The Pattern Recognition Receptor (RAGE) Is a Counterreceptor for Leukocyte Integrins: A Novel Pathway for Inflammatory Cell Recruitment (original) (raw)
Abstract
The pattern recognition receptor, RAGE (receptor for advanced glycation endproducts), propagates cellular dysfunction in several inflammatory disorders and diabetes. Here we show that RAGE functions as an endothelial adhesion receptor promoting leukocyte recruitment. In an animal model of thioglycollate-induced acute peritonitis, leukocyte recruitment was significantly impaired in RAGE-deficient mice as opposed to wild-type mice. In diabetic wild-type mice we observed enhanced leukocyte recruitment to the inflamed peritoneum as compared with nondiabetic wild-type mice; this phenomenon was attributed to RAGE as it was abrogated in the presence of soluble RAGE and was absent in diabetic RAGE-deficient mice. In vitro, RAGE-dependent leukocyte adhesion to endothelial cells was mediated by a direct interaction of RAGE with the β2-integrin Mac-1 and, to a lower extent, with p150,95 but not with LFA-1 or with β1-integrins. The RAGE–Mac-1 interaction was augmented by the proinflammatory RAGE-ligand, S100-protein. These results were corroborated by analysis of cells transfected with different heterodimeric β2-integrins, by using RAGE-transfected cells, and by using purified proteins. The RAGE–Mac-1 interaction defines a novel pathway of leukocyte recruitment relevant in inflammatory disorders associated with increased RAGE expression, such as in diabetes, and could provide the basis for the development of novel therapeutic applications.
Keywords: leukocyte, integrin, RAGE, diabetes, adhesion
Introduction
Leukocyte recruitment is an integral part of inflammatory processes or vascular remodeling and requires multistep adhesive and signaling events, including selectin-mediated rolling, leukocyte activation, and integrin-mediated firm adhesion and diapedesis (1). During firm adhesion of leukocytes to the endothelium, members of the β2-integrin family, LFA-1 (αLβ2, CD11a/CD18), Mac-1 (αMβ2, CD11b/CD18), and p150,95 (αXβ2, CD11c/CD18), as well as β1-integrins on the leukocyte surface, interact with endothelial counterligands such as ICAM-1, surface-associated fibrinogen (FBG), or VCAM-1. However, additional, yet still undefined, counter-receptors might also be engaged (2–4). In ICAM-1–deficient mice the defect in leukocyte recruitment is not complete, therefore, the existence of further β2-integrin–dependent, ICAM-1–independent mechanisms has been proposed (2). Furthermore, the importance of ICAM-1–dependent and ICAM-1–independent mechanisms of leukocyte recruitment in different pathophysiological situations, such as in diabetes, has not yet been established.
The pattern recognition receptor RAGE, (receptor for advanced glycation endproducts), has been implicated in leukocyte migration (5, 6), however, the underlying mechanisms are not yet completely understood. RAGE is a multiligand receptor on vascular cells playing a key role in inflammatory processes. RAGE is expressed at low levels in normal tissues and in the vasculature and becomes up-regulated in the diabetic vasculature or at other sites where its ligands accumulate (5, 7, 8). In particular, through recognition of β-sheet fibrillar structures, RAGE binds amyloid components, proinflammatory cytokine-like mediators of the S100/calgranulin family, or amphoterin (9–11). In diabetic vessels, RAGE ligands also include AGEs (advanced glycation endproducts) of at least two types, (carboxymethyl)-lysine (CML) adducts and hydroimidazolones (5, 12, 13). Binding of these ligands to RAGE initiates sustained cellular activation mediated by receptor-dependent signaling involving the transcription factor NFκB (14). RAGE has been thought to promote the proinflammatory phenotype of the diabetic vasculature (15–17), thereby participating in diabetic vascular complications (18–20).
These diverse observations have prompted us to investigate the role of RAGE in leukocyte recruitment. We demonstrate here the existence of direct interactions between RAGE and the β2-integrin Mac-1 and p150,95 that define RAGE as a new adhesion receptor and provide evidence for a novel pathway of leukocyte recruitment that may be relevant in pathophysiological situations including diabetes.
Materials and Methods
Materials.
The following reagents were provided from these sources: Isolated Mac-1, LFA-1, and ICAM-1 (Dr. Sarah Bodary, Genentech, CA); S100-B (Sigma-Aldrich); blocking mAb against human CD18, 60.3 (IgG2a) (Dr. John Harlan, Seattle, WA); β2-integrin activating mAb, Kim185 (Dr. M. Robinson, Slough Berkshire, UK); mAb L15 against LFA-1 (IgG2a) (Dr. C. Figdor, Nijmegen, The Netherlands); mAb LPM19c against Mac-1 (IgG1) (Dr. A. May, Munich, Germany); mAb K20 against β1-chain (CD29) (IgG2a) and mAb HP2.1 against α4-chain (IgG1), (Immunotech); mAb against ICAM-1 (IgG1) (DakoCytomation); respective isotype-matched control antibodies were from DakoCytomation. The blocking mAb against mouse Mac-1 (M1/70) (IgG2b) and mouse ICAM-1 (3E2) (IgG1), mAb against mouse αvβ3 (RMV-7) (IgG1), as well as respective isotype-matched control antibodies, which were used for in vivo inhibition studies, were from BD Biosciences. MnCl2, FBG and fibronectin (FN) (Sigma-Aldrich), vitamin D3 (BIOMOL Research Laboratories, Inc.), transforming growth factor-β (R&D Systems), high molecular weight kininogen (HK) (Enzyme Research Laboratories). Recombinant soluble mouse RAGE was expressed in Sf 9 cells and purified as described previously (21). In vitro synthesis of CML-albumin was performed as described (14). Anti-RAGE polyclonal antibody was raised in rabbit against KLH-conjugated peptide, AQNITARIGEPLVLKC, that corresponds to the NH2 terminus of mature human RAGE. Mouse RAGE has a highly homologous sequence at the corresponding region, GQNITARIGEPLVLSC, thereby recombinant mouse RAGE is recognized by this polyclonal antibody. Polyclonal anti-RAGE peptide antibody was purified by protein G chromatography.
Cell Culture and Adhesion Assay.
Myelo-monocytic U937 and THP-1 cells, K562 cells and K562 cells transfected with different β2-integrin forms, as well as human umbilical vein endothelial cells (HUVEC) were cultivated as previously described (22–24). K562 cells (nontransfected, or stable LFA-1- or p150,95-transfectants) were kindly provided by Dr. Y. van Kooyk (University of Nijmegen, The Netherlands) and were cultured in a mixture of 75% RPMI-1640 medium containing 10% FCS and 25% ISCOVE's medium supplemented with 5% FCS. K562 cells transfected with Mac-1 were kindly provided by Dr. M. Smith (Celltech, Slough, UK). Comparable expression of LFA-1, p150,95, and Mac-1, respectively, of the transfected K562 cells was confirmed by flow cytometry (22).
Generation of RAGE-transfected CHO Cells.
A full-length human RAGE cDNA was isolated from lung cDNA library by polymerase chain reaction with primers 5′-GATGGCAGCCGGAACAGCAGTT-3′ and 5′-GAGGCCAGAACAGTTCAAGGG-3′. Human RAGE cDNA was subcloned into an expression vector under MPSV LTR promoter and used for transfection into CHO cells as described previously (25). Surface expression of RAGE was confirmed by flow cytometry analysis using mAb raised against purified recombinant human soluble RAGE.
Human neutrophils were isolated from peripheral blood as previously described (26).
Adhesion of U937 cells, THP-1 cells, K562 cells, or neutrophils to immobilized soluble RAGE (10 μg/ml) (and to BSA as control) was performed as previously described (22–24). Briefly, microtiter plates were coated with RAGE in bicarbonate buffer, pH 9.6, and blocked with 3% BSA solution. Isolated human neutrophils, U937 cells, or THP-1 cells, which had been differentiated for 24 h in the presence of vitamin D3 (100 nM) and transforming growth factor-β (2 ng/ml), or K562 cells were washed in serum-free RPMI medium and plated onto the precoated wells (105/well) at 37°C for 60 min in the absence or presence of inhibitors. After the incubation period, the wells were washed and adherent cells were fixed with methanol/acetone (1:1) at 4°C for 30 min. Adherent cells were then stained with crystal violet and quantified by measuring absorbance at 590 nm.
Adhesion of U937 cells, THP-1 cells, or neutrophils to nontransfected CHO cells, RAGE-transfected CHO cells, or cultured HUVEC was tested as previously described (22, 26, 27). Briefly, nontransfected CHO, RAGE-transfected CHO cells, or HUVEC were grown to confluency onto 96-well plates. Fluorescence-labeled neutrophils (105/well), differentiated U937 cells, THP-1 cells, or K562 cells were washed twice, followed by no pretreatment or stimulation with PMA (50 ng/ml) or the β2-integrin stimulating mAb Kim185 (10 μg/ml). Cells were washed and added to CHO cells or HUVEC at 37°C for 60 min in the absence or presence of inhibitors. After washing, adhesion of neutrophils, U937 cells, THP-1 cells, or K562 cells was quantified as the percentage of total cells added using a fluorescence microplate reader (Bio-Tek) (22, 26, 27).
In Vitro Ligand-receptor Interactions.
Binding of biotinylated-FBG or ICAM-1 to immobilized Mac-1 or LFA-1 was performed as previously described (22, 26, 27). Alternatively, binding of soluble RAGE (5 μg/ml) to immobilized Mac-1, LFA-1, or BSA (each 5 μg/ml) was performed in TBS containing 0.3% BSA, 0.05% Tween-20. After incubation for 2 h at 22°C in each case, a polyclonal antibody against RAGE was added, followed by addition of appropriate secondary peroxidase-conjugated antibody (DakoCytomation) and the substrate ABTS, and binding was quantitated at 405 nm. Nonspecific binding to BSA-coated wells was used as blank and was subtracted to calculate specific binding.
Mice.
RAGE−/− mice, tie2-RAGE mice, as well as tie2-RAGE×RAGE−/− mice (progeny of RAGE−/− mice that were crossed with tie2-RAGE transgenic mice, overexpressing RAGE on the endothelium) were described previously (20, 28). Induction of diabetes with streptozotocin in wild-type and RAGE−/− mice was performed as previously described (29) and was approved by the Regional Commission in Giessen, Germany (File No. Gi20/11–40/99).
In Vivo Peritonitis Model.
Thioglycollate-induced peritonitis in diabetic or nondiabetic wild-type or RAGE−/− mice was performed as previously described (22, 27). For inhibition studies, 30 min before the injection of thioglycollate, the following compounds were administered intraperitoneally: 100 μg mouse soluble RAGE, or 100 μg mAb against mouse ICAM-1 in PBS. Control mice were treated with the same volume of PBS, isotype-matched control antibodies (100 μg), or a mAb against mouse αvβ3 (100 μg). To evaluate neutrophil recruitment, mice were killed at 1 and 4 h after injection of thioglycollate, whereas for monocyte/macrophage recruitment, mice were killed at 40 and 72 h after thioglycollate injection. Thereafter, the peritoneal lavage was collected and the number of emigrated neutrophils or macrophages was quantitated (22, 27, 30). There was no difference in the circulating neutrophil count of the wild-type, RAGE−/−, or tie2-RAGE×RAGE−/− mice.
Statistical Analysis.
Data were analyzed by t test or analysis of variance (ANOVA), as appropriate. P values of <0.05 were regarded as significant. Statistical analysis of in vivo data: in Fig. 1, A–D , data are presented as mean ± SD. Fig. 1, A and B, were analyzed by a two-way ANOVA with interaction term and appropriate contrasts to compare the groups. In case of interaction pairwise t tests were performed and the p-values are adjusted according to the Holm procedure. Fig. 1 C was analyzed by a one-way ANOVA and appropriate contrasts to compare the pairs in case of a significant result for the global test. The closed test procedure is used to guarantee the overall error rate of 0.05. Fig. 1 D was analyzed by a t test for unequal variances for 40 and 72 h, respectively. Due to the fact that leukocyte count data are usually not normally distributed, a nonparametric analysis was done for sensitivity analysis.
Figure 1.
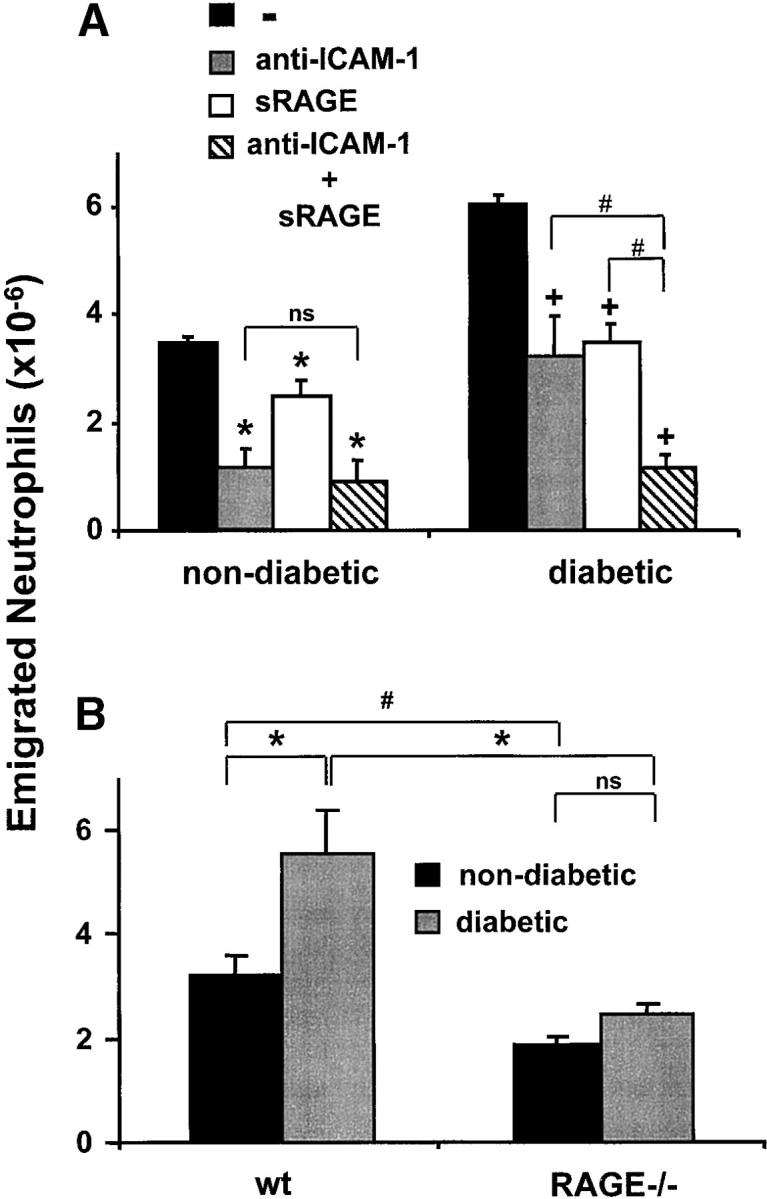
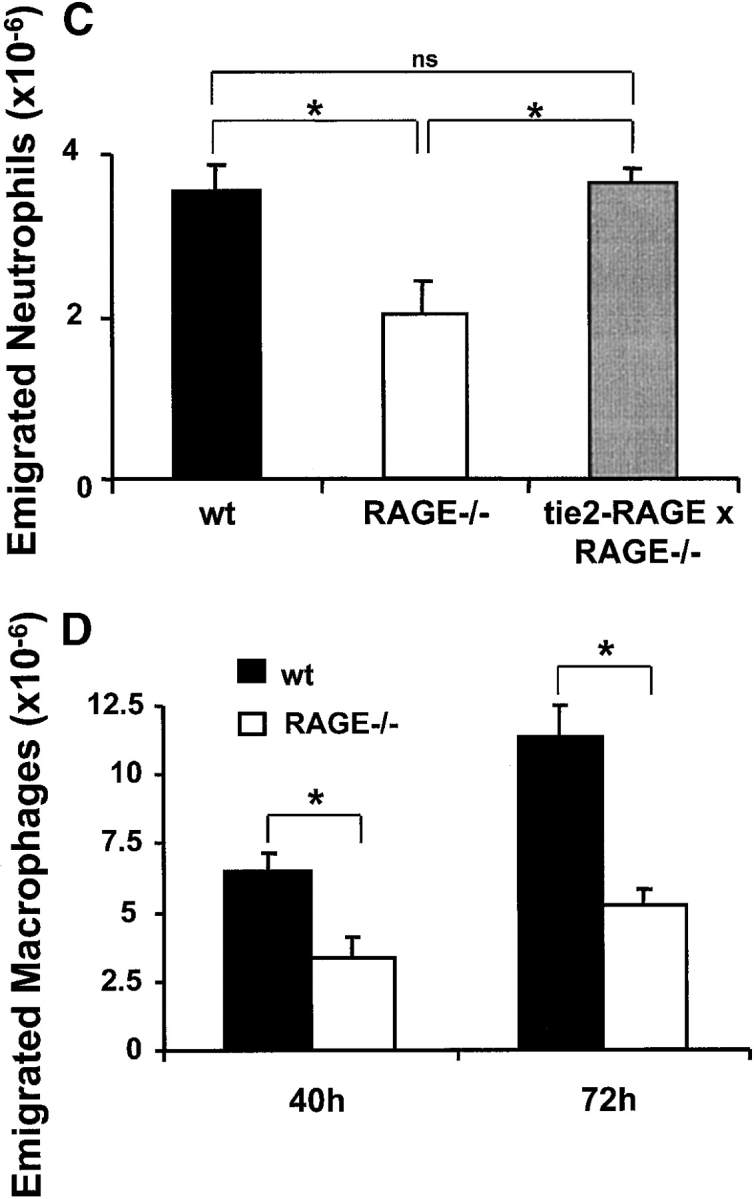
The contribution of RAGE to inflammatory reactions in vivo. After thioglycollate injection into the mouse peritoneum to induce acute inflammation, the number of neutrophils in the peritoneal lavage was analyzed after 4 h. (A) Prior to thioglycollate administration, nondiabetic or diabetic mice were treated by intraperitoneal injection with PBS (black bars), with a blocking mAb against ICAM-1 (gray bars), with soluble RAGE (white bars), or a combination of the blocking mAb against ICAM-1 and soluble RAGE (hatched bars). *, P < 0.0001 as compared with control (nondiabetic mice treated with PBS); +, P < 0.0001 as compared with control (diabetic mice treated with PBS); #, P < 0.001; ns, not significant (P = 0.1169). (B) The number of emigrated neutrophils into the peritoneum of nondiabetic (black bars) or diabetic (gray bars) wild-type or RAGE−/− mice was compared 4 h after thioglycollate injection. *, P < 0.0001; #, P = 0.0059; ns, not significant (P = 0.2656). (C) The number of neutrophils infiltrated into the peritoneum of wild-type mice (black bars), RAGE−/− mice (white bars), or tie2-RAGE×RAGE−/− mice (gray bars) was compared 4 h after thioglycollate injection. *, P < 0.0001; ns, not significant (P = 0.6976). (D) After thioglycollate injection into the mouse peritoneum the number of macrophages in the peritoneal lavage of wild-type (wt, black bars) or RAGE −/− (white bars) mice was analyzed after 40 and 72 h. *, P < 0.0001. Data are mean ± SD (n = 5 mice per treatment) of typical experiments; similar results were obtained in three separate sets of experiments.
Results
RAGE Mediates Leukocyte Recruitment In Vivo.
To test whether RAGE is engaged in leukocyte recruitment in vivo, we studied the role of RAGE in a mouse model of acute inflammation. Peritonitis was induced by thioglycollate injection, and after four hours there was the expected increase in the total leukocyte count in the peritoneum, mostly attributable to emigrated neutrophils. The portion of neutrophils among all leukocytes after four hours was 50–70% as compared with 3–5% at one hour after stimulation (22, 27). This process is Mac-1 dependent, since treatment with a blocking antibody against Mac-1 (administration 30 min before the induction of peritonitis) almost completely abolished neutrophil extravasation into the inflamed peritoneum (27). Neutrophil recruitment to the peritoneum was also significantly reduced in mice that were pretreated with blocking mAb against ICAM-1 (∼65–75% reduction), whereas an isotype-matched control mAb or a mAb against an irrelevant endothelial antigen (αvβ3-integrin) did not inhibit (data not shown). Interestingly, treatment with soluble RAGE also resulted in a partial (25%) inhibition of neutrophil extravasation (Fig. 1 A).
Since RAGE is expressed at low levels in normal tissues including the vasculature, and becomes up-regulated under diabetic conditions, we performed the same model of acute inflammation in diabetic mice. In diabetic mice, a twofold increase in the recruited number of neutrophils to the inflamed peritoneum was observed when compared with nondiabetic mice, whereas basal leukocyte counts (no thioglycollate stimulation) in the peritoneum were similar in both diabetic and nondiabetic mice (data not shown). Again, neutrophil extravasation was almost completely abolished by the use of blocking antibody against Mac-1 (data not shown). In diabetic mice, both soluble RAGE or a blocking antibody against ICAM-1 reduced neutrophil extravasation by ∼50%, and the combination of both inhibitors almost completely prevented neutrophil emigration (Fig. 1 A). These data indicate that in a setting where RAGE is up-regulated, such as in diabetes, RAGE is directly involved in neutrophil recruitment.
To further test this hypothesis, we compared acute thioglycollate-induced peritonitis in wild-type and RAGE−/− mice. There was a significant reduction (∼30–40%) in neutrophils that had migrated into the peritoneum of RAGE−/− mice as compared with wild-type mice (Fig. 1 B). Thioglycollate-elicited neutrophils isolated from the peritoneum of wild-type or RAGE−/− mice, respectively, showed no significant difference in the expression of adhesion receptors, in particular β1- and β2-integrins (data not shown). Moreover, neutrophils isolated from wild-type and RAGE−/− mice showed comparable adhesion to FBG and FN in vitro (data not shown). The increased neutrophil extravasation into the peritoneum that was observed in diabetic wild-type mice was not found in diabetic RAGE−/− mice (Fig. 1 B). There was no difference in the basal (no thioglycollate stimulation) counts of peritoneal leukocytes between wild-type and RAGE−/− mice irrespective of whether the mice were diabetic (data not shown).
The decrease in neutrophil extravasation observed in RAGE−/− mice as compared with wild-type mice could be reversed by restoring the expression of RAGE on endothelial cells. Progeny of RAGE−/− mice that were crossed with tie2-RAGE transgenic mice, which overexpress RAGE on the endothelium, showed comparable neutrophil extravasation as wild-type mice (Fig. 1 C). Therefore, the described difference in neutrophil emigration between wild-type and RAGE−/− mice was indeed due to the absence of RAGE on the endothelium.
Finally, the same peritonitis model was extended for 40 and 72 h following thioglycollate administration, to evaluate the recruitment of monocytes/macrophages into the peritoneum in wild-type and RAGE−/− mice. There was a significant reduction (>50%) in the number of macrophages that had migrated into the peritoneum of RAGE−/− mice as compared with wild-type mice (Fig. 1 D). Together, these data demonstrate that RAGE mediates leukocyte recruitment in vivo.
Integrin-mediated Leukocyte Adhesion to RAGE.
Since these observations suggested a direct participation of RAGE in leukocyte extravasation, we studied whether leukocytes can directly adhere to immobilized RAGE. Human peripheral blood neutrophils adhered strongly to RAGE-coated plates dependent on the presence of divalent cations (Fig. 2 A). Soluble RAGE and a blocking mAb against β2-integrin abolished this adhesion, whereas a blocking mAb against β1-integrin or a mAb against α4-integrin had no effect. Moreover, neutrophil and myelomonocytic cell adhesion to immobilized RAGE was inhibited by a blocking mAb against Mac-1, whereas a blocking mAb against LFA-1, or isotype-matched control mAbs (unpublished data) did not affect cell adhesion (Fig. 2 A). The Mac-1 ligand FBG, but not the β1-integrin ligand FN, interfered with the adhesion of neutrophils to immobilized RAGE (unpublished data). The same interaction pattern between RAGE and Mac-1 was seen when the adhesion of neutrophils to CHO cells transfected with RAGE was tested (Fig. 2 B), whereas no appreciable adhesion to nontransfected CHO cells was observed. Similar results were obtained when the adhesion of THP-1 cells or U937 cells to immobilized RAGE or RAGE-transfected CHO cells (and nontransfected CHO cells as control) was investigated (unpublished data).
Figure 2.

The adhesive properties of RAGE. (A) Adhesion of PMA-stimulated human neutrophils to immobilized RAGE is shown in the absence (-) or presence of EDTA (5 mM), soluble RAGE (10 μg/ml), mAb to β1-integrin, mAb to β2-integrin, mAb to Mac-1, mAb to LFA-1, or mAb to α4-integrin (each antibody at 20 μg/ml). (B) Adhesion of PMA-stimulated human neutrophils to CHO cells (gray bars) or RAGE-transfected CHO cells (black bars) is shown in the absence (-) or presence of mAb to β1-integrin, mAb to β2-integrin, mAb to Mac-1, or mAb to LFA-1 (each antibody at 20 μg/ml), or soluble RAGE (10 μg/ml). Cell adhesion is represented either as absorbance of labeled cells at 590 nm or as percentage of total added cells. All data are mean ± SD (n = 3) of a typical experiment, similar results were obtained in at least three separate experiments.
To further characterize the binding between RAGE and β2-integrins, the adhesion of K562 erythroleukemic cells transfected with either LFA-1, Mac-1, or another β2-integrin, p150,95, to immobilized RAGE was studied (Fig. 3 A). Whereas nontransfected K562 cells or LFA-1 transfectants did not show any appreciable adhesion to RAGE, both under unstimulated or stimulated conditions (using the stimulating β2-integrin antibody Kim185), K562 cells expressing Mac-1 as well as p150,95 adhered to immobilized RAGE in the presence of the β2-integrin stimulating antibody Kim185. Similarly, only Mac-1- or p150,95-transfected K562 cells adhered to RAGE-transfected CHO cells, whereas LFA-1 transfectants or nontransfected cells did not adhere (Fig. 3 B). None of the K562 cells interacted with nontransfected parental CHO cells, indicating that the observed interaction was dependent on the expression of RAGE. Adhesion of Mac-1–transfected cells to immobilized RAGE or RAGE-transfected CHO cells was inhibited by blocking mAb against Mac-1, as well as by soluble RAGE (Fig. 3 C). These data demonstrate for the first time that RAGE acts as a counter-receptor for the β2-integrin Mac-1 as well as p150,95 and is capable of mediating heterophilic cell–cell interactions.
Figure 3.

RAGE is a counter-receptor for β2-integrins. (A) Adhesion of nontransfected K562 cells, LFA-1-transfected K562 cells, Mac-1-transfected K562 cells, or p150,95-transfected K562 to immobilized RAGE is shown in the absence (gray bars) or presence (black bars) of β2-integrin–stimulating antibody Kim185 (10 μg/ml). (B) Adhesion of nontransfected K562 cells, LFA-1–transfected K562 cells, Mac-1–transfected K562 cells, or p150,95-transfected K562 cells, which were preincubated with Kim185 (10 μg/ml), to nontransfected CHO cells (gray bars) or RAGE-transfected CHO cells (black bars) is shown. (C) Adhesion of Mac-1–transfected K562 cells, which were preincubated with Kim185 (10 μg/ml), to RAGE-transfected CHO cells is shown in the absence (-) or presence of mAb to Mac-1 (20 μg/ml) or soluble RAGE (10 μg/ml). Cell adhesion is represented either as absorbance at 590 nm or as percentage of total added cells. All data are mean ± SD (n = 3) of a typical experiment, similar results were observed in at least three separate experiments.
We then tested, whether endothelial RAGE could also mediate leukocyte–endothelial interactions. Both mAb against ICAM-1 and soluble RAGE could partially block Mac-1–dependent adhesion of human neutrophils to endothelial cell monolayer. The combination of ICAM-1 and RAGE blockade almost completely abolished neutrophil adhesion to endothelial cells (Fig. 4) . Isotype-matched control mAbs had no inhibitory effect (data not shown). Taken together, these results clearly demonstrate that RAGE can act as a novel adhesive counter-receptor for β2-integrin–dependent leukocyte–endothelial interactions.
Figure 4.

Contribution of RAGE to neutrophil adhesion to cultured endothelial cells. PMA-stimulated adhesion of human neutrophils to endothelial cells is shown in the absence (-) or presence of mAb to β2-integrin, mAb to Mac-1, mAb to ICAM-1 (each antibody at 20 μg/ml), soluble RAGE (10 μg/ml), or a combination of soluble RAGE and mAb to ICAM-1. Cell adhesion is represented as percentage of total added cells. All data are mean ± SD (n = 3) of a typical experiment, similar results were observed in at least three separate experiments.
Binding Interactions Between Mac-1 and RAGE.
Since the previous observations implicated that there might be a direct binding of RAGE to the β2-integrin Mac-1, the interaction of soluble RAGE with immobilized purified Mac-1 or LFA-1 was analyzed. Soluble RAGE bound directly to Mac-1, and this binding was equivalent in its extent to the binding of ICAM-1 to this integrin (Fig. 5 A). Moreover, binding of soluble RAGE to Mac-1 was dose-dependent and saturable (Fig. 5 B). In contrast, no specific binding of soluble RAGE to immobilized LFA-1 was observed, while the expected binding of ICAM-1 to LFA-1 was apparent.
Figure 5.

The interaction between RAGE and Mac-1. (A) The binding of ICAM-1 or soluble RAGE (each 5 μg/ml) to immobilized LFA-1 (gray bars) or Mac-1 (black bars) is shown. (B) Dose-dependent specific binding of soluble RAGE to immobilized Mac-1 is shown. Specific binding is expressed as absorbance at 405 nm. Data are mean ± SD (n = 3) of a typical experiment; similar results were observed in at least three separate experiments.
To further characterize the interaction between RAGE and Mac-1, we tested whether the major ligand-binding I-domain of Mac-1, as well as other Mac-1 ligands, can compete for the binding of soluble RAGE to Mac-1. Here, the isolated I-domain of Mac-1 completely abolished binding of RAGE to Mac-1 (Fig. 6 A), and binding of soluble RAGE to Mac-1 was blocked by both FBG and HK (IC50: 150 nM or 200 nM, respectively; Fig. 6 B). In contrast, the endothelial Mac-1 ligand, ICAM-1, had no inhibitory effect on binding of soluble RAGE at all (Fig. 6 A). Moreover, RAGE could also block the binding of FBG to Mac-1 (IC50: ∼175 nM; Fig. 6 C). These data indicate that the major ligand-binding I-domain of Mac-1 is involved in RAGE binding and that the binding site for RAGE on Mac-1 overlaps with the binding sites of other ligands, such as FBG and HK (22).
Figure 6.
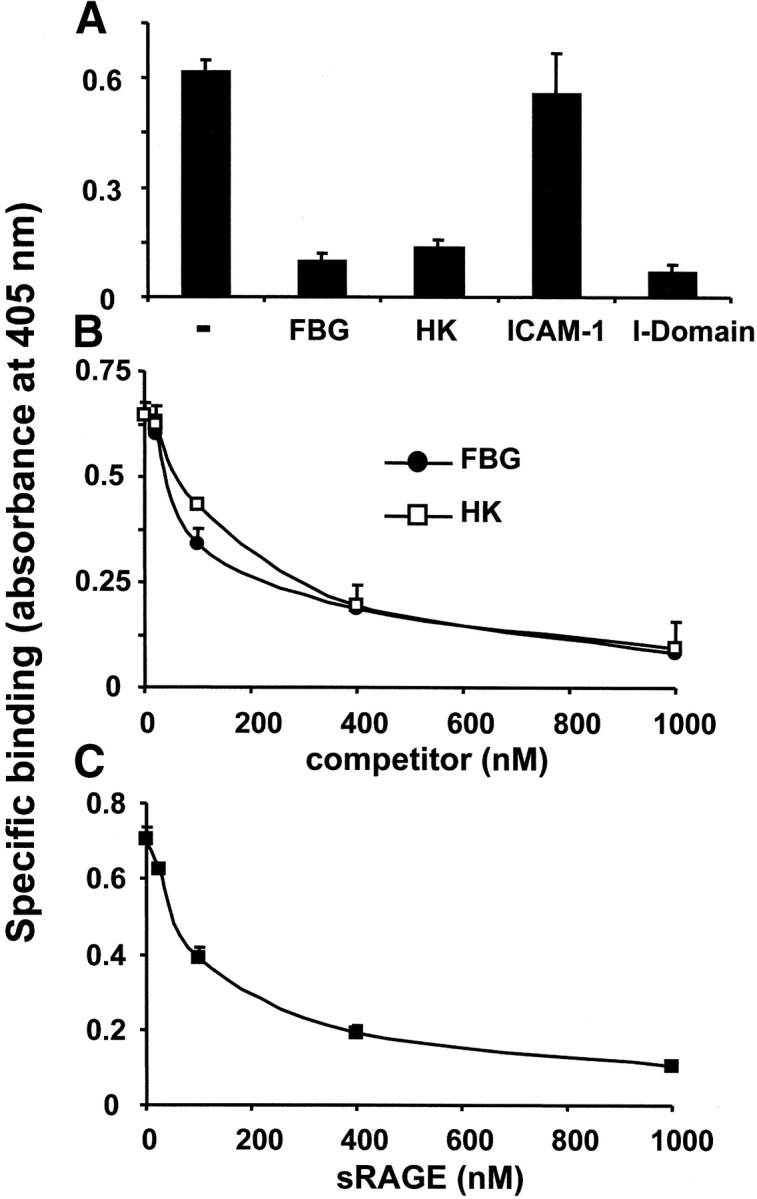
Influence of Mac-1 ligands on the interaction between RAGE and Mac-1. (A) The specific binding of soluble RAGE (100 nM) to immobilized Mac-1 is shown in the absence (-) or presence of FBG, HK, ICAM-1, or the isolated I-domain of Mac-1 (each 500 nM). (B) The specific binding of soluble RAGE (100 nM) to immobilized Mac-1 is shown in the absence or presence of increasing concentrations of FBG (black circles) or HK (white squares). (C) The specific binding of FBG (100 nM) to immobilized Mac-1 is shown in the absence or presence of increasing concentrations of soluble RAGE. Specific binding is expressed as absorbance at 405 nm. Data are mean ± SD (n = 3) of a typical experiment; similar results were observed in at least three separate experiments.
For further characterization of the RAGE–Mac-1 interaction we examined whether two RAGE ligands, CML-modified albumin and S100-B protein, could interfere with the interaction between RAGE and Mac-1. While CML did not affect the binding of RAGE to immobilized Mac-1 at all, S100-B protein augmented this interaction more than twofold in a dose-dependent, saturable manner (Fig. 7 A). A similar approximate twofold increase of Mac-1–dependent adhesion of myelomonocytic U937 cells to RAGE was observed in the presence of S100-B protein (Fig. 7 B). No direct interaction between Mac-1 and S100 was observed (unpublished data). Taken together, these data demonstrate that RAGE directly interacts with Mac-1 and that this interaction is enhanced by another RAGE ligand, S100-B protein, but not by CML-adducts.
Figure 7.

Influence of RAGE ligands on the interaction between RAGE and Mac-1. (A) The specific binding of soluble RAGE (5 μg/ml) to immobilized Mac-1 is shown in the absence or presence of increasing concentrations of S100-B (black circles) or CML (white squares). Specific binding is expressed as absorbance at 405 nm. Data are mean ± SD (n = 3) of a typical experiment; similar results were observed in at least three separate experiments. (B) PMA-stimulated adhesion of U937 cells to immobilized RAGE is presented in the absence (-) or presence of mAb to Mac-1 (20 μg/ml), CML (800 nM), S100-B (800 nM), or the combination of S100-B and mAb to Mac-1. Cell adhesion is represented as percent of control. Data are mean ± SD (n = 3) of a typical experiment; similar results were observed in at least three separate experiments.
Discussion
The present report demonstrates that RAGE serves as a novel counter-receptor for the leukocyte integrin Mac-1, being directly involved in leukocyte recruitment in vitro and in vivo. RAGE is a central player in inflammation (5) and in diabetic complications (5, 20) and our findings help to clarify its functional role. Our data show that RAGE serves as a binding partner for the β2-integrins Mac-1 and p150,95, in addition to the classical counter-receptor ICAM-1. Although ICAM-1 is the major endothelial cell ligand of both Mac-1 and LFA-1, its exclusive expression on the inflammatory endothelium could not account for the entire phenomenon of leukocyte adhesion and extravasation (2, 31). Thus, RAGE is a counter-receptor for leukocyte integrins that contributes to the recruitment of inflammatory cells, particularly under stress or pathological conditions such as in diabetes.
The following features are consistent with a specific interaction of RAGE, particularly with the leukocyte β2-integrin Mac-1. i, Neutrophils and myelomonocytic cells adhered to immobilized RAGE or RAGE-transfected cells, and this interaction was attributable to Mac-1. ii, Adhesion of K562 cells transfected with different β2-integrins to either immobilized RAGE or RAGE-transfected cells further established that RAGE interacts with Mac-1, but not with LFA-1. Interestingly, K562 cells bearing a third β2-integrin, p150,95, also interacted with RAGE, which is not surprising, as p150,95 and Mac-1 share many structural features in ligand binding (26). iii, In a purified system, a direct binding between RAGE and Mac-1 was observed. RAGE interacts with the ligand-binding I-domain of Mac-1. Competition studies indicated that the binding site of RAGE overlaps with those of established Mac-1 ligands, such as FBG and HK, and that the affinity of the RAGE–Mac-1 interaction falls into a similar range as compared with these Mac-1 ligands. Binding of RAGE to Mac-1 was augmented by a RAGE-ligand, S100-B protein, whereas another RAGE ligand, CML, had no influence, suggesting that the formation of a ternary complex between Mac-1, RAGE, and S100 is likely and needs further experimental proof. Moreover, our observations are in agreement with a previous report demonstrating Mac-1 activation by another S100 family member, S100-A9 (32). Based on these data we postulate that RAGE constitutes an adhesive receptor mediating intercellular contacts. The possibility that RAGE also interacts with Mac-1 in a cis fashion on leukocytes and that this interaction is enhanced in the presence of S100 proteins needs to be addressed in future studies.
RAGE mediates leukocyte recruitment in vivo, as we observed in the model of thioglycollate-induced acute peritonitis, and in vitro studies indicate that it likely does so by interacting with Mac-1. RAGE-mediated leukocyte recruitment was more prominent in diabetic mice in which RAGE was up-regulated. While RAGE only partially contributed to leukocyte recruitment in vivo in control mice, in diabetic mice a higher percentage of leukocyte recruitment was inhibited by soluble RAGE. Moreover, the increase in neutrophil extravasation due to the diabetes observed in wild-type mice was not present in RAGE−/− mice. These results support the conclusion that RAGE-mediated leukocyte recruitment becomes more important as RAGE expression increases at the site of inflammation. Finally, in RAGE−/− mice, a significant reduction (>50%) of macrophages migrating into the peritoneum was observed as compared with wild-type mice, indicative for the importance of RAGE in the progression of chronic inflammatory processes related to macroangioapathies.
While RAGE is expressed at low levels in normal tissues and the vasculature, the receptor becomes up-regulated wherever its ligands may accumulate (5). Thus, the mechanism for leukocyte recruitment presented here may be particularly applicable in pathophysiological conditions associated with high RAGE expression, such as in diabetes, chronic immune responses, atherosclerosis, or cancer (5, 10, 11). Simultaneously, an increased RAGE expression will lead to further RAGE-dependent cellular activation and dysfunction (5, 8). Engagement of RAGE by its ligands, such as AGE, thereby increases the permeability of the vascular endothelium and may elevate the expression of adhesion molecules, such as VCAM-1, procoagulant tissue factor, or proinflammatory interleukin-6 (15–17, 33), probably through RAGE–mediated activation of NFκB pathways. Thus, RAGE-mediated cellular activation may explain the increased recruitment of inflammatory cells. However, the data presented here demonstrate that in addition to RAGE-mediated cellular activation, RAGE can directly modulate leukocyte recruitment as it acts as an endothelial cell adhesive receptor attracting leukocytes.
RAGE is implicated in diabetic complications, particularly in accelerated atherosclerosis of the diabetic vessel wall (18–20, 29, 34, 35). Since increased leukocyte recruitment is a prominent feature of the atherosclerotic vasculature, it is possible that the RAGE-dependent leukocyte adhesion defined here could contribute to the proatherogenic phenotype of the diabetic vessel. This is supported by previous observations in which soluble RAGE was shown to suppress the diabetes-associated accelerated atherosclerosis of animals deficient in apoE (18, 29, 35).
Although in previous studies amelioration of the inflammatory response in several pathophysiologies with soluble RAGE was demonstrated and attributed to its propensity to interfere with RAGE-mediated cellular activation, the present findings define soluble RAGE as a potential inhibitor of leukocyte recruitment. The interference of soluble RAGE with such a central interaction in the immune response also provides a further explanation for its broad spectrum of functions. Finally, antagonizing the interaction between RAGE and β2-integrins might provide a novel therapeutic antiinflammatory strategy in diabetes or chronic immune responses associated with high RAGE expression.
Acknowledgments
The excellent technical assistance of Thomas Schmidt-Wöll and Jens Weber is gratefully acknowledged.
This work was supported by grants from the Deutsche Forschungsgemeinschaft to T. Chavakis (Ch279/1-1, Ch279/2-1, and SFB 405), to P.P. Nawroth (Na138/5-3 and SFB 405) and to K.T. Preissner (Pr327/18-1), and to T. Linn (Li353/10-1 and JDRF 1-2002-816).
Abbreviations used in this paper: AGE, advanced glycation endproducts; CML, (carboxymethyl)-lysine; FBG, fibrinogen; FN, fibronectin; HK, high molecular weight kininogen; HUVEC, human umbilical vein endothelial cells; RAGE, receptor for AGE.
References
- 1.Springer, T.A. 1994. Traffic signals for lymphocyte recirculation and leukocyte emigration: The multistep paradigm. Cell. 76:301–314. [DOI] [PubMed] [Google Scholar]
- 2.Carlos, T.M., and J.M. Harlan. 1994. Leukocyte-endothelial adhesion molecules. Blood. 84:2068–2101. [PubMed] [Google Scholar]
- 3.Plow, E.F., T.A. Haas, L. Zhang, J. Loftus, and J.W. Smith. 2000. Ligand binding to integrins. J. Biol. Chem. 275:21785–21788. [DOI] [PubMed] [Google Scholar]
- 4.Chavakis, T., K.T. Preissner, and S. Santoso. 2003. Leukocyte trans-endothelial migration: JAMs add new pieces to the puzzle. Thromb. Haemost. 89:13–17. [PubMed] [Google Scholar]
- 5.Schmidt, A.M., S.D. Yan, S.F. Yan, and D.M. Stern. 2001. The multiligand receptor RAGE as a progression gactor amplifying immune and inflammatory responses. J. Clin. Invest. 108:949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schmidt, A.M., S.D. Yan, J. Brett, R. Mora, R. Nowygrod, and D.M. Stern. 1993. Regulation of human mononuclear phagocyte migration by cell surface binding-proteins for advanced glycation end products. J. Clin. Invest. 91:2155–2168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li, J., and A.M. Schmidt. 1997. Characterization and functional analysis of the promoter of RAGE. J. Biol. Chem. 272:16498–16506. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka, N., H. Yonekura, S. Yamagishi, H. Fujimori, Y. Yamamoto, and H. Yamamoto. 2000. The receptor for advanced glycation end products is induced by the glycation products themselves and tumor necrosis factor-α through nuclear factor-κB, and by 17β-estradiol through Sp-1 in human vascular endothelial cells. J. Biol. Chem. 275:25781–25790. [DOI] [PubMed] [Google Scholar]
- 9.Yan, S.D., X. Chen, J. Fu, M. Chen, H. Zhu, A. Roher, T. Slattery, L. Zhao, M. Nagashima, J. Morser, et al. 1996. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature. 382:685–691. [DOI] [PubMed] [Google Scholar]
- 10.Hofmann, M., S. Drury, C. Fu, W. Qu, A. Taguchi, Y. Lu, C. Avila, N. Kambham, A. Bierhaus, P. Nawroth, et al. 1999. RAGE mediates a novel proinflammatory axis: the cell surface receptor for S100/calgranulin polypeptides. Cell. 97:889–901. [DOI] [PubMed] [Google Scholar]
- 11.Taguchi, A., D.C. Blood, G. del Toro, A. Canet, D.C. Lee, W. Qu, N. Tanji, Y. Lu, E. Lalla, C. Fu, et al. 2000. Blockade of RAGE/amphoterin supresses tumor growth and metastases. Nature. 405:354–360. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt, A.M., M. Vianna, M. Gerlach, J. Brett, J. Ryan, J. Kao, C. Esposito, H. Hegarty, W. Hurley, M. Clauss, et al. 1992. Isolation and characterization of binding proteins for advance glycosylation endproducts from lung tissue which are present on the endothelial cell surface. J. Biol. Chem. 267:14987–14997. [PubMed] [Google Scholar]
- 13.Kislinger, T., C. Fu, B. Huber, W. Qu, A. Taguchi, S. Du Yan, M. Hofmann, S.F. Yan, M. Pischetsrieder, D. Stern, and A.M. Schmidt. 1999. N(epsilon)-(carboxymethyl)lysine adducts of proteins are ligands for RAGE that activate cell signaling pathways and modulate gene expression. J. Biol. Chem. 274:31740–31749. [DOI] [PubMed] [Google Scholar]
- 14.Bierhaus, A., S. Schiekofer, M. Schwaninger, M. Andrassy, P.M. Humpert, J. Chen, M. Hong, T. Luther, T. Henle, I. Kloting, et al. 2001. Diabetes-associated sustained activation of the transcription factor nuclear factor-kappaB. Diabetes. 50:2792–2808. [DOI] [PubMed] [Google Scholar]
- 15.Bierhaus, A., T. Illmer, M. Kasper, T. Luther, P. Quehenberger, H. Tritschler, P. Wahl, R. Ziegler, M. Muller, and P.P. Nawroth. 1997. Advanced glycation end product (AGE)-mediated induction of tissue factor in cultured endothelial cells is dependent on RAGE. Circulation. 96:2262–2271. [DOI] [PubMed] [Google Scholar]
- 16.Wautier, J.L., C. Zoukourian, O. Chappey, M.P. Wautier, P.J. Guillausseau, R. Cao, O. Hori, D. Stern, and A.M. Schmidt. 1996. Receptor-mediated endothelial cell dysfunction in diabetic vasculopathy: soluble RAGE blocks hyperpermeability in diabetic rats. J. Clin. Invest. 97:238–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schmidt, A.M., O. Hori, J.X. Chen, J.F. Li, J. Crandall, J. Zhang, R. Cao, S.D. Yan, J. Brett, and D. Stern. 1995. Advanced glycation endproducts interacting with their endothelial receptor induce expression of VCAM-1 in cultured human endothelial cells and in mice. J. Clin. Invest. 96:1395–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park, L., K.G. Raman, K.J. Lee, Y. Lu, L.J. Ferran, Jr., W.S. Chow, D. Stern, and A.M. Schmidt. 1998. Suppression of accelerated diabetic atherosclerosis by soluble receptor for AGE (sRAGE). Nat. Med. 4:1025–1031. [DOI] [PubMed] [Google Scholar]
- 19.Goova, M., J. Li, T. Kislinger, W. Qu, Y. Lu, L.G. Bucciarelli, S. Nowygrod, B.M. Wolf, X. Caliste, S.F. Yan, et al. 2001. Blockade of RAGE restores effective wound healing in diabetic mice. Am. J. Pathol. 159:513–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wendt, T.M., N. Tanji, J. Guo, T.R. Kislinger, W. Qu, Y. Lu, L.G. Bucciarelli, L.L. Rong, B. Moser, G.S. Markowitz, et al. 2003. RAGE drives the development of glomerulosclerosis and implicates podocyte activation in the pathogenesis of diabetic nephropathy. Am. J. Pathol. 162:1123–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Renard, C., O. Chappey, M.P. Wautier, M. Nagashima, E. Lundh, J. Morser, L. Zhao, A.M. Schmidt, J.M. Scherrmann, and J.L. Wautier. 1997. Recombinant advanced glycation end product receptor pharmacokinetics in normal and diabetic rats. Mol. Pharmacol. 52:54–62. [DOI] [PubMed] [Google Scholar]
- 22.Chavakis, T., S.M. Kanse, R.A. Pixley, A.E. May, I. Isordia-Salas, R.W. Colman, and K.T. Preissner. 2001. Regulation of leukocyte recruitment by polypeptides derived from high molecular weight kininogen. FASEB J. 15:2365–2376. [DOI] [PubMed] [Google Scholar]
- 23.Chavakis, T., S.M. Kanse, F. Lupu, H.P. Hammes, W. Muller-Esterl, R.A. Pixley, R.W. Colman, and K.T. Preissner. 2000. Different mechanisms define the antiadhesive function of high molecular weight kininogen in integrin- and urokinase receptor-dependent interactions. Blood. 96:514–522. [PubMed] [Google Scholar]
- 24.Chavakis, T., A.E. May, K.T. Preissner, and S.M. Kanse. 1999. Molecular mechanisms of zinc-dependent leukocyte adhesion involving the urokinase receptor and β2-integrins. Blood. 93:2976–2983. [PubMed] [Google Scholar]
- 25.Lin, J.H., M. Wang, W.H. Andrews, R. Wydro, and J. Morser. 1994. Expression of efficiency of the human thrombomodulin-encoding gene in various vector and host systems. Gene. 147:287–292. [DOI] [PubMed] [Google Scholar]
- 26.Santoso, S., U.J. Sachs, H. Kroll, M. Linder, A. Ruf, K.T. Preissner, and T. Chavakis. 2002. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counter-receptor for the leukocyte integrin Mac-1. J. Exp. Med. 196:679–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chavakis, T., M. Hussain, S.M. Kanse, G. Peters, R.G. Bretzel, J.I. Flock, M. Herrmann, and K.T. Preissner. 2002. Staphylococcus aureus extracellular adherence protein (Eap) serves as anti-inflammatory factor by inhibiting the recruitment of host leukocytes. Nat. Med. 8:687–693. [DOI] [PubMed] [Google Scholar]
- 28.Constien, R., A. Forde, B. Liliensiek, H.J. Grone, P. Nawroth, G. Hammerling, and B. Arnold. 2001. Characterization of a novel EGFP reporter mouse to monitor Cre recombination as demonstrated by a Tie2 Cre mouse line. Genesis. 30:36–44. [DOI] [PubMed] [Google Scholar]
- 29.Bucciarelli, L.G., T. Wendt, W. Qu, Y. Lu, E. Lalla, L.L. Rong, M.T. Goova, B. Moser, T. Kislinger, D.C. Lee, et al. 2002. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation. 106:2827–2835. [DOI] [PubMed] [Google Scholar]
- 30.Ploplis, V.A., E.L. French, P. Carmeliet, D. Collen, and E.F. Plow. 1998. Plasminogen deficiency differentially affects recruitment of inflammatory cell populations in mice. Blood. 91:2005–2009. [PubMed] [Google Scholar]
- 31.Sligh, J.E., C.M. Ballantyne, S.S. Rich, H.K. Hawkins, C.W. Smith, A. Bradley, and A.L. Beaudet. 1993. Inflammatory and immune responses are impaired in mice deficient in intercellular adhesion molecule 1. Proc. Natl. Acad. Sci. USA. 90:8529–8533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Newton, R.A., and N. Hogg. 1998. The human S100 protein MRP-14 is a novel activator of beta 2 integrin Mac-1 on neutrophils. J. Immunol. 160:1427–1435. [PubMed] [Google Scholar]
- 33.Schmidt, A.M., M. Hasu, D. Popov, J.H. Zhang, J. Chen, S.D. Yan, J. Brett, R. Cao, K. Kuwabara, G. Costache, et al. 1994. RAGE has a central role in vessel wall interactions and gene activation in response to AGEs. Proc. Natl. Acad. Sci. USA. 91:8807–8811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kislinger, T., N. Tanji, T. Wendt, W. Qu, Y. Lu, L.J. Ferran, Jr., A. Taguchi, K. Olson, L. Bucciarelli, M. Goova, et al. 2001. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 21:905–910. [DOI] [PubMed] [Google Scholar]
- 35.Sakaguchi, T., S.F. Yan, S.D. Yan, D. Belov, L.L. Rong, M. Sousa, M. Andrassy, S.P. Marso, S. Duda, B. Arnold, et al. 2003. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J. Clin. Invest. 111:959–972. [DOI] [PMC free article] [PubMed] [Google Scholar]