Prediction of Saccharomyces cerevisiae replication origins (original) (raw)
Short abstract
This study presents the first algorithm, Oriscan, to predict eukaryotic replication origins and examines the structure of the origins and evolutionary conservation of origin sequences at the nucleotide level in several Saccharomyces species.
Abstract
Background
Autonomously replicating sequences (ARSs) function as replication origins in Saccharomyces cerevisiae. ARSs contain the 17 bp ARS consensus sequence (ACS), which binds the origin recognition complex. The yeast genome contains more than 10,000 ACS matches, but there are only a few hundred origins, and little flanking sequence similarity has been found. Thus, identification of origins by sequence alone has not been possible.
Results
We developed an algorithm, Oriscan, to predict yeast origins using similarity to 26 characterized origins. Oriscan used 268 bp of sequence, including the T-rich ACS and a 3' A-rich region. The predictions identified the exact location of the ACS. A total of 84 of the top 100 Oriscan predictions, and 56% of the top 350, matched known ARSs or replication protein binding sites. The true accuracy was even higher because we tested 25 discrepancies, and 15 were in fact ARSs. Thus, 94% of the top 100 predictions and an estimated 70% of the top 350 were correct. We compared the predictions to corresponding sequences in related Saccharomyces species and found that the ACSs of experimentally supported predictions show significant conservation.
Conclusions
The high accuracy of the predictions indicates that we have defined near-sufficient conditions for ARS activity, the A-rich region is a recognizable feature of ARS elements with a probable role in replication initiation, and nucleotide sequence is a reliable predictor of yeast origins. Oriscan detected most origins in the genome, demonstrating previously unrecognized generality in yeast replication origins and significant discriminatory power in the algorithm.
Background
Every growing cell must faithfully copy its genome prior to cell division. This process must be tightly controlled so that every part of the genome is replicated once and only once. Forty years ago, Jacob, Brenner, and Cuzin proposed the first such control scheme: the replicon model [1]. The replicon was defined as the fundamental unit of replication, much like the operon or regulon in transcription. The initiator protein - DnaA in bacteria - binds a sequence within a replicon called a replicator, and then DNA synthesis initiates from a nearby, well-defined origin. Thus, through synthesis or activation of the initiator protein, the cell can direct the start of replication and couple it to other events in the cell cycle.
The replicon hypothesis is a useful description of replication in prokaryotes, but the situation in eukaryotes has proven to be more complex. Eukaryotic chromosomes contain many origins of replication, and the apparent importance of conserved replicator sequences varies substantially among organisms. The unicellular fungus S. cerevisiae occupies a middle ground, in that initiation occurs at discrete origins as in bacteria, but, as in other eukaryotes, not all origins are used in every cell cycle [2]. Thanks to the advantages of yeast as a model organism in replication studies, more is known about its origins than those of any other eukaryote. The conserved, recognizable features of origin sequences are the subject of this report.
Yeast replication origins are autonomously replicating sequences (ARS), defined operationally as sequences that support the maintenance of a plasmid in growing yeast cells [3]. ARS elements do so by directing initiation of DNA replication, resulting in replication intermediates that can be detected by techniques such as two-dimensional gel electrophoresis [4,5]. However, some ARS sequences do not act as origins on the chromosome, or do so only inefficiently, perhaps due to the earlier firing of a nearby origin or repressive chromatin. We will refer to any _cis-_acting element that leads to replication initiation on a plasmid or chromosome as an origin.
At the core of every yeast replication origin is a replicator sequence containing a conserved 17 bp stretch known as the ARS consensus sequence (ACS) [6]. One of the strands of the ACS is T-rich; by convention, we will always refer to this strand when describing origin sequences. The ACS is required for binding of the initiator protein, the origin recognition complex (ORC) [7,8]. ORC is a multifunctional heterohexameric ATPase, conserved among eukaryotes [9,10], with roles in transcriptional silencing [11,12] and cytokinesis [13,14] in addition to replication initiation. ORC subunits bind origins throughout the cell cycle and recruit other components of the pre-replicative complex (pre-RC), notably the minichromosome maintenance (MCM) protein hexamer [15].
ARS sequences are composed of two or three domains. The most important is the central, ACS-containing A domain, which is absolutely necessary but not sufficient for origin function [16]. ARSs also have a B domain 3' to the ACS that contains individual elements important, but not essential, for activity [17]. The B1 element, found in every ARS, is adjacent to the ACS and is part of the ORC binding site [7,8]. B2 elements, present in most but not all ARSs, function in pre-RC assembly [18,19] and frequently overlap with DNA unwinding elements (DUEs) [20,21]. DUEs are unwound by (-) superhelical tension and presumably during replication initiation. B3 is a transcription factor binding site found in some ARSs that influences nucleosome positioning [22]. Several origins also require a 5'-located C domain containing a transcription factor binding site or sites for full function [23-25]. It was recently shown that the MCM1 protein binds several ARSs on either or both sides of the ACS; this probably contributes to origin function as well [26].
The ACS is not unique, as many positions are degenerate, including five positions that may be either A or T. Additionally, exact matches are rare; most origins match 14 or more of the 17 positions, but a handful match only 11 to 13. In the yeast genome, there are about 17,500 matches to 14 positions of the ACS and 89,000 matches to 13 positions, orders of magnitude more than the 300-400 origins indicated by experiments [27-29]. Despite their common functions, B and C domains from different origins do not have clearly recognizable sequence similarity [17]. Therefore, it has not been possible to distinguish a functional ACS from the vast excess of inactive occurrences without labor-intensive experiments such as ARS assays, two-dimensional gels, or microarray-based detection of origin activity or ORC/MCM binding. Moreover, these techniques have resolutions ranging from about 300 bp to several kb. A sequence-based capability to recognize an ARS element would identify the exact location of ORC binding; this resolution can be attained experimentally only through site-directed mutagenesis or replication initiation point mapping [30], neither of which is practical on a genomic scale. This accuracy would facilitate advanced analyses of origin components and potential interactions with surrounding genomic elements.
Here, we report the development of an algorithm we call Oriscan to predict the exact location of yeast replication origins based solely on sequence information. The algorithm searched for sequences similar to a training set, or group of known examples, consisting of 26 yeast origins that were pinpointed by site-directed mutagenesis. In addition to the ACS, the Oriscan algorithm uses 251 bp of flanking sequence, including a region of elevated, strand-biased adenine content in the B domain against a background of generally increased A+T content. A total of 94 out of the 100 top predictions match evidence of origin activity, including a number of ARSs not detected in previous studies. This extremely high accuracy demonstrates that high-ranking predictions made by Oriscan have sequence attributes sufficient for origin function. These attributes have substantial generality as well, as the top 350 predictions of Oriscan identified 58% of about 340 probable origins identified by ARS assay or ORC/MCM binding. Furthermore, we found that ACS conservation extends beyond the genome of S. cerevisiae; using alignments to related yeasts, we demonstrate that ACSs show significantly increased conservation. Thus, despite the apparent heterogeneity of sequences flanking the ACS of yeast origins, the success of Oriscan demonstrates that these sequences possess subtle but recognizable uniformity.
Results
Analysis of known origin sequences and construction of Oriscan
We used 26 known yeast replication origin sequences (Additional data file 1) to build a profile with which to search the genome. The origins were aligned by their ACS with no gaps. We observed that sequences flanking the ACS differed significantly from the rest of the genome; in particular, the region 3' to the ACS contains a high proportion of A residues (approximately 44%; Figure 1a). In the ACS, there are 3.0 Ts for every A, and this ratio changes to 0.6 in this A-rich area. To assess sequence conservation quantitatively, we used nucleotide frequencies to calculate the information content [31] at each position of the aligned origin sequences (Figure 1b). We used the formula I = Σ_i_ f _i_log2(f i/p i), where f i is the observed frequency of a base in a single position, p i is that base's frequency in the whole genome, the summation is over the four bases, and I is the information content in bits. A bit represents a two-fold reduction in variability. Because the A+T content of the yeast genome is 61.7%, a perfectly conserved A or T residue contains 1.7 bits instead of exactly 2, and a perfectly conserved G or C would contain 2.4 bits. As expected, the ACS has the highest information content in the region we analyzed, averaging 0.88 bits per position. The area 25 to 105 bp 3' of the center of the ACS is also enriched in information, visible in Figure 1a as the broad peak of high adenine content. The mean information content here is 0.18 ± 0.01 bits/position, significantly greater (p < 0.001) than 23 randomly chosen yeast genomic sequences, which showed an information content of 0.10 bits/position.
Figure 1.
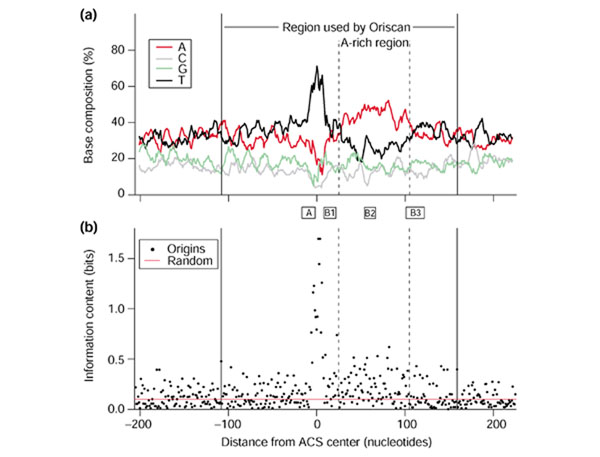
Yeast replication origin profile and information content. In both panels, solid vertical lines at coordinates -108 and +159 indicate the 268 nucleotide region used by Oriscan. (a) Yeast origins were aligned by ACS with no gaps. The frequency of each base in the ACS T-rich strand in a 9 nucleotide window is plotted by distance from the ACS center. The ACS is visible as the high central peak in T frequency; the nearby A-rich region is enclosed in dashed vertical lines. Solid vertical lines enclose the region used in the Oriscan algorithm. (b) Information content in bits is shown for each position of the aligned origins. The ACS appears as the high central peak. The A-rich region to the right also shows elevated information content. The red line indicates the average information content for an alignment of randomly chosen sequences. Between (a) and (b), the positions of A and B elements in ARS1 [48] are shown for reference.
The 100 nucleotides immediately 5' to the ACS (-108 to -9 bp relative to the ACS) and the 53 nucleotides 3' to the A-rich region (+107 to +159 bp) had a significantly higher A+T content (68%) than bulk sequence (62%; p < 0.001). Inclusion of these sequences improved the performance of Oriscan, but use of sequences further from the ACS degraded performance. In our search for replication origins, therefore, we chose to use sequences from -108 to +159 bp, including both the A-rich region and both areas of increased AT content.
Predictions and evaluation
We compared Oriscan predictions to an evaluation set of all origins identified using ARS assays and two-dimensional gel electrophoresis plus the set of proposed ARS elements (pro-ARSs) identified via ORC and MCM binding [28]. This list totaled 408 probable origins, although some of these are false positives, as discussed below. Each member of the evaluation set and the experimental evidence for that member is detailed in Additional data file 1. We did not include the chromosomal origins identified by microarray [27] in the evaluation set, henceforth referred to as array origins, for several reasons. This set has lower precision (±4 kb) than the ORC/MCM data (±0.5-1 kb) as indicated by comparison to precisely localized origins. It is also subject to chromosomal context effects, including passive replication by nearby, earlier origins and poorly understood effects on the efficiency of firing.
Before evaluation, we removed the training set from consideration. Training set members generally scored extremely well but are not a fair test of Oriscan's performance. An Oriscan prediction was scored as a match if it fell in a region with demonstrated origin activity or within 250 nucleotides of a region identified as an ORC/MCM binding site. We considered the latter to be a conservative expansion of the pro-ARS locations, since this set was shown to mislocalize several known origins by up to 600 bp [28].
The Oriscan algorithm consists of steps that sequentially discard inactive sequences to separate origins away from the rest of the genome, followed by a ranking procedure to sort the predictions in order of similarity to known origins (Figure 2). We demanded that candidates pass three successive thresholds based on position-weight matrices generated from the training set (Additional data file 2). In the first step, Oriscan analyzed the 17 bp ACS. We empirically set the threshold to select the best 12,000 matches (Additional data file 3), as this included matches to 87%, or 354, of the 408-member evaluation set (ACS; Figure 2). Further relaxation of stringency caused inclusion of many more non-origin sequences, thereby degrading performance. Our threshold allowed up to four mismatches within the 17 bp consensus (WWWWTTTAYRTTTWGTT, where W = A or T, Y = C or T, and R = A or G), and imposed larger penalties for mismatches to more conserved positions. Thus, some candidates with only two mismatches were rejected.
Figure 2.
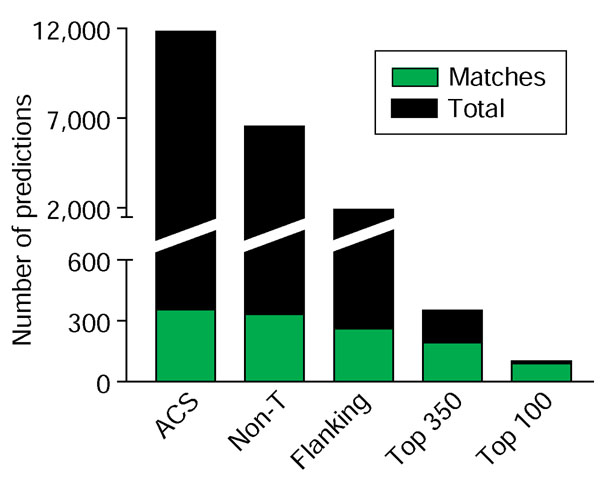
Refinement of Oriscan predictions. The number of matching (green) and total (black) predictions at different stages in the algorithm are shown. From the 12 million positions in the yeast genome, the best 11,800 matches to the core ACS were selected, and these matched 354 members of the ORC/MCM evaluation set (ACS). Selection against poly-T sequences removed 5,268 predictions, leaving 6,532, including 332 matches to the ORC/MCM set (non-T). Further selection using the 268 nucleotide matrix containing flanking sequence removed 4,632 predictions, leaving 1,900, including 257 matches (flanking). These predictions were then ranked; the top 350 contained 179 matches, and the top 100 contained 84 matches.
Thymine matches 14 of the 17 positions in the ACS, so many poly-T sequences passed the first step even though such a sequence has never been identified as an ORC binding site. Therefore, in the second step, we examined the three non-T positions - A, R, and G - of the ACS independently of the rest. We rejected about 5,000 sequences that mismatched at least two of the three positions. Only 22 matches were lost, leaving 332 matches (non-T; Figure 2). The third step in the Oriscan algorithm was to analyze the flanking sequences from 108 nucleotides 5' to 159 nucleotides 3' to the central nucleotide of the ACS. We selected 1,882 sequences, including matches to 257 members of the evaluation set, that scored ≥2.4 standard deviations better than mean bulk sequence. As with the ACS, this cutoff was chosen to pass as many probable origins as possible without degrading performance.
The final phase of the algorithm consisted of ranking the 1,882 matches in order of their likelihood of activity (Additional data file 4). This was done by deriving two profiles: one corresponding to active origins based on the training set, and the other to inactive candidates. Candidates with scores near histogram peaks in the active profile, but not the inactive profile, received high ranks, and the inverse cases received low ranks. Distributions of ACS and flanking scores as Oriscan progressively whittled down origin candidates are shown in Additional data file 5.
We found that origin predictions ranking in the top 350 frequently coincided with evidence of origin activity (Top 350; Figure 2). This was almost always the case for predictions with a rank of 100 or better (Top 100; Figure 2). The breakdown of matching and non-matching predictions by rank is shown in Figure 3a. The high concentration of matches among the top predictions is easily visible. Figure 3b shows the specificity and sensitivity of Oriscan predictions, where specificity is the fraction of predictions that match, and sensitivity is the fraction of the evaluation set that was predicted. The specificity of the strongest predictions is very high; it is nearly as high for the top 100 as for the top 50, but then declines as predictions of lower rank are added. Sensitivity rises as more predictions are added, but less so after the top 350; specificity declines a bit more steeply after this point. We therefore chose 100 and 350 predictions as useful cutoffs. A total of 84 of the top 100 predictions, training set excluded, matched a member of the evaluation set. The top 350 predictions had 56% specificity and 42% sensitivity against the evaluation set. For reasons discussed below, these values underestimate the true performance of the algorithm.
Figure 3.
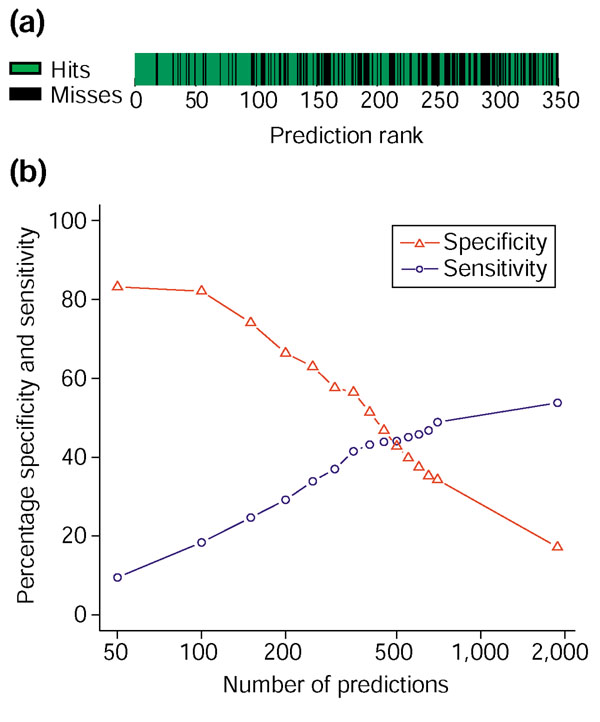
Specificity and sensitivity of ranked predictions. The training set was removed from consideration before generation of this figure. (a) Prediction accuracy is depicted visually as a function of rank. Each prediction was plotted in rank order and coded green if it matched a member of the evaluation set of probable origins or black if it did not, and plotted in rank order from left to right. The high concentration of matches in the top predictions is visible as large blocks of green on the left. (b) Specificity, defined as 100% minus the false positive rate, and sensitivity, 100% minus the false negative rate, are plotted for ranked groups of predictions in cumulative increments of 50 for the first 700 predictions and then for the total ranked list of 1,900 predictions. The ORC/MCM set was used for evaluation. Sensitivity gradually increases, and specificity decreases, as predictions of lower rank are included.
New origins found by Oriscan
We wished to test whether some predictions that did not match the evaluation set were actually origins. The predictions were assayed for the ability to promote replication of an otherwise originless plasmid [32]. Of the 16 predictions in the top 100 that did not match the evaluation set, ten had ARS activity (Figure 4a and Additional data file 6). Thus, 94 of the top 100 predicted origins are supported by experimental evidence, showing that Oriscan effectively recognizes sequences that are sufficient for origin function. We calculated the probability of ARS activity in a randomly selected genomic fragment to be 1.4%, based on the comprehensive screen of chromosome VI [33] and the mean length of the fragments we used (434 bp). Our 10 out of 16 success rate is highly significant by the exact binomial test (p < 0.0001).
Figure 4.
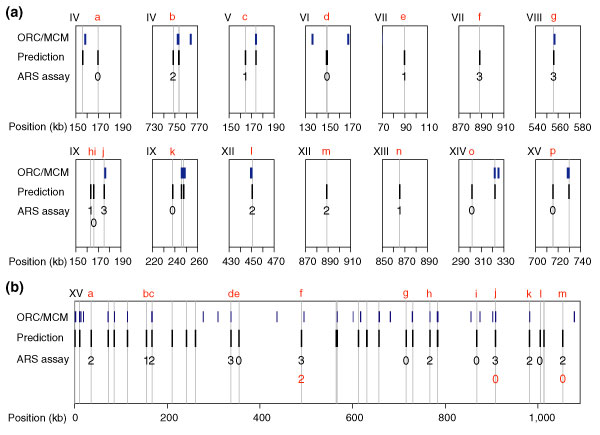
Predictions and ARS assay results compared to probable origin locations. (a) Shown are predictions in the top 100 that did not match the evaluation set along with their ARS activities. Likely origins in the evaluation set are in blue (ORC/MCM), and Oriscan predictions are in black. The width of the bars is not to scale. Vertical gray lines drawn through predictions show whether there is overlap with an evaluation set member. ARS assay results are scored on a scale of 0 to 3 for origin strength; 0 indicates inactivity, 1 indicates weak activity, and 2 and 3 indicate increasingly strong activity. Chromosomes are identified in Roman numerals at the top left of each plot, and positions in kb are given beneath the axis. Each prediction assayed is given a lowercase letter in red for reference in the text. For legibility, ARS assay results are offset for the pair of closely spaced predictions on chromosome IX. (b) All predictions and ARS assay results on chromosome XV. Plotting conventions are as in (a), except that origins which were tested after mutation of the ACS (f, j, and m) have a number indicating the ARS activity of the mutant in red under the original number. There are two very closely spaced predictions at 715 kb (g); neither was active, and this is denoted with a single 0.
Of the 16 predictions, three were within 600 nucleotides of an ORC/MCM binding site (g, j, and l in Figure 4a), and all three were ARSs. We presume that these are cases of positional errors in the ORC/MCM set. Furthermore, four of these 16 (a, f, k, and m in Figure 4a) were within 4 kb of an array origin found by Raghuraman et al. [27], despite not having matched an ORC/MCM binding site; two of these four (f and m) showed ARS activity.
Buoyed by these results, we performed ARS assays on a sample of predictions from chromosome XV that included many with ranks between 101 and 350 (Figure 4b and Additional data files 6 and 7). We tested ten of the 18 predictions that did not match the evaluation set, and five were active (a, b, f, k, and m in Figure 4b). Of these five, one (k) was 300 nucleotides from the edge of an ORC/MCM site and is probably another case of a positional error, and another (m) matched an array origin. As above, the five out of ten success rate is highly significant (p < 0.0001). As a control, we tested four predictions that agreed with ORC/MCM binding sites (c, d, h, and j in Figure 4b), and all four were active.
This sample indicates that 56% is an underestimate of the accuracy of Oriscan's top 350 predictions. Based on extrapolation from chromosome XV to the rest of the genome, there should be at least 50 active Oriscan predictions not detected previously, including about ten positional errors. Inclusion of these 50 ARSs raised the specificity of Oriscan to about 70% and sensitivity to about 50% for the top 350 predictions. This is a lower bound because we assumed that no more predictions were active on chromosome XV. We made this assumption because the ten predictions we tested were not chosen randomly.
The evaluation set contains false positives, as testing of ORC/MCM binding sites showed that about one in five was not an ARS [28]. This extrapolates to about 70 unspecified members of the evaluation set. Reduction of the evaluation set size by 70 increases the sensitivity of Oriscan's top 350 predictions to 58%, or 73% for the 1,882 predictions that passed the first three steps of Oriscan. Specificity was not affected. Thus, Oriscan is capable of detecting most origins in the genome, indicating that the description of origins that underpins the algorithm has considerable generality. As a way of checking our statistical adjustments to sensitivity, we used Oriscan to analyze its own training set. Because there were no false positives or negatives, no adjustments are necessary. To avoid bias, we analyzed each training set member with a version of the algorithm that excludes that member; without such exclusion, known as jackknifing, most of the training set would have outscored the rest of the genome by several standard deviations. We found that 15 of the 26 training set members ranked in the top 350, giving a sensitivity of 58%. This matched the final sensitivity of the top 350 predictions, supporting the validity of our statistical corrections.
Oriscan pinpoints the ORC binding site
To demonstrate the single nucleotide precision of Oriscan, we selected three predictions on chromosome XV (f, j, and m in Figure 4b) shown to have ARS activity, for site-directed mutagenesis. One (j) was the strongest prediction Oriscan made and matched an ORC/MCM binding site; a second (m) matched an array origin but not an ORC/MCM binding site; and a third (f) was near but outside an evaluation set member. In each case, four bases near the center of the ACS were mutated to give a BamHI restriction site, which is GC-rich and easily verified by restriction analysis. We then repeated the ARS assay. In the first two cases (j and m), the mutations completely abolished ARS activity. In the last case (f), ARS activity was noticeably weakened, as indicated by both colony growth rate and an ade2-1 colony color assay indicative of the copy number of the ARS plasmid [34] (Additional data file 6). The compromised but not abolished ARS activity may indicate that the original ARS contains multiple ORC binding sites, including the one predicted by Oriscan. To assay ARS activity of (f), we had cloned a 486 bp fragment that contained a total of five matches to ≥13 of the 17 ACS positions, including ≥9 of 11 central positions. One of the other four matches may also bind ORC.
Evolutionary conservation of the ACS
The genomes of several related Saccharomyces sensu stricto species have recently been sequenced [35,36]. We sought to determine whether core ACSs predicted by Oriscan were highly conserved in these species, which would suggest conservation of origin function. We used high-quality multiple alignments of segments of intergenic sequences from S. cerevisiae with S. paradoxus, S. mikatae, S. kudriavzevii, and S. bayanus that were computed using the T-Coffee program [37] (D.Y. Chiang and M.B. Eisen, unpublished observations).
We analyzed the sequences that aligned to the core ACSs of all known origins and experimentally supported top 350 predictions; we found 75 alignments with sequence from all five species. We used these to calculate the rate of evolution at each position using maximum parsimony [38] (Figure 5a). As a control, we performed the same calculations on all aligned ACSs that passed the Non-T step of Oriscan (Figure 2) and were not located within an ORC/MCM binding site or known origin (N = 1,580). Ten positions in the ACS match a single base; of these, eight showed higher conservation than the control. The mean decrease in rate of evolution at these positions was 38% ± 9%. No degenerate positions showed significant conservation, consistent with the reduced constraint on these bases. The two positions at the 5' end that are predominantly A or T actually showed above-background mutation rates. However, it was particularly common for an ACS that contained an A at one of these positions in S. cerevisiae to have a T in one or more of the other species, and vice versa, despite the rarity of transversion (pyrimidine to purine or purine to pyrimidine) mutations. Thus, it seems likely that these positions maintain their preference to be A or T in closely related genomes.
Figure 5.
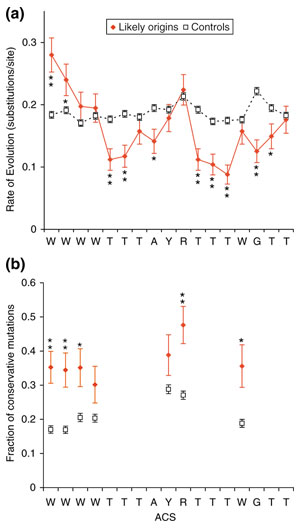
Conservation of the ACS across species. (a) The rate of evolution was calculated for the ACSs of 75 experimentally supported predictions and known origins (red solid diamonds, solid lines) using alignments to sequence of four other yeasts (see text). As a control, we performed the same analysis on 1,580 alignments of ACSs that passed the non-T step of Oriscan but did not match an ORC/MCM or known origin locus (black open squares, dashed lines). Substitutions per site were estimated by maximum parsimony, and error bars indicate the standard error of a Poisson distribution. Statistical significance is indicated by asterisks (* indicates p < 0.02; ** indicates p < 0.001). (b) The fraction of mutations that were conservative, that is, between the two allowed bases at a degenerate position, was calculated for each degenerate nucleotide of the ACS using the same probable active and control ACS alignments as in (a). Symbols and asterisks are as in (a).
To analyze this preference for matching residues quantitatively, we examined mutation bias in all degenerate ACS positions. We define conservative mutations as changing between the two acceptable nucleotides at a degenerate position, such as A to T or T to A at the 5' positions discussed above. Once again, we compared probable active ACSs to the control set (Figure 5b). We found that the fraction of conservative mutations is higher for the probably active loci at every position; this difference was statistically significant in five of seven positions. Overall, the fraction of conservative mutations increased from 21% in the controls to 37% in the set of probable active ACSs.
Thermodynamic analysis of predictions
Elevated AT content results in easily melted DNA. This has been previously linked to origin function independently of exact sequence, particularly in the case of DUEs [21,39]; these generally overlap with the A-rich region and the B2 element. We analyzed the melting free energy [21,40] of origin candidates selected by Oriscan to determine whether thermodynamic characteristics might be useful in refining predictions and also to see whether the predictions have a similar melting free energy to that of known origins. Specifically, we considered the sequence from -108 to +159 - the same area analyzed by the large position-weight matrix - and the A-rich subset of this region, +25 to +105. For both of these regions, known origins have lower melting free energies than intergenic sequences, which in turn show less stability than bulk sequence. We found that predictions ranking above 350 had melting free energies indistinguishable from known origins in both of these regions, but inclusion of helical stability analysis in the Oriscan algorithm did not improve performance (not shown). The position-weight matrix, which accounts for the A versus T strand bias in the A-rich region, contributes to prediction accuracy more effectively than melting free energy. This is consistent with a role for the A-rich region beyond being easily melted.
Discussion
We developed the Oriscan algorithm to predict the exact location of replication origins in the S. cerevisiae genome based entirely on the similarity of their sequences to previously identified ARS elements. Oriscan uses both the ORC binding site and its flanking regions to identify candidates, and it then ranks potential origins by their likelihood of activity. Starting from an initial selection of 12,000 ACS matches, of which the vast majority (97%) are inactive, Oriscan picked the 100 most similar to known origins. All but six match sites with evidence of origin activity, including ten previously undetected ARS elements. Thus, the algorithm can recognize many origins with near-perfect specificity, indicating that we have defined sufficient conditions for origin function in yeast. The top 350 predictions have 70% specificity and 58% sensitivity, showing considerable generality. This specificity value means that Oriscan has a false positive rate of only one per 115 kb of sequence. In comparison, current eukaryotic promoter prediction algorithms have false positive rates of one per 12 kb with 52% sensitivity [41], or one per 1.1 kb with 53% sensitivity [42]. Oriscan's performance is the first time that eukaryotic replication origins have been accurately identified by sequence alone.
Oriscan selects matches to the ACS that are flanked by sequence broadly similar to known origins. The most striking feature outside the ACS is an A-rich region 25-105 nucleotides to the 3' side. The rest of the flanking sequence used by Oriscan was enriched in both A and T residues. The A-rich region encompasses the area where DUEs are found [21] and where the first RNA primers are synthesized [43]. Thus, it is similar to the AT-rich boxes in Escherichia coli oriC, which are likewise situated adjacent to the replicator sequence [44] and are a DUE [45]. The role of the strand-bias is unknown, but A-tracts influence nucleosome positioning in vivo [46,47].
The addition of 208 experimentally supported Oriscan predictions (Additional data file 8) to the 26-member training set allowed us to reexamine the ACS and its flanking sequences. Immediately 5' to the ACS, we found a T-rich region (Figure 6), mirroring the A-rich region on the 3' side. This enrichment had not been previously appreciated amid the noise of low-level conservation and generally elevated AT content. Nucleotide frequencies became considerably smoother elsewhere in flanking regions when considering the larger sample (Figure 6).
Figure 6.
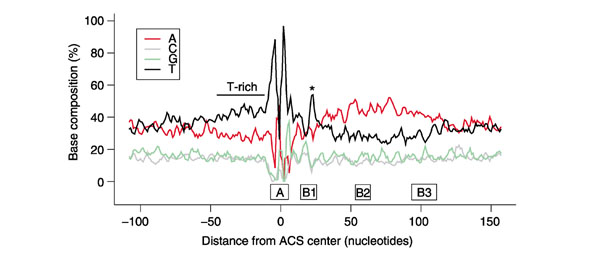
Augmented sequence profile of known and predicted yeast replication origins. The 26-member training set and 208 experimentally supported predictions were combined, and their nucleotide frequencies were moving-averaged in a 3 nucleotide window. We used a 3 nucleotide window because it was the minimum needed to produce a relatively smooth plot. Shown is the 268 nucleotide region analyzed by Oriscan; the positions of A and B elements in ARS1 [48] are indicated below the horizontal axis. A peak in the frequency of T residues between the ACS and the A-rich region corresponding to the WTTT consensus within the B1 element is indicated by an asterisk, and a T-rich region is noted 5' to the ACS.
Interestingly, we also found conserved sequence within the B1 region (Figure 6 and Additional data files 9 and 10). In ARS1, the B1 element lies from 14 to 27 nucleotides downstream of the ACS center [48]. We found that positions 22-25 showed the moderately conserved consensus WTTT, visible in Figure 6 as the spike in T content between the ACS and the A-rich region. A small amount of conservation in this region was noted previously [6].
The addition of the predicted sequences caused small adjustments in the ACS itself (Additional data files 9 and 10). A large change was not expected because this sequence is well conserved and the new ACSs were selected on the basis of their similarity to the original 26.
The ACS and B1 elements have been described as short sequences that could be inactivated with a small number of mutations [2], whereas the B2 element was frequently associated with DUEs and was sometimes short and sequence specific, as in ARS1 [48], but in other cases, such as ARS305 [49], it extended over a longer region and was not easily inactivated by point mutations. When present, the B3 element was also sequence specific, although binding site sequences for other transcription factors can be substituted [48]. Our results clearly agree with the conception of the ACS and B1 elements as short, discrete sequences. Meanwhile, the size of the A-rich region and the overlap of its location with that of B2 elements suggests that most yeast origins contain a long B2 element. We find no indication of a short, highly conserved stretch within the A-rich region (Figure 6 and Additional data file 9), although it may have escaped our notice if its position is variable. Nonetheless, a MEME search [50] of the known and predicted origin sequences listed in Additional data file 8 for conserved sequence motifs did not reveal any such candidates (data not shown). B3 elements were not analyzed by Oriscan because of their variability in sequence and position, as well as their complete absence from some origins.
We found that ACSs likely to be part of origins show highly significant conservation across species in a position-specific manner. Increased conservation has also been found for another important class of non-coding DNA: transcription factor binding sites [51]. That we found a high degree of conservation at predicted and known origins is an additional verification of Oriscan's ability to identify functional origins amid a sea of other sequence. The lower rate of evolution at non-degenerate positions in probably active ACSs and the preference for conservative mutations at degenerate positions strongly suggests that many origin loci have been conserved during the 20 million years since these species diverged. It has been shown that the most slowly evolving positions generally participate in the most important contacts with the protein that binds them [52]. We expect therefore the primary sequence-specific contacts between ORC and the ACS to occur at the highly conserved, nondegenerate nucleotides between positions -4 and +7. Surprisingly, two of these positions were not included in the original, 11 bp version of the ACS [6].
Chromatin-bound ORC complexes that do not associate with MCM may be involved in functions independent of replication, particularly control of chromatin silencing [11,53]. We analyzed the data of Wyrick et al. [28] and found 60 sites not designated pro-ARSs which bound ORC (p < 0.05) but not MCM (_p_ > 0.05 within 2 kb). We compared these 60 sites to the top 350 Oriscan predictions and found only a single match. In contrast, Oriscan found 180 of the 408 evaluation set members, which were ORC/MCM binding sites and known origins. Thus, Oriscan specifically recognized ORC binding sites that associate with MCM and therefore probably function as origins.
Conversely, there are 88 sites that were reported to bind MCM but not ORC [28]. Of these, ten were predicted by Oriscan; two of the sites are on chromosome XV, and both showed ARS activity (a and b in Figure 4b). It is unlikely that these loci do not, in fact, bind ORC, since Oriscan recognized an ACS. ORC binding probably failed to be detected by chromatin immunoprecipitation.
Oriscan made a number of predictions that did not agree with the evaluation set. We tested 25 of these discrepancies and found that 15 had ARS activity. Extrapolating from the results on chromosome XV, we expect that there are at least 50 predictions overall in Oriscan's top 350 that are origins but do not match the evaluation set. Surely, many of these origins were missed earlier because of experimental error. For example, we found three probable positional errors, where Oriscan predicted an origin within a few hundred nucleotides of an ORC/MCM binding signal. We also found that in eight of the 15 new origins, binding of replication proteins was detected but at a statistically insignificant level, such that the location was not designated a pro-ARS [28]. Of the new origins we found, four did not correspond to any ORC/MCM or array origin signal. We speculate that these origins may be used under different conditions than those used in the binding assay. Perhaps they become more active in a transcription factor or chromatin modification-dependent manner.
We found that 55% of the top 100 Oriscan predictions match array origins [27] within 4 kb. This is a poorer match than with the evaluation set, but is greater than the 44% overlap of the training set with the array origins, and about the same as the 52% agreement between ORC/MCM sites and the array origins. The incomplete overlap of these three sets with array origins is probably caused by chromosomal context effects, including failure of an origin to fire because a nearby origin fires first as well as the difficulty of detecting weak origins using microarrays.
While Oriscan efficiently recognized most origins, it was unable to detect all of them, as indicated by the sensitivity of 58% for the top 350 predictions. Origins are missed at both the ACS and flanking sequence recognition steps of Oriscan. Even four training set members (ARS121, ARS304, ARS601, and ARS1413) are not recognized by Oriscan after jackknifing because they have unusual ACSs. Relaxing the algorithm to retain these four allows too many non-origin sequences through and degrades performance. The 12,000 ACS matches selected by the first step of Oriscan include matches to 87% of the evaluation set; the remaining 13% are most probably missed because they have unusual ACSs like the four training set members.
A similar situation exists for the flanking sequence, as seven training set members fail this step, and application of the flanking sequence matrix reduces sensitivity by about 10%. These missed origins may simply be outliers, or they may differ more fundamentally from most ARSs. Finally, the ranking step at the end of Oriscan uses the scores from previous steps and is subject to the same sources of insensitivity. Thus, the incomplete sensitivity of Oriscan results from a combination of factors rooted in the heterogeneity of origins that previously prevented their systematic identification entirely.
Conclusions
The Oriscan algorithm's strategy of searching for a specific, well-conserved sequence within a broader region with more loosely defined characteristics was highly effective in identifying yeast replication origins. Thus, we demonstrated that the majority of origins in yeast have a subtle, recognizable consistency beyond the ACS. This paradigm, particularly the idea of a broad region of low-level conservation, will probably be applicable elsewhere, such as in the search for origins in other organisms. _Cis_-acting sequences seem to function in metazoan origin determination at some stages of the life cycle [54,55]. However, a detailed understanding of their structure has been elusive because of the greater size and complexity of these origins [56]. If enough similar origins are identified at high resolution, Oriscan might serve as a model for building an algorithm to recognize them.
Materials and methods
Oriscan
The training set consisted of 26 known yeast origins (Additional data file 1) for which the ACS had been rigorously identified by site-directed mutagenesis. Three of the origins are compound, having more than one active ACS [33,57]. In these cases, we did not incorporate the flanking sequence, which may have structural organization different from the more common simple origins. Therefore, from simple origins only, we extracted the sequence from -108 to +159 relative to the ACS T-rich strand center. These bounds were initially chosen by eye, based on the plots in Figure 1; Oriscan was not sensitive to small changes (<10 nucleotides) in the bounds, and greater changes either gave no change or a decrease in performance. The seven active ACSs from compound origins were added, and these sequences were aligned manually by the ACS with no gaps. We constructed a position-weight matrix, designated the 268 nucleotide matrix, consisting of one column for each position and one row for each of the four bases. We also constructed the ACS submatrix, which covered only the ACS and consisted of the natural logarithm of each original matrix element divided by the appropriate genomic frequency (0.3085 for A and T, and 0.1915 for G and C). To avoid infinite values, immediately before log transformation, zeroes were substituted with (2Nseq)-1, where Nseq is the training set size (30 for the ACS). A third log-transformed submatrix, the non-T matrix, consisted only of the non-T positions in the ACS (wwwwtttAyRtttwGtt). Log transformation improved the performance of the ACS and non-T submatrices; extremely rare bases are penalized to a greater extent, due to the deformation of log-space as frequency approaches zero. Thus, log-transformed matrices are more effective when analyzing a highly conserved sequence such as the ACS. Conversely, the 268 nucleotide matrix performed better without log transformation due to the more variable, less conserved sequence it covers.
We used the three matrices to analyze the yeast genome. PERL and R [58] scripts were written to scan the genome for matches to the ACS and further analyze those matches. Matrix scores were adjusted such that lower values indicate better matches. The three matrix scores were subjected to the following cutoffs: ACS matrix score of -6 or better, corresponding to 2-4 mismatches out of 17, depending on the conservation of the mismatched position; non-T matrix score of 0.5 or better, corresponding to no more than one mismatch; and 268 nucleotide matrix score of 38 or better, which was 2.4 standard deviations better than the mean for bulk sequence. These scores, along with the number of adenine residues between 25 and 105 nucleotide 3' of the ACS center, were saved for all positions (approximately 2,000) that passed the cutoffs.
We then calculated overall similarity of each candidate sequence to the training set as follows. For each of the four analyses, we generated smoothed histograms showing the score distributions for known origins and the about 2,000 candidates. These histograms essentially describe the characteristics of the populations of known origins and of the set of candidates that passed the cutoffs described above. The histograms of known origins in the ACS and 268 nucleotide analyses, intended as a reference for where other origins should score, were constructed using jackknifing. This is a treatment in which each individual sequence is evaluated using matrices that were constructed from only the other members of the training set. Without jackknifing, training set members score disproportionately well.
In histogram smoothing, 15 equally spaced breaks were calculated for each analysis such that the extremes matched the maximum and minimum scores. Normalized histograms were calculated for these intervals, and also for a 16-break set of intervals with the same spacing but shifted such that the extremes were half an interval beyond the minimum and maximum. These results were pooled, resulting in histograms that counted each observation twice, and the counts were smoothed by a three-point moving average. This procedure was designed to suppress the jaggedness that is common to histograms based on sample sizes in the range of our training set. In order to avoid overtraining, it was never optimized to increase performance.
We then derived an estimate of the population characteristics of inactive predictions by scaling the known histograms by a factor of 0.1 and subtracting them from the prediction histograms; the difference was then renormalized. The value of 0.1 was chosen as an estimate of the proportion of active sequences in the candidate population. The performance of Oriscan was insensitive to variation of this parameter between 0.05 and 0.2. The inactive and known histograms were interpolated using cubic splines. To reduce sampling error in low-density regions, we set minimum values; for the splines representing inactive predictions, the minimum was 0.9(2_N_ p)-1 where N p is the number of predictions that passed the initial cutoffs. The minimum value for the known histograms was the lesser of the bestfit Gaussian distribution or (2_N_ k)-1 where N k is the number of sequences in the training set. We used these cubic spline interpolations to estimate the likelihood of finding an origin on one hand, or an inactive sequence on the other, given the characteristics of every candidate; we reasoned that origins would probably have scores in high-density regions, or peaks, of the known histograms, whereas inactive predictions would generally score in high-density regions of the inactive histograms. The splines are a way of estimating the histogram height, or population density, at every score in a given range.
For each candidate, both sets of splines were evaluated at its four scores. A final value p describing similarity to the known and wrong populations was calculated as p = 0.1_f_1(0.9_f_0 + 0.1_f_1)-1 where _f_1 is the product of the four known spline evaluations, and _f_0 is the product of the four 'wrong' spline evaluations. The candidates were then ranked by p, with values near one indicative of the greatest similarity to known origins.
Predictions were evaluated against experimental evidence as follows. The training set was always excluded. After the prediction run, every position matching a member of the training set was stricken. Then, each remaining prediction was checked to see if it fell within bounds of a region suggested or shown to have ARS or origin activity. The evaluation set had 408 members and was constructed by combining all ARS elements, chromosomal origins supported by two-dimensional gels, and the set of pro-ARS loci [28]. The pro-ARS coordinates were extended by 250 nucleotides in each direction because Wyrick et al. [28] showed that these coordinates have imperfect precision. As described in Results, this extension was sometimes insufficient, but a larger extension would have further increased the likelihood of an incorrect prediction spuriously falling within a pro-ARS site.
To evaluate a set of predictions, the number of predictions that did not match the evaluation set was divided by the number of predictions analyzed (for example, the top 100 or top 350) to give the raw false positive rate. This rate was then adjusted upward for the expected number of wrong predictions that match an active locus by chance. The adjustment consisted of dividing the false positive rate by 1-x spec, where x spec is the expected apparent specificity of random predictions, equivalent to the fraction of the genome covered by the evaluation set. Specificity was then calculated by subtracting this adjusted false positive rate from one.
We calculated sensitivity in a similar manner, with the raw false negative rate calculated as the number of members of the evaluation set not matched by a prediction divided by the total size. This was then adjusted upward by dividing by 1-x sens, where x sens is the expected apparent sensitivity for random predictions, calculated as the fraction of the genome covered by the set of predictions being evaluated (each prediction was treated as covering a window of sequence the size of the average member of the evaluation set). Sensitivity was then calculated by subtracting the false negative rate from one.
ARS assays
To test predictions for ARS activity, we designed PCR primers (Qiagen, Inc.) with flanking BamHI sites to amplify a 300-550 nucleotide fragment containing the 268 nucleotide region detected by the algorithm. The PCR product was digested with BamHI (NEB, Inc.) and ligated into the BamHI site of pRS326 [32]. The ligation reactions were transformed into CaCl2-competent E. coli DH5α, and correct products were verified by restriction mapping and sequencing. Site directed mutagenesis was performed as described [59]; in each case, the central 11 bp of the ACS were mutated to contain the GC-rich BamHI restriction site. To assay ARS activity, 100 ng of plasmid was transformed into S. cerevisiae W303-1a (relevant genotype ura3-1 ade2-1) using the lithium acetate procedure [60]. Transformants were plated on SC URA-10 μg/ml ADE plates and incubated at least six days at 30°C, alongside an empty vector control. An insert was considered to have ARS activity if it promoted growth of colonies able to be propagated at a frequency 103 times greater than the empty vector. We categorized ARS elements according to growth rate and color, which is indicative of plasmid copy number [34] (Additional data files 6 and 7).
Evolutionary conservation of the ACS
Alignments of the ACS of predicted and known origins and control ACSs were extracted from T-Coffee alignments of 1 kb stretches upstream of S. cerevisiae ORFs generously provided by D.Y. Chiang and M.B. Eisen. The number of substitutions in each position of each alignment were calculated using an algorithm in which substitutions or deletions relative to S. cerevisiae were used to determine the maximum parsimony cost (number of substitutions) based on the comb-shaped phylogenetic tree that describes the relationship between the five species [61]. Substitutions per position, plotted in Figure 5a, were calculated by dividing the total cost at each position by the number of species and the number of alignments. Statistical significance was calculated empirically; we randomly selected 75 alignments from the control set and calculated the substitutions per site as above. This was repeated 10,000 times, and the p value was calculated as the frequency at which an evolutionary rate occurred that was equal to or less than the rate found for the probable origins.
The number of conservative mutations was determined by recalculating the total cost at each position after setting synonymous bases (for example, C and T for the central pyrimidine position) equal to each other, and then measuring the difference in cost; this difference was divided by the total original cost to give the fractions plotted in Figure 5b. Statistical significance was calculated using Student's two-tailed _t_-test.
Additional data files
The following additional information is provided with the online version of this article: Locations of known origins and ORC/MCM binding sites used in the evaluation and training sets (Additional data file 1); position weight matrices used by Oriscan (Additional data file 2); ACS matches identified by Oriscan (Additional data file 3); the ranked list of 1,882 candidate origins that passed the first three steps of Oriscan (Additional data file 4); ACS and flanking score distributions (Additional data file 5); the ARS assay results (Additional data file 6); representative ARS assays (Additional data file 7); sequences of the training set and experimentally supported predictions ranking 350 or better (Additional data file 8); the frequency matrix calculated from the sequences in Additional data file 8, and used to generate Figure 6 and Additional data file 10 (Additional data file 9); and finally, a graphical representation of ACS and B1 nucleotide frequencies in all experimentally supported Oriscan predictions and known origins (Additional data file 10).
Supplementary Material
Additional data file 1
Locations of known origins and ORC/MCM binding sites used in the evaluation and training sets
Additional data file 2
Position weight matrices used by Oriscan
Additional data file 3
ACS matches identified by Oriscan
Additional data file 4
The ranked list of 1,882 candidate origins that passed the first three steps of Oriscan
Additional data file 5
ACS and flanking score distributions
Additional data file 6
The ARS assay results
Additional data file 7
Representative ARS assays
Additional data file 8
Sequences of the training set and experimentally supported predictions ranking 350 or better
Additional data file 9
The frequency matrix calculated from the sequences in Additional data file 8, and used to generate Figure 6 and Additional data file 10
Additional data file 10
A graphical representation of ACS and B1 nucleotide frequencies in all experimentally supported Oriscan predictions and known origins
Acknowledgments
Acknowledgements
This work was supported by NIH grant GM 31655 to N.R.C. The authors wish to thank Jasper Rine and Michael S. Kobor for strains, plasmids, and advice on ARS assays, Derek Y. Chiang and Michael B. Eisen for advice and unpublished alignments of multiple yeast genomes, Hunter B. Fraser and Sophie Dumont for critical reading of the manuscript, and Lior Pachter for helpful discussions. A.M.B. is a Howard Hughes Medical Institute predoctoral fellow.
References
- Jacob F, Brenner S, Cuzin F. On the regulation of DNA replication in bacteria. Cold Spring Harb Symp Quant Biol. 1963;28:329–438. [Google Scholar]
- Newlon CS, Collins I, Dershowitz A, Deshpande AM, Greenfeder SA, Ong LY, Theis JF. Analysis of replication origin function on chromosome III of Saccharomyces cerevisiae. Cold Spring Harb Symp Quant Biol. 1993;58:415–423. doi: 10.1101/sqb.1993.058.01.048. [DOI] [PubMed] [Google Scholar]
- Stinchcomb DT, Struhl K, Davis RW. Isolation and characterisation of a yeast chromosomal replicator. Nature. 1979;282:39–43. doi: 10.1038/282039a0. [DOI] [PubMed] [Google Scholar]
- Brewer BJ, Fangman WL. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell. 1987;51:463–471. doi: 10.1016/0092-8674(87)90642-8. [DOI] [PubMed] [Google Scholar]
- Huberman JA, Spotila LD, Nawotka KA, el-Assouli SM, Davis LR. The in vivo replication origin of the yeast 2 microns plasmid. Cell. 1987;51:473–481. doi: 10.1016/0092-8674(87)90643-x. [DOI] [PubMed] [Google Scholar]
- Theis JF, Newlon CS. The ARS309 chromosomal replicator of Saccharomyces cerevisiae depends on an exceptional ARS consensus sequence. Proc Natl Acad Sci USA. 1997;94:10786–10791. doi: 10.1073/pnas.94.20.10786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diffley JF, Cocker JH. Protein-DNA interactions at a yeast replication origin. Nature. 1992;357:169–172. doi: 10.1038/357169a0. [DOI] [PubMed] [Google Scholar]
- Bell SP, Stillman B. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature. 1992;357:128–134. doi: 10.1038/357128a0. [DOI] [PubMed] [Google Scholar]
- Chesnokov I, Remus D, Botchan M. Functional analysis of mutant and wild-type Drosophila origin recognition complex. Proc Natl Acad Sci USA. 2001;98:11997–12002. doi: 10.1073/pnas.211342798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin KA, Hidaka M, Stillman B. Conserved initiator proteins in eukaryotes. Science. 1995;270:1667–1671. doi: 10.1126/science.270.5242.1667. [DOI] [PubMed] [Google Scholar]
- Foss M, McNally FJ, Laurenson P, Rine J. Origin recognition complex (ORC) in transcriptional silencing and DNA replication in S. cerevisiae. Science. 1993;262:1838–1844. doi: 10.1126/science.8266071. [DOI] [PubMed] [Google Scholar]
- Pak DT, Pflumm M, Chesnokov I, Huang DW, Kellum R, Marr J, Romanowski P, Botchan MR. Association of the origin recognition complex with heterochromatin and HP1 in higher eukaryotes. Cell. 1997;91:311–323. doi: 10.1016/s0092-8674(00)80415-8. [DOI] [PubMed] [Google Scholar]
- Chesnokov IN, Chesnokova ON, Botchan M. A cytokinetic function of Drosophila ORC6 protein resides in a domain distinct from its replication activity. Proc Natl Acad Sci USA . 2003;100:9150–9155. doi: 10.1073/pnas.1633580100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasanth SG, Prasanth KV, Stillman B. Orc6 involved in DNA replication, chromosome segregation, and cytokinesis. Science . 2002;297:1026–1031. doi: 10.1126/science.1072802. [DOI] [PubMed] [Google Scholar]
- Aparicio OM, Weinstein DM, Bell SP. Components and dynamics of DNA replication complexes in S. cerevisiae: redistribution of MCM proteins and Cdc45p during S phase. Cell . 1997;91:59–69. doi: 10.1016/S0092-8674(01)80009-X. [DOI] [PubMed] [Google Scholar]
- Celniker SE, Sweder K, Srienc F, Bailey JE, Campbell JL. Deletion mutations affecting autonomously replicating sequence ARS1 of Saccharomyces cerevisiae. Mol Cell Biol. 1984;4:2455–2466. doi: 10.1128/mcb.4.11.2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newlon CS, Theis JF. The structure and function of yeast ARS elements. Curr Opin Genet Dev. 1993;3:752–758. doi: 10.1016/s0959-437x(05)80094-2. [DOI] [PubMed] [Google Scholar]
- Wilmes GM, Bell SP. The B2 element of the Saccharomyces cerevisiae ARS1 origin of replication requires specific sequences to facilitate pre-RC formation. Proc Natl Acad Sci USA. 2002;99:101–106. doi: 10.1073/pnas.012578499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Stillman B. Assembly of a complex containing Cdc45p, replication protein A, and Mcm2p at replication origins controlled by S-phase cyclin-dependent kinases and Cdc7p-Dbf4p kinase. Mol Cell Biol. 2000;20:3086–3096. doi: 10.1128/MCB.20.9.3086-3096.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto K, Ishimi Y. Single-stranded-DNA-binding protein-dependent DNA unwinding of the yeast ARS1 region. Mol Cell Biol. 1994;14:4624–4632. doi: 10.1128/mcb.14.7.4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natale DA, Umek RM, Kowalski D. Ease of DNA unwinding is a conserved property of yeast replication origins. Nucleic Acids Res . 1993;21:555–560. doi: 10.1093/nar/21.3.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipford JR, Bell SP. Nucleosomes positioned by ORC facilitate the initiation of DNA replication. Mol Cell. 2001;7:21–30. doi: 10.1016/s1097-2765(01)00151-4. [DOI] [PubMed] [Google Scholar]
- Sharma K, Weinberger M, Huberman JA. Roles for internal and flanking sequences in regulating the activity of mating-type-silencer-associated replication origins in Saccharomyces cerevisiae. Genetics . 2001;159:35–45. doi: 10.1093/genetics/159.1.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker SS, Francesconi SC, Eisenberg S. A DNA replication enhancer in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1990;87:4665–4669. doi: 10.1073/pnas.87.12.4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri S, Byers R, Upton T, Eisenberg S. Functional analysis of a replication origin from Saccharomyces cerevisiae: identification of a new replication enhancer. Nucleic Acids Res. 1997;25:5057–5064. doi: 10.1093/nar/25.24.5057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang VK, Fitch MJ, Donato JJ, Christensen TW, Merchant AM, Tye BK. Mcm1 binds replication origins. J Biol Chem. 2003;278:6093–6100. doi: 10.1074/jbc.M209827200. [DOI] [PubMed] [Google Scholar]
- Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, Lockhart DJ, Davis RW, Brewer BJ, Fangman WL. Replication dynamics of the yeast genome. Science. 2001;294:115–121. doi: 10.1126/science.294.5540.115. [DOI] [PubMed] [Google Scholar]
- Wyrick JJ, Aparicio JG, Chen T, Barnett JD, Jennings EG, Young RA, Bell SP, Aparicio OM. Genome-wide distribution of ORC and MCM proteins in S. cerevisiae: high-resolution mapping of replication origins. Science. 2001;294:2357–2360. doi: 10.1126/science.1066101. [DOI] [PubMed] [Google Scholar]
- Yabuki N, Terashima H, Kitada K. Mapping of early firing origins on a replication profile of budding yeast. Genes Cells. 2002;7:781–789. doi: 10.1046/j.1365-2443.2002.00559.x. [DOI] [PubMed] [Google Scholar]
- Bielinsky AK, Gerbi SA. Discrete start sites for DNA synthesis in the yeast ARS1 origin. Science. 1998;279:95–98. doi: 10.1126/science.279.5347.95. [DOI] [PubMed] [Google Scholar]
- Schneider TD, Stormo GD, Gold L, Ehrenfeucht A. Information content of binding sites on nucleotide sequences. J Mol Biol. 1986;188:415–431. doi: 10.1016/0022-2836(86)90165-8. [DOI] [PubMed] [Google Scholar]
- Theis JF, Newlon CS. Domain B of ARS307 contains two functional elements and contributes to chromosomal replication origin function. Mol Cell Biol. 1994;14:7652–7659. doi: 10.1128/mcb.14.11.7652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirahige K, Iwasaki T, Rashid MB, Ogasawara N, Yoshikawa H. Location and characterization of autonomously replicating sequences from chromosome VI of Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:5043–5056. doi: 10.1128/mcb.13.8.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shero JH, Koval M, Spencer F, Palmer RE, Hieter P, Koshland D. Analysis of chromosome segregation in Saccharomyces cerevisiae. Methods Enzymol. 1991;194:749–773. doi: 10.1016/0076-6879(91)94057-j. [DOI] [PubMed] [Google Scholar]
- Kellis M, Patterson N, Endrizzi M, Birren B, Lander ES. Sequencing and comparison of yeast species to identify genes and regulatory elements. Nature. 2003;423:241–254. doi: 10.1038/nature01644. [DOI] [PubMed] [Google Scholar]
- Cliften P, Sudarsanam P, Desikan A, Fulton L, Fulton B, Majors J, Waterston R, Cohen BA, Johnston M. Finding functional features in Saccharomyces genomes by phylogenetic footprinting. Science. 2003;301:71–76. doi: 10.1126/science.1084337. [DOI] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J. T-Coffee: a novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Durbin R, Eddy S, Krogh A, Mitchison G. Biological Sequence Analysis: Probabilistic Models of Proteins and Nucleic Acids. Cambridge, UK: Cambridge University Press; 1998. [Google Scholar]
- Natale DA, Schubert AE, Kowalski D. DNA helical stability accounts for mutational defects in a yeast replication origin. Proc Natl Acad Sci USA. 1992;89:2654–2658. doi: 10.1073/pnas.89.7.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allawi HT, SantaLucia J., Jr Thermodynamics and NMR of internal G•T mismatches in DNA. Biochemistry. 1997;36:10581–10594. doi: 10.1021/bi962590c. [DOI] [PubMed] [Google Scholar]
- Ohler U, Liao GC, Niemann H, Rubin GM. Computational analysis of core promoters in the Drosophila genome. Genome Biol. 2002;3:research0087.1–0087.12. doi: 10.1186/gb-2002-3-12-research0087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese MG. Application of a time-delay neural network to promoter annotation in the Drosophila melanogaster genome. Comput Chem. 2001;26:51–56. doi: 10.1016/S0097-8485(01)00099-7. [DOI] [PubMed] [Google Scholar]
- Bielinsky AK, Gerbi SA. Chromosomal ARS1 has a single leading strand start site. Mol Cell. 1999;3:477–486. doi: 10.1016/s1097-2765(00)80475-x. [DOI] [PubMed] [Google Scholar]
- Kornberg A, Baker T. DNA Replication. New York: WH Freeman and Company; 1992. [Google Scholar]
- Kowalski D, Eddy MJ. The DNA unwinding element: a novel, cis-acting component that facilitates opening of the Escherichia coli replication origin. EMBO J. 1989;8:4335–4344. doi: 10.1002/j.1460-2075.1989.tb08620.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu M, Mori T, Sakurai T, Shindo H. Destabilization of nucleosomes by an unusual DNA conformation adopted by poly(dA)•poly(dT) tracts in vivo. EMBO J. 2000;19:3358–3365. doi: 10.1093/emboj/19.13.3358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter B, Schnappauf G, Thoma F. Poly(dA•dT) sequences exist as rigid DNA structures in nucleosome-free yeast promoters in vivo. Nucleic Acids Res. 2000;28:4083–4089. doi: 10.1093/nar/28.21.4083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marahrens Y, Stillman B. A yeast chromosomal origin of DNA replication defined by multiple functional elements. Science. 1992;255:817–823. doi: 10.1126/science.1536007. [DOI] [PubMed] [Google Scholar]
- Huang RY, Kowalski D. Multiple DNA elements in ARS305 determine replication origin activity in a yeast chromosome. Nucleic Acids Res. 1996;24:816–823. doi: 10.1093/nar/24.5.816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
- Hardison RC. Conserved noncoding sequences are reliable guides to regulatory elements. Trends Genet. 2000;16:369–372. doi: 10.1016/S0168-9525(00)02081-3. [DOI] [PubMed] [Google Scholar]
- Moses AM, Chiang DY, Kellis M, Lander ES, Eisen MB. Position specific variation in the rate of evolution in transcription factor binding sites. BMC Evol Biol. 2003;3:19. doi: 10.1186/1471-2148-3-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrenhofer-Murray AE, Kamakaka RT, Rine J. A role for the replication proteins PCNA, RF-C, polymerase epsilon and Cdc45 in transcriptional silencing in Saccharomyces cerevisiae. Genetics. 1999;153:1171–1182. doi: 10.1093/genetics/153.3.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin RJ, Orr-Weaver TL, Bell SP. Drosophila ORC specifically binds to ACE3, an origin of DNA replication control element. Genes Dev. 1999;13:2639–2649. doi: 10.1101/gad.13.20.2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladenburger EM, Keller C, Knippers R. Identification of a binding region for human origin recognition complex proteins 1 and 2 that coincides with an origin of DNA replication. Mol Cell Biol. 2002;22:1036–1048. doi: 10.1128/MCB.22.4.1036-1048.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spradling AC. ORC binding, gene amplification, and the nature of metazoan replication origins. Genes Dev. 1999;13:2619–2623. doi: 10.1101/gad.13.20.2619. [DOI] [PubMed] [Google Scholar]
- Theis JF, Newlon CS. Two compound replication origins in Saccharomyces cerevisiae contain redundant origin recognition complex binding sites. Mol Cell Biol. 2001;21:2790–2801. doi: 10.1128/MCB.21.8.2790-2801.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The R Project for Statistical Computing http://www.r-project.org
- methodbook.net http://www.methodbook.net/pcr/pcrmut.html
- Gietz RD, Woods RA. Transformation of yeast by the lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002;350:87–96. doi: 10.1016/s0076-6879(02)50957-5. [DOI] [PubMed] [Google Scholar]
- Cliften PF, Hillier LW, Fulton L, Graves T, Miner T, Gish WR, Waterston RH, Johnston M. Surveying Saccharomyces genomes to identify functional elements by comparative DNA sequence analysis. Genome Res. 2001;11:1175–1186. doi: 10.1101/gr.182901. [DOI] [PubMed] [Google Scholar]
- Dimock K, James AP, Seligy VL. Molecular cloning of the ADE1 gene of Saccharomyces cerevisiae and stability of the transformants. Gene. 1984;27:233–237. doi: 10.1016/0378-1119(84)90144-6. [DOI] [PubMed] [Google Scholar]
- Bouton AH, Smith MM. Fine-structure analysis of the DNA sequence requirements for autonomous replication of Saccharomyces cerevisiae plasmids. Mol Cell Biol. 1986;6:2354–2363. doi: 10.1128/mcb.6.7.2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Button LL, Astell CR. The Saccharomyces cerevisiae chromosome III left telomere has a type X, but not a type Y', ARS region. Mol Cell Biol. 1986;6:1352–1356. doi: 10.1128/mcb.6.4.1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newlon CS, Lipchitz LR, Collins I, Deshpande A, Devenish RJ, Green RP, Klein HL, Palzkill TG, Ren RB, Synn S, et al. Analysis of a circular derivative of Saccharomyces cerevisiae chromosome III: a physical map and identification and location of ARS elements. Genetics . 1991;129:343–357. doi: 10.1093/genetics/129.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vujcic M, Miller CA, Kowalski D. Activation of silent replication origins at autonomously replicating sequence elements near the HML locus in budding yeast. Mol Cell Biol. 1999;19:6098–6109. doi: 10.1128/mcb.19.9.6098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis JF, Yang C, Schaefer CB, Newlon CS. DNA sequence and functional analysis of homologous ARS elements of Saccharomyces cerevisiae and S. carlsbergensis. Genetics. 1999;152:943–952. doi: 10.1093/genetics/152.3.943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houten JV, Newlon CS. Mutational analysis of the consensus sequence of a replication origin from yeast chromosome III. Mol Cell Biol. 1990;10:3917–3925. doi: 10.1128/mcb.10.8.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poloumienko A, Dershowitz A, De J, Newlon CS. Completion of replication map of Saccharomyces cerevisiae chromosome III. Mol Biol Cell. 2001;12:3317–3327. doi: 10.1091/mbc.12.11.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Micklem G, Nasmyth K. A yeast silencer contains sequences that can promote autonomous plasmid replication and transcriptional activation. Cell. 1987;51:709–719. doi: 10.1016/0092-8674(87)90094-8. [DOI] [PubMed] [Google Scholar]
- Rivier DH, Ekena JL, Rine J. HMR-I is an origin of replication and a silencer in Saccharomyces cerevisiae. Genetics. 1999;151:521–529. doi: 10.1093/genetics/151.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearsey S. Structural requirements for the function of a yeast chromosomal replicator. Cell. 1984;37:299–307. doi: 10.1016/0092-8674(84)90326-x. [DOI] [PubMed] [Google Scholar]
- Celniker SE, Campbell JL. Yeast DNA replication in vitro: initiation and elongation events mimic in vivo processes. Cell. 1982;31:201–213. doi: 10.1016/0092-8674(82)90420-2. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Tanaka Y, Isono K. Systematic mapping of autonomously replicating sequences on chromosome V of Saccharomyces cerevisiae using a novel strategy. Yeast. 1996;12:101–113. doi: 10.1002/(SICI)1097-0061(199602)12:2<101::AID-YEA885>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Feldmann H, Olah J, Friedenreich H. Sequence of a yeast DNA fragment containing a chromosomal replicator and a tRNA Glu 3 gene. Nucleic Acids Res. 1981;9:2949–2959. doi: 10.1093/nar/9.12.2949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson BM, Brewer BJ, Reynolds AE, Fangman WL. A yeast origin of replication is activated late in S phase. Cell. 1991;65:507–515. doi: 10.1016/0092-8674(91)90468-e. [DOI] [PubMed] [Google Scholar]
- Eisenberg S, Civalier C, Tye BK. Specific interaction between a Saccharomyces cerevisiae protein and a DNA element associated with certain autonomously replicating sequences. Proc Natl Acad Sci USA. 1988;85:743–746. doi: 10.1073/pnas.85.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman KL, Brewer BJ, Fangman WL. Replication profile of Saccharomyces cerevisiae chromosome VI. Genes Cells. 1997;2:667–678. doi: 10.1046/j.1365-2443.1997.1520350.x. [DOI] [PubMed] [Google Scholar]
- Iraqui I, Vissers S, Cartiaux M, Urrestarazu A. Characterisation of Saccharomyces cerevisiae ARO8 and ARO9 genes encoding aromatic aminotransferases I and II reveals a new aminotransferase subfamily. Mol Gen Genet. 1998;257:238–248. doi: 10.1007/s004380050644. [DOI] [PubMed] [Google Scholar]
- Chan SMC. PhD thesis. Ithaca: Cornell University; 1985. Chromosome structure of yeast: replication origins and telomeres. [Google Scholar]
- Atcheson C. PhD thesis. Chicago: University of Chicago; 1991. Meiosis-specific regulation of the SPO11 gene of the yeast Saccharomyces cerevisiae. [Google Scholar]
- Hsiao CL, Carbon J. Characterization of a yeast replication origin (ars2) and construction of stable minichromosomes containing cloned yeast centromere DNA (CEN3). Gene. 1981;15:157–166. doi: 10.1016/0378-1119(81)90125-6. [DOI] [PubMed] [Google Scholar]
- Walker SS, Francesconi SC, Tye BK, Eisenberg S. The OBF1 protein and its DNA-binding site are important for the function of an autonomously replicating sequence in Saccharomyces cerevisiae. Mol Cell Biol. 1989;9:2914–2921. doi: 10.1128/mcb.9.7.2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman KL, Diller JD, Ferguson BM, Nyland SV, Brewer BJ, Fangman WL. Multiple determinants controlling activation of yeast replication origins late in S phase. Genes Dev. 1996;10:1595–1607. doi: 10.1101/gad.10.13.1595. [DOI] [PubMed] [Google Scholar]
- Sasnauskas KV, Giadvilaite AA, Ianulaitis AA. [Cloning of the ADE2 gene of Saccharomyces cerevisiae and localization of the ARS-sequence]. Genetika. 1987;23:1141–1148. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional data file 1
Locations of known origins and ORC/MCM binding sites used in the evaluation and training sets
Additional data file 2
Position weight matrices used by Oriscan
Additional data file 3
ACS matches identified by Oriscan
Additional data file 4
The ranked list of 1,882 candidate origins that passed the first three steps of Oriscan
Additional data file 5
ACS and flanking score distributions
Additional data file 6
The ARS assay results
Additional data file 7
Representative ARS assays
Additional data file 8
Sequences of the training set and experimentally supported predictions ranking 350 or better
Additional data file 9
The frequency matrix calculated from the sequences in Additional data file 8, and used to generate Figure 6 and Additional data file 10
Additional data file 10
A graphical representation of ACS and B1 nucleotide frequencies in all experimentally supported Oriscan predictions and known origins