Type I Interferon Sensitizes Lymphocytes to Apoptosis and Reduces Resistance to Listeria Infection (original) (raw)
Abstract
Infection with Listeria monocytogenes causes lymphocyte apoptosis that is mediated by the actions of the pore-forming virulence factor listeriolysin O (LLO). Previous work showed that activated lymphocytes were highly sensitive to LLO-induced apoptosis, whereas resting lymphocytes were less susceptible. We now show that mice deficient in the type I interferon (IFN) receptor were more resistant to Listeria infection and had less apoptotic lesions than wild-type counterparts. Furthermore, treatment of resting splenic lymphocytes with recombinant IFN-αA enhanced their susceptibility to LLO-induced apoptosis. Together, these data suggest that type I IFN signaling is detrimental to handling of a bacterial pathogen and may enhance the susceptibility of lymphocytes undergoing apoptosis in response to bacterial pore-forming toxins.
Keywords: apoptosis, cytokines, Listeria, inflammation, T lymphocytes
Introduction
Previous studies with Listeria monocytogenes have exposed the role of multiple cytokines at different stages of the infection using gene-ablated mice or neutralizing monoclonal antibodies (for review see references 1 and 2). IL-12, TNF, and the IL-1 family of proteins participate in activating different cells and controlling Listeria growth. These early cytokines contribute to the induction of IFN-γ, a product of NK and T cells, and activation of macrophages.
Much is known about the role of type I IFNs (IFN-α and IFN-β) in the antiviral response and type II IFN (IFN-γ) in the antiviral and antibacterial response (3–6). However, there exists a paucity of data on the role of type I IFNs during bacterial infection. Here we report that the absence of the shared IFN-α/β receptor (IFN-αβR−/−) provides mice with an advantage during Listeria infection. This resistance was accompanied by an amelioration of the lymphocyte apoptotic process that took place during the early exponential growth of Listeria in tissues.
We and others have described an early transitory phase of lymphocyte apoptosis in infective foci that peaks at 48 h after infection (7, 8). Experiments ex vivo indicated that listeriolysin O (LLO), a pore-forming molecule and major virulence factor, caused lymphocyte apoptosis particularly in cells that were replicating (9). Moreover, injection of pure LLO subcutaneously led to lymphocyte apoptosis in the T cell–rich zones of draining lymph nodes. Of note is that during the in vivo infection the apoptotic lesions were either not affected or were increased in mice in which IL-1, IL-12, or IFN-γ were either neutralized or not produced (7). The lesions were unrelated to activation-induced cell death, to Fas–Fas ligand interactions, or to the production of reactive oxygen or nitrogen intermediates. In toto the evidence points to the release of soluble LLO during the early robust growth of Listeria as the mechanism leading to the apoptotic death of lymphocytes in infective foci. In fact, neutralization of LLO by injection of monoclonal antibody to LLO controlled the infection as well as the intensity of the apoptotic lesions (10). The data presented herein suggests that type I IFN sensitizes lymphocytes to undergo apoptosis during Listeria infection, and that this has a negative effect on bacterial handling in the mouse.
Materials and Methods
Mice and Listeria Infection.
Wild-type and IFN-αβR−/− mice on the 129Sv/Ev genetic background were provided by H.W. Virgin IV (Washington University School of Medicine, St. Louis, MO; reference 3). L. monocytogenes strain EGD was used as before for intraperitoneal infection (7). Histological examination of hematoxylin and eosin– and TdT-mediated dUTP nick-end labeling (TUNEL)-stained sections was performed on the spleens from infected mice (7). Cytometric bead array (CBA) analysis was performed with the Mouse Inflammation Kit and used according to the manufacturer's instructions with 50 μL serum per assay (BD Biosciences). CBA data was analyzed on a FACSCalibur™ with CELLQuest™ software and the CBA analysis software package (BD Biosciences). For flow cytometric staining, portions of spleens from infected mice were disrupted to generate single cell suspensions. Cells were then stained for CD69 and either CD4 or CD8 using conventional techniques. All antibodies were obtained from BD Biosciences.
Cell Cultures.
A CD4 T cell line reactive to ovalbumin was generated from normal 129Sv/J mice (The Jackson Laboratory) immunized with an ovalbumin emulsion in complete Freund's adjuvant using previously described techniques (9). The line was passaged every 10–12 d by the addition of irradiated 129Sv/J splenocytes (3,000 rads), 10 μM ovalbumin, and 50 U/ml IL-2. The usual behavior was that of proliferation that slowly stopped by about day 8. On day 10 after stimulation, the T cells were harvested and were not in cell cycle. Assays with the T cell line were performed as described previously (9). Whole splenocytes were isolated from 129Sv/Ev or IFN-αβR−/− mice. Single cell suspensions were made and cells were plated at a density of 5 × 106 cells/ml in DMEM and 10% FCS, and treated with recombinant mouse IFN-αA at 1, 10, or 100 U/ml (specific activity: 4.8 × 107 U/mg; PBL Biomedical Laboratories). Cells were incubated for 24 h, and then the nonadherent cells were removed and purified. Both the T cell line and the splenocytes were purified over a Histopaque 1119 gradient (Sigma-Aldrich). After Histopaque, cells were resuspended in DMEM containing 1% FCS, and then treated with 250 ng/ml of purified recombinant LLO (4.4 nM) for 6 h. Cells were stained with annexin V-PE and 7-AAD (BD Biosciences), and analyzed by flow cytometry. For assays involving splenocytes, cells were also stained with anti–CD3-APC (BD Biosciences) to identify T cells.
Results and Discussion
Listeria Infection.
Infection of 129Sv/Ev mice with Listeria showed the expected increase in the number of organisms in the spleen and liver as a function of time (Fig. 1, A and B). IFN-αβR−/− mice had the same number of Listeria as wild-type mice 24 h after infection. Therefore, there was no difference between the two strains during the initial innate resistance to infection, which was mediated by neutrophils and macrophages (1). However, at 48 h after infection, there was about a 20-fold increase in CFUs in both the spleens and livers of wild-type versus IFN-αβR−/− mice. By 96 h after infection, IFN-αβR−/− mice had no further Listeria growth and showed ∼1,200-fold fewer CFUs per organ (Fig. 1, A and B).
Figure 1.

Bacterial burden and cytokine profile of 129Sv/Ev and IFN-αβR−/− mice infected with L. monocytogenes. Mice were infected with 2.5 × 104 Listeria intraperitoneally and at the indicated days after infection (P.I.). CFUs were assessed in the spleen (A) and liver (B). Serum CBA analysis was performed on the same mice to yield serum concentrations of IL-6 (C), TNF-α (D), IFN-γ (E), and IL-12 (F). Bars represent the means ± the SEM of at least two independent experiments with a total number of five to seven mice per group.
At 48 h after infection, the peak time at which lymphocyte apoptosis was evident, histological examination showed a marked difference between both sets of mice (Fig. 2). The difference in lymphocyte apoptosis was seen at three different infectious doses (Table I). Wild-type mice showed infective foci around the periarteriolar lymphoid sheath, characterized by a paucity of lymphocytes and the presence of macrophages and neutrophils. Abundant TUNEL+ round nuclei were evident (Fig. 2 B). (Previous histochemical and ultrastructural studies have shown these TUNEL+ cells to be lymphocytes [7]). At the highest infectious dose tested (5 × 104), lesions extended into the periphery of the follicles (Fig. 2 B). These lesions were identical to those previously reported for several strains of mice (7). In contrast, IFN-αβR−/− mice showed infective lesions with a considerably reduced number of TUNEL+ nuclei (Fig. 2 D and Table I). Examination at 24 h after infection showed minor changes in the periarteriolar lymphoid sheath and only occasional TUNEL+ cells in both strains of mice.
Figure 2.
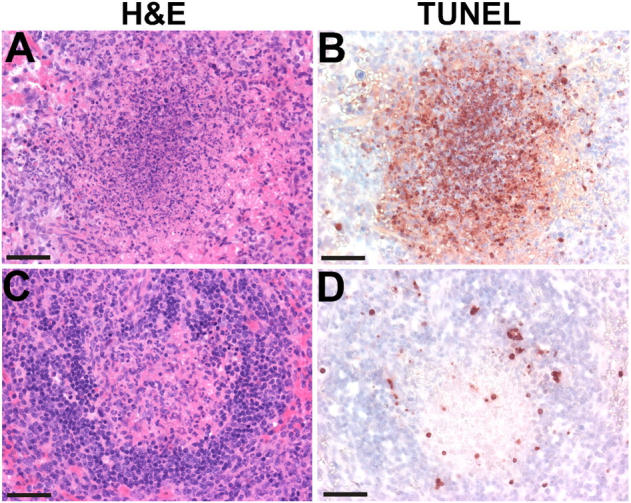
Histologic analysis of 129Sv/Ev and IFN-αβR−/− mice infected with L. monocytogenes. 129Sv/Ev (A and B) and IFN-αβR−/− (C and D) mice were infected with 5 × 104 Listeria intraperitoneally and spleens were removed and stained by either hematoxylin and eosin (H&E) (A and C) or by TUNEL (B and D) 48 h later. Each panel shows one white pulp profile stained by hematoxylin and eosin and the corresponding profile stained by TUNEL. Pictures are representative profiles from five mice per group. Bars, 50 μm.
Table I.
Spleens Examined 48 h after the Indicated Dose of Listeria
| _L. monocytogenes_dose | Mice | n | Pathology(48 h) | Apoptosis(48 h) |
|---|---|---|---|---|
| 5 × 103 | WT | 4 | 30–50% - severe | 3+ |
| 5 × 103 | IFN-αβR−/− | 5 | 10% - mild | 1+ |
| 2.5 × 104 | WT | 4 | 50–100% - severe | 4+ |
| 2.5 × 104 | IFN-αβR−/− | 5 | 10% - mild | 1+ |
| 5 × 104 | WT | 5 | 75–100% - severe | 4+ |
| 5 × 104 | IFN-αβR−/− | 6 | 20% - mild | 1–2+ |
Analysis of cytokines in the serum of infected mice at 24 h after infection, a time when there was no difference in Listeria load, showed an equal increase in both IL-6 (Fig. 1 C) and IFN-γ (Fig. 1 E) in both strains. However, the concentration of IL-12 was markedly higher in the IFN-αβR−/− mice (Fig. 1 F). The elevated level of IL-12 is consistent with published data showing that type I IFN signaling resulted in the down-regulation of IL-12 expression (11). The cytokine profiles at 48 and 96 h shown in Fig. 1, C–F, likely reflect the extent of infection in each strain (i.e., wild-type mice showed continued Listeria growth, whereas IFN-αβR−/− mice showed a limited Listeria burden).
During early Listeria infection, one sees a nonspecific stage of T lymphocyte activation, made evident by the up-regulation of CD69 cell surface expression (8). It has also been shown that type I IFN signaling can induce CD69 expression on T cells (12). Moreover, in vivo Listeria infection induces type I IFN production (13, 14) and, in vitro, _Listeria_-infected macrophages produce type I IFN after detecting the presence of an intracellular pathogen (15). Therefore, we assayed _Listeria_-infected wild-type and IFN-αβR−/− mice for the percentage of CD4 and CD8 T cells expressing CD69. Infected wild-type mice showed a three- to sixfold increase in the number of CD69+ T cells in their spleens, whereas the increase in infected IFN-αβR−/− mice was only two- to threefold. In uninfected 129Sv/Ev mice, 4% of the CD8+ T cells were CD69+. This number increased to 13 and 25% at days 1 and 2 after infection, respectively. In uninfected IFN-αβR−/− mice, 4% of the CD8+ T cells were CD69+, which increased to 8 and 12% at days 1 and 2, respectively. The picture was similar for CD4+ T cells, where in 129Sv/Ev mice, 10% of the CD4+ cells were CD69+ before infection, 18% were CD69+ at day 1, and 30% were CD69+ at day 2 after infection. In IFN-αβR−/− mice, 8% of the CD4+ cells were CD69+ in the uninfected mouse, with 10% CD69+ at day 1 and 15% CD69+ at day 2 after infection. This difference in CD69 may reflect a higher level of activation of T lymphocytes in wild-type mice, a step which could enhance their susceptibility to _Listeria_-induced apoptosis.
Response of T Cells to LLO in Culture.
We demonstrated previously that treatment with sublytic doses of LLO induced apoptosis in T cells (9). In that study, we observed a difference in the extent of apoptosis between resting lymphocytes and those recently activated by exposure to antigen. Because type I IFN is produced early during Listeria infection (13–15), we hypothesized that the early burst of IFN could enhance the susceptibility of lymphocytes to apoptosis.
We chose to monitor apoptosis in lymphocytes treated with type I IFN by flow cytometry using annexin V that binds to phosphatidylserine exposed on the outer leaflet of the plasma membrane, and 7-AAD, which measures nuclear permeability. This assay distinguishes apoptotic cells, which stain with annexin V but not 7-AAD, from late apoptotic or necrotic cells, which stain positive for both markers.
We first tested CD4+ T lymphocytes from ex vivo culture 10 d after antigen stimulation by incubating them in the presence or absence of IFN-αA for 24 h, and then exposing them to 250 ng/ml LLO for 6 h. We selected this time point on the basis of previous kinetic studies on cultured T cells treated with LLO (9). In the absence of LLO, 8–10% of cells underwent apoptosis (i.e., became annexin V+/7-AAD−). 6 h of treatment with LLO increased this number to ∼22%, in accordance with our previous study (Fig. 3, A, D, and J; reference 9). The set of cells staining with both annexin V and 7-AAD also increased upon LLO treatment. Our previous study demonstrated that LLO-treated cells shifted over time from the single positive (annexin V+/7-AAD−) to the double positive (annexin V+/7-AAD+), the latter of which contains late apoptotic as well as necrotic cells (9).
Figure 3.
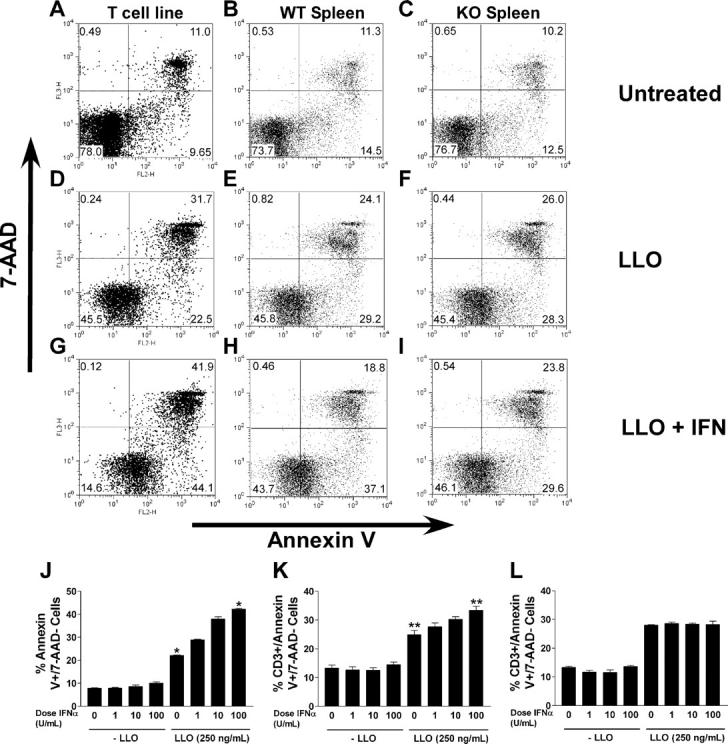
Annexin V/7-AAD analysis of LLO-treated lymphocytes. A cultured, resting T cell line (A, D, G, and J) and 129Sv/Ev splenocytes (B, E, H, and K) or IFN-αβR−/− splenocytes (C, F, I, and L) were either untreated (A–C), treated with 250 ng/ml LLO (D–F), or treated with 250 ng/ml LLO after 24 h of treatment with 100 U/ml IFN-αA (G–I). Cells were stained for annexin V/7-AAD. For the spleen, data is gated on CD3+ events only. The flow cytometry plots shown in A–I are representative of two to three experiments. J–L show the average result of two experiments with the cell line and three with the splenocytes, each performed in duplicate or triplicate. J–L show the percentage of annexin V+/7-AAD− cultured cells after the indicated treatments. Although their number is related with responsiveness to IFN-αA, the double positive cells taken from the spleen are not, probably because they represent a mixture of cells varying in their in vitro susceptibility. J, T cell line; K, wild-type spleen cells; L, IFN-αβR−/− spleen cells. Asterisks indicates statistical significance at a p-value of <0.0001 (*) or <0.0041 (**) by unpaired t test.
Treatment with IFN-αA alone did not significantly increase the number of apoptotic cells. Annexin V+/7-AAD− cells constituted, on average, 8, 8, and 10% of the cells after treatment with 1, 10, and 100 U/ml IFN-αA, respectively (Fig. 3 J). However, treatment with IFN-αA augmented the number of cells undergoing LLO-induced apoptosis, in a dose-dependent manner. Cells treated with 1, 10, and 100 U/ml IFN-αA and then exposed to 250 ng/ml LLO had annexin V+/7-AAD− populations of 29, 38, and 43%, respectively (Fig. 3, G and J).
To extend our results to primary cells, we isolated splenocytes, cultured them for 24 h in increasing doses of IFN-αA, treated them with LLO, and assayed them for apoptosis by annexin V/7-AAD staining. As an average, 13% of untreated CD3+ cells were annexin V+/7-AAD− (Fig. 3, B and K). This number increased to 24% upon LLO treatment (Fig. 3, E and K). The addition of type I IFN increased the response of T cells to LLO-induced apoptosis in a dose-dependent manner. At the highest IFN-αA dose tested (100 U/ml), 37% of cells CD3+ became annexin V+/7-AAD− (Fig. 3, H and K). This increase, albeit modest, was highly reproducible in three experiments. In contrast, type I IFN did not increase LLO-induced apoptosis in splenocytes isolated from IFN-αβR−/− mice. The percentage increase in annexin V+/7-AAD− cells taken from IFN-αβR−/− spleen after treatment with LLO was the same in the absence of IFN-αA (Fig. 3, F and L) and at all doses of IFN-αA tested (Fig. 3, I and L).
(Spleen cells from wild-type mice cultured for 2, 4, and 6 h with LLO resulted in 26, 29, and 24% of annexin V+/7-AAD− cells, respectively. The addition of IFN-αA increased the number of cells at the same time points to 35, 42, and 37%. Spleen cells from the IFN-αR−/− mice cultured for 2, 4, and 6 h with LLO resulted in 30, 32, and 30%, respectively. However, this number did not increase after culture in IFN-αA: 29, 31, and 30%. Both untreated spleen cell populations were 16% annexin V+/7-AAD− cells after 6 h of culture. Thus, there were no major changes in kinetics during the 2–6-h time period. Fig. 3 shows the 6 h results.)
Comments.
Our experiments with IFN-αβR−/− mice indicate a deleterious effect on early resistance to Listeria infection by type I IFNs. Fehr et al. (16) reported that IFN-αβR−/− mice were not impaired in their resistance to Listeria infection. Moreover, their results suggested an increased resistance to Listeria at day 5 after infection when compared with wild-type controls. They hypothesized that this was a function of type I and II IFN receptor competition for components of signaling pathways. However, that study, which focused on the role of transcription factors and nitric oxide in resistance to Listeria, did not fully explain the increased resistance of IFN-αβR−/− mice.
Our report expands on previous findings in the following ways. In the absence of a response to type I IFNs (i.e., in the receptor null mice), the infective foci contained fewer TUNEL+ lymphocytes as well as fewer activated T cells. Type I IFN unresponsive mice were also more resistant to infection. We believe that early type I IFN production during Listeria infection results in nonspecific activation of T cells. The provocative finding is that this early activation also sensitizes these cells to the apoptogenic actions of LLO. Furthermore, macrophages treated with type I IFN were shown to be susceptible to Listeria killing in vitro (17). These results suggest that type I IFN could enhance the susceptibility to death of multiple leukocyte subsets during Listeria infection.
The immunological consequences of cellular death during Listeria infection of the mouse must be further examined, but could involve the development of responses that are inhibitory for bacterial clearance. Clearance of apoptotic bodies by macrophages is generally thought to down-regulate inflammation through macrophage release of TGF-β, prostaglandin E2, and platelet-activating factor (18–20). Apoptotic lymphocytes themselves may also down-regulate inflammation through the release of preformed TGF-β upon apoptosis induction (21). We suggest an inverse relationship between death by apoptosis, in part enhanced by type I IFNs and the growth of Listeria. In the absence of type I IFN signaling, apoptosis is limited, and less inhibition of phagocyte antimicrobial processes takes place. In support of these hypotheses, we have observed that SCID mice, lacking T and B lymphocytes, do not show apoptotic lesions after Listeria infection, and are more resistant than wild-type mice during the first 2–4 d after infection (22 and unpublished data).
The cellular source of type I IFN during Listeria infection and the Listeria component(s) inducing its production remain unknown at this time. Candidate ligands would include Toll-like receptor (TLR)4 or TLR9 agonists such as lipoteichoic acid or CpG DNA motifs, which have been shown to induce type I IFN expression by dendritic cells (23). Analysis of TLR4−/−, TLR9−/−, and MyD88−/− mice or cells may yield insight into the generation of type I IFN in response to Listeria infection. Macrophages recognizing cytosolic Listeria also generate IFN-β (15), suggesting this cell type as a source for the cytokine. Alternatively, Listeria could generate proteins that promote expression of type I IFN to enhance its virulence and dissemination.
A classical role ascribed to type I IFN signaling is the enhancement of cancer cell and virus-infected cell apoptosis. One possible mechanism for type I IFN's sensitization of T cells to undergo apoptosis would be the accumulation of the p53 tumor suppressor gene (24). After p53 expression, cellular stress caused by LLO disruption of the plasma membrane might then lead to apoptosis induction. Further studies will examine the role of type I IFNs in LLO-induced apoptosis.
Acknowledgments
We thank the members of the Unanue laboratory, particularly Craig A. Byersdorfer, Dr. Brian T. Edelson, Scott B. Lovitch, and Dr. Anish Suri for many helpful discussions and for critical reading of the manuscript, and Jeremy Herzog for technical assistance. We thank Kathy Fredericks for animal husbandry and technical assistance. We thank Dr. Herbert W. Virgin IV for his kind gift of mice.
This work was supported by grants from the National Institutes of Health.
The authors have no conflicting financial interests.
References
- 1.Unanue, E.R. 1997. Studies in listeriosis show the strong symbiosis between the innate cellular system and the T-cell response. Immunol. Rev. 158:11–25. [DOI] [PubMed] [Google Scholar]
- 2.Edelson, B.T., and E.R. Unanue. 2000. Immunity to Listeria infection. Curr. Opin. Immunol. 12:425–431. [DOI] [PubMed] [Google Scholar]
- 3.Muller, U., U. Steinhoff, L.F.L. Reis, S. Hemmi, J. Pavlovic, R.M. Zinkernagel, and M. Aguet. 1994. Functional role of type I and type II IFNs in antiviral defense. Science. 264:1918–1921. [DOI] [PubMed] [Google Scholar]
- 4.Dalton, D.K., S. Pitts-Meek, S. Keshav, I.S. Figari, A. Bradley, and T.A. Stewart. 1993. Multiple defects of immune cell function in mice with disrupted IFN-γ genes. Science. 259:1739–1742. [DOI] [PubMed] [Google Scholar]
- 5.Huang, S., W. Hendriks, A. Althage, S. Hemmi, H. Bluethmann, R. Kamijo, J. Vilcek, R.M. Zinkernagel, and M. Aguet. 1993. Immune response in mice that lack the IFN-γ receptor. Science. 259:1742–1745. [DOI] [PubMed] [Google Scholar]
- 6.van den Broek, M.F., U. Muller, S. Huang, R.M. Zinkernagel, and M. Aguet. 1995. Immune defence in mice lacking type I and/or type II IFN receptors. Immunol. Rev. 148:5–18. [DOI] [PubMed] [Google Scholar]
- 7.Merrick, J.C., B.T. Edelson, V. Bhardwaj, P.E. Swanson, and E.R. Unanue. 1997. Lymphocyte apoptosis during early phase of Listeria infection in mice. Am. J. Pathol. 151:785–792. [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang, J., L.L. Lau, and H. Shen. 2003. Selective depletion of nonspecific T cells during the early stage of immune responses to infection. J. Immunol. 171:6032–6038. [DOI] [PubMed] [Google Scholar]
- 9.Carrero, J.A., B. Calderon, and E.R. Unanue. 2004. Listeriolysin O from Listeria monocytogenes is a lymphocyte apoptogenic molecule. J. Immunol. 172:4866–4874. [DOI] [PubMed] [Google Scholar]
- 10.Edelson, B.T., P. Cossart, and E.R. Unanue. 1999. Cutting edge: paradigm revisited: antibody provides resistance to Listeria infection. J. Immunol. 163:4087–4090. [PubMed] [Google Scholar]
- 11.Cousens, L.P., R. Peterson, S. Hsu, A. Dorner, J.D. Altman, R. Ahmed, and C.A. Biron. 1999. Two roads diverged: interferon α/β– and interleukin 12–mediated pathways in promoting T cell interferon γ responses during viral infection. J. Exp. Med. 189:1315–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun, S., X. Zhang, D.F. Tough, and J. Sprent. 1998. Type I IFN–mediated stimulation of T cells by CpG DNA. J. Exp. Med. 188:2335–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Serbina, N.V., T.P. Salazar-Mather, C.A. Biron, W.A. Kuziel, and E.G.P. Am. 2003. TNF/iNOS-producing dendritic cells mediate innate immune defense against bacterial infection. Immunity. 19:59–70. [DOI] [PubMed] [Google Scholar]
- 14.Havell, E.A. 1986. Augmented induction of interferons during Listeria monocytogenes infection. J. Infect. Dis. 153:960–969. [DOI] [PubMed] [Google Scholar]
- 15.O'Riordan, M., C.H. Yi, R. Gonzales, K.D. Lee, and D.A. Portnoy. 2002. Innate recognition of bacteria by a macrophage cytosolic surveillance pathway. Proc. Natl. Acad. Sci. USA. 99:13861–13866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fehr, T., G. Schoedon, B. Odermatt, T. Holtschke, M. Schneeman, M.F. Bachmann, T.W. Mak, I. Horak, and R.M. Zinkernagel. 1997. Crucial role of IFN consensus sequence binding protein, but neither of IFN regulatory factor 1 nor of nitric oxide synthesis for protection against murine listeriosis. J. Exp. Med. 185:921–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stockinger, S., T. Materna, D. Stoiber, L. Bayr, R. Steinborn, T. Kolbe, H. Unger, T. Chakraborty, D.E. Levy, M. Muller, et al. 2002. Production of type I IFN sensitizes macrophages to cell death induced by Listeria monocytogenes. J. Immunol. 169:6522–6529. [DOI] [PubMed] [Google Scholar]
- 18.Fadok, V.A., D.L. Bratton, A. Konowal, P.W. Freed, J.Y. Westcott, and P.M. Henson. 1998. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J. Clin. Invest. 101:890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Freire-de-Lima, C.G., D.O. Nascimento, M.B. Soares, P.T. Bozza, H.C. Castro-Faria-Neto, F.G. de Mello, G.A. DosReis, and M.F. Lopes. 2000. Uptake of apoptotic cells drives the growth of a pathogenic trypanosome in macrophages. Nature. 403:199–203. [DOI] [PubMed] [Google Scholar]
- 20.Savill, J., I. Dransfield, C. Gregory, and C. Haslett. 2002. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat. Rev. Immunol. 2:965–975. [DOI] [PubMed] [Google Scholar]
- 21.Chen, W., M.E. Frank, W. Jin, and S.M. Wahl. 2001. TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity. 14:715–725. [DOI] [PubMed] [Google Scholar]
- 22.Bancroft, G.J., M.J. Bosma, G.C. Bosma, and E.R. Unanue. 1986. Regulation of macrophage Ia expression in mice with severe combined immunodeficiency: induction of Ia expression by a T cell-independent mechanism. J. Immunol. 137:4–9. [PubMed] [Google Scholar]
- 23.Hoshino, K., T. Kaisho, T. Iwabe, O. Takeuchi, and S. Akira. 2002. Differential involvement of IFN-β in Toll-like receptor-stimulated dendritic cell activation. Int. Immunol. 14:1225–1231. [DOI] [PubMed] [Google Scholar]
- 24.Takaoka, A., S. Hayakawa, H. Yanai, D. Stoiber, H. Negishi, H. Kikuchi, S. Sasaki, K. Imai, T. Shibue, K. Honda, et al. 2003. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature. 424: 516–523. [DOI] [PubMed] [Google Scholar]