loading, PMN were spun and washed in buffer without BSA or Ca2+. Cells were spun and resuspended in cuvettes in BSA/Ca2+-free media. S1P was applied at 30 s. Sr2+ and Ca2+ were added at 300 and 450 s, respectively. S1P causes dose-dependent entry of each cation in relative proportions similar to those seen after TG, ionomycin, and PAF stimulation in our prior studies (24). Representative traces are shown, n = 3–4 per condition.
Sensitivity of SIP-induced Ca2+ Entry to Inorganic Channel Blockers
To ensure that the influx of Ca2+ seen after S1P stimulation was due to entry through channel mechanisms, we studied the effects of the inorganic calcium channel blockers lanthanum (La3+), nickel (Ni2+), zinc (Zn2+), and gadolinium (Gd3+) on S1P-induced influx. PMN were treated with 1 mm La3 +, Ni2+, or Zn2+ prior to S1P stimulation (Fig. 7). La3+ and Zn2+ abolished all PMN calcium influx in response to S1P, but Ni2+ was ineffective. This pattern of sensitivity is identical to that of PMN responses to GPC agonists and thapsigargin we described in prior studies (30).
Fig. 7. Inhibition of S1P-induced calcium entry by inorganic channel blockers.
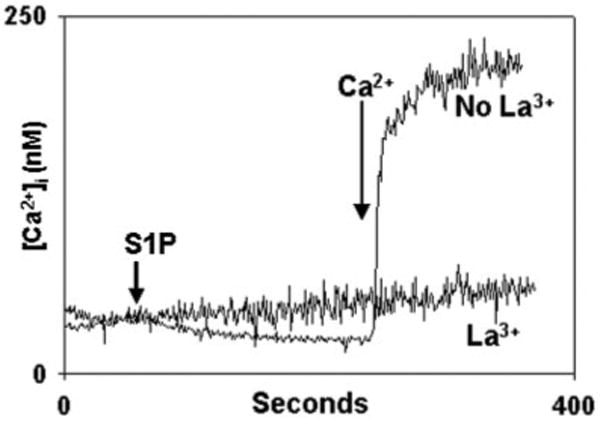
PMN were stimulated with S1P in the presence of the heavy metal and lanthanide channel-blockers La3+, Ni2+, and Zn2+ (all 1 mm), and the media were re-calcified at the times shown. La3+ abolishes Ca2+ entry. Experiments performed in the presence of Zn2+ show traces overlapping those done in the presence of La3+ (data not shown). In the presence of Ni2+, [Ca2+]i traces were indistinguishable from those seen in the absence of any blockers (data not shown). Representative traces are shown, n = 3–4 per condition.
Gadolinium (Gd3+) is another lanthanide inhibitor of calcium entry channels, but at sub-micromolar concentrations Gd3+ is thought to be specific for the store-operated and mechanically sensitive calcium channels that are likely to be composed of TRP proteins (1, 30, 35–38). We therefore performed more detailed studies of PMN calcium entry responses to S1P in the presence of Gd3+ (Fig. 8). S1P-initiated calcium influx was inhibited by nanomolar concentrations of Gd3+ with an EC50 of ∼250 nm and complete inhibition at 1 _μ_m. Again, this pattern was very similar to that we have previously reported for store-operated calcium entry channels in the PMN (30).
Fig. 8. Gadolinium (Gd3+) inhibits S1P-induced calcium entry.
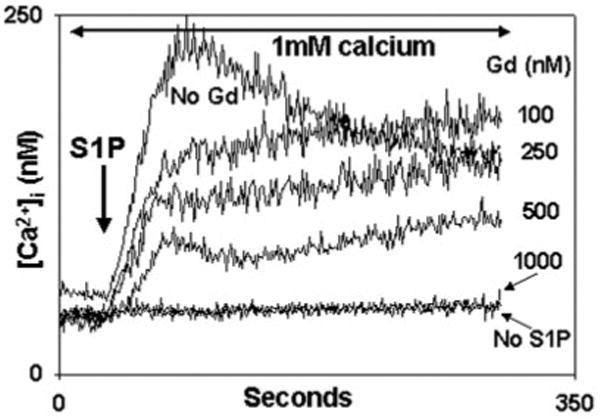
PMN were treated with the concentrations of Gd3+ shown for 1 min in the presence of calcium prior to study. 10 _μ_m S1P was added at t = 30 s. Gd3+ caused dose-dependent suppression of S1P-induced Ca2+ entry. Complete blockade of Ca2+ entry was seen at ∼1 _μ_m. Representative traces are shown, n = 3 for each concentration.
Direct Depletion of ER Calcium Stores Activates S1P Synthesis
The stimulation of S1P production from sphingosine by GPC agonists is well known (39, 40), but if S1P links store depletion to SOCE, then direct store depletion should elicit S1P synthesis. This has not been examined. We therefore studied PMN synthesis of [3H]S1P from [3H]sphingosine after thapsigargin (TG) stimulation using thin-layer chromatography (31). TG depletes ER stores directly by inhibition of the ER calcium ATPase. We found that TG initiated rapid incorporated of [3H]sphingosine into [3H]S1P (Fig. 9). The kinetics of [3H]S1P formation seen after TG store depletion in PMN were very similar to those seen in HL-60 cells stimulated by formyl peptide (39), with rapid synthesis noted over the first 2 min and subsequent formation of a plateau lasting more than 10 min. Moreover, the conversion of [3H]sphingosine to [3H]S1P in response to TG was completely inhibited by the SphK inhibitor DMS (13, 14).
Fig. 9. Direct store depletion causes PMN S1P synthesis.
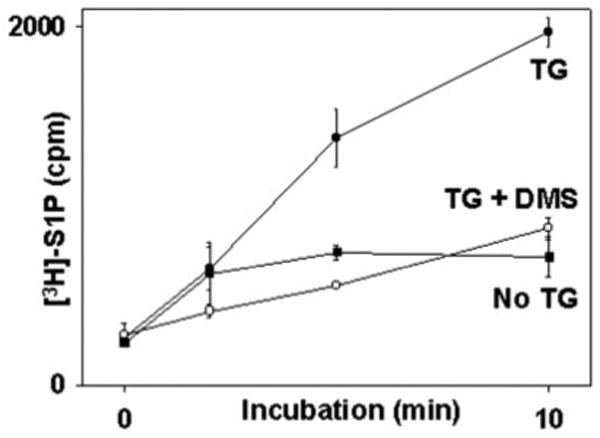
PMN (2 × 106) were incubated with DMS or vehicle as indicated for 10 min. Reactions were started (t = 0) by adding [3H]sphingosine with or without TG (see text). The reactions were stopped at 0, 2, 5, and 10 min. Total [3H]S1P was collected and assayed by TLC. The mean ± S.E. of three observations at each time point is shown. TG initiated rapid synthesis of S1P. No significant S1P synthesis was seen in the absence of TG. DMS abolished S1P production in TG-treated cells to the level of unstimulated cells.
Inhibition of S1P Synthesis Uncouples Store Depletion from SOCE
A wide variety of external stimuli can cause cells to metabolize sphingosine to S1P via the actions of SphK (reviewed in Olivera and Spiegel (41)). This conversion is inhibited by DMS (13, 14). If direct store depletion initiates S1P synthesis and thus SOCE, then inhibition of S1P synthesis by DMS should inhibit SOCE resulting from direct store depletion. We therefore assessed whether SOCE initiated by TG (500 nm) or ionomycin (100 nm) could be inhibited by DMS. Store depletion by TG (Fig. 10A) as well as ionomycin (Fig. 10B) proceeded normally in calcium-free media in the presence of DMS. Upon re-addition of calcium to the medium, however, we saw dose-dependent inhibition of SOCE by DMS with 100% inhibition at about 15 _μ_m. These data clearly demonstrate that blockade of SphK and the resultant loss of cellular S1P synthesis abolishes the link between ER calcium store depletion and calcium entry into the PMN.
Fig. 10. DMS inhibits PMN SOCE initiated by direct store depletion.
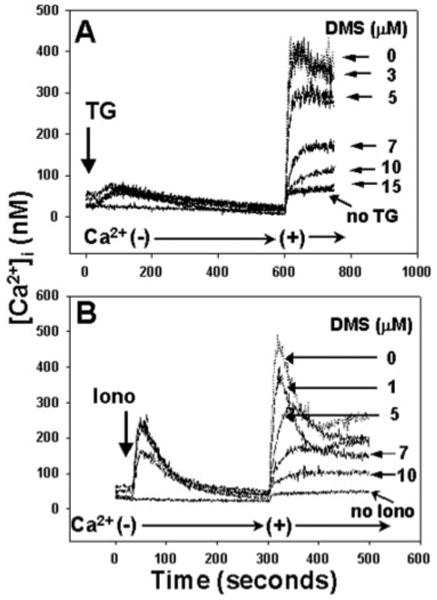
PMN were incubated for 1 min with increasing doses of DMS in calcium-free media. In A, 500 nm TG was added at 30 s. 1 mm Ca2+ was added after the completion of store release. In B, 100 nm ionomycin (Iono) was added at 30 s. Again, 1 mm calcium was added after the completion of store release. In each case, store release was unaffected by DMS, but SOCE was suppressed in a dose-dependent manner. In the presence of 15 _μ_m DMS, calcium entry was suppressed to the level of the “leak” current. Representative traces are shown, n = 3–4 for each concentration of DMS.
We also examined this issue in HL60 cells (Fig. 11) and found that TG-initiated Ca2+ store release was unaffected by DMS and completely independent of the state of cell differentiation. Conversely, DMS inhibited TG-initiated SOCE completely. The SOCE response to TG, which, unlike responses to cell-surface agonists, is GPCR-independent, was noted to be markedly increased by differentiation of the cells.
Fig. 11. DMS inhibits HL60 SOCE initiated by direct store depletion.
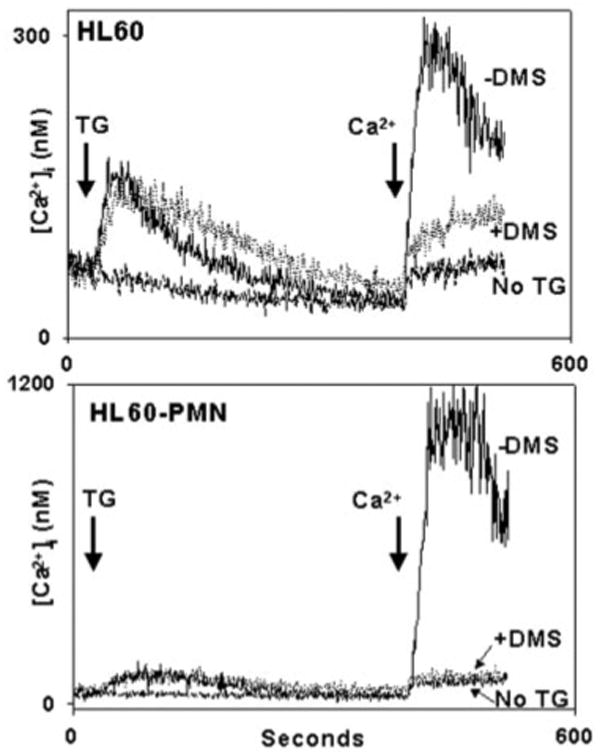
HL60 (upperpanel) and HL60-PMN (lowerpanel) cells were studied in a like manner to the PMN studied in Fig. 10. Quantitative Ca2+ store release was unaffected by the differentiation of the cells (note the difference in scale) or by the presence of DMS. Again though, 10 _μ_m DMS was found to inhibit TG-initiated SOCE to the level of “leak.” SOCE response to TG (which is GPCR-independent) was markedly increased by differentiation of the cells.
DMS-inhibitable SOCE Is [Ca2+]i-independent
TG induces both S1P synthesis and DMS-inhibitable calcium entry. To determine whether such DMS-inhibitable calcium entry truly represents SOCE, it is important to know whether it occurs in a manner that is independent of elevations in [Ca2+]i. We therefore studied the effects of DMS on store depletion-dependent entry of manganese (Mn2+) into 1,2-bis(2-aminophenoxy) ethane-_N, N, N′, N′_-tetraacetic acid (BAPTA)-loaded PMN (Fig. 12). Under these conditions, ER stores are depleted without elevations of [Ca2+]i, and store-operated divalent cation entry can be assessed as the quenching of Fura by Mn2+ entry (24). As expected, in the absence of prior store depletion by TG (A), PMN showed no sign of Mn2+ entry. Cells that have undergone prior store depletion by TG, however (B), allow rapid entry of Mn2+ from the media. SuchMn2+ entry was blocked by prior PMN incubation with DMS (C). Thus DMS inhibits store-operated influx of divalent cations in a [Ca2+]_i_-independent fashion.
Fig. 12. DMS inhibits SOCE in a calcium-independent manner.
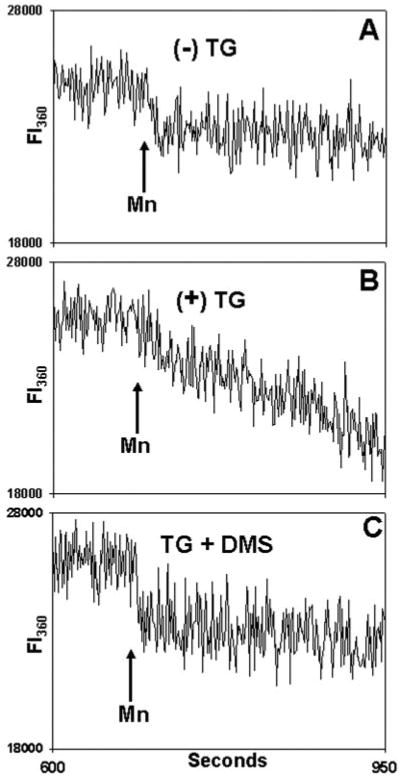
Fura-loaded PMN were incubated in 20 _μ_m BAPTA for 9 min at room temperature. A, cells were exposed to vehicle only (Me2SO) at 30 s. Samples were then observed in the cuvette for 10 min. At that time, a mixture of 500 _μ_m Mn2+ and 1.5 mm Ca2+ was added where indicated to quench Fura. In B, the cells were exposed to TG (500 nm) at 30 s. Again, 500 _μ_m Mn2+ and 1.5 mm Ca2+ were added at the time indicated. In C, cells were pretreated for 1 min with 10 _μ_m DMS and then exposed to TG (500 nm) at 30 s. Again, after allowing 10 min for store depletion, Mn2+ and Ca2+ were added. Fluorescent intensity (FI) was monitored at both 340/380 nm (not shown) and at 360 nm, as shown (FI360). No 340/380 nm [Ca2+]i release transient was seen at any time (data not shown). On Mn2+ addition, a small initial drop in total fluorescence at 360 nm is noted due to quenching of Fura in the media, but no Mn2+ entry into the cells was detected in the absence of prior store depletion (A). In contrast, prior TG treatment (B) caused rapid Mn2+ entry. TG-dependent Mn2+ entry was blocked by prior treatment with DMS (C). Linear curve-fitting (SigmaPlot) was then performed on the FI360 after Mn2+ addition (n = 3–4 independent experiments per condition). This revealed a mean slope of −2.3 ± 0.6 units/s after treatment with vehicle only. The FI360 slope decreased to −14.9 ± 0.9 units/s in TG-treated cells but was returned to unstimulated levels (−3.1 ± 0.4 units/s) by DMS pretreatment (p < 0.01, analysis of variance/Tukey's test). The data show that DMS inhibits the calcium entry response to TG store depletion in a [Ca2+]i -independent fashion.
Inhibition of S1P Synthesis Uncouples GPC Receptors from SOCE
Stimulation of PMN by GPC agonists is known to cause S1P synthesis (21) as well as store depletion. As seen in Figs. 10 and 11, blockade of S1P synthesis by DMS uncouples pharmacologic store depletion from SOCE. We therefore evaluated the dependence of SOCE after physiologic GPC store depletion upon S1P synthesis. To accomplish this, we stimulated PMN with 100 nm PAF in the presence and absence of DMS. DMS had no discernable effect on store depletion by PAF (Fig. 13, A–C). When calcium was added to the media, brisk normal SOCE was seen in the absence of DMS (Fig. 13A). After PMN pretreatment with 10 _μ_m DMS, however, the SOCE response to PAF was abolished (Fig. 13B). Finally (Fig. 13C), when DMS was added after PAF-induced store depletion only minimal inhibition of SOCE was seen. Thus inhibition of PAF-induced SOCE by DMS requires the presence of the inhibitor at the time of store depletion and SphK activation. These findings confirm that the activation of SOCE by physiologic GPC store depletion depend on S1P synthesis by SphK. These experiments also address the issue of whether S1P receptors linked to Gq (i.e. S1PR2 and S1PR3) might activate cation channels directly in the PMN, as has been suggested in some cell types (42, 43). PAF receptors (PAFrs) do indeed activate Gq/11 (44), but the inhibition of PAF-mediated calcium entry by DMS (Fig. 13B) suggests that PAFr/Gq-dependent calcium entry requires S1P synthesis. Moreover, the data in Fig. 9C show that calcium entry after PAFr/Gq activation requires SphK activity subsequent to ER store depletion.
Fig. 13. DMS inhibits G-protein-coupled receptor-initiated SOCE.
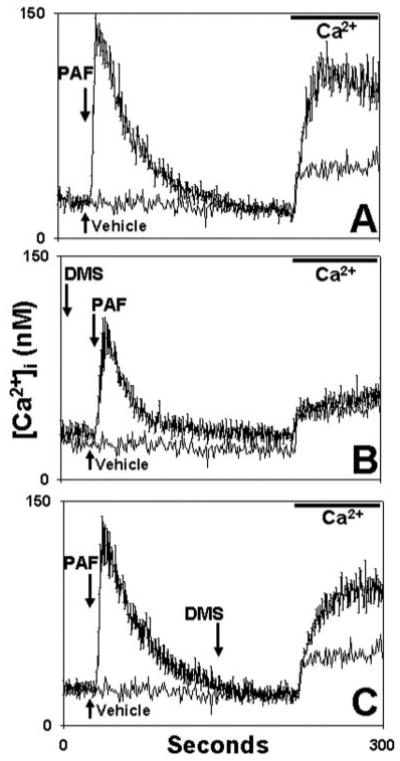
PMN were exposed to 100 nm PAF at t = 30 s in calcium-free medium. In A (control) no DMS was used. The DMS vehicle (Me2SO) was applied at 150 s. In B, DMS was applied 100 s prior to PAF. In C, DMS was added at 150 s, allowing >1 min for full DMS effect. All media were re-calcified at t = 220 s to measure SOCE. The traces represent the mean [Ca2+]i ± S.E. of three experiments. Each experimental trace is shown compared with an identical “blank” control trace where only PAF vehicle (3 _μ_l of ethanol) was used at t = 30. This “blank” trace demonstrates the nonspecific calcium “leak.” DMS given prior to store depletion (B) completely eliminated PAF-initiated SOCE. DMS given after store depletion (C) caused only minimal inhibition.
DMS Does Not Inhibit Calcium Entry
Prior studies observing inhibition of calcium flux by DMS have proposed that it might inhibit calcium entry channels directly (45). This is unlikely in the light of the findings presented in Fig. 13. If DMS were to act by direct inhibition of calcium channels, its inhibition of Ca2+ entry should be independent of whether it was given before or after store depletion, as long as it was present at the time of calcium entry. Such was clearly not the case. Conversely, if DMS acts by inhibiting S1P synthesis at the time of store depletion, its effect should be diminished if given after store depletion and SphK activation have begun, and such was indeed the case (Fig. 13, B versus C). Moreover, as seen in Fig. 9, DMS clearly inhibits the synthesis of S1P stimulated by TG.
To further discount any possibility that inhibition of calcium entry by DMS could reflect channel blockade, we evaluated the effects of DMS on calcium entry initiated directly by exogenous S1P (Fig. 14). If DMS were to inhibit Ca2+ entry through SOCE channels, it should act like La3+ or Gd3+ (Fig. 5) and block the calcium entry responses elicited by S1P. In fact, we found that DMS caused no inhibition of S1P-initiated Ca2+ or Sr2+ entry at any concentration. Rather, DMS enhanced cation entry responses to S1P in a dose-dependent fashion. Because DMS increases rather than decreases S1P-induced increases in [Ca2+]i, it clearly cannot act by blocking S1P-induced cation entry. Nonetheless, the synergistic ability of exogenous DMS and S1P to increase [Ca2+]i is unexplained, and several possible mechanisms might be advanced. DMS might interfere with S1P degradation as well as its synthesis. Or, exogenous DMS and S1P might interact to produce cell-membrane changes allowing cationic influx. This latter explanation seems less likely, because the influx seen was dose-responsive and formed typical “plateaus” at all concentrations, this suggesting regulated rather than pathologic cation entry. Last, entry of Sr2+ and Ca2+ always occurred in the same relative proportions as those seen when normal PMN are stimulated with thapsigargin (30). Importantly, none of these events can underlie the actions of DMS other conditions. Where used to inhibit endogenous S1P formation, DMS uniformly acts to decrease calcium entry, even at high concentrations (Fig. 10A).
Fig. 14. DMS does not block calcium influx.
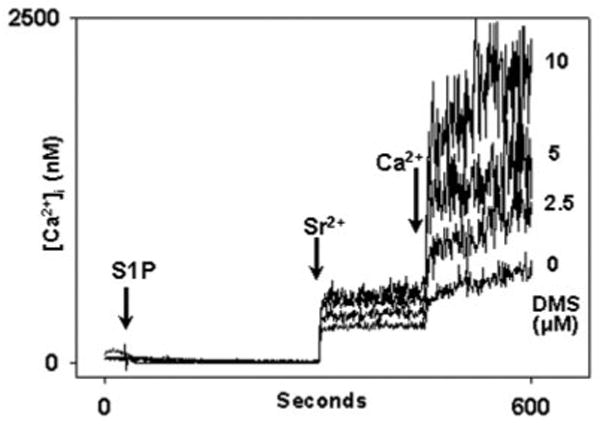
In this experiment PMN were incubated with DMS at the concentrations shown or with vehicle (3 _μ_l of Me2SO) for 1 min prior to recordings in Ca2+-free, BSA-free media. At t = 30 s, 10 _μ_m S1P was applied. Sr2+ (1 mm) was added at 300 s, and 1 mm Ca2+ was added at 450 s. DMS caused dose-dependent enhancement of the entry of each cation. Control experiments using DMS alone demonstrated no calcium entry above the normal “leak” seen on cation re-addition (data not shown). The data show that DMS does not inhibit Ca2+ entry into PMN. Rather, it suggests that a previously undescribed synergism may exist between the cellular effects of exogenous S1P and DMS. These effects remain to be studied. Representative traces are shown, n = 3 independent experiments.
Discussion
In 1993, Randriamampita and Tsien (3) described a soluble messenger molecule in cellular extracts that could elicit SOCE, which they termed calcium influx factor (CIF). In partially characterizing this molecule, they suggested it was released from organelles both into the cytoplasm and into the extracellular medium. They also suggested CIF would be a phosphorylated molecule with a molecular mass less than 500 Da, bearing hydroxyl and amino groups on adjacent carbons. Although other mechanisms of inducing SOCE have been proposed, the possibility that SOCE, at least in some forms, might require the action of a CIF has never been discounted. The identity of that CIF, however, has remained unknown.
S1P is an important extracellular signaling molecule and growth factor that conforms to all the characteristics initially suggested for CIF (3). S1P is found in yeast, plant, insect, and mammalian cells. It is synthesized rapidly from sphingosine by SphK in response to a wide variety of extracellular signals. Once synthesized, S1P can either pass outward through plasma membranes to act upon the many cell surface GPC receptors of the Edg/S1PR class, or it can act as an intracellular second messenger to mobilize cell calcium (46, 47). S1P may also be an intermediary in the activation of [Ca2+]i by other second messengers, such as lysophosphatidic acid (48).
The multiple routes by which S1P production is linked to calcium mobilization have complicated analyses of its mechanisms of action in other cell types. Because PMN had been shown to mobilize Ca2+ through an SphK-mediated pathway (14) we hypothesized S1P might be an important PMN activator. Our initial experiments showed that S1P did indeed mobilize Ca2+ in PMN, but we saw no trace of the transient store release spike typically expected when stimulating neutrophil GPCR. We therefore hypothesized that S1P might act primarily as a second messenger in PMN by eliciting SOCE.
We have shown here that direct exposure of normal human PMN to S1P activates calcium influx pathways with a wide variety of characteristics typical of PMN SOCE. Exogenous S1P-mediated Ca2+ entry occurs rapidly without measurable depletion of calcium stores and is independent of Edg/S1PR signaling through G_α_i, Gq, or phospholipase C. Rapid activation of Ca2+ entry by exogenous S1P appears to occur over a dose range of approximately one order magnitude of extracellular S1P concentrations that may be attainable in normal plasma and are likely to be present in disease states (17, 26). S1P was fully sufficient to initiate SOCE de novo in PMN, and its synthesis was necessary for SOCE under all conditions tested. In vivo, S1P acts downstream from the afferent, store depletion arm of physiologic SOCE. The amplitude and morphology Ca2+ entry in PMN treated with S1P is similar to that seen in PMN treated with TG or ionomycin (30) and indistinguishable from physiologic PMN SOCE responses to PAF (24).
We also studied the linkage between endogenous S1P production and activation of PMN SOCE. Isolated store depletion by TG was shown to induce the rapid and DMS-inhibitable formation of S1P from sphingosine; S1P synthesis occurred over 1–2 min with the subsequent formation of a plateau of S1P concentration, and TG-initiated, DMS-inhibitable SOCE was shown to be [Ca2+]i -independent. We found that DMS had no effect at all upon the degree of direct Ca2+ store depletion by TG or ionomycin or upon G-protein-coupled store depletion by PAF. Yet DMS blocked Ca2+ entry responses to TG, ionomycin, and PAF quantitatively and in a dose-dependent fashion. This inhibition seen was not due to channel blockade, because it was dependent upon the inhibitor being present at the time of store depletion rather than at the time of calcium entry. Moreover, DMS enhances Ca2+ entry responses to exogenous S1P. Rather, the inhibitory effects of DMS were all compatible with its observed inhibition of store depletion-initiated S1P synthesis. These findings all demonstrate a clear, direct linkage between ER store depletion and the activation of SphK and SOCE.
Thus, in summary, the synthesis of S1P from sphingosine by SphK is a necessary step, downstream from store depletion either by direct or G-protein-coupled mechanisms, and upstream from channel activation and calcium entry. In effect, S1P synthesis occurs in direct response to store depletion and is both necessary and sufficient for the initiation of store-operated calcium entry in the PMN.
The characteristics of PMN calcium entry seen after stimulation by S1P are also clearly those of SOCE. The pronounced sensitivity to Gd3+ inhibition and the ionic selectivity of the conductance was typical of cation entry through SOCE channels in the PMN (30). These findings are also compatible with those of Su et al. (49), who noted that lymphoid cells have two SOCE channels, one of which is also nonselective and may require diacylglycerol production as well as store depletion for activation. Thus multiple linkage mechanisms must clearly exist to connect store depletion to total “stimulated calcium entry” (7).
The mechanisms by which S1P activates SOCE channels remain unknown. Despite being a diffusible messenger molecule with the characteristics of a “CIF,” S1P may only activate some store depletion-stimulated calcium entry mechanisms. Other similar messengers may exist. Moreover, S1P may act directly on channels or via downstream mechanisms that require the interaction of membrane channels with the cytoskeleton. In this regard, S1P has been shown to regulate myosin light chain phosphatase (50). Also, it has been suggested that membrane sphingolipids may play a role in the assembly of oligomeric proteins (such as calcium channel proteins) that are located in lipid rafts (51). This gives rise to the speculation that conformational events based upon membrane lipid composition could also play a role in S1P-mediated calcium entry. Thus the finding that S1P acts as a soluble CIF in this instance in no way discounts the potential involvement and importance of direct physical interactions between membrane channels and intracellular structures. Rather, it remains to be determined whether SOCE requires elements of both hypotheses, and whether the soluble calcium influx factor S1P may link store depletion to downstream conformational events.
Acknowledgments
We thank Drs. K. B. Kannan, Zoltan Fekete, Eleonora Feketeova, and Qi Lu for their assistance with cell preparations and flow cytometry and Linda Vetrecin for her assistance in the preparation of the manuscript.
Footnotes
*
This work was supported by a grant from the Foundation for UMDNJ (to K. I.) and by Grant GM-59179 from the National Institutes of Health (to C. J. H.). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “_advertisement_” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
1
The abbreviations used are: SOCE, store-operated calcium entry; S1P, sphingosine 1-phosphate; CIF, calcium influx factor; PMN, polymorphonuclear leukocytes; DMS, d-_erythro-N,N_-dimethylsphingosine; SphK, sphingosine kinase; IP3, inositol 1,4,5-trisphosphate; ER, endoplasmic reticulum; GPCR, G-protein-coupled receptor; BSA, bovine serum albumin; PAF, platelet-activating factor; fMLP, formyl-Met-LeuPhe; PTX, pertussis toxin; PLC, phospholipase C; GPC, G-protein-coupled; GPCR, GPC receptor agonist; PAFr, PAF receptor.
References
- 1.Putney JW, Jr, Broad LM, Braun FJ, Lievremont JP, Bird GS. J Cell Sci. 2001;114:2223–2229. doi: 10.1242/jcs.114.12.2223. [DOI] [PubMed] [Google Scholar]
- 2.Putney JW., Jr Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- 3.Randriamampita C, Tsien RY. Nature. 1993;364:809–814. doi: 10.1038/364809a0. [DOI] [PubMed] [Google Scholar]
- 4.Fasolato C, Hoth M, Penner R. J Biol Chem. 1993;268:20737–20740. [PubMed] [Google Scholar]
- 5.Yao Y, Ferrer-Montiel AV, Montal M, Tsien RY. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]
- 6.Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Nature. 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- 7.Irvine RF. FEBS Lett. 1990;263:5–9. doi: 10.1016/0014-5793(90)80692-c. [DOI] [PubMed] [Google Scholar]
- 8.Csutora P, Su Z, Kim HY, Bugrim A, Cunningham KW, Nuccitelli R, Keizer JE, Hanley MR, Blalock JE, Marchase RB. Proc Natl Acad Sci U S A. 1999;96:121–126. doi: 10.1073/pnas.96.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trepakova ES, Csutora P, Hunton DL, Marchase RB, Cohen RA, Bolotina VM. J Biol Chem. 2000;275:26158–26163. doi: 10.1074/jbc.M004666200. [DOI] [PubMed] [Google Scholar]
- 10.Liu H, Sugiura M, Nava VE, Edsall LC, Kono K, Poulton S, Milstien S, Kohama T, Spiegel S. J Biol Chem. 2000;275:19513–19520. doi: 10.1074/jbc.M002759200. [DOI] [PubMed] [Google Scholar]
- 11.Kohama T, Olivera A, Edsall L, Nagiec MM, Dickson R, Spiegel S. J Biol Chem. 1998;273:23722–23728. doi: 10.1074/jbc.273.37.23722. [DOI] [PubMed] [Google Scholar]
- 12.Zhang H, Desai NN, Olivera A, Seki T, Brooker G, Spiegel S. J Cell Biol. 1991;114:155–167. doi: 10.1083/jcb.114.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Olivera A, Kohama T, Edsall L, Nava V, Cuvillier O, Poulton S, Spiegel S. J Cell Biol. 1999;147:545–558. doi: 10.1083/jcb.147.3.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Aas V, Algeroy S, Sand KL, Iversen JG. Cell Adhes Commun. 2001;8:125–138. doi: 10.3109/15419060109080712. [DOI] [PubMed] [Google Scholar]
- 15.Goetzl EJ, An S. FASEB J. 1998;12:1589–1598. [PubMed] [Google Scholar]
- 16.Spiegel S, Milstien S. Biochim Biophys Acta. 2000;1484:107–116. doi: 10.1016/s1388-1981(00)00010-x. [DOI] [PubMed] [Google Scholar]
- 17.Pyne S, Pyne NJ. Biochem J. 2000;349:385–402. doi: 10.1042/0264-6021:3490385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Siehler S, Manning DR. Biochim Biophys Acta. 2002;1582:94–99. doi: 10.1016/s1388-1981(02)00142-7. [DOI] [PubMed] [Google Scholar]
- 19.Le Stunff H, Galve-Roperh I, Peterson C, Milstien S, Spiegel S. J Cell Biol. 2002;158:1039–1049. doi: 10.1083/jcb.200203123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edsall LC, Pirianov GG, Spiegel S. J Neurosci. 1997;17:6952–6960. doi: 10.1523/JNEUROSCI.17-18-06952.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Brocklyn JR, Lee MJ, Menzeleev R, Olivera A, Edsall L, Cuvillier O, Thomas DM, Coopman PJ, Thangada S, Liu CH, Hla T, Spiegel S. J Cell Biol. 1998;142:229–240. doi: 10.1083/jcb.142.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.MacKinnon AC, Buckley A, Chilvers ER, Rossi AG, Haslett C, Sethi T. J Immunol. 2002;169:6394–6400. doi: 10.4049/jimmunol.169.11.6394. [DOI] [PubMed] [Google Scholar]
- 23.Birchwood CJ, Saba JD, Dickson RC, Cunningham KW. J Biol Chem. 2001;276:11712–11718. doi: 10.1074/jbc.M010221200. [DOI] [PubMed] [Google Scholar]
- 24.Hauser CJ, Fekete Z, Adams JM, Garced M, Livingston DH, Deitch EA. J Leukocyte Biol. 2001;69:63–68. [PubMed] [Google Scholar]
- 25.Van Veldhoven PP, Foglesong RJ, Bell RM. J Lipid Res. 1989;30:611–616. [PubMed] [Google Scholar]
- 26.Murata N, Sato K, Kon J, Tomura H, Yanagita M, Kuwabara A, Ui M, Okajima F. Biochem J. 2000;352:809–815. [PMC free article] [PubMed] [Google Scholar]
- 27.Gallagher R, Collins S, Trujillo J, McCredie K, Ahearn M, Tsai S, Metzgar R, Aulakh G, Ting R, Ruscetti F, Gallo R. Blood. 1979;54:713–733. [PubMed] [Google Scholar]
- 28.Studzinski GP, Reddy KB, Hill HZ, Bhandal AK. Cancer Res. 1991;51:3451–3455. [PubMed] [Google Scholar]
- 29.Grynkiewicz G, Poenie M, Tsien RY. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 30.Itagaki K, Kannan KB, Livingston DH, Deitch EA, Fekete Z, Hauser CJ. J Immunol. 2002;168:4063–4069. doi: 10.4049/jimmunol.168.8.4063. [DOI] [PubMed] [Google Scholar]
- 31.Meyer zu Heringdorf D, Lass H, Kuchar I, Alemany R, Guo Y, Schmidt M, Jakobs KH. FEBS Lett. 1999;461:217–222. doi: 10.1016/s0014-5793(99)01463-5. [DOI] [PubMed] [Google Scholar]
- 32.M'Rabet L, Coffer PJ, Wolthuis RM, Zwartkruis F, Koenderman L, Bos JL. J Biol Chem. 1999;274:21847–21852. doi: 10.1074/jbc.274.31.21847. [DOI] [PubMed] [Google Scholar]
- 33.Tiffany HL, Lavigne MC, Cui YH, Wang JM, Leto TL, Gao JL, Murphy PM. J Biol Chem. 2001;276:23645–23652. doi: 10.1074/jbc.M101031200. [DOI] [PubMed] [Google Scholar]
- 34.He R, Sang H, Ye RD. Blood. 2003;101:1572–1581. doi: 10.1182/blood-2002-05-1431. [DOI] [PubMed] [Google Scholar]
- 35.Trebak M, Bird GS, McKay RR, Putney JW., Jr J Biol Chem. 2002;277:21617–21623. doi: 10.1074/jbc.M202549200. [DOI] [PubMed] [Google Scholar]
- 36.Halaszovich CR, Zitt C, Jungling E, Luckhoff A. J Biol Chem. 2000;275:37423–37428. doi: 10.1074/jbc.M007010200. [DOI] [PubMed] [Google Scholar]
- 37.Liu X, Ambudkar IS. J Biol Chem. 2001;276:29891–29898. doi: 10.1074/jbc.M103283200. [DOI] [PubMed] [Google Scholar]
- 38.Luo D, Broad LM, Bird GS, Putney JW., Jr J Biol Chem. 2001;276:5613–5621. doi: 10.1074/jbc.M007524200. [DOI] [PubMed] [Google Scholar]
- 39.Alemany R, Meyer zu Heringdorf D, van Koppen CJ, Jakobs KH. J Biol Chem. 1999;274:3994–3999. doi: 10.1074/jbc.274.7.3994. [DOI] [PubMed] [Google Scholar]
- 40.Meyer zu Heringdorf D, Lass H, Alemany R, Laser KT, Neumann E, Zhang C, Schmidt M, Rauen U, Jakobs KH, van Koppen CJ. EMBO J. 1998;17:2830–2837. doi: 10.1093/emboj/17.10.2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olivera A, Spiegel S. Prostaglandins. 2001;64:123–134. doi: 10.1016/s0090-6980(01)00108-3. [DOI] [PubMed] [Google Scholar]
- 42.Matthews G, Neher E, Penner R. J Physiol. 1989;418:105–130. doi: 10.1113/jphysiol.1989.sp017830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kuriyama H, Kitamura K, Itoh T, Inoue R. Physiol Rev. 1998;78:811–920. doi: 10.1152/physrev.1998.78.3.811. [DOI] [PubMed] [Google Scholar]
- 44.Shi LC, Wang HY, Horwitz J, Friedman E. J Neurochem. 1996;67:1478–1484. doi: 10.1046/j.1471-4159.1996.67041478.x. [DOI] [PubMed] [Google Scholar]
- 45.Mathes C, Fleig A, Penner R. J Biol Chem. 1998;273:25020–25030. doi: 10.1074/jbc.273.39.25020. [DOI] [PubMed] [Google Scholar]
- 46.Spiegel S, Milstien S. J Biol Chem. 2002;277:25851–25854. doi: 10.1074/jbc.R200007200. [DOI] [PubMed] [Google Scholar]
- 47.Le Stunff H, Peterson C, Liu H, Milstien S, Spiegel S. Biochim Biophys Acta. 2002;1582:8–17. doi: 10.1016/s1388-1981(02)00132-4. [DOI] [PubMed] [Google Scholar]
- 48.Young KW, Bootman MD, Channing DR, Lipp P, Maycox PR, Meakin J, Challiss RA, Nahorski SR. J Biol Chem. 2000;275:38532–38539. doi: 10.1074/jbc.M006631200. [DOI] [PubMed] [Google Scholar]
- 49.Su Z, Csutora P, Hunton D, Shoemaker RL, Marchase RB, Blalock JE. Am J Physiol. 2001;280:C1284–C1292. doi: 10.1152/ajpcell.2001.280.5.C1284. [DOI] [PubMed] [Google Scholar]
- 50.Essler M, Retzer M, Ilchmann H, Linder S, Weber PC. Cell Signal. 2002;14:607–613. doi: 10.1016/s0898-6568(02)00013-x. [DOI] [PubMed] [Google Scholar]
- 51.Ait Slimane T, Hoekstra D. FEBS Lett. 2002;529:54–59. doi: 10.1016/s0014-5793(02)03183-6. [DOI] [PubMed] [Google Scholar]