Targeted disruption of Nrf2 causes regenerative immune-mediated hemolytic anemia (original) (raw)
Abstract
A basic leucine zipper transcription factor, NF-E2-related factor 2 (Nrf2), plays a critical role in the cellular defense mechanism by mediating a coordinate up-regulation of antioxidant responsive element-driven detoxification and antioxidant genes. Here, we report that targeted disruption of Nrf2 causes regenerative immune-mediated hemolytic anemia due to increased sequestration of damaged erythrocytes. Splenomegaly and spleen toxicity in Nrf2-/- mice raised a possibility of hemolytic anemia and splenic extramedullary hematopoiesis in Nrf2-/- mice. In support of this, hematology analysis revealed that Nrf2-/- mice suffer from anemia with abnormal red cell morphologies (i.e., Howell-Jolly bodies, acantocytes, and schistocytes). In addition, Nrf2-/- erythrocytes were more sensitive to H2O2-induced hemolysis, and erythrocyte-bound IgG levels were markedly increased in Nrf2-/- mice compared with Nrf2+/+ mice. Because IgG bound to erythrocytes in the presence of oxidative damage in erythrocytes (regardless of Nrf2 genotype), these data support that Nrf2-/- erythrocytes have higher levels of damage compared with Nrf2+/+ cells. Finally, Nrf2-/- mice showed increased levels of erythrocyte-bound IgG compared with Nrf2+/+ mice after H2O2 injection in vivo, suggesting that the decreased glutathione and increased H2O2 render the Nrf2-/- mice more susceptible to toxicity. Taken together, these observations indicate that a chronic increase in oxidative stress due to decreased antioxidant capacity sensitizes erythrocytes and causes hemolytic anemia in Nrf2-/- mice, suggesting a pivotal role of Nrf2-antioxidant responsive element pathway in the cellular antioxidant defense system.
The antioxidant responsive element (ARE) (1) is a cis-acting regulatory element that plays an important role in the gene expression of phase II detoxification enzymes and antioxidant proteins such as GSTs, NAD(P)H:quinone oxidoreductase-1 (NQO1), hemeoxygenase-1 (HO-1), and glutamate-cysteine ligase (GCL). Many studies have reported an important role of the ARE in protecting cells from oxidative stress. For example, _tert_-butylhydroquinone pretreatment, which activates the ARE and subsequently increases the expression of many ARE-driven genes, protected neuroblastoma cells from oxidative cell death (2). Overexpression of one or two ARE-driven genes, however, did not protect cells from glutamate-induced cytotoxicity (3), suggesting that a coordinate up-regulation of ARE-driven genes is more efficient in protecting cells compared with induction of one or two protective genes. Because of this characteristic, the transcription factor that binds to and activates the ARE sequence has been studied extensively.
A basic leucine zipper (b-Zip) transcription factor, NF-E2-related factor 2 (Nrf2), was originally cloned during the search for proteins that bind to the NF-E2/AP-1 motif in the hyper-sensitive site 2 of the locus control region of the β-globin gene cluster (4). Interestingly, Nrf2 appears to regulate ARE-driven genes and not the β-globin gene (5-8). Many studies have also demonstrated that Nrf2 plays a central role in the ARE-driven cellular protection. For example, Nrf2-/- (knockout) mice were more susceptible to butylated hydroxytoluene-induced acute pulmonary injury (9), acetaminophen-induced liver toxicity (10), and hyperoxia-induced lung injury (11) due to decreased levels of ARE-driven cellular protection systems (i.e., glutathione synthesis and detoxification process). Primary neural cells derived from Nrf2-/- mice were also more sensitive to oxidative stress (6, 7), whereas overexpression of Nrf2 protected primary neurons from glutamate-induced oxidative cell death (12, 13). Furthermore, the _tert_-butylhydroquinone-mediated protection of primary neurons depends on activation of Nrf2 in astrocytes (12). Recently, it has been reported that Caenorhabditis elegans protein SKN-1, which is similar to mammalian Nrf2, may function to increase resistance to oxidative stress in C. elegans (14). In addition to protection conferred by Nrf2-dependent ARE-driven genes, Nrf2 is also directly involved in apoptosis signaling pathways. One study actually implies that Nrf2 is a substrate for caspase-3-like proteases (15), and another indicates that Nrf2 inhibits Fas-mediated apoptosis pathway (16). Moreover, Nrf2 is an important effector of PERK-mediated cell survival (17) and regulates the sensitivity of death receptor signals (18). These data suggest that the Nrf2-ARE pathway promotes cell survival by modulating both cellular antioxidant potentials and apoptosis signaling pathways.
Although Nrf2 is expressed widely and is important for cellular antioxidant potential, Nrf2 knockout mice develop and grow normally (5). Young Nrf2-/- mice are not anemic (5), whereas targeted disruption of either NF-E2 or Nrf1 (binding factors of locus control region of β-globin) resulted in anemia (19, 20). Interestingly, we observed signs of anemia in old Nrf2-/- mice presented by splenomegaly and spleen toxicity. Because anemia can reactivate splenic extramedullary hematopoiesis and subsequently induce splenomegaly, we hypothesized that old Nrf2-/- mice suffer from anemia. We also considered that erythrocytes might be a sensitive indicator of in vivo oxidative stress as mature erythrocytes lack an adaptive response to external stimuli due to insufficient genetic material. This study, therefore, was designed to investigate the role of Nrf2 in erythrocyte maintenance, and the mechanism by which Nrf2-/- mice develop anemia.
Materials and Methods
Mice. Nrf2-/- mice were generated as described earlier (5). Mice were killed by CO2, and blood was collected into EDTA-coated tubes for the hematological analysis and erythrocytes morphology examination (Wright's staining). Animals were perfused with PBS, and organs were weighed and frozen.
Cytotoxicity. Spleens were sectioned (10 μm), fixed (4% paraformaldehyde, 20 min), and stained for terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (Roche Molecular Biochemicals) and cyclooxygenase-2 (Santa Cruz Biotechnology) (6, 7). For cytotoxicity, primary splenocytes were prepared as described by Gal et al. (21). Briefly, spleens were isolated (from 6-mo-old mice), and a single cell suspension was prepared by mincing and repeated pipetting in PBS. Cells were washed and plated into 96-well plates (105 cells per well). After 24 h, the cells were treated (for 24 h) with H2O2 (Sigma), and the viability was measured by using the MTS assay [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (Promega)].
Western Blot, NQO1 Activity, and RT-PCR Analysis. Whole-cell extracts (10 μg protein) from spleens were resolved on a SDS/PAGE (10%), and Western blot analysis for HO-1 (Stressgen Biotechnologies, Victoria, Canada), GCL catalytic subunit (GCLC), and the GCL modulatory subunit (GCLM) was performed as described (6, 7). Blots were quantified by using the nih image program. NQO1 enzymatic activity was determined by a colorimetric method for whole cell extracts by using menadione (Sigma) as a substrate (22). For RT-PCR analysis, total RNA was isolated by TRIzol reagent (Invitrogen), and cDNA was synthesized (Promega) according to the manufacturers' protocols. Aliquots of cDNA were used for PCR amplification by using a taq DNA polymerase (Promega). PCR primers specific to each gene are as follows: Nrf2, 5′-TCTCCTCGCTGGAAAAAGAA-3′ and 5′-AATGTGCTGGCTGTGCTTTA-3′; HO-1, 5′-TACACATCCAAGCCGAGAAT-3′, and 5′-GTTCCTCTGTCAGCATCACC-3′; NQO1, 5′-CATTCTGAAAGGCTGGTTTGA-3′ and 5′-CTAGCTTTGATCTGGTTGTCAG-3′; GST A4, 5′-GCCAAGTACCCTTGGTTGAA-3′ and 5′-CAATCCTGACCACCTCAACA-3′; GCLM, 5′-ACCTGGCCTCCTGCTGTGTG-3′ and 5′-GGTCGGTGAGCTGTGGGTGT-3′; GCLC, 5′-ACAAGCACCCCCGCTTCGGT-3′ and 5′-CTCCAGGCCTCTCTCCTCCC-3′; ferritin light chain, 5′-GTGGAAGCTGCCGTGAAC-3′ and 5′-CAGTCTGCGCTGGTTGTG-3′; ferritin heavy chain, 5′-AAGTGCGCCAGAACTACCAC-3′ and 5′-TCTTGCGTAAGTTGGTCACG-3′; thioredoxin reductase-1, 5′-GGGAGAAAAAGGTCGTCTA-3′ and 5′-ACATTGGTCTGCTCTTCATC-3′; peroxiredoxin 1, 5′-TGCCAGATGGACAATTCAAA-3′ and 5′-CAGCTGGACACACTTCACCA-3′; and β-actin, 5′-AGAGCATAGCCCTCGTAGAT-3′ and 5′-CCCAGAGCAAGAGAGGTATC-3′.
In Vitro Hemolysis. Blood was collected into EDTA-coated tubes and centrifuged (600 × g, 5 min), and plasma was removed. Erythrocytes were washed (PBS), resuspended (PBS plus 20 mM glucose) to make 2% packed cell volume, and incubated with H2O2 in a CO2 incubator (37°C, 20% O2 and 5% CO2). After 12 h, cells were centrifuged (600 × g, 10 min, 4°C), and absorbance (541 nm) was measured from supernatant. Lactate dehydrogenase activity was also measured (Sigma) from supernatant as an indicator of hemolysis.
Flow Cytometry Analysis. Isolated erythrocytes (≈5 × 106 cells) were incubated with H2O2 and plasma as described in the legends for Figs. 1, 2, 3, 4, 5. After washing (PBS), erythrocytes were incubated with FITC-conjugated anti-mouse IgG secondary antibody (Vector, 4°C, overnight), and fluorescence intensity (20,000 cells per sample) was measured (FACSCalibur, Becton Dickinson).
Fig. 1.

Morphological changes in Nrf2-/- mice. Two distinctive morphological changes, such as white teeth (A) and splenomegaly (B), were observed in Nrf2-/- mice. (C) The organ to body-weight ratio data showed a dramatically increased spleen weight (14 mo, male, n = 7). S, spleen; K, kidney; L, liver; b, body. (D) Terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling staining and (E) cyclooxygenase-2 immunostaining showed that Nrf2-/- spleen has more cell death and inflammation compared with Nrf2+/+ counterparts. (F) Primary splenocytes were incubated with H2O2 (24 h), and cell viability was measured by MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium salt (Promega)] cytotoxicity assay (n = 6). Each data bar or point represents mean ± SE. *, P < 0.05 by Student's t test.
Fig. 2.

Decreased expression levels of Nrf2-dependent ARE-driven genes in Nrf2-/- mice. Whole cell extracts (spleens) were used for Western blot analysis of HO-1 (A) and GCLC (B) and for NQO1 activity measurement (C) (n = 3). (D) RT-PCR analysis showed decreased levels of many ARE-driven genes in Nrf2-/- splenocyte culture. GCLM, glutamate-cysteine ligase modulatory subunit; FTL, ferritin light chain; FTH, ferritin heavy chain; TXNRD1, thioredoxin reductase-1; PRDX1, peroxiredoxin 1. PCR cycles are listed to the right of the gels.
Fig. 3.

Sensitized Nrf2-/- erythrocytes. Peripheral blood smear (A, Nrf2+/+; B, Nrf2-/-) showed that Nrf2-/- have morphological abnormalities such as Howell-Jolly bodies (C), schistocytes (D), and acantocytes (E), suggesting sensitized erythrocytes and hemolytic anemia in Nrf2-/- mice. For in vitro hemolysis, erythrocytes were incubated with H2O2 (37°C, 12 h). After centrifugation (600 × g, 10 min), Hb contents (F) and lactate dehydrogenase activity (G) in supernatant were measured as indicators of hemolysis (n = 4). *, P < 0.05 by Student's t test.
Fig. 4.

Immune-mediated hemolytic anemia resulting from oxidative damages. (A) Erythrocytes (≈5 × 106 cells) from Nrf2+/+ and Nrf2-/- mice were incubated (4°C, overnight) with FITC-conjugated anti-mouse IgG antibody. (B and C) After washing, fluorescent intensity was measured by flow cytometry. Each data bar represents mean ± SE (n = 5). (D) Erythrocytes (≈5 × 106 cells) from Nrf2+/+ (E, purple in G, and H) or Nrf2-/- mice (F, green in G, and I) were incubated (37°C, 1 h) with plasma (50 μl) from either Nrf2+/+ (green in H and in I) or Nrf2-/- mice (red in H and in I). (J) Nrf2+/+ erythrocytes were incubated (15°C, 10 h) with PBS (K)orH2O2 (L) to induce oxidative damage (L, echinocyte-like erythrocytes). After washing, Nrf2+/+ erythrocytes were incubated with plasma (50 μl) from either Nrf2+/+ (M, and purple in O), or Nrf2-/- mice (N, and green in O), and labeled with FITC-conjugated anti-mouse IgG antibody for flow cytometry analysis.
Fig. 5.

Sensitive Nrf2-/- erythrocytes due to decreased antioxidant potential and increased oxidative stress. (A) Peripheral blood was drawn from Nrf2+/+ (purple in D) and Nrf2-/- mice (green in D) before and after H2O2 injection (0.25 μmol/g body weight, tail vein, 1 h). (B) Erythrocytes (≈5 × 106 cells) were incubated with FITC-conjugated anti-mouse IgG antibody for flow cytometry analysis. (C) Fluorescence intensity of each mouse was corrected by subtracting fluorescence intensity of control erythrocytes (before H2O2 injection) from fluorescence intensity of H2O2-treated erythrocytes (n = 4). Nrf2-/- mice showed decreased total GSH (D) and increased H2O2 levels (E). Each data bar represents mean ± SE (n = 4). *, P < 0.05 by Student's t test.
Glutathione (GSH) and H2O2 Levels. Total GSH levels were determined from serum by measuring the reaction of GSH and dithionitrobenzoic acid coupled to the recycling of oxidized GSH to GSH by GSH reductase (23). Serum H2O2 was quantified by a colorimetric method according to the manufacturer's protocol (Assay Designs, Ann Arbor, MI). Briefly, diluted serum and standards were incubated with color reagent mixture (containing xylenol orange, sorbitol, and ammonium iron sulfate) for 30 min, and optical density (550 nm) was measured.
Results
White Teeth, Splenomegaly, and Spleen Toxicity in Nrf2-/- Mice. Initially, we observed two distinctive phenotype changes in Nrf2-/- mice, such as white teeth (Fig. 1_A_) and splenomegaly (Fig. 1_B_). Interestingly, only spleen weight elevated dramatically (9.1-fold increase), whereas other organs (i.e., liver and kidney) did not (Fig. 1_C_). Nrf2-/- spleens also showed increased levels of cell death (terminal deoxynucleotidyltransferase-mediated dUTP nick-end labeling staining, Fig. 1_D_) and inflammation (cyclooxygenase-2 immunohistochemistry, Fig. 1_E_) compared with Nrf2+/+ spleens. Similarly, Nrf2-/- primary splenocytes were more sensitive to H2O2-induced cytotoxicity compared with Nrf2+/+ cells (Fig. 1_F_).
Decreased Levels of ARE-Driven Genes in Nrf2-/- Mice. To understand the underlying mechanism of increased sensitivity in Nrf2-/- spleens, we measured expression levels of ARE-driven antioxidant genes. Nrf2-/- spleens showed decreased protein levels of HO-1 (56.2% decrease, Fig. 2_A_), GCLC (46.7% decrease, Fig. 2_B_), and GCLM (data not shown). NQO1 activity was also significantly lower in Nrf2-/- spleens compared with Nrf2+/+ counterparts (Fig. 2_C_). Furthermore, RT-PCR analysis showed the expression levels of many Nrf2-dependent ARE-driven genes were higher in Nrf2+/+ splenocytes compared with Nrf2-/- cells (Fig. 2_D_), suggesting a negative correlation between susceptibility of splenocytes to oxidative stress and expression levels of Nrf2-dependent ARE-driven genes.
Anemia in Nrf2-/- Mice. Because hemolytic anemia and subsequent splenic extramedullary hematopoiesis can induce splenomegaly, and Hb or heme from hemolysis can cause spleen toxicity, we thus hypothesized that the Nrf2-/- were experiencing hemolytic anemia. Indeed, hematological parameters showed that Nrf2-/- mice had decreased red blood cell number, hematocrit, Hb concentration, and markedly increased reticulocytes (Table 1), implying that the Nrf2-/- mice suffered from anemia. It is interesting to note that the anemic condition was mild in young Nrf2-/- mice (6 mo) and was more severe in older Nrf2-/- mice (14 mo). The anemia found in the Nrf2-/- mice appears to be a result of a decrease in survival of erythrocytes and not that of a decrease in reticulocyte production (regenerative). This can be said because reticulocyte production is actually enhanced, most likely to compensate for decreased circulating erythrocytes. We also found that old Nrf2-/- mice (14 mo) have significantly decreased circulating platelets (thrombocytopenia; Nrf2+/+, 1,520.9 ± 87.9; Nrf2-/-, 989.8 ± 110.9). The mean corpuscular volume (Nrf2+/+, 50.7 ± 0.5; Nrf2-/-, 52.6 ± 2.1), mean corpuscular Hb (Nrf2+/+, 14.9 ± 0.2; Nrf2-/-, 15.9 ± 0.2), and mean corpuscular Hb concentration values (Nrf2+/+, 29.5 ± 0.3; Nrf2-/-, 30.3 ± 1.0) were not dramatically different (14 mo; mean ± SE). Similar effects were seen in a small sampling of female Nrf2-/- animals (data not shown).
Table 1. Anemia in Nrf2−/− mice.
| Age | Genotype | RBC (× 106 cells per μl) | Hemoglobin, g/dl | Hematocrit, % | Reticulocyte, % |
|---|---|---|---|---|---|
| 6 mo | Nrf2+/+ (n = 7) | 10.4 ± 0.3 | 15.9 ± 0.3 | 53.7 ± 0.9 | 2.8 ± 0.3 |
| Nrf2−/− (n = 7) | 8.8 ± 0.2* | 14.1 ± 0.4* | 46.8 ± 1.1* | 4.2 ± 0.4* | |
| % of control | 84.2 | 89.0 | 87.2 | 152.3 | |
| 14 mo | Nrf2+/+ (n = 7) | 10.3 ± 0.2 | 15.3 ± 0.3 | 51.9 ± 1.8 | 2.0 ± 0.4 |
| Nrf2−/− (n = 7) | 7.0 ± 0.6* | 11.1 ± 0.9* | 36.5 ± 3.1* | 10.8 ± 4.1* | |
| % of control | 67.9 | 72.3 | 70.3 | 541.7 |
Morphological Abnormalities and Increased Sensitivity in Nrf2-/- Erythrocytes. Peripheral blood smears (Fig. 3 A and B) showed Nrf2-/- erythrocytes have morphological abnormalities such as Howell-Jolly body (Fig. 3_C_), schistocytes (Fig. 3_D_), and acantocytes (Fig. 3_E_), suggesting that Nrf2-/- erythrocytes are more sensitive, and anemia in Nrf2-/- mice is due to hemolysis of damaged erythrocytes. Peripheral blood smears from young Nrf2-/- mice (6 mo) showed mild poikilocytes and anisocytes (data not shown), implying an age-dependent increase of damage in Nrf2-/- erythrocytes, which is similarly observed in the hematological parameters (Table 1). We also observed that Nrf2-/- erythrocytes are highly sensitive to H2O2-induced hemolysis in vitro (Fig. 3 F and G), further supporting that Nrf2-/- erythrocytes are highly susceptible to stress. In addition, Nrf2-/- erythrocytes showed an increased methemoglobin formation after incubation with high concentrations of H2O2 (data not shown), suggesting that Hb in Nrf2-/- erythrocytes is more easily oxidized compared with Nrf2+/+ erythrocytes.
Oxidative Damages Cause Immune-Mediated Hemolytic Anemia. Because it appeared that anemia in Nrf2-/- mice is caused by decreased erythrocyte survival with normal erythrocyte production, we hypothesized that the sequestration rate of Nrf2-/- erythrocytes is higher than that of Nrf2+/+ erythrocytes. To test this hypothesis, we measured erythrocyte-bound IgG and IgM levels using flow cytometry (see scheme in Fig. 4_A_), because IgG and IgM mediate phagocytosis of damaged erythrocytes in splenic macrophages. As shown in Fig. 4 B and C, Nrf2-/- erythrocytes have significantly increased levels of IgG (120% increase) and also IgM (20% increase, data not shown) compared with Nrf2+/+ erythrocytes, implying an increased sequestration rate of Nrf2-/- erythrocytes. To further investigate the mechanism of increased erythrocyte-bound IgG levels in Nrf2-/- mice, we tested two possibilities: (i) autoimmune-mediated antibody binding to Nrf2-/- erythrocytes, and (ii) damage-induced antibody binding to erythrocytes. To test the “autoimmune” hypothesis, we mixed erythrocytes from Nrf2-/- or Nrf2+/+ mice with plasma from either Nrf2+/+ and Nrf2-/- mice and measured the erythrocyte-bound IgG levels (see scheme in Fig. 4_D_). The IgG levels of Nrf2+/+ erythrocytes (Fig. 4_E_, purple in Fig. 4_G_) incubated with plasma from Nrf2+/+ (green in Fig. 4_H_), or Nrf2-/- mice (red in Fig. 4_H_) were not different from those of control Nrf2+/+ erythrocytes (purple in Fig. 4_H_). Similarly, IgG levels of control Nrf2-/- erythrocytes (Fig. 4_F_, green in Fig. 4_G_, purple in Fig. 4_I_) were the same as those of Nrf2-/- erythrocytes incubated with plasma from Nrf2+/+ (green in Fig. 4_I_) or Nrf2-/- mice (red in Fig. 4_I_), arguing against the “autoimmune” hypothesis. To test the “damage-induced antibody binding” hypothesis, we incubated Nrf2+/+ erythrocytes with H2O2 to induce oxidative damage, followed by incubation with plasma from either Nrf2+/+ (Fig. 4_M_) or Nrf2-/- mice (Fig. 4_N_) (see scheme in Fig. 4_J_). As shown in Fig. 4 K and L, H2O2 induced oxidative damage resembling echinocyte-like cells. The IgG levels of damaged Nrf2+/+ erythrocytes incubated with plasma from Nrf2+/+ (Fig. 4_M_) or Nrf2-/- (Fig. 4_N_) mice were increased in a dose-dependent manner. Interestingly, the IgG levels of damaged Nrf2+/+ erythrocytes incubated with Nrf2+/+ plasma were virtually the same as those incubated with Nrf2-/- plasma (Fig. 4_O_), implying antibody binding depends on the levels of oxidative damages in erythrocytes, not Nrf2 genotype. Taken together, these observations rule out the possibility of an autoimmune-mediated antibody binding to erythrocytes in Nrf2-/- mice and strongly support that increased levels of oxidative damages in Nrf2-/- erythrocytes recruit antibodies, finally resulting in phagocytosis and anemia.
Decreased Antioxidant Potential and Increased Oxidative Stress Sensitize Nrf2-/- Erythrocytes. To further test damage-induced antibody binding in vivo, we tail vein injected H2O2 into Nrf2+/+ and Nrf2-/- mice (5 mo) and measured erythrocyte-bound IgG levels as an indicator of oxidative damages in erythrocytes (see scheme in Fig. 5_A_). As shown in Fig. 5 B and C, erythrocyte-bound IgG levels of H2O2-injected Nrf2-/- mice were significantly higher than those of H2O2-injected Nrf2+/+ mice, implying that Nrf2-/- erythrocytes are more vulnerable to reactive oxygen species (ROS) and that oxidative damage recruits antibodies to erythrocytes. In addition, sera from Nrf2-/- mice have decreased basal GSH (Fig. 5_D_) and increased basal H2O2 levels (Fig. 5_E_) compared with Nrf2+/+ mice, suggesting that Nrf2-/- erythrocytes are more sensitive to ROS.
Discussion
In this study, we report that targeted disruption of Nrf2 causes regenerative immune-mediated hemolytic anemia. Oxidative damages in Nrf2-/- erythrocytes induced hemolytic anemia with morphological abnormal erythrocytes (i.e., Howell-Jolly bodies, acantocytes, and schistocytes). Subsequently, hemolytic anemia resulted in a compensatory reticulocytosis and spleen toxicity (possibly by Hb or heme) in Nrf2-/- mice, suggesting that splenomegaly in Nrf2-/- mice is contributed by both splenic hematopoiesis and hyperactivation of splenic phagocytosis system. In support of this, splenectomy surgery decreased reticulocyte numbers greatly in Nrf2-/- mice compared with Nrf2+/+ mice (data not shown). A correlation also existed between spleen sizes, severity of anemia, and erythrocyte-bound IgG levels (data not shown). In addition to increased hemolysis, decreased expression levels of ARE-driven genes in Nrf2-/- mice may have further increased the sensitivity of Nrf2-/- splenocytes. Finally, we showed that Nrf2-/- erythrocytes have higher levels of bound IgG, and that Nrf2-/- mice are more susceptible to 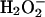 -induced erythrocyte damage. Similar to the splenocytes, sensitivity of Nrf2-/- erythrocytes may have been a result of decreased antioxidant potential (i.e., GSH), followed by increased oxidative stress. Previous findings also have demonstrated that Nrf2 plays a role in an orchestrated regulation of gene clusters that maintain GSH levels and detoxify H2O2 (6). This strongly suggests that Nrf2-/- mice have higher levels of circulating H2O2. Interestingly, Nrf2-/- mice have extremely white teeth, which may be explained by increased H2O2 levels in Nrf2-/- mice, because H2O2 is known to have a strong tooth-whitening effect. Together, these observations support our hypothesis that decreased antioxidant potential together with increased reactive oxygen species induces oxidative damages in Nrf2-/- erythrocytes, leading to attachment of antibodies and, finally, macrophage-mediated phagocytosis in spleen.
-induced erythrocyte damage. Similar to the splenocytes, sensitivity of Nrf2-/- erythrocytes may have been a result of decreased antioxidant potential (i.e., GSH), followed by increased oxidative stress. Previous findings also have demonstrated that Nrf2 plays a role in an orchestrated regulation of gene clusters that maintain GSH levels and detoxify H2O2 (6). This strongly suggests that Nrf2-/- mice have higher levels of circulating H2O2. Interestingly, Nrf2-/- mice have extremely white teeth, which may be explained by increased H2O2 levels in Nrf2-/- mice, because H2O2 is known to have a strong tooth-whitening effect. Together, these observations support our hypothesis that decreased antioxidant potential together with increased reactive oxygen species induces oxidative damages in Nrf2-/- erythrocytes, leading to attachment of antibodies and, finally, macrophage-mediated phagocytosis in spleen.
Three b-Zip proteins, NF-E2, Nrf1, and Nrf2, were identified as binding factors to the locus control region in the β-globin gene cluster. Although it is highly suspected that targeted disruption of one of these factors can impair β-globin synthesis and result in β-thalassemia, it has been demonstrated that each of these genes is not essential for β-globin gene expression in vivo. Interestingly, targeted disruption of these genes resulted in different types of hematological disorders in mice. NF-E2 knockout mice showed severe thrombocytopenia and anemia with dysmorphic red cell changes (19). In contrast to in vitro data (24), the expression levels of β-globin and other putative NF-E2-regulated genes were not decreased in NF-E2 knockout mice (19). A subsequent study demonstrated that NF-E2 regulates the expression of thromboxane synthase in megakaryocytes and addressed a possible role of thromboxane synthase in platelet development (25). It was also shown that thrombocytopenia in NF-E2 knockout mice is due to a defect in megakaryocyte growth and differentiation into platelets (26). Nrf1 knockout mice also have been reported to develop anemia in early stages of embryo development, and they die in utero (20). Nrf1 knockout mice have an abnormal fetal liver erythropoiesis as a result of a defect in the fetal liver microenvironment specific for erythroid cells (20). Chan et al. (20) showed a persistent presence of yolk sac-derived primitive nucleated erythrocytes in Nrf1 knockout embryos, addressing that switching of erythropoiesis (from yolk sac to liver) is abnormal in Nrf1 knockout mice. An important role of ARE-driven genes also has been implicated in the fetal liver microenvironment (20). Further investigation demonstrated that Nrf1 is essential for the hepatocyte lineage, and lack of Nrf1 induces apoptosis of embryonic hepatocytes during development, primarily due to failure in maintenance of redox balance (27). Similarly to NF-E2 knockout mice (19), β-globin gene expression appeared to be normal in Nrf1 knockout mice (20), suggesting anemia in Nrf1 knockout mice is due to decreased antioxidant potential rather than decreased β-globin synthesis. Currently, it is not known whether Nrf1 knockout embryos have thrombocytopenia. In this study, we observed that Nrf2 knockout mice develop regenerative immune-mediated hemolytic anemia. Unlike that seen in NF-E2 or Nrf1 knockout mice, the severity of anemia is age-dependent in Nrf2-/- mice, and anemia appeared to be caused by an increased sequestration of erythrocytes with normal erythrocyte production. In addition, Nrf2-/- mice have decreased circulating platelets, raising the possibility of Evan's syndrome in these mice. Although the cause of decreased circulating platelets in Nrf2-/- mice is not clear, we speculate that: (i) ARE-driven antioxidant potential can play a role in platelet survival; or (ii) thromboxane synthase (28) can play a role in platelet development. The levels of β-globin gene expression in Nrf2 knockout mice were similar to those of wild-type mice (data not shown). Furthermore, hematological parameters and an osmotic fragility test (data not shown) ruled out the possibility of thalassemia in Nrf2-/- mice. Taken together, these observations exclude the possibility of β-thalassemia in Nrf2 knockout mice and strongly support an important role of b-Zip proteins in erythrocyte maintenance during conditions of oxidative stress. It still remains to be elucidated, however, whether concerted knockout of these b-Zip factors induces thalassemia due to possible overlapping functions between b-Zip proteins (29).
Previously, it has been reported that Nrf2-/- mice develop lupus-like autoimmune nephritis (30). Recent work in our laboratory has demonstrated that aged female Nrf2-/- mice develop lupus-like phenotypes with multiple organ pathologies that closely resemble human systemic lupus erythematosus (J. Li, T. Stein, and J.A.J., unpublished data). These female mice are prone to develop antibodies against double-stranded DNA and the Smith antigen as well as IgG, IgM, and C3 deposition in kidney, liver, heart, and brain. In our flow cytometry analysis (Fig. 4), erythrocytes from Nrf2-/- mice have increased IgG and IgM levels compared with those from Nrf2+/+ mice. Furthermore, before the development of autoimmune antibodies and organ pathology, oxidative damage occurs in the liver and kidney, as indicated by the increased levels of the DNA oxidation marker 8-hydroxydeoxyguanosine and the later increase in the lipid peroxidation product malondialdehyde (J. Li, T. Stein, and J.A.J., unpublished data). This suggests that lack of Nrf2 accelerates the development of lupus-like phenotypes, and that the regenerative immune-mediated hemolytic anemia documented herein may be indicative of the subsequent onset of lupus-like phenotypes. Subsequent work is focused specifically on determining whether the regenerative immune-mediated hemolytic anemia in Nrf2 knockout mice is a contributing factor to the selective development of lupus-like phenotypes in female mice.
A genome-wide search for genes involved in human systemic lupus erythematosus (SLE) demonstrated that multiple genes influence susceptibility to human SLE. Interestingly, one of those (2q21-33) includes the Nrf2 locus (2q31) (31), implying a possible function of Nrf2 in an antilupus pathway. It is not clear whether autoantibodies in Nrf2-/- mice are generated spontaneously (autoimmune) or secondarily to oxidative cellular damages. This study, however, argues against the development of spontaneous autoimmunity in Nrf2-/- mice, demonstrated by the lack of binding of IgG in plasma from Nrf2-/- mice to normal healthy Nrf2+/+ erythrocytes. Furthermore, IgG from Nrf2-/- mice did bind to damaged Nrf2+/+ erythrocytes, again arguing against an autoimmune process. These findings indicate that IgG from Nrf2-/- mice is damage-specific IgG, which is mediating phagocytosis of damaged erythrocytes in normal situations. Therefore, the present study strongly supports that increased oxidative damage in Nrf2-/- erythrocytes initiates an immune reaction. The question then becomes how Nrf2-/- mice develop autoantibodies such as antinuclear and antidouble-stranded DNA antibody. One possibility could be that elevated oxidative stress can increase cell death in Nrf2-/- mice, which may then result in a release of antigenic components (i.e., nuclear materials and double-stranded DNA) and thus trigger an antigenic response (i.e., autoantibodies). Taken together, it appeared that lack of Nrf2 increases the susceptibility to lupus by modulating antioxidant potential in the cells, not directly inducing spontaneous autoimmune development. A link between a genetic polymorphism of Nrf2 and increased susceptibility of human systemic lupus erythematosus is an interesting question that has yet to be determined.
Conclusion
We addressed a pivotal role of Nrf2 in protecting cells from oxidative stress by showing that targeted disruption of Nrf2 results in an immune-mediated hemolytic anemia due to increased oxidative damages in Nrf2-/- erythrocytes. One unique feature about the Nrf2-ARE pathway (named the programmed cell life pathway) (2) is that it coordinately up-regulates many protective detoxification and antioxidant genes, which can synergistically increase the efficiency of the cellular defense system. Because the Nrf2-ARE pathway functions as a master regulator of antioxidant genes, the programmed cell life pathway may serve as a therapeutic target for prevention of neurodegenerative diseases and carcinogenesis, in which oxidative stress is involved.
Acknowledgments
We thank Delinda Johnson for reading this manuscript and for helpful suggestions. We also thank the Clinical Pathology Laboratory and Flow Cytometry Facility at the University of Wisconsin, Madison. This study was supported by National Institutes of Environmental Health Sciences Grants ES08089 and ES10042 (to J.A.J.) and National Institutes of Health Grant DK16666 (to Y.W.K.).
Abbreviations: Nrf2, NF-E2-related factor 2; ARE, antioxidant responsive element; NQO1, NAD(P)H:quinone oxidoreductase-1; HO-1, hemeoxygenase-1; GCL, glutamate-cysteine ligase; b-Zip, basic leucine zipper; GCLC, GCL catalytic subunit; GSH, glutathione.
References
- 1.Rushmore, T. H., Morton, M. R. & Pickett, C. B. (1991) J. Biol. Chem. 266**,** 11632-11639. [PubMed] [Google Scholar]
- 2.Li, J., Lee, J. M. & Johnson, J. A. (2002) J. Biol. Chem. 277**,** 388-394. [DOI] [PubMed] [Google Scholar]
- 3.Duffy, S., So, A. & Murphy, T. H. (1998) J. Neurochem. 71**,** 69-77. [DOI] [PubMed] [Google Scholar]
- 4.Moi, P., Chan, K., Asunis, I., Cao, A. & Kan, Y. W. (1994) Proc. Natl. Acad. Sci. USA 91**,** 9926-9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chan, K., Lu, R., Chang, J. C. & Kan, Y. W. (1996) Proc. Natl. Acad. Sci. USA 93**,** 13943-13948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee, J. M., Calkins, M. J., Chan, K., Kan, Y. W. & Johnson, J. A. (2003) J. Biol. Chem. 278**,** 12029-12038. [DOI] [PubMed] [Google Scholar]
- 7.Lee, J. M., Shih, A. Y., Murphy, T. H. & Johnson, J. A. (2003) J. Biol. Chem. 278**,** 37948-37956. [DOI] [PubMed] [Google Scholar]
- 8.Thimmulappa, R. K., Mai, K. H., Srisuma, S., Kensler, T. W., Yamamoto, M. & Biswal, S. (2002) Cancer Res. 62**,** 5196-5203. [PubMed] [Google Scholar]
- 9.Chan, K. & Kan, Y. W. (1999) Proc. Natl. Acad. Sci. USA 96**,** 12731-12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan, K., Han, X. D. & Kan, Y. W. (2001) Proc. Natl. Acad. Sci. USA 98**,** 4611-4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cho, H. Y., Jedlicka, A. E., Reddy, S. P., Kensler, T. W., Yamamoto, M., Zhang, L. Y. & Kleeberger, S. R. (2002) Am. J. Respir. Cell Mol. Biol. 26**,** 175-182. [DOI] [PubMed] [Google Scholar]
- 12.Kraft, A. D., Johnson, J. A. & Johnson, J. A. (2004) J. Neurosci. 24**,** 1101-1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shih, A. Y., Johnson, D. A., Wong, G., Kraft, A. D., Jiang, L., Erb, H., Johnson, J. A. & Murphy, T. H. (2003) J. Neurosci. 23**,** 3394-3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.An, J. H. & Blackwell, T. K. (2003) Genes Dev. 17**,** 1882-1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohtsubo, T., Kamada, S., Mikami, T., Murakami, H. & Tsujimoto, Y. (1999) Cell Death Differ. 6**,** 865-872. [DOI] [PubMed] [Google Scholar]
- 16.Kotlo, K. U., Yehiely, F., Efimova, E., Harasty, H., Hesabi, B., Shchors, K., Einat, P., Rozen, A., Berent, E. & Deiss, L. P. (2003) Oncogene 22**,** 797-806. [DOI] [PubMed] [Google Scholar]
- 17.Cullinan, S. B., Zhang, D., Hannink, M., Arvisais, E., Kaufman, R. J. & Diehl, J. A. (2003) Mol. Cell. Biol. 23**,** 7198-7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Morito, N., Yoh, K., Itoh, K., Hirayama, A., Koyama, A., Yamamoto, M. & Takahashi, S. (2003) Oncogene 22**,** 9275-9281. [DOI] [PubMed] [Google Scholar]
- 19.Shivdasani, R. A. & Orkin, S. H. (1995) Proc. Natl. Acad. Sci. USA 92**,** 8690-8694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chan, J. Y., Kwong, M., Lu, R., Chang, J., Wang, B., Yen, T. S. & Kan, Y. W. (1998) EMBO J. 17**,** 1779-1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gal, A., Tamir, S., Tannenbaum, S. R. & Wogan, G. N. (1996) Proc. Natl. Acad. Sci. USA 93**,** 11499-11503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Prochaska, H. J. & Santamaria, A. B. (1988) Anal. Biochem. 169**,** 328-336. [DOI] [PubMed] [Google Scholar]
- 23.Tietze, F. (1969) Anal. Biochem. 27**,** 502-522. [DOI] [PubMed] [Google Scholar]
- 24.Kotkow, K. J. & Orkin, S. H. (1995) Mol. Cell. Biol. 15**,** 4640-4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Deveaux, S., Cohen-Kaminsky, S., Shivdasani, R. A., Andrews, N. C., Filipe, A., Kuzniak, I., Orkin, S. H., Romeo, P. H. & Mignotte, V. (1997) EMBO J. 16**,** 5654-5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Levin, J., Peng, J. P., Baker, G. R., Villeval, J. L., Lecine, P., Burstein, S. A. & Shivdasani, R. A. (1999) Blood 94**,** 3037-3047. [PubMed] [Google Scholar]
- 27.Chen, L., Kwong, M., Lu, R., Ginzinger, D., Lee, C., Leung, L. & Chan, J. Y. (2003) Mol. Cell. Biol. 23**,** 4673-4686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yaekashiwa, M. & Wang, L. H. (2003) DNA Cell Biol. 22**,** 479-487. [DOI] [PubMed] [Google Scholar]
- 29.Leung, L., Kwong, M., Hou, S., Lee, C. & Chan, J. Y. (2003) J. Biol. Chem. 278**,** 48021-48029. [DOI] [PubMed] [Google Scholar]
- 30.Yoh, K., Itoh, K., Enomoto, A., Hirayama, A., Yamaguchi, N., Kobayashi, M., Morito, N., Koyama, A., Yamamoto, M. & Takahashi, S. (2001) Kidney Int. 60**,** 1343-1353. [DOI] [PubMed] [Google Scholar]
- 31.Gaffney, P. M., Kearns, G. M., Shark, K. B., Ortmann, W. A., Selby, S. A., Malmgren, M. L., Rohlf, K. E., Ockenden, T. C., Messner, R. P., King, R. A., et al. (1998) Proc. Natl. Acad. Sci. USA 95**,** 14875-14879. [DOI] [PMC free article] [PubMed] [Google Scholar]