A primordial RNA modification enzyme: the case of tRNA (m1A) methyltransferase (original) (raw)
Abstract
The modified nucleoside 1-methyladenosine (m1A) is found in the T-loop of many tRNAs from organisms belonging to the three domains of life (Eukaryota, Bacteria, Archaea). In the T-loop of eukaryotic and bacterial tRNAs, m1A is present at position 58, whereas in archaeal tRNAs it is present at position(s) 58 and/or 57, m1A57 being the obligatory intermediate in the biosynthesis of 1-methylinosine (m1I57). In yeast, the formation of m1A58 is catalysed by the essential tRNA (m1A58) methyltransferase (MTase), a tetrameric enzyme that is composed of two types of subunits (Gcd14p and Gcd10p), whereas in the bacterium Thermus thermophilus the enzyme is a homotetramer of the TrmI polypeptide. Here, we report that the TrmI enzyme from the archaeon Pyrococcus abyssi is also a homotetramer. However, unlike the bacterial site-specific TrmI MTase, the P.abyssi enzyme is region-specific and catalyses the formation of m1A at two adjacent positions (57 and 58) in the T-loop of certain tRNAs. The stabilisation of P.abyssi TrmI at extreme temperatures involves intersubunit disulphide bridges that reinforce the tetrameric oligomerisation, as revealed by biochemical and crystallographic evidences. The origin and evolution of m1A MTases is discussed in the context of different hypotheses of the tree of life.
INTRODUCTION
All types of cellular RNAs contain modified nucleosides, but the largest number and greatest variety are found in transfer RNAs (tRNAs) (1). Modifications consist of simple chemical alterations of nucleosides (e.g. methylation of base or ribose, base isomerisation, reduction, thiolation or deamination) or more complex hypermodifications. The type of chemical alteration of a nucleoside as well as the pattern of tRNA modification depends on the origin of the tRNA molecule (2). Nevertheless, few modified nucleosides are commonly found in tRNAs from all three biological domains (Eukaryota, Bacteria and Archaea) (3,4), suggesting a very ancient origin of the corresponding modification enzymes (5).
Only a limited number of RNA modification enzymes have been biochemically characterised, and most of these are from Escherichia coli and Saccharomyces cerevisiae. Moreover, of those enzymes characterised, only a fraction have been studied in enough detail to reveal the specificity and mechanism of the corresponding reaction (reviewed in 6). Besides, for some known enzymes, the corresponding genes remain unknown (for a recent review, see 7). From the emerging genomic sequencing data, homologues of known RNA modification enzymes can be predicted, and the evolutionary history and emergence of the RNA maturation machinery can be inferred (see for example 8–10). Significantly, some theoretical predictions of enzymatic activities or specificities have turned out to be inaccurate or even wrong, causing mis-annotations in the database, which has led to confusion and further proliferation of erroneous homology-based functional predictions (11). Therefore, in silico predictions of new putative RNA-modification enzymes (and especially of their molecular and cellular functions) have to be carefully validated by in vivo and in vitro characterisation of the respective proteins.
Among enzymes that are expected to be representative of the most ancient tRNA-modification enzymes, are those that catalyse the formation of universally encountered methylated nucleosides m1G, m5U, m1A and each of the three 2′-_O_-methylated nucleosides Um, Cm and Gm. It was found that the archaeal and eukaryotic tRNA (m1G37) MTase (Trm5p) is unrelated to the bacterial iso-specific enzyme (TrmD). Trm5p belongs to the ‘classical’ Rossmann fold MTases (RFM) superfamily (9), while TrmD belongs to evolutionarily and structurally distinct SPOUT superfamily (8). Moreover, the tRNA (m1G9) MTase (Trm10p) from S.cerevisiae, catalysing the same chemical reaction but at another position of the tRNA molecule, was found to share similarity neither with Trm5p nor TrmD (12). Therefore, it seems that the three tRNA (m1G) MTases (Trm5p, TrmD and Trm10p) evolved their common function by convergence rather than by divergence from a common ancestor.
Clouet d'Orval et al. (13) reported that the formation of two 2′-_O_-ribose methylated nucleosides in the anticodon stem and loop of archaeal tRNATrp is carried out by a C/D-box RNA-guided MTase (a protein without intrinsic specificity). However, the 2′-_O_-methylation of two nucleosides in the anticodon loop of yeast tRNATrp (positions 32 and 34) is catalysed by the region-specific, non-guided MTase Trm7p, a member of the RFM superfamily (14). Likewise, 2′-_O_-methylation of a guanosine at position 18 in bacterial (E.coli and Thermus thermophilus) and eukaryotic (S.cerevisiae) tRNAs are catalysed by non-guided, site-specific tRNA MTases (TrmH and Trm3p, respectively) which belong to the SPOUT superfamily (9). Surprisingly, it was found that RrmJ-like site-specific MTases and C/D-box RNA-guided MTases evolved from a common RFM ancestor (10). They share the same overall structure and possess very similar active sites.
From the few examples selected above of enzymes catalysing what was thought to be the primordial set of modified nucleosides (5), it now appears that the present-day tRNA modification machinery is more diverse and complex than initially thought. On the one hand, the same modification can be carried out by unrelated enzymes, whereas, on the other hand, orthologous enzymes (i.e. ‘the same enzymes in different species’) can exhibit different specificity. The elucidation of the origin and evolution of the RNA modification machinery will obviously require detailed evolutionary and functional studies of many modification enzymes (orthologues and paralogues, and even completely unrelated proteins) from the three domains of life.
In the present work, we focus our attention on the enzyme that catalyses the formation of 1-methyladenosine (m1A) in the T-loop of tRNA. m1A is found at seven different positions (8, 9, 14, 22 and 58) in tRNAs sequenced so far (2). However, only m1A58 in the T-loop has been found in tRNAs from organisms belonging to the three domains of life. In archaeal tRNAs, m1A is only found at position 58 but is also formed at position 57, m1A57 being the obligatory intermediate in the two-step biosynthesis of 1-methylinosine (m1I57; 15). In S.cerevisiae, the formation of m1A58 is catalysed by the essential tRNA (m1A58) MTase, a tetrameric enzyme that is composed of two types of evolutionary related subunits (Gcd10p and Gcd14p) (16). One subunit (Gcd10p) is essential for the binding of the tRNA substrate while the other subunit (Gcd14p) is responsible for AdoMet-binding and catalysis of the methyltransfer reaction (17).
Recently, we cloned, expressed and biochemically characterised a Gcd14p orthologue from the hyperthermophilic bacterium T.thermophilus (18). The purified recombinant enzyme (called TrmI) is a homotetramer and catalyses the site-specific formation of m1A at position 58 of the T-loop of tRNA in the absence of any other complementary protein. In this work, we report the identification of the archaeal Gcd14p/TrmI orthologue. We characterised key features that distinguish this enzyme from its homologues from the other two biological domains. These results will be discussed in the framework of the evolutionary origin of tRNA (m1A) MTases as well as the strategy used by the archaeal TrmI protein to resist heat inactivation at extreme temperatures.
MATERIALS AND METHODS
Strains, media, growth conditions and general procedures
Restriction endonucleases and T4-DNA ligase were purchased from Roche Diagnostics, except when otherwise indicated. Oligonucleotides were synthesised by Invitrogen. The Pyrococcus furiosus Vc1 (DSM 3638) strain was kindly provided by K.O.Stetter (Regensburg, Germany). Pyrococcus abyssi GE5 genomic DNA was a kind gift of R.Cunin (Brussels). Escherichia coli (strain MC1061) and T.thermophilus (strains HB27 and RD1) total tRNA was prepared as described (19). Protein concentrations were measured using the Bio-Rad protein assay, using bovine serum albumin as a standard.
Cloning of the P.abyssi PAB0283 open reading frame (ORF)
The P.abyssi trmI gene was amplified by PCR from P.abyssi GE5 genomic DNA (20) using the ML-82 (tatcatatgataagggaaggggataaggtagtt) and ML-99 (tatctcgagaattctccttgcgaaagttatgtaacca) oligonucleotides and Pwo DNA polymerase (Roche Diagnostics). The 774 bp amplified product was cloned into the SmaI site of pUC18 vector, giving the pML46 plasmid. The 761 bp NdeI–XhoI fragment of pML46 was then cloned between the NdeI and XhoI restriction sites of the pET30b expression vector (Novagen) resulting in the pML52 plasmid. This plasmid allowed T7 expression in E.coli of the P.abyssi TrmI protein bearing a C-terminal His-tag.
Expression and purification of the recombinant P.abyssi PAB0283 protein
The His-tagged P.abyssi TrmI protein was expressed in E.coli and purified essentially as described for the T.thermophilus TrmI protein (18). The E.coli strain Rosetta (DE3) (Novagen) was transformed by the pML52 plasmid. Transformed cells were grown at 37°C in 2 l of LB supplemented with kanamycin to an optical density at 660 nm (OD660) of 0.5. At this stage, IPTG (isopropylthiogalactopyranoside; Roche Diagnostics) was added up to a final concentration of 1 mM to induce recombinant protein expression. Cells were harvested after 3 h incubation at 37°C and resuspended in 100 ml of buffer A (50 mM Tris–HCl, pH 8.5, 500 mM KCl). The cells were lysed by a 30 min sonication at 4°C using a Vibracell 75041 sonicator (40% amplitude). Cell debris was removed by centrifugation (20 000 g during 30 min) and the cleared lysate was applied to a column of Chelating Sepharose Fast Flow (1× 30 cm; Amersham Biosciences) charged with Ni2+. The column was washed with buffer A and the adsorbed material was eluted with a linear gradient (750 ml; from 0 to 1.0 M) of imidazole in buffer A. The fractions containing the P.abyssi TrmI protein were pooled and dialysed against buffer A supplemented with 200 mM imidazole to keep the protein soluble at high concentration (5 mg/ml). The final yield was estimated to 50 mg of purified protein per litre of culture. Aliquots (200 µl) of the resulting preparation (5 mg protein/ml) were flash-frozen in liquid nitrogen and stored at –80°C.
Site-directed mutagenesis of the PAB0283 ORF
The C196S and C233S mutants of the PAB0283 ORF were obtained with the QuickChange site-directed mutagenesis kit (Stratagene) using pML46 as a template. The mutants were sequenced to confirm that they only contain the desired substitutions and no other mutations. The mutated genes were then subcloned into the pET30b vector to allow expression of the corresponding proteins in E.coli.
Pyrococcus furiosus S30 extract
One litre of P.furiosus Vc1 (DSM 3638) cells cultivated to ∼2 × 108 cells/ml in the complex medium described by Legrain et al. (21) were harvested and resuspended in 10 ml buffer S (10 mM Tris–acetate, pH 8.0, 14 mM Mg acetate, 60 mM K acetate, 0.1 mM DTT). Cells were crushed in a mortar in the presence of cold alumine (twice the weight of the cells). Broken cells were resuspended in 1.5 ml buffer S per gram of cells and centrifugated during 30 min at 30 000 g. The supernatant was recentrifugated in the same conditions and then dialysed during 4 h against cold buffer S. The protein concentration in the extract was 50 mg/ml. The extract was stored at –80°C in 500 µl aliquots.
T7 in vitro transcription of tRNA genes
The general procedure for generating in vitro transcripts of tRNA genes is based on the method described previously (22). Plasmids pML1 and pHG1 allowing T7 transcription of respectively T.thermophilus tRNAAsp and yeast tRNAAsp were described previously (18,23). Radioactive (32P) in vitro transcripts were obtained using MvaI-digested plasmids as templates. [α-32P]ATP, [α-32P]GTP and [α-32P]UTP were purchased from ICN Biomedicals and T7-RNA polymerase was purchased from Roche Diagnostics. Radioactive transcripts were purified by 10% polyacrylamide gel electrophoresis.
tRNA MTase assays
The two types of tRNA MTase assays used in this work were described by Droogmans et al. (18). The first method consisted measuring the amount of 14C transferred to total E.coli or T.thermophilus tRNA using [methyl-14C]AdoMet as the methyl donor. The reaction mixture (300 µl) consisted of 50 mM Tris–HCl, 10 mM MgCl2, 100 µg total tRNA, 25 nCi [methyl-14C]AdoMet (50 mCi/mmol; Amersham Biosciences) and enzyme. The second type of tRNA MTase assay involved in vitro transcribed, 32P-labelled tRNAs as substrates (24). Modified nucleotides were analysed by 2D-thin layer chromatography (TLC) on cellulose plates (Merck). First dimension developed with solvent A (isobutyric acid/concentrated NH4OH/water; 66/1/33; v/v/v); second dimension developed with solvent B [0.1 M sodium phosphate pH 6.8/(NH4)2SO4/_n_-propanol; 100/60/2; v/w/v]. The nucleotides were identified using a reference map (25).
Crystallisation, data collection and structure determination
The purified His-tagged P.abyssi TrmI protein samples were concentrated to ∼10 mg/ml by ultrafiltration (YM30, Amicon) and the protein concentration was estimated by UV absorption. Crystallisation trials were performed at 20°C by the sitting-drop vapour diffusion method using 24-well tissue-culture VDX plates (Hampton Research). Initial searches for crystallisation conditions were performed using the standard sparse-matrix crystal screens (26) from Hampton Research (Crystal Screen I & Crystal Screen II). Crystals appeared in the presence of ammonium sulfate and at acidic pH. These conditions were refined and the best crystals were obtained in 200 mM ammonium sulfate, 100 mM sodium acetate pH 4.6, 25% (w/v) polyethylene glycol 4000. In each trial, a sitting drop of 4 µl of protein solution (in 50 mM Tris–HCl, pH 8.5, 500 mM KCl, 200 mM imidazole) mixed with 4 µl of well solution was equilibrated against a reservoir containing 500 µl of well solution. The crystals grew to their full size (∼0.25 × 0.10 × 0.10 mm) in about 3 days.
A suitable crystal was flash frozen and diffraction data were collected on a MAR CCD (165 mm) detector (Marresearch) on beam BM30 at the European Synchrotron Radiation Facilities (Grenoble, France). A complete data set was collected at a wavelength of 0.9793 Å to a maximum resolution of 2.66 Å. Data were processed with the programs DENZO and SCALEPACK (27). The crystal displays orthorhombic symmetry, with unit-cell parameters a = 126.98 Å, b = 65.81 Å, c = 151.45 Å. The structure was solved by molecular replacement and refined with CNS (28) against the data set at 2.66 Å using cross-validated maximum likelihood as the target function. The structure was inspected using Turbo-Frodo (29). Refinement of the structure is under way and will be presented in detail elsewhere.
RESULTS
The P.abyssi PAB0283 ORF encodes an MTase involved in the formation of m1A 58 in tRNA
The P.abyssi PAB0283 ORF encoding a 30 kDa Gcd14p/TrmI homologue (30) was PCR-amplified and cloned into the pET30b expression vector, allowing the production of a C-terminal His-tagged protein in E.coli. The His-tagged protein was purified to quasi-homogeneity by immobilised metal ion affinity chromatography (Fig. 1A).
Figure 1.
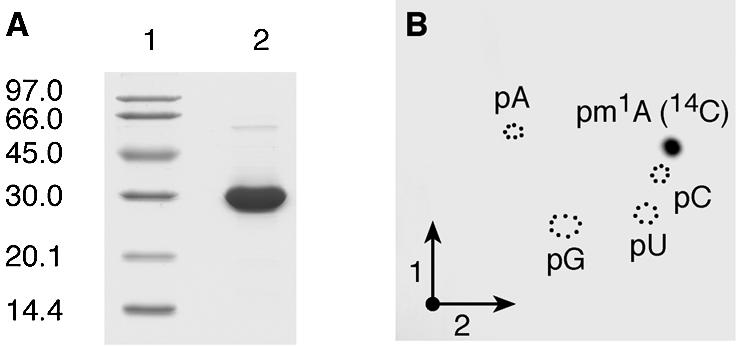
Affinity-purified P.abyssi PAB0283 protein catalyses the formation of m1A in E.coli tRNA in vitro. (A) SDS–PAGE analysis under reducing conditions of the purified P.abyssi PAB0283 protein. Lane 1, molecular weight marker (Pharmacia-Biotech). Lane 2, 5 µg of purified protein. (B) Autoradiography of a 2D-chromatogram of 5′-phosphate nucleosides on thin layer cellulose plate. Total (bulk) E.coli tRNA (50 µg) was incubated in the presence of [methyl-14C]AdoMet and 5 µg of the purified P.abyssi PAB0283 protein as described in Materials and Methods. After 30 min incubation at 60°C, the tRNA was recovered, digested by nuclease P1 and the resulting nucleotides were analysed by 2D-TLC on a cellulose plate (see Materials and Methods). Circles in dotted lines show the migration of the four canonical nucleotides used as u.v. markers.
Unfractionated (bulk) E.coli tRNA was used as substrate to test the tRNA (m1A) MTase activity of the P.abyssi PAB0283 protein in vitro. Escherichia coli tRNAs do not contain m1A and the universally conserved A58 is not known to be modified in the tRNAs from this organism (2). The purified recombinant protein was incubated at 60°C for 30 min with [methyl-14C]AdoMet and unfractionated E.coli tRNA. After incubation, the tRNA was recovered by phenol extraction and ethanol precipitation and completely hydrolysed into 5′-phosphate nucleosides by nuclease P1. The resulting hydrolysate was analysed by 2D-TLC followed by autoradiography. The result shown in Figure 1B revealed the presence of a single radioactive compound with migration characteristics identical to that of 1-methyladenosine 5′-phosphate (pm1A) according to the reference map published by Keith (25). Moreover, unfractionated tRNA from the T.thermophilus RD1 strain lacking TrmI (and thus lacking m1A58 in tRNA) (18) was a very good substrate of the purified P.abyssi enzyme whereas tRNA obtained from the T.thermophilus wild type (WT) strain accepted ∼20–30% of the methyl groups compared to the tRNA extracted from the mutant strain. This contrasts with the T.thermophilus TrmI enzyme which does not incorporate any significant amounts of methyl groups in tRNA extracted from WT T.thermophilus (18). These results indicate that the P.abyssi enzyme is involved in the formation of m1A58 and suggest that the enzyme could also act on (an)other position(s) in the tRNA (see below).
Further evidence for the formation of m1A58 catalysed by the PAB0283 protein was obtained using a second type of experiments. The purified enzyme was incubated under identical experimental conditions as above but with either an [α-32P]ATP or an [α-32P]GTP labelled precursor tRNA substrate obtained after in vitro transcription by T7-RNA polymerase of a synthetic T.thermophilus tRNAAsp gene. This transcript was already successfully used in a previous work as substrate of the T.thermophilus TrmI MTase (18). After the incubation with purified TrmI, the formation of m1A in the T.thermophilus tRNAAsp was analysed by 2D-TLC. The [α-32P]ATP-labelled tRNAAsp was completely hydrolysed with nuclease P1 to generate 5′-phosphate nucleosides with the 32P-phosphate present only in 5′-phosphate adenosine and 5′-phosphate adenosine derivatives (Fig. 2A; panel ATP/P1), while the [α-32P]GTP-labelled tRNA was digested with RNase T2, thus generating the different 3′-phosphate nucleosides of which only those that were 5′-adjacent to G in the tRNA sequence harboured a 32P-radiolabelled phosphate (nearest neighbours analysis; Fig. 2A; panel GTP/T2). The results show the presence of m1A, 5′-adjacent to a G in the T.thermophilus tRNAAsp sequence after incubation with purified PAB0283 protein. Note that hydrolysis by RNase T2 resulted in the accumulation of 1-methyladenosine 2′-3′ cyclic phosphate (m1A>p; an intermediate in the hydrolysis reaction) because the presence of this modification inhibits the action of RNAse T2, as has already been reported (18). Quantification of the relative amount of 32P in the different radioactive spots on the TLC plates, revealed that ∼1 mol of m1A is formed per mole of tRNA after 60 min incubation at 60°C.
Figure 2.

Characterisation of the tRNA MTase activity of recombinant PAB0823 protein using different tRNA substrates. Radiolabelled T7 in vitro transcripts of WT T.thermophilus tRNAAsp (A), mutant T.thermophilus tRNAAsp (G57A) (B) and WT yeast tRNAAsp (C) were incubated for 1 h at 60°C in the presence of 20 µg of the purified recombinant PAB0283 protein. After the incubation, the different tRNA transcripts were digested by nuclease P1 or RNAse T2 and the resulting nucleotides were analysed by 2D-TLC on cellulose plates and autoradiography. The nature of the labelled triphosphate nucleoside and the enzyme used to hydrolyse the transcripts are given above each chromatogram. Circles in dotted lines show the migration of the canonical nucleotides used as u.v. markers. m1A>p is for 1-methyladenosine 2′-3′ cyclic phosphate. A schematic representation of the T-loop of the different tRNA substrates is given on the left of each series of chromatograms.
These results show that the P.abyssi PAB0283 protein catalyses the formation of m1A at position 58 in the T-loop of tRNAs in the absence of any other polypeptide. Thus the PAB0283 protein has been renamed P.abyssi TrmI and the corresponding gene trmI.
The P.abyssi TrmI MTase displays region specificity
The modified nucleoside m1I is found at position 57 of archaeal tRNAs (2). The biosynthesis of m1I at this position of tRNA occurs via a two-step enzymatic process: a first step in which m1A57 is formed by an AdoMet-dependent tRNA MTase followed by the deamination of the 6-amino group of the adenosine moiety (15). To determine whether the P.abyssi TrmI enzyme catalyses the formation of m1A57 in addition to m1A58, a T.thermophilus tRNAAsp mutant in which G57 is mutated to A [T.thermophilus tRNAAsp(G57A)] was constructed by site-directed mutagenesis. The T7 transcript of T.thermophilus tRNAAsp(G57A) was radiolabelled with [α-32P]ATP or [α-32P]GTP. The labelled transcripts were incubated at 60°C in the presence of the purified P.abyssi TrmI enzyme and AdoMet, after which they were digested by nuclease P1 or RNase T2. The resulting 32P-labelled nucleotides were analysed by 2D-TLC as above and individual nucleotides quantified by scintillation counting. The results show that 1.25 mol of m1A was formed per mol of tRNA under these experimental conditions (Fig. 2B, panel ATP/P1). Interestingly, RNase T2 digestion of the incubated [α-32P]ATP or [α-32P]GTP-labelled T.thermophilus tRNAAsp (G57A) revealed that m1A is formed at two positions, one 5′-adjacent to a G (position 58) and another 5′-adjacent to an A (Fig. 2B, panels GTP/T2 and ATP/T2). We reasoned that since m1A formation is not observed after RNase T2 digestion of the TrmI-modified, [α-32P]ATP-labelled WT T.thermophilus tRNAAsp (Fig. 2A, panel ATP/T2), the presence of m1A in the RNAse T2 hydrolysate of [α-32P]ATP-labelled mutant tRNAAsp (G57A) indicates that A57 is being modified in the mutant T.thermophilus tRNAAsp by the P.abyssi TrmI enzyme. Interestingly, the efficiency of m1A formation at position 57 is considerably higher (1.0 mol per mol tRNA) than at position 58 (0.25 mol per mol tRNA).
To obtain further evidence that the P.abyssi TrmI enzyme can methylate A57, a radiolabelled yeast tRNAAsp was tested as a substrate for the purified enzyme. This particular tRNA was chosen because it was previously reported that it is a good substrate for the enzymes involved in m1I57 formation present in a crude P.furiosus extract (31). The T7 transcript of yeast tRNAAsp was radiolabelled with [α-32P]ATP or [α-32P]UTP and the labelled transcripts were incubated as above with the purified P.abyssi TrmI enzyme and AdoMet. The incubated transcripts were then digested by nuclease P1 or RNase T2 and the resulting 32P-labelled nucleotides were analysed by 2D-TLC. The results show that 1.0 mol m1A is formed per mol tRNA under these conditions (Fig. 2C, panel ATP/P1). RNase T2 digestion of the incubated [α-32P]UTP or [α-32P]ATP-labelled yeast tRNAAsp revealed that m1A is efficiently formed at a position 5′-adjacent to an A (Fig. 2C, panel ATP/T2), while only trace amounts of m1A are formed at a position 5′-adjacent to a U (Fig. 2C, panel UTP/T2). Again, these results provide evidence that the P.abyssi TrmI enzyme catalyses the formation of m1A57 in tRNA.
In order to determine whether the m1A formed by the P.abyssi TrmI enzyme in yeast tRNAAsp is an intermediate in m1I57 biosynthesis, [α-32P]ATP-labelled yeast tRNAAsp, modified by P.abyssi TrmI was incubated in a P.furiosus crude extract for different periods of time and subsequently tested for the conversion of m1A57 into m1I57. The results presented in Figure 3 (A and C) show that m1I is formed in the TrmI-premethylated yeast tRNAAsp incubated in the P.furiosus extract, at the same time the amount of m1A is being reduced. As a negative control, WT T.thermophilus tRNAAsp (with A58 and G57) premodified by the P.abyssi TrmI enzyme (thus containing m1A only at position 58; see Fig. 2A) was incubated in the P.furiosus crude extract. Even after 60 min of incubation at 60°C, no m1I formation was observed (see Fig. 3B and D).
Figure 3.

A deaminase present in a crude P.furiosus extract transforms m1A57 preformed in yeast tRNAAsp by the P.abyssi TrmI enzyme into m1I57 and does not deaminate the m1A58 preformed in T.thermophilus tRNAAsp. [α-32P]ATP-labelled yeast tRNAAsp (A and C) and [α-32P]ATP- labelled T.thermophilus tRNAAsp (B and D) were incubated for 1 h at 60°C in the presence of the purified P.abyssi TrmI enzyme and of AdoMet. The transcripts were then recovered and incubated in a crude P.abyssi S30 extract for different periods of time as shown. At the end of the incubation time the transcripts were recovered, digested by nuclease P1 and analysed by 1D-TLC using solvent B (see Materials and Methods) on cellulose plates followed by autoradiography (A and B). (C) and (D) correspond to the autoradiograms of 2D-TLC of the samples incubated for 60 min in the presence of the P.furiosus extract.
These data demonstrate that the P.abyssi TrmI enzyme can methylate adenosines at positions 57 and 58 in tRNA. Furthermore, m1A57 formed in yeast tRNAAsp by P.abyssi TrmI served as substrate for the P.furiosus deaminase that converts m1A57 to m1I57.
The tetramerisation of the P.abyssi TrmI MTase involves intersubunit disulphide bridges
Sequence analysis of the P.abyssi TrmI protein and its homologues revealed a pattern of characteristic MTase motifs and significant similarity to the known structure of Rv2118c from Mycobacterium tuberculosis (32). A 3D homology model of the P.abyssi TrmI tetramer was constructed, based on the structure of Rv2118c (data not shown). The model revealed that two residues located near the dimer–dimer interface of the Rv2118c tetramer (T205 and A241), were substituted by Cys196 and Cys233 residues in the P.abyssi protein. Moreover, these two Cys residues are conserved among the three Pyrococcus species sequenced so far (P.abyssi, P.furiosus and Pyrococcus horikoshii; Fig. 4). In the 3D homology model of the P.abyssi TrmI tetramer residues C196 and C233 from different TrmI monomers are spatially close (<5 Å, data not shown), which has led us to speculate that they may form a disulphide.
Figure 4.

Sequence alignment of a few selected tRNA (m1A) MTases from Archaea, Bacteria and Eukaryota (for more extensive alignment with many other tRNA (m1A) MTases, see 30). The size of insertions in S.cerevisiae Gcd14p omitted for clarity is indicated in parentheses. Highly conserved residues are shown on a black background and residues with a similar physico-chemical character are on a grey background. The most conserved motifs among those typical for the RFM superfamily of MTases (51) are indicated.
Experimental evidence for intersubunit disulphide bridges in recombinant P.abyssi TrmI was obtained by SDS–PAGE analysis performed under non-reducing conditions and by comparing the molecular mass of the protein to that observed under reducing conditions (Fig. 5). In the absence of a reducing agent, the molecular mass observed for the TrmI protein is ∼60 kDa (lane 1). This exactly corresponds to twice the molecular mass of the protein analysed under reducing conditions (30 kDa; lane 6). Based on the theoretical model described above, the presence of such a dimer in non-reducing SDS–PAGE analysis reflects the involvement of disulphide bridges in tetramerisation.
Figure 5.
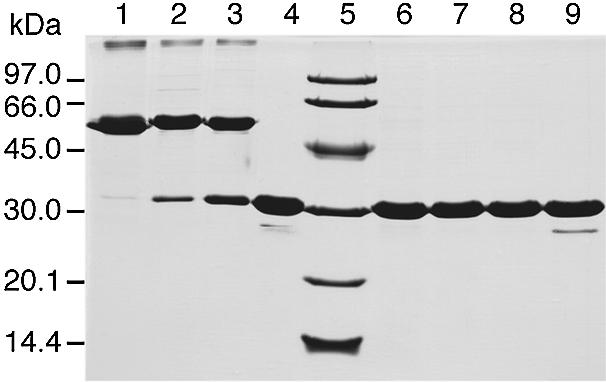
SDS–PAGE analysis under reducing or non-reducing conditions of the P.abyssi TrmI protein demonstrates the existence of intersubunit disulphide bridges. Before loading, protein samples corresponding to the WT TrmI protein (lanes 1 and 6), C196S mutant (lanes 2 and 7), C233S mutant (lanes 3 and 8) and C196S+C233S double mutant (lanes 4 and 9) were incubated in the presence (lanes 6–9) or absence (lanes 1–4) of 100 mM β-mercaptoethanol for 5 min at 100°C. The molecular mass markers (Pharmacia-Biotech) were loaded into lane 5.
Further evidence for intersubunit disulphide bridges in the P.abyssi TrmI protein was provided by X-ray crystallographic analysis of the protein. The protein was crystallised under the conditions described in Materials and Methods. The homology model of tetramer of P.abyssi TrmI (see above) served as a search model for molecular replacement and led to a reasonable starting structure for analysis of the crystallographic data. The presence of one tetramer in the asymmetric unit is consistent with a calculated Matthews coefficient of 2.74 (solvent content = 54.8 %) assuming four tetramers in the unit cell [a = 126.98 Å, b = 65.81 Å, c = 151.45 Å and P222 symmetry (P212121 space group)]. The initial electron density maps obtained after rigid body refinement and simulated annealing were very clean for the large catalytic C-terminal domain of the protein, which is responsible for binding the AdoMet cofactor and transferring the methyl group from AdoMet to the tRNA. The small N-terminal extremity (residues 1–70) was not well defined in the electron density. This probably reflects reorganisation of this domain compared to the quaternary structure of Rv2118c from M.tuberculosis. The quality of the present model derived from the crystal of TrmI is sufficient to unambiguously define the geometry of the intersubunit interfaces and interactions between residues C196 and C233 from different TrmI monomers (C196 and C233 are located in the well-resolved C-terminal domain).
The catalytic domain of each subunit (residues 70–250) is a modified Rossmann fold consisting of a central seven-stranded β-sheet, flanked by α-helices on both sides (Fig. 6). The first five strands of the β-sheet are parallel. The last two strands are antiparallel. Those two strands and the loop regions connecting them protrude out of the structure to form an oligomerisation interface. Indeed, the extended strand β6, that contains Cys233, packs against the same strand from a second subunit, making an antiparallel β-sheet structure. This dimeric assembly of the protruded region further packs with the two C-terminal antiparallel β-sheets formed by two other subunits forming a central β-barrel structure (Fig. 6). This quaternary arrangement of the subunits that consists of a dimer of tight dimers is mainly stabilised by hydrophobic interactions between monomers that contribute significantly to the stability of the dimer. Additional stabilisation of the tetrameric form is obtained by four disulphide bridges between residues Cys196 and Cys233 from different subunits (Fig. 6).
Figure 6.
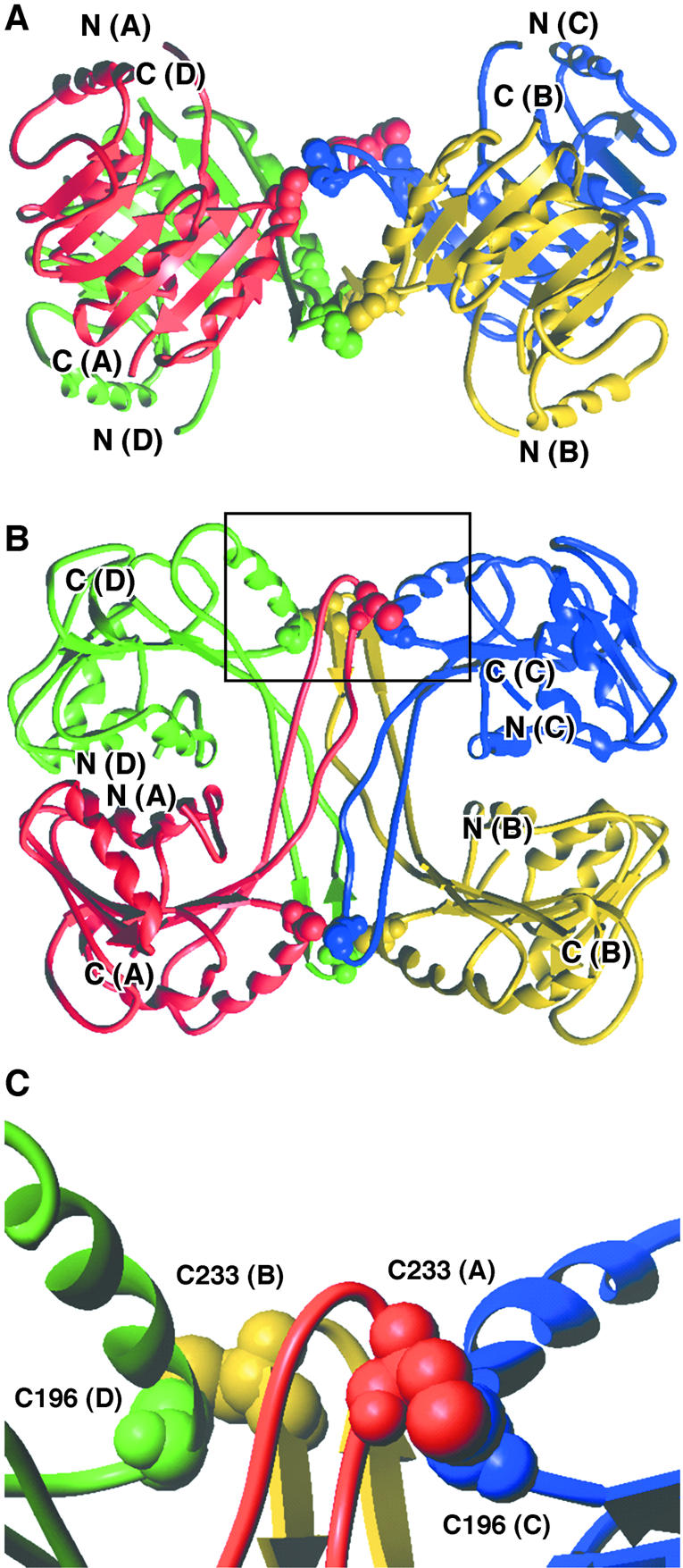
Crystal structure of the catalytic domains (residues 70–250) of the P.abyssi TrmI tetramer. The different monomers are colour-coded as follows: chain A, red; chain B, yellow; chain C, blue; chain D, green. Cysteine residues are shown in CPK representation. (A and B) Two perpendicular ribbon diagrams, showing secondary structure elements. (C) Detailed view of the area delimited in (B) showing disulphide bridges existing between C196 and C233 of different monomers. These disulphide bridges connect C196(C) to C233(A) and C196(D) to C233(B). Another set of symmetry- related disulphide bridges connect C196(A) to C233(C) and C196(B) to C233(D).
Intersubunit disulphide bridges stabilise the P.abyssi TrmI MTase at extreme temperatures
To determine whether the intersubunit disulphide bridges involving Cys196 and Cys233 are important for stability and/or function of the P.abyssi TrmI enzyme, mutants were constructed in which the Cys residues at positions 196 and 233 were replaced by Ser, whose side-chain is isosteric to Cys, but does not form covalent bonds. The single mutants C196S and C233S and the double mutant (C196S+C233S) were constructed by site-directed mutagenesis. Each TrmI mutant was purified following the same procedure used to purify the WT enzyme. The migration profiles of the TrmI mutants were analysed by SDS–PAGE under reducing and non-reducing conditions and compared to the WT enzyme. Under reducing conditions, the three enzymes migrated as monomers on SDS–PAGE (see Fig. 5, lanes 7–9). Under non-reducing conditions, both single mutants appear as a mixture of dimers and monomers (Fig. 5, lanes 2 and 3). Interestingly, while the electrophoretic behaviour of the individually mutated TrmI proteins was sensitive to the presence of the reducing agent, the purified double mutant TrmI migrated as a monomer under reducing and non-reducing conditions on SDS–PAGE (see Fig. 5, lanes 4 and 9). This last result is consistent with the absence of intersubunit disulphide bridges in the double mutant TrmI.
The native states of the mutant P.abyssi TrmI enzymes were analysed by molecular sieving and compared to the elution profile of the WT enzyme. Compared to the tetrameric state of the WT (Fig. 7A), the double mutant behaved predominantly as a dimer (Fig. 7D). The single mutants, however, appeared to form high molecular weight aggregates (Fig. 7B and C). This could explain their sensitivity towards the reducing agent during SDS–PAGE analysis as reported above. Since the single mutants form aggregates they were not analysed further.
Figure 7.

Estimation of the apparent molecular mass of the purified WT P.abyssi TrmI protein (A) and of the C196S (B), C233S (C) and C196S+C233S (D) mutants by gel filtration chromatography on a Superdex 200 prep grade 16/60 column (Pharmacia Biotech). The samples consisted of 2.5 mg of purified WT or mutant TrmI proteins in 50 mM Tris–HCl pH 8.5, 500 mM KCl, 200 mM imidazole. Elution was performed with the same buffer. The molecular masses of the proteins were calculated using a standard consisting of carbonic anhydrase from bovine erythrocytes (29 kDa), bovine serum albumin (66 kDa), bovine serum albumin dimer (132 kDa) and β-amylase from sweet potato (200 kDa).
The activity of the WT P.abyssi TrmI enzyme and of the double C196S+C233S mutant were compared. At 70°C (the optimal temperature of the WT enzyme in these assay conditions) both enzymes showed an equal specific activity (∼80 nmol of methyl groups transferred to E.coli total tRNA per hour per mg of pure enzyme). To measure the resistance to irreversible thermodenaturation of WT and C196S+C233S mutant P.abyssi TrmI enzymes, reflecting their thermostability, these enzymes were preincubated for different periods of time at 80 and 85°C. The residual MTase activity of each enzyme after the heat treatment was measured at 70°C, using total E.coli tRNA as substrate and [14C-methyl]AdoMet as methyl donor. The preincubation at 80°C for 1 h did not affect the activity of the WT or the mutant enzymes (Fig. 8A). However, increasing the preincubation temperature by only 5°C drastically affected the activity of the C196S+C233S double mutant (see Fig. 8B). These results show that the intersubunit disulphide bridges formed between Cys196 and Cys233 are important for the stability of the enzyme at extreme temperatures.
Figure 8.

Resistance of the WT P.abyssi TrmI enzyme and of the C196S+C233S mutant to thermodenaturation. The WT (open circles) and mutant (open squares) enzymes (400 µg/ml) were heated for different periods of time at 80 (A) or 85°C (B) in Tris–HCl 50 mM, MgCl2 10 mM. The remaining activity was measured at 70°C using total E.coli tRNA as substrate, [14C-methyl]AdoMet as methyl donor, and 10 µl of the preheated protein solution.
DISCUSSION
Systematic comparative sequence analysis of archaeal proteins with their bacterial and eukaryotic counterparts reveals that archaeal proteins involved in central metabolism, metabolite uptake and cell wall biosynthesis are generally similar to their bacterial homologues, whereas proteins involved in DNA replication and gene expression are often more closely related to their eukaryotic homologues (for reviews see 33,34). In particular, many components of the RNA modification apparatus are often more similar between Archaea and Eukaryota (9).
Considering the tRNA-substrates themselves, some structural features are shared between Archaea and Eukaryota, while others are shared between Archaea and Bacteria. Apparently, no tRNA features common to Eukaryota and Bacteria seem to exist that are not also shared by Archaea (35). From this observation and also from the systematic comparison of tRNA genes from organisms representing all three domains of life, where the genome has been completely sequenced (36), it appears that life might have arisen from the archaeal domain. Alternatively, as proposed by Cavalier-Smith (37), Bacteria might be an ancestral group, from which an ancient Neomuran (a common ancestor of Archaea and Eukaryota) evolved. According to this last scenario, Archaea and Eukaryota inherited many enzymes involved in tRNA metabolism from their common bacterial (neomuran) ancestor, but also a number of unique enzymes arose. Some of these new enzymes conferred new functions, while others replaced the older, non-orthologous bacterial-like enzymes to fulfil a similar cellular and molecular function (as probably in the case of Trm5p/TrmD).
In this context, the main interest is to compare enzymes catalysing the formation of a modified nucleoside in the tRNAs from organisms belonging to the three domains of life. The MTase catalysing the formation of m1A in the T-loop of tRNA appears to be an excellent model enzyme for such a study, since A58 is the most conserved nucleoside in tRNA of the three biological domains (35) and this universally conserved nucleoside bears one of the few universally encountered methylations (m1A). A site-specific tRNA (m1A) MTase has already been characterized in a eukaryote (the yeast S.cerevisiae) and in the bacterium T.thermophilus.
In the present work, we have characterised the MTase responsible for the formation of m1A in the T-loop of the tRNAs of the archaeon P.abyssi.
A new case of region-specific RNA modification enzyme
A unique characteristic of the P.abyssi TrmI enzyme is its region specificity towards its tRNA substrate. In contrast with the eukaryotic and bacterial enzymes, which are site-specific for position 58, the archaeal enzyme can methylate adenosine at position(s) 57 and/or 58 of some tRNAs. Moreover, in the two tRNA substrates containing A57 and A58 used in this study, the methylation of A57 appears to be more efficient than the methylation of A58. Therefore, one could speculate that the formation of m1A58 could be dependent on the previous formation of m1A57. However, the fact that WT T.thermophilus tRNAAsp containing G57 is efficiently methylated at position 58 by the P.abyssi TrmI methyltransferase does not support this hypothesis. Alternatively, since inosine is more structurally related to guanine than to adenosine, the deamination of m1A57 to m1I57 could influence the efficiency of m1A formation at position 58. The cloning and expression of the tRNA (m1A57) deaminase will help to address this problem.
Region specificity has already been observed in the case of ribosomal RNA modification enzymes: the KsgA and Dim1p MTases methylate two adjacent adenosines in the small subunit rRNA of E.coli and S.cerevisiae, respectively (38,39). In the case of tRNA modification enzymes, the bacterial pseudouridine synthase TruA acting in the anticodon stem and loop of tRNAs modifies uridines in positions 38, 39 and 40 (40), while the S.cerevisiae Pus3p enzyme modifies uridines in positions 38 and 39 only (41).
The modified nucleoside m1I57 is found exclusively in archaeal tRNAs. Contrary to the biosynthesis of m1I37 in the anticodon loop of S.cerevisiae tRNAs, which proceeds by a deamination followed by a methylation step, the biosynthesis of m1I57 in archaeal tRNAs proceeds by the methylation of A57 into m1A57, followed by a deamination leading to m1I57 (reviewed in 42). In this work, we have shown that the P.abyssi TrmI enzyme catalyses the first step in the formation of m1I57 and verified that prior formation of m1A57 is required for the subsequent deamination step catalysed by a still uncharacterised deaminase. Our identification of the enzyme that catalyses the preceding obligatory methylation will greatly facilitate the identification of the m1A57 deaminase enzyme, the final step in m1I57 formation.
Disulphide bridges appear as an important stabilising factor of hyperthermophilic proteins
Due to the easy oxidation and instability of sulphur-containing amino acid residues at high temperatures, proteins from thermophilic organisms usually contain a decreased number of cysteine residues compared to their mesophilic counterparts. From this observation, it has been postulated that proteins from hyperthermophilic organisms should contain very few disulphide bonds (discussed in 43). Moreover, the relatively high reducing potential of the intracellular environment favours the reduced form of cysteine residues. In this context, it was a surprise that structural data showed the existence of disulphide bridges in several proteins from hyperthermophilic organisms (44 and references therein). In support of this finding, a recent computational genomics approach indicated that the intracellular proteins of certain hyperthermophilic Archaea are rich in disulphide bonds (45). Our discovery that disulphide bonds exist in the P.abyssi TrmI enzyme is consistent with this emerging theme.
Crystallographic data presented in this paper showed that the tetrameric structure of the P.abyssi TrmI enzyme involves a large number of hydrophobic interactions between the subunits. Such interactions are known to contribute to the thermostability of hyperthermophilic proteins. At extreme temperatures however, intersubunit disulphide bridges are crucial for the stability of this enzyme. Although several proteins from hyperthermophilic organisms contain disulphide bridges, only a few examples exist where these bridges were demonstrated to confer thermostability to the protein (46,47). Moreover, the disulphide bridges reported in these cases were always intramolecular, in contrast to the intermolecular disulphides we report here.
tRNA (m1A) MTase is a primordial modification enzyme
Analysis of the Protein Data Bank with the DALI program (48) revealed another protein with a quaternary structure very similar to the TrmI MTase (32). The putative precorrin-C15 MTase CbiT (MT0146, 1l3i) (49) forms a tetramer with subunits related by 222 symmetry. We found that the monomer structures of CbiT and TrmI are also more similar to each other than to any other protein in PDB (Z-score 20.3 according to DALI), which indicates their evolutionary relationship. The tetramerisation interface of both the CbiT and TrmI is formed by a strikingly similar assembly of elongated β-hairpins from each monomer, which together form a β-sandwich (almost a β-barrel) comprising two four-stranded β-sheets (each formed by a dimer of hairpins). However, in all crystal forms of CbiT (P21212, P1, C2 short and long cell, and in complex with AdoHcy in C2), the interactions between the monomers are much weaker than in the TrmI structures. Not only is the interior of the sandwich/barrel in CbiT more hydrophilic than in TrmI, but also the tight monomer–monomer interactions observed in the ‘primary dimer’ of TrmI are absent from CbiT. Interestingly, in the AdoHcy-bound crystal form of CbiT a disulphide bond was observed between the monomers in the ‘primary dimer’. This disulphide, however, is not present in other crystal forms of CbiT and the authors suggested that it is not essential for protein stability, but was formed due to spontaneous oxidation during several months of storage of the protein used to make this crystal. The disulphides in CbiT and archaeal TrmI are found in non-homologous positions and between different subunits. Nonetheless, it would be interesting to determine if the disulphide in CbiT may influence the stability of the MTase at high temperature.
Taking the structural and sequence comparisons into account (30; this work), we can propose a phylogenetic framework for the emergence of ‘molecular functions’ determined experimentally for the eukaryotic (17), bacterial (18) and archaeal (this work) tRNA (m1A) MTases (Fig. 9). We first suggest that CbiT is a sister group of tRNA (m1A) MTases, and that their last common ancestor was a homotetrameric protein with weak monomer–monomer interactions, stabilised by contacts between elongated β-hairpins protruding from the catalytic domain. Evolution of a primordial tRNA (m1A) MTase involved fusion with an N-terminal domain and tightening up of monomer–monomer interactions in the ‘primary dimer’. We speculate that these structural innovations were directly connected to the development of an extensive tRNA-binding surface and increased stability of the protein. In the hyperthermophilic Pyrococcus genus, further stabilisation of the tRNA (m1A) MTase homotetramer was achieved by the development of disulphides between two dimers. In the eukaryotic lineage, the tRNA (m1A) MTase gene underwent duplication followed by accelerated divergence, which allowed rapid subfunctionalisation of the two daughter lineages. The Gcd14p copy retained the cofactor-binding and catalytic function, while the Gcd10p copy became specialised in tRNA binding (17), at the expense of the now ‘idle’ AdoMet-binding site and the catalytic residues, which were inactivated by non-conservative substitutions. Thus, the archaeal/bacterial homotetrameric tRNA (m1A) MTase has evolved into a eukaryotic heterotetrameric structure (Fig. 9).
Figure 9.
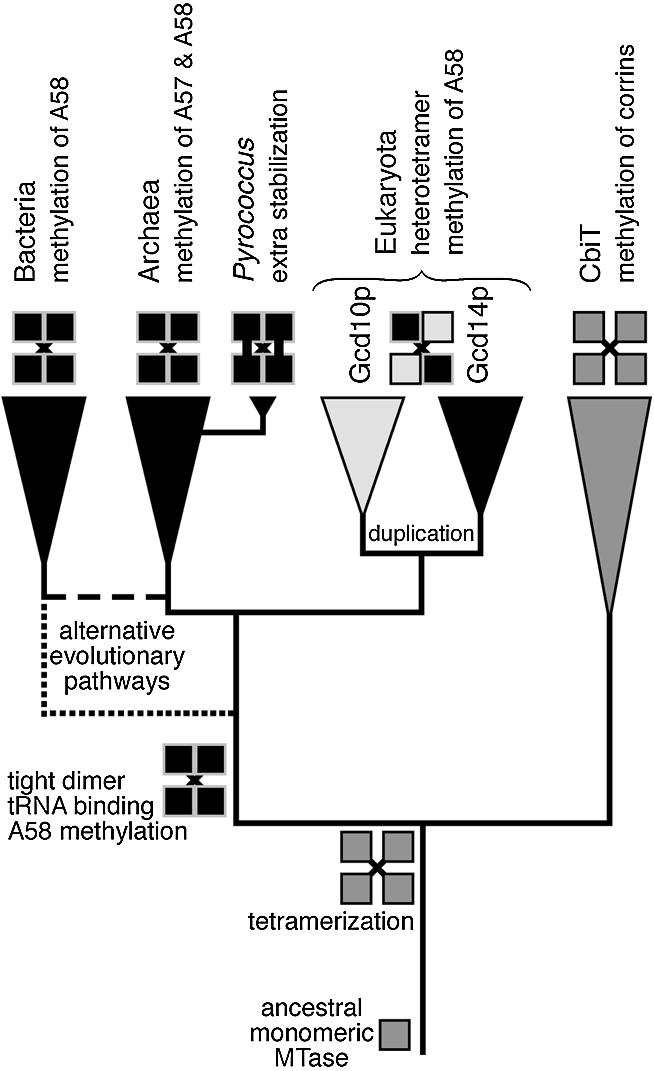
A hypothetical evolutionary scenario of origin and divergence of tRNA (m1A) MTases in the three domains of life. Alternative evolutionary routes (in the prokaryotic part of the tree) have been indicated by broken lines.
Our conclusions contrast with the result of a previous study focused on another tRNA modification enzyme, the tRNA (m1G37) MTase, where it was postulated that the archaeal MTase is more closely related to the corresponding S.cerevisiae enzyme than to its bacterial counterpart (50). Indeed, the bacterial enzyme (TrmD) appears to be structurally and evolutionarily unrelated to the eukaryotic and archaeal enzymes (Trm5p). With this hypothesis, TrmD and Trm5p appear to have evolved independently to probably fulfil the same important function. Note that in Eukaryota and Archaea but not in Bacteria, a distinct family of tRNA (m1G) MTases (Trm10p-like) catalysing the formation of m1G9 in tRNAs has also emerged (12).
As far as the tRNA (m1A) MTases are concerned, these orthologous enzymes are present in all major archaeal and eukaryotic lineages, but only in a few bacterial taxons, mostly limited to Actinobacteria and a small number of species such as Thermotoga, Aquifex, or Thermus, of which many, but not all, are thermophilic (30). Due to sequence divergence and uneven rates of evolution in different lineages, the topology of the prokaryotic part of the gene/protein tree could not be resolved with confidence (data not shown). Assuming the traditional topology of the tree of life, the phylogenetic pattern and those parts of the tree, which could be resolved, suggest one of the following scenarios of origin of the primordial tRNA (m1A) MTase: (i) it evolved in the common ancestor of Archaea and Eukaryota (37) and was horizontally transferred from Archaea to ancient Actinobacteria and to a few other bacterial lineages or (ii) it evolved in the last common ancestor of all extant life forms and was lost from the majority of bacterial lineages. The alternative tree of life proposed by Cavalier-Smith (37) offers a more parsimonious scenario (with only a few horizontal gene transfers and gene losses), in which the primordial tRNA (m1A) MTase evolved in thermophilic Bacteria and was vertically inherited by the neomuran ancestor of Archaea and Eukaryota (Fig. 9). The ultimate validation of one of these two scenarios would require cloning and functional characterisation of more prokaryotic members of the TrmI family and identification of synapomorphies (derived characters shared by individual lineages), which may help to resolve the branching order. The disulphide links in the pyrococcal tRNA (m1A) MTases (Fig. 6) may be regarded as an example of such synapomorphy. Arguably, comparative studies of tRNA (m1A) MTases, as well as of other RNA modification enzymes may provide the key to determination of relationships between Bacteria, Archaea and Eukaryota, and to the ultimate inference of the true tree of life.
Acknowledgments
ACKNOWLEDGEMENTS
We are grateful to J. Anderson (Marquette University, Milwaukee, USA) for critical reading of the manuscript and to J.-P. ten Have for the art work. We thank R. Cunin (Vrije Universiteit Brussel, Belgium) for providing us with P.abyssi GE5 genomic DNA and R. Giegé (CNRS-IBMC Strasbourg, France) for the original plasmid carrying the wild type yeast tRNAAsp. L.D. is a Research Associate of the FNRS (Fonds National de la Recherche Scientifique). J.M.B. is supported by an EMBO/HHMI Young Investigator award and by a fellowship from the Foundation for Polish Science. This work was supported in Belgium by grants from the FRFC (Fonds pour la Recherche Fondamentale Collective), from the French Community of Belgium (Actions de Recherches Concertées) and from the Université Libre de Bruxelles (Fonds E. Defay). In France, H.G. was supported by grants from the CNRS (Centre National de la Recherche Scientifique) and from the French Ministry of Scientific Research (Geomex Program, period 2002–2003).
REFERENCES
- 1.McCloskey J.A. and Crain,P.F. (1998) The RNA modification database-1998. Nucleic Acids Res., 26, 196–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sprinzl M., Horn,C., Brown,M., Ioudovitch,A. and Steinberg,S. (1998) Compilation of tRNA sequences and sequences of tRNA genes. Nucleic Acids Res., 26, 148–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grosjean H., Sprinzl,M. and Steinberg,S. (1995b) Post-transcriptionally modified nucleosides in transfer RNA: their locations and frequencies. Biochimie, 77, 139–141. [DOI] [PubMed] [Google Scholar]
- 4.Motorin Y. and Grosjean,H. (1998) Chemical structures and classification of posttranscriptionally modified nucleosides in RNA. In Grosjean,H. and Benne,R. (eds), Modification and editing of RNA. ASM Press, Washington, DC, pp. 543–549. [Google Scholar]
- 5.Cermakian N. and Cedergren,R. (1998) Modified nucleosides always were: an evolutionary model. In Grosjean,H. and Benne,R. (eds), Modification and editing of RNA. ASM Press, Washington, DC, pp. 535–541. [Google Scholar]
- 6.Garcia G.A. and Goodenough-Lashua,D.M. (1998) Mechanisms of RNA-modifying and -editing enzymes. In Grosjean,H. and Benne,R. (eds), Modification and editing of RNA. ASM Press, Washington, DC, pp. 135–168. [Google Scholar]
- 7.Hopper A.K. and Phizicky,E.M. (2003) tRNA transfers to the limelight. Genes Dev., 17, 162–180. [DOI] [PubMed] [Google Scholar]
- 8.Anantharaman V., Koonin,E.V. and Aravind,L. (2002) SPOUT: a class of methyltransferases that includes SpoU and trmD RNA methylase superfamilies and novel superfamilies of predicted prokaryotic RNA methylases. J. Mol. Microbiol. Biotechnol., 4, 71–75. [PubMed] [Google Scholar]
- 9.Anantharaman V., Koonin,E.V. and Aravind,L. (2002) Comparative genomics and evolution of proteins involved in RNA metabolism. Nucleic Acids Res., 30, 1427–1464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feder M., Pas,J., Wyrwicz,L.S. and Bujnicki,J.M. (2003) Molecular phylogenetics of the RrmJ/fibrillarin superfamily of ribose 2′-_O_-methyltransferases. Gene, 302, 129–138. [DOI] [PubMed] [Google Scholar]
- 11.Devos D. and Valencia,A. (2001) Intrinsic errors in genome annotation. Trends Genet., 17, 429–431. [DOI] [PubMed] [Google Scholar]
- 12.Jackman J.E., Montange,R.K., Malik,H.S. and Phizicky,E.M. (2003) Identification of the yeast gene encoding the tRNA m1G9 methyltransferase responsible for modification at position 9. RNA, 9, 574–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clouet d'Orval B., Bortolin,M.-L., Gaspin,C. and Bachellerie,J.-P. (2001) Box C/D RNA guides for the ribose methylation of archaeal tRNAs. The tRNATrp intron guides the formation of two ribose-methylated nucleosides in the mature tRNATrp. Nucleic Acids Res., 29, 4518–4529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pintard L., Lecointe,F., Bujnicki,J.M., Bonnerot,C., Grosjean,H. and Lapeyre,B. (2002) Trm7p catalyses the formation of two 2′-_O_-methylriboses in yeast tRNA anticodon loop. EMBO J., 21, 1811–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grosjean H., Constantinesco,F., Foiret,D. and Benachenhou,N. (1995) A novel enzymatic pathway leading to 1-methylinosine modification in Haloferax volcanii tRNA. Nucleic Acids Res., 23, 4312–4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anderson J., Phan,L., Cuesta,R., Carlson,B.A., Pak,M., Asano,K., Björk,G.R., Tamame,M. and Hinnebusch,A.G. (1998) The essential Gcd10p-Gcd14p nuclear complex is required for 1-methyladenosine modification and maturation of initiator methionyl-tRNA. Genes Dev., 12, 3650–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson J., Phan,L. and Hinnebusch,A.G. (2000) The Gcd10p/Gcd14p complex is the essential two-subunit tRNA (1-methyladenosine) methyltransferase of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA, 97, 5173–5178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Droogmans L., Roovers,M., Bujnicki,J., Tricot,C., Hartsch,T., Stalon,V. and Grosjean,H. (2003) Cloning and characterization of tRNA (m1A58) methyltransferase (TrmI) from Thermus thermophilus HB27, a protein required for cell growth at extreme temperatures. Nucleic Acids Res., 31, 2148–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buck M., Connick,M. and Ames,B.N. (1983) Complete analysis of tRNA-modified nucleosides by high-performance liquid chromatography: the 29 modified nucleosides of Salmonella typhimurium and Escherichia coli tRNA. Anal. Biochem., 129, 1–13. [DOI] [PubMed] [Google Scholar]
- 20.Cohen G.N., Barbe,V., Flament,D., Galperin,M., Heilig,R., Lecompte,O., Poch,O., Prieur,D., Querellou,J., Ripp,R., Thierry,J.C., Van der Oost,J., Weissenbach,J., Zivanovic,Y. and Forterre,P. (2003) An integrated analysis of the genome of the hyperthermophilic archaeon Pyrococcus abyssi. Mol. Microbiol., 47, 1495–1512. [DOI] [PubMed] [Google Scholar]
- 21.Legrain C., Demarez,M., Glansdorff,N. and Piérard,A. (1995) Ammonia-dependent synthesis and metabolic channelling of carbamoyl phosphate in the hyperthermophilic archaeon Pyrococcus furiosus. Microbiol., 141, 1093–1099. [DOI] [PubMed] [Google Scholar]
- 22.Reyes V.M. and Abelson,J. (1987) A synthetic substrate for tRNA splicing. Anal. Biochem., 166, 90–106. [DOI] [PubMed] [Google Scholar]
- 23.Perret V., Garcia,A., Puglisi,J., Grosjean,H., Ebel,J.P., Florentz,C. and Giegé,R. (1990) Conformation in solution of yeast tRNAAsp transcripts deprived of modified nucleotides. Biochimie, 72, 735–743. [DOI] [PubMed] [Google Scholar]
- 24.Droogmans L. and Grosjean,H. (1991) 2′-_O_-methylation and inosine formation in the wobble position of anticodon-substituted tRNAPhe in a homologous yeast in vitro system. Biochimie, 73, 1021–1025. [DOI] [PubMed] [Google Scholar]
- 25.Keith G. (1995) Mobilities of modified ribonucleotides on two-dimensional cellulose thin layer chromatography. Biochimie, 77, 142–144. [DOI] [PubMed] [Google Scholar]
- 26.Jancarik J. and Kim,S.-H. (1991) Sparse matrix sampling: a screening method for crystallisation of proteins. J. Appl. Crystallogr., 24, 409–411. [Google Scholar]
- 27.Otwinowski Z. and Minor,W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol., 276, 307–326. [DOI] [PubMed] [Google Scholar]
- 28.Brunger A.T., Adams,P.D., Clore,G.M., DeLano,W.L., Gros,P., Grosse-Kunstleve,R.W., Jiang,J.S., Kuszewski,J., Nilges,M., Pannu,N.S., Read,R.J., Rice,L.M., Simonson,T. and Warren,G.L. (1998) Crystallography & NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr. D, 54, 905–921. [DOI] [PubMed] [Google Scholar]
- 29.Roussel A. and Cambillau,C (1992) TURBO-FRODO. Biographics, Architecture et Fonction des Macromolécules Biologiques, Marseille, France.
- 30.Bujnicki J.M. (2001) In silico analysis of the tRNA:m1A58 methyltransferase family: homology-based fold prediction and identification of new members from Eubacteria and Archaea. FEBS Lett., 507, 123–127. [DOI] [PubMed] [Google Scholar]
- 31.Constantinesco F., Motorin,Y. and Grosjean,H. (1999) Transfer RNA modification enzymes from Pyrococcus furiosus: detection of the enzymatic activities in vitro. Nucleic Acids Res., 27, 1308–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gupta A., Kumar,H., Dineshkumar,T.K., Varshney,U. and Subramanya,H.S. (2001) Crystal structure of Rv2118c: an AdoMet-dependent methyltransferase from Mycobacterium tuberculosis H37Rv. J. Mol. Biol., 312, 381–391. [DOI] [PubMed] [Google Scholar]
- 33.Olsen G.J. and Woese,C.R. (1997) Archaeal genomics: an overview. Cell, 89, 991–994. [DOI] [PubMed] [Google Scholar]
- 34.Doolittle W.F. and Logsdon,J.M.Jr (1998) Archaeal genomics: do archaea have a mixed heritage? Curr. Biol., 8, R209–R211. [DOI] [PubMed] [Google Scholar]
- 35.Marck C. and Grosjean,H. (2002) tRNomics: analysis of tRNA genes from 50 genomes of Eukarya, Archaea and Bacteria reveals anticodon-sparing strategies and domain-specific features. RNA, 8, 1189–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xue H., Tong,K.-L., Marck,C., Grosjean,H. and Wong,J.T.-F. (2003) Transfer RNA paralogs: evidence for genetic code-amino acid biosynthesis coevolution and an archaeal root of life. Gene, 310, 59–66. [DOI] [PubMed] [Google Scholar]
- 37.Cavalier-Smith T. (2002) The neomuran origin of archaebacteria, the negibacterial root of the universal tree and bacterial megaclassification. Int. J. Syst. Evol. Microbiol., 52, 7–76. [DOI] [PubMed] [Google Scholar]
- 38.Helser T.L., Davies,J.E. and Dahlberg,J.E. (1972) Mechanism of kasugamycin resistance in Escherichia coli. Nature New Biol., 235, 6–9. [DOI] [PubMed] [Google Scholar]
- 39.Lafontaine D., Delcour,J., Glasser,A.L., Desgres,J. and Vandenhaute,J. (1994) The DIM1 gene responsible for the conserved m62Am62A dimethylation in the 3′-terminal loop of 18S rRNA is essential in yeast. J. Mol. Biol., 241, 492–497. [DOI] [PubMed] [Google Scholar]
- 40.Kammen H.O., Marvel,C.C., Hardy,L. and Penhoet,E.E. (1988) Purification, structure and properties of Escherichia coli tRNA pseudouridine synthase I. J. Biol. Chem., 263, 2255–2263. [PubMed] [Google Scholar]
- 41.Lecointe F., Simos,G., Sauer,A., Hurt,E.C., MotorinY. and Grosjean,H. (1998) Characterization of yeast protein Deg1 as pseudouridine synthase (Pus3) catalyzing the formation of ψ38 and ψ39 in tRNA anticodon loop. J. Biol. Chem., 273, 1316–1323. [DOI] [PubMed] [Google Scholar]
- 42.Grosjean H., Auxilien,S., Constantinesco,F., Simon,C., Corda,Y., Becker,H., Foiret,D., Morin,A., Jin,Y.X., Fournier,M. and Fourrey,J.L. (1996) Enzymatic conversion of adenosine to inosine and to _N_1-methylinosine in transfer RNA: a review. Biochimie, 78, 488–501. [DOI] [PubMed] [Google Scholar]
- 43.Ren B. and Ladenstein,R. (2001) Protein disulfide oxidoreductase from Pyrococcus furiosus: structural properties. Methods Enzymol., 334, 74–88. [DOI] [PubMed] [Google Scholar]
- 44.Appleby T.C., Mathews,I.I., Porcelli,M., Cacciapuoti,G. and Ealick,S.E. (2001) Three-dimensional structure of a hyperthermophilic 5′-deoxy-5′-methylthioadenosine phosphorylase from Sulfolobus solfataricus. J. Biol. Chem., 276, 39232–39242. [DOI] [PubMed] [Google Scholar]
- 45.Mallick P., Boutz,D.R., Eisenberg,D. and Yeates,T.O. (2002) Genomic evidence that the intracellular proteins of archaeal microbes contain disulfide bonds. Proc. Natl Acad. Sci. USA, 99, 9679–9684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Toth E.A., Worby,C., Dixon,J.E., Goedken,E.R., Marqusee,S. and Yeates,T.O. (2000) The crystal structure of adenylosuccinate lyase from Pyrobaculum aerophilum reveals an intracellular protein with three disulfide bonds. J. Mol. Biol., 301, 433–450. [DOI] [PubMed] [Google Scholar]
- 47.Meyer J., Clay,M.D., Johnson,M.K., Stubna,A., Münck,E., Higgins,C. and Wittung-Stafshede,P. (2002) A hyperthermophilic plant-type [2Fe-2S] ferredoxin from Aquifex aeolicus is stabilized by a disulfide bond. Biochemistry, 41, 3096–3108. [DOI] [PubMed] [Google Scholar]
- 48.Holm L. and Sander,C. (1993) Protein structure comparison by alignment of distance matrices. J. Mol. Biol., 233, 123–138. [DOI] [PubMed] [Google Scholar]
- 49.Keller J.P., Smith,P.M., Benach,J., Christendat,D., deTitta,G.T. and Hunt,J.F. (2002) The crystal structure of MT0146/CbiT suggests that the putative precorrin-8w decarboxylase is a methyltransferase. Structure, 10, 1475–1487. [DOI] [PubMed] [Google Scholar]
- 50.Björk G.R., Jacobsson,K., Nilsson,K., Johansson,M.J.O., Byström,A.S. and Persson,O.P. (2001) A primordial tRNA modification required for the evolution of life. EMBO J., 20, 231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fauman E.B., Blumenthal,R.M. and Cheng,X. (1999) Structure and evolution of AdoMet-dependent MTases. In Cheng,X. and Blumenthal,R.M. (eds), S-adenosylmethionine-dependent methyltransferases: structure and functions. World Scientific Inc., Singapore, pp. 1–38. [Google Scholar]