QM/MM Study of Energy Storage and Molecular Rearrangements Due to the Primary Event in Vision (original) (raw)
Abstract
The energy storage and the molecular rearrangements due to the primary photochemical event in rhodopsin are investigated by using quantum mechanics/molecular mechanics hybrid methods in conjunction with high-resolution structural data of bovine visual rhodopsin. The analysis of the reactant and product molecular structures reveals the energy storage mechanism as determined by the detailed molecular rearrangements of the retinyl chromophore, including rotation of the φ(C11–C12) dihedral angle from −11° in the 11-cis isomer to −161° in the all-trans product, where the preferential sense of rotation is determined by the steric interactions between Ala-117 and the polyene chain at the C13 position, torsion of the polyene chain due to steric constraints in the binding pocket, and stretching of the salt bridge between the protonated Schiff base and the Glu-113 counterion by reorientation of the polarized bonds that localize the net positive charge at the Schiff-base linkage. The energy storage, computed at the ONIOM electronic-embedding approach (B3LYP/6-31G*:AMBER) level of theory and the _S_0 → _S_1 electronic-excitation energies for the dark and product states, obtained at the ONIOM electronic-embedding approach (TD-B3LYP/6-31G*//B3LYP/6-31G*:AMBER) level of theory, are in very good agreement with experimental data. These results are particularly relevant to the development of a first-principles understanding of the structure-function relations in prototypical G-protein-coupled receptors.
INTRODUCTION
Understanding the structure-function relationship of G-protein-coupled receptors (GPCRs) at the detailed molecular level is a subject of great interest, since GPCRs are the central components of a variety of biological signal transmission pathways (Ji et al., 1998; Gether and Kobilka, 1998; Marinissen and Gutkind, 2001). The membrane glycoprotein rhodopsin is a prototypical GPCR present in the rod cells of the retina (Meng and Bourne, 2001; Hamm, 2001), responsible for turning on the signaling transmission cascade in the vertebrate vision process (Ebrey and Koutalos, 2001; Menon et al., 2001; Okada et al., 2001). Rhodopsin is composed of a bundle of seven transmembrane _α_-helices surrounding the covalently bound retinyl chromophore. The chromophore is the protonated Schiff base of 11-_cis_-retinal, which isomerizes to the all-trans form during the primary event (Goldschmidt et al., 1976; Honig, 1978; Birge, 1981) after photoexcitation of the system (see Fig. 1). The reaction produces bathorhodopsin in the ground-electronic state, with a high quantum yield (∼0.67) that is wavelength-dependent (Schoenlein et al., 1991; Wang et al., 1994) within 200 fs, making it one of the fastest and most efficient photoreactions in nature (Ji et al., 1998; Gether and Kobilka, 1998; Marinissen and Gutkind, 2001). The formation of product bathorhodopsin is endothermic and stores ∼50% of the photon energy (Honig et al., 1979; Cooper, 1979a,b; Shick et al., 1987). The stored energy is subsequently used to promote thermal reactions in the protein bleaching sequence and in the subsequent transducin cycle.
FIGURE 1.

Photoisomerization of the retinyl chromophore in rhodopsin.
The detailed molecular mechanism responsible for the storage of the photon energy through photoisomerization of the retinyl chromophore in rhodopsin has been the subject of intense research and remains an open problem (Kim et al., 2003; Furutani et al., 2003; Ferre and Olivucci, 2003; Rohrig et al., 2002; Birge, 2001; Kandori et al., 2001; Sugihara et al., 2002a; Ben-Nun et al., 2002; Gonzalez-Luque et al., 2000; Blomgren and Larsson, 2003). Intriguingly, the isomerization of the 11-cis retinal in solutions is much slower (e.g., involves a 10-ps reaction time in methanol, see Kandori et al., 1995) and has a much lower efficiency (e.g., quantum yield ∼0.2, independent of wavelength in at least four different solvents, see Becker and Freedman, 1985). Furthermore, the wavelength of maximum absorbance of the retinyl chromophore in rhodopsin is significantly shifted relative to the absorption of retinal in solutions and approximately coincides with the wavelength of maximum intensity of the solar spectrum (∼500 nm). These findings suggest that the molecular environment in rhodopsin has been highly optimized by natural selection for absorption of visible light and energy storage through photoisomerization of the retinyl chromophore. For many years, however, progress in understanding the influence of the protein environment on the reaction speed and efficiency has been hindered by the lack of high-resolution structural data.
Theoretical studies of the primary photochemical event in rhodopsin have a long history (Warshel and Karplus, 1974; Warshel, 1976; Weiss and Warshel, 1979; Warshel and Barboy, 1982; Birge and Hubbard, 1980; Tallent et al., 1992). Most of them were performed much before the crystallographic structure of rhodopsin was available. The pioneer studies by Warshel and co-workers (Warshel, 1976; Weiss and Warshel, 1979; Warshel and Barboy, 1982) were based on the semiempirical QCFF/PI method for the description of the chromophore and a simplified representation of the protein-chromophore interaction based on a surface of closed-packed spheres described by a model potential with adjustable parameters. More recently, the QCFF/PI surfaces have been recalibrated (Warshel and Chu, 2001) to reproduce the trend of ab initio studies for the isolated chromophore (Vreven et al., 1997; Garavelli et al., 1998) but applied only in studies of bacteriorhodopsin (Warshel and Chu, 2001). Birge and co-workers (Birge and Hubbard, 1980; Tallent et al., 1992) have implemented MNDO/AM1 and INDO-PSDCI procedures and have described the photoisomerization dynamics in terms of a one-dimensional potential model with an arbitrary rate constant for the dissipation of the internal energy. These early theoretical studies proposed energy storage mechanisms based on a mixture of charge separation and protein strain (Birge and Hubbard, 1980; Warshel and Barboy, 1982) as alternatives to the existing charge separation models (Rosenfeld et al., 1977; Honig et al., 1979). However, rigorous calculations of the strain and electrostatic contributions have not been possible since the protein structure was not known.
The recently reported x-ray crystal structures of bovine rhodopsin (Palczewski et al., 2000; Teller et al., 2001; Okada et al., 2002), although at a resolution of 2.6–2.8 Å, have already motivated theoretical studies that focused on the analysis of the geometry and electronic excitation energy of the _S_0 → _S_1 transition of the retinyl chromophore. These studies have included density-functional theory calculations based on the self-consistent-charge tight-binding approximation (Sugihara et al., 2002a,b), classical molecular dynamics simulations (Rohrig et al., 2002), ab initio restricted-Hartree-Fock calculations of reduced-model systems (Yamada et al., 2002), and quantum mechanics/molecular mechanics computations at the CASPT2//CASSCF/AMBER level of theory (Ferre and Olivucci, 2003). In particular, the ab initio studies provided a rigorous description of the chromophore in the dark state. However, the detailed conformational changes responsible for the storage of energy through 11-cis/all-trans isomerization still remain largely elusive to first-principles examinations.
In this article, two-layer quantum mechanics/molecular mechanics (molecular orbitals: molecular mechanics) hybrid methods (QM/MM(MO:MM) are used to describe the retinyl chromophore rearrangements due to the 11-cis/all-trans isomerization as influenced by the relaxation of the surrounding protein environment. This study includes an explicit treatment of the protein environment and assumes that the formation of the product bathorhodopsin involves complete relaxation of both nuclear and electronic degrees of freedom, after isomerization of the retinyl chromophore. This assumption is subsequently validated by comparing the energy storage, predicted at the ONIOM electronic-embedding approach (ONIOM-EE) (B3LYP/6-31G*:AMBER) level of theory, to the corresponding values obtained by calorimetry. Furthermore, the electronic excitation energies for the dark and isomerization product states are computed at the ONIOM-EE (TD-B3LYP/6-31G*:AMBER) level of theory and compared to the corresponding experimental values. It is important to note that the studies reported in this article are only focused on the analysis of relaxed molecular structures for reactants and products in the ground electronic state, obtained under the assumption that the protein relaxation occurs only after isomerization of the retinyl chromophore is complete (i.e., the protein relaxation time is much longer than the 200-fs reaction time). A description of intermediate structures, however, would require modeling the photoexcitation and excited state non-adiabatic processes in vibronically coupled potential energy surfaces. These processes have been the subject of recent studies in reduced dimensional model systems (Vreven et al., 1997; Garavelli et al., 1998, 1999; Ben-Nun and Martinez, 1998; La Penna et al., 1998; Molteni et al., 1999; Hahn and Stock, 2000; Flores and Batista, 2004) although the complete description of reaction dynamics with an explicit treatment of the protein environment has yet to be reported.
The specific two-layer QM/MM(MO:MM) hybrid method applied in this article is the ONIOM hybrid approach (Maseras and Morokuma, 1995; Svensson et al., 1996; Humbel et al., 1996; Dapprich et al., 1999; Vreven and Morokuma, 2000a; Vreven et al., 2001) as implemented in the Gaussian package (Frisch et al., 2003), which has been extensively tested against the standard complete active-space self-consistent field (CASSCF) method. It has been demonstrated that ONIOM can accurately reproduce the standard CASSCF(10e/10o) (i.e., 10 active electrons in 10 orbitals) results for the first singlet excited state (_S_1) photoisomerization pathways in protonated Schiff bases (Vreven and Morokuma, 2000b) and _S_0 → _S_1 vertical excitation in bacteriorhodopsin (Vreven and Morokuma, 2003). The computational results reported in this article show direct evidence of molecular rearrangements responsible for storage of ∼50% of the photon energy through isomerization of the retinyl chromophore, the molecular origin of a preferential sense of rotation of the φ(C11–C12) dihedral angle, and the detailed chromophore-protein interactions responsible for energy storage. These results are particularly relevant to the development of a first-principles understanding of the structure-function relationship of G-protein-coupled receptors at the molecular level.
The article is organized as follows. The section titled Structural Models describes the preparation of the system. The sections titled ONIOM Hybrid Methods and Reaction Path outline the computational approach, including a description of the ONIOM hybrid methods and the optimization approach implemented for computing intermediate structures along the reaction pathway after electronic and partial-nuclear relaxation. The section titled Structural Integrity addresses the structural integrity of the computational approach. Results are presented next, as organized in five subsections: Energy Storage reports QM/MM(MO:MM) results for the reaction energetics, including the computations of the energy storage and its comparison to experimental values; Molecular Rearrangements discusses the underlying molecular changes due to 11-cis/all-trans isomerization; Chromophore-Protein Interactions addresses the detailed interactions responsible for the energy storage mechanism; and finally, Electronic Excitations reports the electronic excitations for rhodopsin and bathorhodopsin as well as a comparison with the corresponding experimental values. Last section summarizes our findings.
METHODS
Structural models
The computational models investigated in this article are based on the refinement of the crystal structure of bovine rhodopsin (Protein Data Bank, i.e., PDB, accession code 1F88, monomer A), solved at 2.8 Å resolution (Palczewski et al., 2000). Model systems based on the crystal structure with PDB accession code 1L9H, monomer A, solved at 2.6 Å resolution (Okada et al., 2002), have also been prepared. A crucial difference between the two crystal structures is the observation of two water molecules next to the retinyl chromophore in the 1L9H structure, which are not observed at the lower resolution. Our calculations show that these two molecules are very important in the stabilization of the chromophore since they are bound within the cavity by a hydrogen-bond network that extends from Glu-181 to Glu-113, next to the Schiff-base linkage. Such a hydrogen-bond network is consistent with a recently postulated proton-transfer mechanism in a subsequent step of the photobleaching sequence (Birge and Knox, 2003; Yan et al., 2003). These two water molecules are therefore included in our computational models.
Starting from the 1F88 PDB crystal structures, hydrogen atoms are added by using the molecular modeling program TINKER (Ponder, 2001). The protonation of all titratable groups is standard. The rhodopsin cavity is set neutral, consistent with experiments (Fahmy et al., 1993). The Schiff-based linkage between Lys-296 and the chromophore bears a net positive charge NH+, compensated by the Glu-113 counterion (forming a salt bridge; Sakmar et al., 1989; Zhukovsky and Oprian, 1989). Amino acids Glu-122, Asp-83, and Glu-181, within the protein core, are assumed to be neutral as indicated by FTIR experiments (Fahmy et al., 1993) and UV-vis spectroscopic measurements of site-directed mutants (Yan et al., 2002). Finally, the regular ends of the protein and the artificial ends due to the missing or incomplete amino acids from the x-ray structures in the third cytoplasmic loop (236–239) and in the C-terminal tail (328–348) are capped with 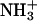 and
and 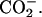 Thus the model system in the present calculations contains 5170 atoms with a total charge of +4_e_.
Thus the model system in the present calculations contains 5170 atoms with a total charge of +4_e_.
ONIOM hybrid methods
The QM/MM(MO:MM) calculations reported in this article are based on the ONIOM two-layer hydrogen link-atom scheme (Maseras and Morokuma, 1995; Svensson et al., 1996; Humbel et al., 1996; Dapprich et al., 1999; Vreven and Morokuma, 2000a, 2003; Vreven et al., 2001). The full system of 5170 atoms is partitioned into two layers by placing a frontier at the C_δ_–C_ε_ bond of the Lys-296 side chain (i.e., two bonds beyond the C–NH+ linkage). The quantum-mechanical (QM) layer corresponds to a reduced system with 54 atoms, including 48 atoms of the retinyl chromophore, five atoms of Lys-296 (NH+,CH2), and a link hydrogen atom that saturates the extra valence of the terminal -C-H2 at the boundary. The molecular-mechanics (MM) layer corresponds to the remainder of the protein. For the sake of comparison, model systems were also prepared with the frontier displaced by an additional bond (i.e., including the C_δ_–C_ε_ bond of the Lys-296 side chain in the reduced system) and no significant differences were observed in any of the results.
The total energy E of the system can be obtained according to the so-called ONIOM molecular-embedding (ONIOM-ME) approach from three independent calculations,
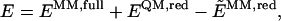 |
(1) |
|---|
where _E_MM,full is the energy of the full system computed at the molecular-mechanics level of theory, including the electrostatic interaction between all atomic charges; _E_QM,red is the energy of the reduced system computed at the quantum-mechanics level of theory; and 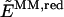 is the energy of the reduced system computed at the molecular-mechanics (MM) level of theory. Therefore, according to Eq. 1 the electrostatic interactions between the two layers are considered at the MM level of theory, where the polarization of the reduced system due to the influence of the protein environment is completely neglected.
is the energy of the reduced system computed at the molecular-mechanics (MM) level of theory. Therefore, according to Eq. 1 the electrostatic interactions between the two layers are considered at the MM level of theory, where the polarization of the reduced system due to the influence of the protein environment is completely neglected.
To include the polarization of the reduced system in the description of the reaction energetics, the so-called ONIOM electronic-embedding approach (ONIOM-EE) is used. The method is also based on the three independent calculations introduced in Eq. 1 but, contrary to the ONIOM-MM method, it includes the electrostatic interactions between the two layers in the calculation of both _E_QM,red and 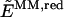 . Therefore, the electrostatic interactions included at the MM level in both the
. Therefore, the electrostatic interactions included at the MM level in both the 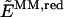 and
and 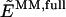 terms cancel; and the resulting energy description includes the polarization of the reduced system induced by the surrounding protein environment.
terms cancel; and the resulting energy description includes the polarization of the reduced system induced by the surrounding protein environment.
All calculations reported in this article involve a description of the MM layer as modeled by the standard AMBER force field (Cornell et al., 1995) setting to zero the charges of atoms closer than four bonds from any atom in the reduced system to avoid overpolarization of the reduced system. This approximation is subsequently validated in terms of the analysis of electrostatic chromophore-protein interactions (see Chromophore-Protein Interactions, below). The distribution of atomic charges in the retinyl chromophore is based on the electrostatic-potential (ESP) atomic charges, obtained at the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory.
The _S_0 → _S_1 electronic-excitation energies are computed at the time-dependent density-functional theory ONIOM-EE (TD-B3LYP/6-31G*:AMBER) level of theory (Stratmann et al., 1998) (i.e., the ONIOM-EE (TD-B3LYP/6-31G*:AMBER) level of theory after geometry optimization of the complete system at the ONIOM-EE (B3LYP/6-31G*:AMBER) level). Full geometry optimizations are carried out with the Gaussian 03 (Frisch et al., 2003) and Tinker programs (Ponder, 2001), without boundary conditions or inclusion of solvent.
Structural integrity
To validate the structural integrity of the computational approach implemented in this article, Fig. 2 quantifies the deviations between the rhodopsin molecular structure optimized at the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory and the x-ray crystallographic structure. Structure optimization was done in vacuo. Despite this approximation, geometry minimization does not result in any significant structural change around the active site of this membrane-bound protein, probably due to the fact that the retinyl chromophore is surrounded by a robust structure of seven transmembrane _α_-helices.
FIGURE 2.

Deviations of the C-α atom coordinates in the rhodopsin molecular structure optimized at the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory from the corresponding C-α atom coordinates in the x-ray crystal structure. The dotted line indicates the RMS ∼0.83 Å.
The comparison includes the C-α atoms located within a 20 Å radius from each atom of the retinyl chromophore and the Schiff-based linkage to Lys-296, a region that accounts for ∼70% of the whole protein. Floppy side chains and residues beyond the 20 Å radius were excluded to facilitate the comparison. Fig. 2 shows that the RMS ∼0.83 Å, with deviations larger than twice the RMS for only 4% of the atoms (all of them located at ∼15 Å from the chromophore). In addition, Fig. 2 shows that the residues with predominant interactions with the retinyl chromophore, including Glu-113, Lys-296, and Ala-117, have even smaller deviations from their corresponding coordinates in the crystal x-ray structure (e.g., Glu-113, RMS ∼0.66 Å; Lys-296, RMS ∼0.64 Å; Ala-292, RMS ∼0.52 Å; and the chromophore, RMS ∼0.22 Å). Therefore, Fig. 2 indicates that the molecular structure obtained by geometry optimization at the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory is in good agreement with the experimental x-ray crystal structure despite the negligence of the environment beyond the protein (including the solvent and the lipid bilayer) and the missing amino-acid fragments in the third cytoplasmic loop. The observed robustness of the molecular structure is probably due to the intrinsic stability of the seven transmembrane helical bundle that surrounds the covalently bound retinyl chromophore. The small deviations of the ab initio coordinates relative to the crystallographic data also suggest that higher resolution structures should have very similar coordinates, at least for the C-α backbone within 20 Å from the retinyl chromophore.
Reaction path
To investigate the molecular rearrangements due to the primary photochemical event, it is assumed that the isomerization dynamics of the retinyl chromophore is much faster than protein relaxation. This assumption is consistent with the experimental 200-fs reaction time (Schoenlein et al., 1991; Wang et al., 1994) as well as with the observation that the isomerization coordinate is mainly coupled to the vibrational modes of the retinyl chromophore (Lin et al., 1998). It is also assumed that the molecular structure of the all-trans retinyl chromophore in the ground-electronic state, produced by the underlying curve-crossing dynamics after photoexcitation of the system, relaxes to the same minimum energy configuration as the system where the retinyl chromophore is isomerized along the ground-state minimum-energy path (MEP) subject to the constraint of fixed protein environment. These two simplifying assumptions allow the implementation of a practical computational approach to obtain the relaxed molecular structure of the product bathorhodopsin. The method involves full-geometry relaxation of the complete system in the ground electronic state, after 11-cis/all-trans isomerization of the retinyl chromophore along the ground-state MEP defined by a fixed protein environment.
The protein binding pocket imposes steric constraints over molecular rearrangements associated with the 11-cis/all-trans isomerization of the retinyl chromophore. The analysis of the rhodopsin molecular structure, obtained by full geometry optimization at the ONIOM-EE (B3LYP/6-31G*:AMBER) level, reveals that Ala-117 sterically hinders the rotation of the φ(C11–C12) dihedral angle toward positive angles. These steric interactions establish a negative sense of rotation followed upon photoisomerization—i.e., clockwise rotation of the φ(C11–C12) dihedral angle around the rotational axis defined by the C11–C12 vector (see Fig. 3). The steric constraints imposed by Ala-117 are also responsible for an initial −10° twist of the φ(C11–C12) dihedral angle in the reactant molecular structure reported in Molecular Rearrangements, below. Other steric constraints for chromophore rearrangements in the rhodopsin binding pocket are responsible for blocking the motion of the _β_-ionone ring during isomerization, inducing torsional strain in the polyene chain. This computational model for retinyl chromophore isomerization in rhodopsin is consistent with recent experimental studies of circular dichroism (CD) (Fukuda et al., 1984; Ito et al., 1992), ab initio calculations of the CD spectrum (Buss, 2001), and molecular dynamics simulations (Sugihara et al., 2002a,b; Saam et al., 2002).
FIGURE 3.

Definition of positive and negative φ(C11–C12) dihedral angles. The retinyl chromophore and the protein chain beyond the Schiff linkage are represented by brown and gray tubes, respectively. The Schiff linkage is highlighted in blue and the C11–C12 bond in magenta. The rotational axis defined by the C11–C12 bond is represented by dashes. Note that the steric interaction of the C13-methyl substituent group with the Ala-117 amino acid hinders the rotation toward positive angles.
The optimized molecular structure of bathorhodopsin is obtained according to the following schemes:
- AMBER-MM optimization. Starting from the molecular structure of rhodopsin, prepared as described in Structural Models, and fully optimized as described in ONIOM Hybrid Methods, intermediate structures along the ground-state MEP are generated by geometry optimization subject to the constraints of fixed φ(C11–C12) dihedral angle and protein environment, after incrementally rotating φ(C11–C12) in the 0° to −180° range. During this first part of the bathorhodopsin preparation, the geometry optimization is implemented at the AMBER-MM level of theory allowing relaxation of all degrees of freedom of the retinyl chromophore, the bound-water molecules, and 20 atoms more beyond the Schiff-based linkage to Lys-296. A realistic description of chromophore-protein electrostatic interactions is implemented by self-consistently updating the atomic charges in the retinyl chromophore according to the electrostatic potential obtained at the ONIOM-EE level of theory. The underlying self-consistent approach is initialized in terms of the ESP atomic charges obtained in a previously optimized molecular structure and iterated until convergence, which is usually reached within four iterations.
- ONIOM-EE optimization. The unrelaxed bathorhodopsin product obtained according to the AMBER-MM optimization is then fully optimized at the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory, including the retinyl chromophore, bound water molecules, and all residues within a 20 Å radius from the chromophore.
The energy storage due to 11-cis/all-trans isomerization is computed in terms of the energy difference between the minimum energy structures of rhodopsin and bathorhodopsin, obtained at the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory. These structures also provide the _S_0 → _S_1 vertical excitations that are obtained at the ONIOM-EE (TD-B3LYP/6-31G*:AMBER) level of theory.
RESULTS
Energy storage
Table 1 presents the computational results of energy storage, due to 11-cis/all-trans isomerization in rhodopsin, as predicted by the ONIOM-EE(B3LYP/6-31G*:AMBER) level of theory and the comparison to the theoretical predictions to experimental measurements. Table 1 also presents the decomposition of the total energy storage into electrostatic, bonded, and Van der Waals energy contributions. The computed energy stored is 34 kcal/mol. This result is in excellent agreement with calorimetry measurements, reported in the 32–35 kcal/mol energy range (Cooper, 1979a,b; Shick et al., 1987). The decomposition of the total energy storage, presented in Table 1, shows that 50% of the storage energy is due to electrostatic interactions, 32% is due to strain energy, and the remaining 18% corresponds to Van der Waals interactions. The energy storage decomposition has been computed at the ONIOM-EE (B3LYP/6-31G*:AMBER) level by setting to zero the corresponding term in the expression of the total energy of the system (e.g., switching to zero the atomic charges, the Van der Waals interactions, etc.). To elucidate the molecular origin of the individual contributions to the energy storage, the following section analyzes the underlying molecular rearrangements due to 11-cis/all-trans isomerization in rhodopsin.
TABLE 1.
Energy storage due to 11-cis/all-trans isomerization in rhodopsin as predicted by the ONIOM-EE(B3LYP/6-31G*:AMBER) level of theory
| Energy storage | |
|---|---|
| Total energy storage | 34.1 kcal/mol |
| Electrostatic contribution | 17.1 kcal/mol (50%) |
| Strain contribution | 11.1 kcal/mol (32%) |
| Van der Waals contribution | 5.9 kcal/mol (18%) |
| Experimental value | 32.2 ± 0.9 kcal/mol* |
| 34.7 ± 2.2 kcal/mol† |
Molecular rearrangements
Fig. 4 shows the underlying conformational changes due to the 11-cis/all-trans isomerization in rhodopsin. The top and bottom panels show the molecular structure for the 11-cis reactant and the all-trans product, respectively, optimized at the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory. The isomerization reaction changes the φ(C11-C12) dihedral angle (highlighted in magenta) from −11° in the 11-cis isomer to −161° in the all-trans isomer, where the negative sense of rotation is determined by the steric Van der Waals interactions between Ala-117 and the polyene chain at the C13 position. Note that the _β_-ionone ring, the Schiff linkage (highlighted in blue), and the protein chain beyond the linkage (highlighted in gray) remain mostly undisplaced after isomerization of the retinyl chromophore. The steric constraints, responsible for blocking the displacement of the chromophore in the cavity and the rotation of the _β_-ionone ring, for the molecular structure beyond the Schiff linkage induce strain and torsion of the bonds at the Schiff-based linkage due to a reorientation of the C13-methyl substituent group. These results for molecular rearrangements predicted at the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory partially agree with classical predictions based on steered molecular dynamics simulations (Saam et al., 2002).
FIGURE 4.
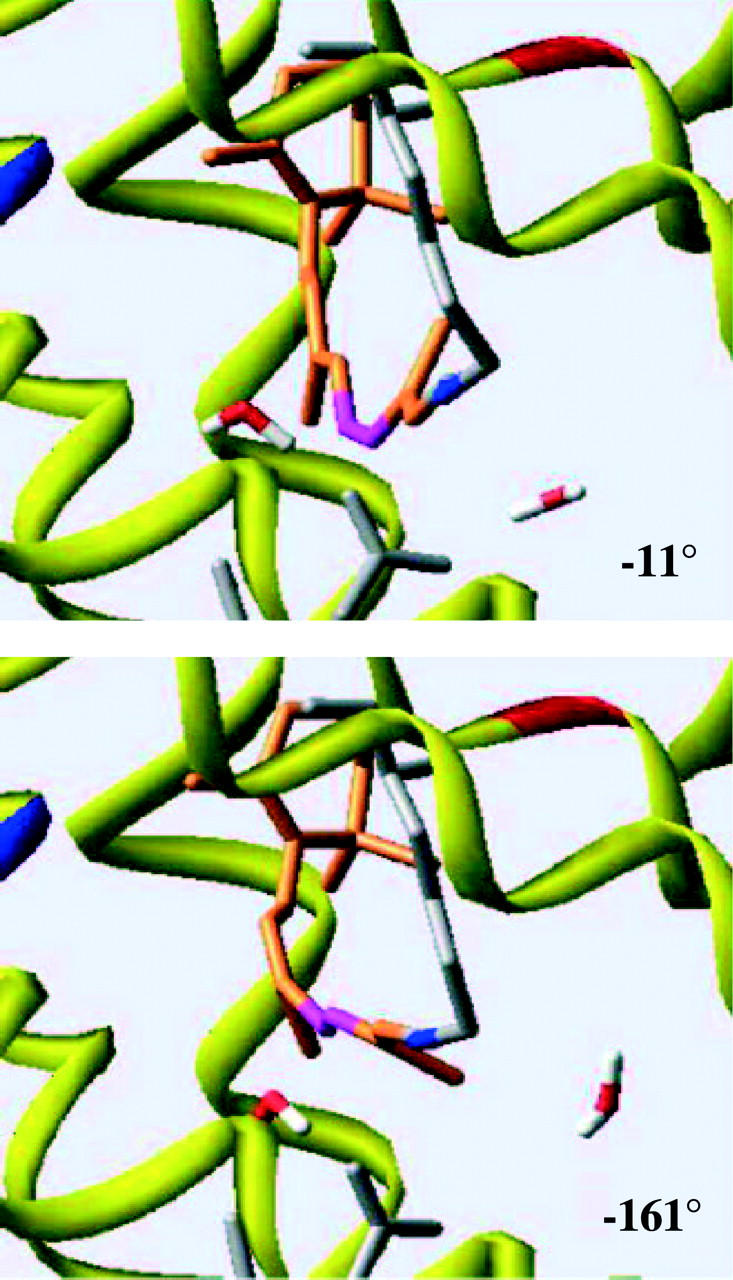
Molecular structures of 11-cis rhodopsin (top panel), with φ(C11–C12) = −11° and all-trans bathorhodopsin (bottom panel), with φ(C11–C12) = −161°.
Table 2 describes a quantitative analysis of the detailed molecular rearrangements along the chromophore polyene chain, including torsions that range from C6 to C_δ_ in the Lys-296 side chain. The comparative analysis of dihedral angles, reported in Table 2, indicates that the polyene chain in the product all-trans isomer is significantly bent and twisted, with out-of-the-plane distortions >20° in the dihedral angles along the C9–C10, C12–C13, C14–C15, and C15–NH+ chromophore bonds.
TABLE 2.
Rearrangement of dihedral angles along the chromophore polyene chain, due to 11-cis rhodopsin/all-trans isomerization in rhodopsin, predicted by the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory
| Dihedral angle | 11-cis | all-trans | Δ |
|---|---|---|---|
| C5–C6–C7–C8 | −44 | −32 | 12 |
| C1–C6–C7–C8 | 136 | 144 | 8 |
| C6–C7–C8–C9 | −172 | −155 | 17 |
| C7–C8–C9–C(methyl 1) | −10 | −10 | 0 |
| C7–C8–C9–C10 | 167 | 162 | 5 |
| C8–C9–C10–C11 | 179 | −152 | 29 |
| C9–C10–C11–C12 | 173 | 171 | 2 |
| C10–C11–C12–C13 | −11 | −161 | 150 |
| C11–C12–C13–C14 | 166 | −174 | 20 |
| C11–C12–C13–C(methyl 2) | −13 | 11 | 24 |
| C12–C13–C14–C15 | 179 | −168 | 13 |
| C13–C14–C15–N | 168 | −164 | 28 |
| C14–C15–N–CE | 171 | −158 | 31 |
| C15–N–CE–CD | 100 | 113 | 13 |
Fig. 5 shows the superposition of the optimized molecular structures for the reactant 11-cis rhodopsin (brown) and the product all-trans bathorhodopsin (white). The molecular superposition indicates that the most significant rearrangements due to the 11-cis/all-trans isomerization are localized within the retinyl chromophore and the Schiff linkage. Fig. 5 shows that the constraints for molecular rearrangements in the rhodopsin binding pocket are due to the steric interactions at both ends of the retinyl chromophore, including motion restrictions due to interactions between the three methyl-substituent groups in the _β_-ionone ring and the surrounding residues, and the almost 90° turn of the protein backbone at the C-α atom of Lys-296 right beyond the Schiff linkage. These constraints are responsible for fixing both ends of the chromophore during the isomerization reactions, inducing torsional structural rearrangements in the polyene chain that extends from C6 to the Schiff linkage. Furthermore, Fig. 5 shows that the 11-cis/all-trans isomerization induces a reorientation of the C13-methyl substituent group and the consequent torsion of the polyene chain at the protonated NH+ Schiff-based linkage to Lys-296. The linkage torsion induces a reorientation of polarized bonds, including the C15–H and N–H+ bonds with partial positive charges on the H atoms (Fig. 6). Note that the polarized bonds NH+ and C15H are rotated in the all-trans bathorhodopsin isomer, relative to their corresponding orientations in the reactant 11-cis rhodopsin. The analysis of ESP atomic charges indicates that these two polarized bonds account for most of the net positive charge at the linkage. Therefore, due to the reorientation of polarized bonds, the net positive charge of retinyl chromophore is farther away from the Glu-113 counterion in bathorhodopsin than in rhodopsin, even when the linkage itself remains in place.
FIGURE 5.

Superposition of molecular structures of 11-cis rhodopsin (brown) and all-trans bathorhodopsin (white), optimized at the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory. The protein chain beyond the Schiff linkage is represented by gray tubes, the Schiff linkage is highlighted in blue and the C11–C12 bond in magenta.
FIGURE 6.

Superposition of rhodopsin (brown) and bathorhodopsin (white) molecular structure at the Schiff linkage, including the explicit visualization of the distribution of surrounding residues responsible for the most significant electrostatic contributions to the total energy storage (see Fig. 7). The protein chain beyond the Schiff linkage is represented by gray tubes and the Schiff linkage is highlighted in blue. The red-dashed arrows indicate the reorientation of polarized bonds in the retinyl chromophore, responsible for the displacement of the net positive charge at the linkage relative to the Glu-113 counterion, including the N–H+ and C15–H bonds (only the hydrogen atoms of these two bonds are represented by white spheres). The negative charge density is highlighted in blue.
The molecular rearrangements described in Figs. 4–6 and Table 2 indicate that the 11-cis/all-trans isomerization reaction produces a highly distorted all-trans retinyl chromophore (e.g., significantly bent and twisted, relative to the linear molecular structure of the all-trans retinal isomer in the gas phase). Therefore, although the seven _trans_membrane helical bundle isolates the retinyl chromophore from interactions with the solvent, the 11-cis/all-trans isomerization reaction does not proceed in the usual sense as expected in the gas phase. In contrast to the analogous isomerization reaction in the gas phase, significant distortions are necessary to fit the isomerized retinyl in the binding pocket.
The underlying molecular rearrangements predicted by the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory can be compared to the molecular rearrangements in the “bicycle-pedal” pathway proposed (Warshel, 1976) and refined (Warshel and Barboy, 1982) by Warshel and co-workers before the rhodopsin crystal structure was available, where the 180° rotation around the C11–C12 bond is compensated by a 40° twist around the C9–C10 and C15–N bonds. As described earlier in this section, the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory predicts that the isomerization reaction involves a 150° rotation around the C11–C12 bond, compensated by distortions larger than 20° in the dihedral angles along the C9–C10, C12–C13, C14–C15, and C15–NH+ bonds. Remarkably, however, the larger distortions predicted by the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory correspond approximately to 30° twists around the C9–C10, C14–C15, and C15–N bonds, in partial agreement with the bicycle-pedal model. The torsion around the C15–N bond, however, is crucial for the reorientation of polarized bonds responsible for the storage of electrostatic energy.
Chromophore-protein interactions
The molecular rearrangements described in the previous section indicate that the storage of strain energy is due to the highly distorted conformation of the all-trans retinyl chromophore in bathorhodopsin. In addition, we showed that the isomerization reaction stores 25% of the photon energy in terms of electrostatic energy by moving the positive charge of the Schiff-base linkage away from the negatively charged Glu-113 counterion. At this point, it is important to mention that the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory predicts almost identical ESP atomic charges for the protonated Schiff-based retinyl chromophore in 11-cis rhodopsin or all-trans bathorhodopsin. Therefore, the electrostatic contribution to the total energy storage due to changes in atomic charges within the polyene chain is negligible. In contrast, the reorientation of the polarized bonds C15–H and N–H+ accounts for a significant electrostatic contribution due to the displacement of the net positive charge relative to the Glu-113 counterion. Since the charge separation mechanism is solely based on rotation of the polarized bonds C15–H and N–H+, with only a minor displacement of the C15–N linkage itself relative to the Glu-113 counterion, the predicted molecular rearrangements responsible for storage of electrostatic energy is significantly different from charge separation processes that rely upon displacement of the polyene chain linkage, away from the carboxylate counter ion toward a nonpolar environment (Warshel, 1976; Weiss and Warshel, 1979; Honig et al., 1979; Warshel and Barboy, 1982).
As mentioned in a previous section, the energy storage is largely due to electrostatic contributions, which account for 50% of the total storage. Although the predominant electrostatic contribution to the energy storage results from stretching of the salt bridge formed by the Schiff linkage and the Glu-113 counterion, other polar residues in the binding pocket also have significant contributions and participate in the energy storage mechanism. To analyze the electrostatic contribution of individual residues, the energy storage has been recomputed at the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory after switching off the atomic charges of all residues except for the residue of interest. This analysis of individual contributions has been systematically applied to all residues and bound-water molecules in the system. Fig. 7 shows the quantitative analysis of electrostatic contributions of individual residues to the total energy storage. Note that the predominant electrostatic contributions to the total energy storage result from the interactions between the retinyl chromophore and a few highly conserved polar residues in the rhodopsin binding pocket, including Glu-113, Ser-186, Ala-117, and also Wat2b. Fig. 7 also indicates that the largest electrostatic contribution to the energy storage indeed results from the separation of positive charge from the Glu-113 counterion. In addition, Fig. 7 shows that the electrostatic interactions due to Ala-117, Ser-186, and Wat2b stabilize the product bathorhodopsin relative to reactant 11-cis rhodopsin and therefore reduce the total energy storage. In contrast, the electrostatic interactions due to Glu-113, Cys-187, and Ala-292 are responsible for increasing the total energy storage. These important residues are spatially distributed in the rhodopsin binding pocket as displayed in Fig. 6, where the distribution of negative charge density is highlighted in blue.
FIGURE 7.

Electrostatic contribution of each residue to the total energy storage.
Electronic excitations
Table 3 reports _S_0 → _S_1 vertical electronic-excitation energies, computed at the ONIOM-EE (TD-B3LYP/6-31G*:AMBER) level of theory, for the molecular structures of 11-cis rhodopsin and all-trans bathorhodopsin optimized at the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory. In addition, Table 3 compares the _S_0 → _S_1 vertical electronic-excitation energies computed at the ONIOM-EE (TD-B3LYP//B3LYP/6-31G*:AMBER) level of theory to the corresponding experimental values. Furthermore, Table 3 compares the _S_0 → _S_1 electronic excitation of 11-cis rhodopsin to recently reported QM/MM (CASPT2//CASSCF/6-31G*:AMBER) calculations by Ferre and Olivucci (2003). It is shown that the ONIOM-EE (TD-B3LYP//B3LYP/6-31G*:AMBER) level of theory predicts vertical excitation energies 59.4 kcal/mol and 59.0 kcal/mol for rhodopsin and bathorhodopsin, respectively, in very good agreement with the corresponding experimental values. For the sake of completeness, Table 3 also reports the vertical excitation energy for retinal in methanol as obtained in terms of an implicit solvent model at the B3LYP/6-31G* level of theory (Foresman et al., 1996). The predicted opsin shift, defined as the vertical excitation energy shift for the chromophore in solution and inside the protein, is only in good qualitative agreement with experimental measurements probably due to the simplifying approximation inherent in the implicit solvent model. It is important to note that although all of these QM/MM studies have considered the polarization of the reduced system, the self-consistent polarization of the protein environment has been neglected. Considering that electrostatic contributions constitute a significant percentage of the overall chromophore-protein interactions, such an approximation can be partially responsible for the observed 3% and 9% errors observed in the calculations of vertical excitations in rhodopsin and bathorhodopsin, respectively (Houjou et al., 2001). A complete study of the influence of the self-consistent protein polarization on the chromophore electronic excitation energies is the subject of work in progress in our group.
TABLE 3.
Electronic-excitation energies, in kcal/mol, for _S_0 → _S_1 transitions in rhodopsin, bathorhodopsin, and retinal in methanol
| Δ_E_rhod. | Δ_E_batho. | Retinal (in methanol) | |
|---|---|---|---|
| ONIOM-EE (TD-B3LYP/6-31G*:AMBER) | 59.4 | 59.0 | 66.8 |
| QM/MM (CASPT2//CASSCF/6-31G*:AMBER) | 64.1† | ||
| Experimental values | 57.4* | 54.0* | 76.2‡ |
CONCLUSIONS
We have implemented a practical computational method that combines QM/MM(MO:MM) hybrid methods with high-resolution structural data of bovine rhodopsin to investigate the primary photochemical event in vision. The approach is based on two fundamental assumptions, partially validated by the favorable comparison of predicted storage energy with the corresponding calorimetry measurements. First, it is assumed that the 11-cis/all-trans isomerization of the retinyl chromophore in rhodopsin is much faster than the protein relaxation. Second, it is assumed that the bathorhodopsin molecular structure formed in the ground electronic state as a result of photoexcitation and subsequent excited state curve-crossing processes relaxes to the same minimum energy configuration as the system with the retinyl chromophore isomerized along the ground-state MEP with a fixed protein environment.
The analysis of the relaxed molecular structures of rhodopsin and bathorhodopsin have revealed the detailed molecular rearrangements of the retinyl chromophore in the rhodopsin binding pocket environment; the preferential rotational direction of the φ(C11–C12) dihedral angle; and the detailed chromophore-protein interactions responsible for both the initial rotational torque and the energy storage mechanism.
We have predicted that the φ(C11–C12) dihedral angle changes from −11° in the 11-cis isomer to −161° in the all-trans isomer. The preferential sense of rotation of φ(C11–C12) is along the direction of increasingly negative angles, since the rotation in the opposite direction is hindered by the steric Van der Waals interactions between Ala-117 and the polyene chain at the C13 position.
We have shown that the all-trans isomer is formed with a twisted and bent polyene chain, due to spatial constraints in the rhodopsin binding pocket, including out-of-the-plane distortions larger than 20° in the dihedral angles along the C9–C10, C12–C13, C14–C15, and C15–NH+ chromophore bonds. The spatial constraints include the steric interactions of the three methyl-substituent groups of the _β_-ionone ring with the surrounding residues, the steric interaction of Ala-117 with the polyene chain at the C13 position, and the steric constraints beyond the linkage to Lys-296 due to the almost perpendicular orientation of the protein chain with respect to the C11–C12 rotational axis in the retinyl chromophore.
We have shown that the energy storage predicted at the ONIOM-EE (B3LYP/6-31G*:AMBER) level of theory is 34 kcal/mol (i.e., ∼50% of the photon energy), in excellent agreement with experimental measurements (32–35 ± 0.9–2.2 kcal/mol) (Cooper, 1979a,b; Shick et al., 1987). We have shown that 50% of the stored energy is electrostatic, mainly due to stretching of the salt bridge between the protonated Schiff base and the Glu-113 counterion. The remaining 50% of the energy stored is due to the steric constraints of the all-trans isomer in the rhodopsin binding pocket. We have indicated that the salt-bridge stretching mechanism involves solely reorientation of the positively charged polarized bonds due to torsion of the polyene chain at the linkage to Lys-296. Therefore, we have shown that the storage mechanism does not involve any significant displacement of the polyene chain at the Schiff-based linkage relative to the Glu-113 counterion, or redistribution of charges within the chromophore. In addition, we have shown that a hydrogen-bonded water molecule (Wat2b), consistently found by x-ray crystallographic studies, can assist the salt-bridge stretching process by stabilizing the reorientation of positively charged groups.
We have shown that the ONIOM-EE (TD-B3LYP/6-31G*//B3LYP/6-31G*:AMBER) level of theory predicts _S_0 → _S_1 electronic-excitation energies for both the reactant 11-cis rhodopsin and the isomerization product all-trans bathorhodopsin (59.4 kcal/mol and 59.0 kcal/mol, respectively) in very good agreement with the corresponding experimental values (57.4 kcal/mol and 54.0 kcal/mol, respectively). It has also been concluded that due to the importance of electrostatic contributions to the total chromophore-protein interaction, further progress in the description of the system requires addressing the influence of the self-consistent polarization of the protein. The development of a practical approach for including the self-consistent polarization of the protein, at the ONIOM-EE level of theory, is the subject of work in progress in our research group.
We have demonstrated that the predominant electrostatic contributions to the total energy storage result from the interaction of the protonated Schiff-based retinyl chromophore with three surrounding residues, including the Glu-113, Ser-186, and Ala-117, and also the hydrogen-bonded water molecule Wat2b. These findings suggest that selective artificial mutations of any of these four amino acids could have significant implications on the efficiency of the underlying phototransduction mechanism. This observation also indicates that expanding the reduced 54-atom system to include these important polar residues should account for most of the influence of protein polarization on the primary photochemical event.
Acknowledgments
The authors are grateful to Mr. Sabas Abuabara for proofreading the manuscript and to Dr. Thom Vreven for many helpful discussions on the ONIOM method as implemented in Gaussian03.
V.S.B. acknowledges a generous allocation of supercomputer time from the National Energy Research Scientific Computing Center, and financial support from a Research Innovation Award from Research Corporation; a Petroleum Research Fund Award PRF 37789-G6 from the American Chemical Society; a junior faculty award from the F. Warren Hellman Family; a National Science Foundation Career Program Award CHE-0345984; a National Science Foundation Nanoscale Exploratory Research Award ECS-0404191; and startup package funds from the Provost's office at Yale University.
References
- Anarboldi, M., M. G. Motto, K. Tsujimoto, V. Balogh-Nair, and K. Nakanishi. 1979. Hydroretinals and hydrorhodopsins. J. Am. Chem. Soc. 101:7082–7084. [Google Scholar]
- Becker, R., and K. Freedman. 1985. A comprehensive investigation of the mechanism and photophysics of isomerization of a protonated and unprotonated Schiff-base of 11-_cis_-retinal. J. Am. Chem. Soc. 107:1477–1485. [Google Scholar]
- Ben-Nun, M., and T. Martinez. 1998. Electronic energy funnels in _cis_-trans photoisomerization of retinal protonated Schiff base. J. Phys. Chem. A. 102:9607–9617. [Google Scholar]
- Ben-Nun, M., F. Molnar, K. Schulten, and T. Martinez. 2002. The role of intersection topography in bond selectivity of _cis_-trans photoisomerization. Proc. Natl. Acad. Sci. USA. 99:1769–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birge, R. 1981. Photophysics of light transduction in rhodopsin and bacteriorhodopsin. Annu. Rev. Biophys. Bioeng. 10:315–354. [DOI] [PubMed] [Google Scholar]
- Birge, R. 2001. Conformation and orientation of the retinyl chromophore in rhodopsin: a critical evaluation of recent NMR data on the basis of theoretical calculations results in a minimum energy structure consistent with all experimental data. Biochemistry. 40:4201–4204. [DOI] [PubMed] [Google Scholar]
- Birge, R. R., and L. M. Hubbard. 1980. Molecular dynamics of _cis_-trans isomerization in rhodopsin. J. Am. Chem. Soc. 102:2195–2205. [Google Scholar]
- Birge, R. R., and B. E. Knox. 2003. Perspectives on the counterion switch-induced photoactivation of the G-protein-coupled receptor rhodopsin. Proc. Natl. Acad. Sci. USA. 100:9105–9107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomgren, F., and S. Larsson. 2003. Initial step of the photoprocess leading to vision only requires minimal atom displacements in the retinal molecule. Chem. Phys. Lett. 376:704–709. [Google Scholar]
- Buss, V. 2001. Inherent chirality of the retinal chromophore in rhodopsin—a nonempirical theoretical analysis of chiroptical data. Chirality. 13:13–23. [DOI] [PubMed] [Google Scholar]
- Cooper, A. 1979a. Energetics of rhodopsin and isorhodopsin. FEBS Lett. 100:382–384. [DOI] [PubMed] [Google Scholar]
- Cooper, A. 1979b. Energy uptake in the first step of visual excitation. Nature. 282:531–533. [DOI] [PubMed] [Google Scholar]
- Cornell, W. D., P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell, and P. A. Kollman. 1995. A second generation force-field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 117:5179–5197. [Google Scholar]
- Dapprich, S., K. Komaromi, K. Byun, K. Morokuma, and M. Frisch. 1999. A new ONIOM implementation in Gaussian98. I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struct. 1:461–462. [Google Scholar]
- Ebrey, T., and Y. Koutalos. 2001. Vertebrate photoreceptors. Prog. Retin. Eye Res. 20:49–94. [DOI] [PubMed] [Google Scholar]
- Fahmy, K., F. Jäger, M. Beck, T. A. Zvyaga, T. P. Sakmar, and F. Siebert. 1993. Protonation states of membrane-embedded carboxylic-acid groups in rhodopsin and metarhodopsin-II—a Fourier-transform infrared-spectroscopy study of site-directed mutants. Proc. Natl. Acad. Sci. USA. 90:10206–10210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre, N., and M. Olivucci. 2003. Probing the rhodopsin cavity with reduced retinal models at the CASPT2//CASSCF/AMBER level of theory. J. Am. Chem. Soc. 125:6868–6869. [DOI] [PubMed] [Google Scholar]
- Flores, S., and V. Batista. 2004. Model study of coherent-control of the femtosecond primary event of vision. J. Phys. Chem. B. 108:6745–6749. [Google Scholar]
- Foresman, J. B., T. A. Keith, K. B. Wiberg, J. Soonian, and M. J. Frisch. 1996. Solvent effects. V. Influence of cavity shape, truncation of electrostatics, and electron correlation ab initio reaction field calculations. J. Phys. Chem. 100:16098–16104. [Google Scholar]
- Frisch, M. J., G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr., T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, and J. A. Pople. 2003. Gaussian 03, Rev. A.1. Gaussian, Inc., Pittsburgh, PA.
- Fukuda, Y., Y. Shichida, T. Yohizawa, M. Ito, A. Kodama, and K. Tsukida. 1984. Studies on structure and function of rhodopsin by use of cyclopentatrienylidene 11-_cis_-locked-rhodopsin. Biochemistry. 23:5826–5832. [DOI] [PubMed] [Google Scholar]
- Furutani, Y., Y. Shichida, and H. Kandori. 2003. Structural changes of water molecules during the photoactivation processes in bovine rhodopsin. Biochemistry. 42:9619–9625. [DOI] [PubMed] [Google Scholar]
- Garavelli, M., F. Bernardi, M. Robb, and M. Olivucci. 1999. The short-chain acroleiniminium and pentadieniminium cations: towards a model for retinal photoisomerization. A CASSCF/PT2 study. J. Mol. Struct. Theor. Chem. 463:59–64. [Google Scholar]
- Garavelli, M., T. Vreven, P. Celani, F. Bernardi, M. Robb, and M. Olivucci. 1998. Photoisomerization path for a realistic retinal chromophore model: the nonatetraeniminium cation. J. Am. Chem. Soc. 120:1285–1288. [Google Scholar]
- Gether, U., and B. Kobilka. 1998. G-protein-coupled receptors. II. Mechanism of agonist activation. J. Biol. Chem. 273:17979–17982. [DOI] [PubMed] [Google Scholar]
- Goldschmidt, C., M. Ottolenghi, and T. Rosenfeld. 1976. Primary processes in photochemistry of rhodopsin at room-temperature. Nature. 263:169–171. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Luque, R., M. Garavelli, F. Bernardi, M. Merchan, M. Robb, and M. Olivucci. 2000. Computational evidence in favor of a two-state, two-mode model of the retinal chromophore photoisomerization. Proc. Natl. Acad. Sci. USA. 97:9379–9384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, S., and G. Stock. 2000. Quantum-mechanical modeling of the femtosecond isomerization in rhodopsin. J. Phys. Chem. B. 104:1146–1149. [Google Scholar]
- Hamm, H. 2001. How activated receptors couple to G-proteins. Proc. Natl. Acad. Sci. USA. 98:4819–4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig, B. 1978. Light energy transduction in visual pigments and bacteriorhodopsin. Annu. Rev. Phys. Chem. 29:31–57. [Google Scholar]
- Honig, B., T. Ebrey, R. Callender, U. Dinur, and R. Callender. 1979. Photoisomerization, energy storage, and charge separation—model for light energy transduction in visual pigments and bacteriorhodopsin. Proc. Natl. Acad. Sci. USA. 76:2503–2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houjou, H., Y. Inoue, and M. Sakurai. 2001. Study of the opsin shift of bacteriorhodopsin: insight from QM/MM calculations with electronic polarization effects of the protein environment. J. Phys. Chem. B. 105:867–879. [Google Scholar]
- Humbel, S., S. Sieber, and K. Morokuma. 1996. The IMOMO method: integration of different levels of molecular orbital approximations for geometry optimization of large systems. Test for _n_-butane conformation and _S_N2 reaction: RCl+ Cl−. J. Chem. Phys. 16:1959–1967. [Google Scholar]
- Ito, M., Y. Katuta, Y. Imamoto, Y. Shichida, and T. Yoshizawa. 1992. Conformational analysis of the rhodopsin chromophore using bicyclic retinal analogues. Photochem. Photobiol. 56:915–919. [Google Scholar]
- Ji, T., M. Grossmann, and I. Ji. 1998. G protein-coupled receptors. I. Diversity of receptor-ligand interactions. J. Biol. Chem. 273:17299–17302. [DOI] [PubMed] [Google Scholar]
- Kandori, H., Y. Katsuta, M. Ito, and H. Sasabe. 1995. Femtosecond fluorescence study of the rhodopsin chromophore in solution. J. Am. Chem. Soc. 117:2669–2670. [Google Scholar]
- Kandori, H., Y. Shichida, and T. Yoshizawa. 2001. Photoisomerization in rhodopsin. Biochemistry. 66:1197–1209. [DOI] [PubMed] [Google Scholar]
- Kim, J., M. Tauber, and R. Mathies. 2003. Analysis of the mode-specific excited-state energy distribution and wavelength-dependent photoreaction quantum yield in rhodopsin. Biophys. J. 84:2492–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Penna, G., F. Buda, A. Bifone, and H. de Groot. 1998. The transition state in the isomerization of rhodopsin. Chem. Phys. Lett. 294:447–453. [Google Scholar]
- Lin, S. W., M. Groesbeek, I. van der Hoef, P. Verdegem, J. Lugtenburg, and R. A. Mathies. 1998. Vibrational assignment of torsional normal modes of rhodopsin: probing excited-state isomerization dynamics along the reactive C11=C12 torsion coordinate. J. Phys. Chem. B. 102:2787–2806. [Google Scholar]
- Marinissen, M., and J. Gutkind. 2001. G-protein-coupled receptors and signaling networks: emerging paradigms. Trends Pharmacol. Sci. 22:368–376. [DOI] [PubMed] [Google Scholar]
- Maseras, M., and K. Morokuma. 1995. IMOMM—a new integrated ab-initio plus molecular mechanics geometry optimization scheme of equilibrium structures and transition states. J. Comp. Chem. 16:1170–1179. [Google Scholar]
- Meng, E., and H. Bourne. 2001. Receptor activation: what does the rhodopsin structure tell us? Trends Pharmacol. Sci. 22:587–593. [DOI] [PubMed] [Google Scholar]
- Menon, S., M. Han, and T. Sakmar. 2001. Rhodopsin: structural basis of molecular physiology. Physiol. Rev. 81:1659–1688. [DOI] [PubMed] [Google Scholar]
- Molteni, C., I. Frank, and M. Parrinello. 1999. An excited state density functional theory study of the rhodopsin chromophore. J. Am. Chem. Soc. 121:12177–12183. [Google Scholar]
- Okada, T., O. Ernst, K. Palczewski, and K. Hofmann. 2001. Activation of rhodopsin: new insights from structural and biochemical studies. Trends Biochem. Sci. 26:318–324. [DOI] [PubMed] [Google Scholar]
- Okada, T., Y. Yoshinori, M. Silow, J. Navarro, J. Landau, and Y. Schichida. 2002. Functional role of internal water molecules in rhodopsin revealed by x-ray crystallography. Proc. Natl. Acad. Sci. USA. 99:5982–5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palczewski, K., T. Kumasaka, T. Hori, C. Behnke, H. Motoshima, B. Fox, I. Le Trong, D. Teller, T. Okada, R. Stenkamp, M. Yamamoto, and M. Miyano. 2000. Crystal structure of rhodopsin: a G-protein-coupled receptor. Science. 289:739–745. [DOI] [PubMed] [Google Scholar]
- Pan, D., and R. A. Mathies. 2001. Chromophore structure in lumirhodopsin and metarhodopsin I by time-resolved resonance Raman microchip spectroscopy. Biochemistry. 40:7929–7936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponder, J. W. 2001. TINKER, Ver. 3.9. Washington University School of Medicine, St. Louis, MO.
- Rohrig, U., L. Guidoni, and U. Rothlisberger. 2002. Early steps of the intramolecular signal transduction in rhodopsin explored by molecular dynamics simulations. Biochemistry. 41:10799–10809. [DOI] [PubMed] [Google Scholar]
- Rosenfeld, T., B. Honig, M. Ottolenghi, J. Hurley, and T. Ebrey. 1977. _Cis_-trans isomerization in photochemistry of vision. Pure Appl. Chem. 49:341–351. [Google Scholar]
- Saam, J., E. Tajkhorshid, S. Hayashi, and K. Schulten. 2002. Molecular dynamics investigation of primary photoinduced events in the activation of rhodopsin. Biophys. J. 83:3097–3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmar, T., R. Franke, and H. Khorana. 1989. Glutamic acid-113 serves as the retinylidene Schiff-base counterion in bovine rhodopsin. Proc. Natl. Acad. Sci. USA. 86:8309–8313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenlein, R., L. Peteanu, R. Mathies, and C. Shank. 1991. The first step in vision—femtosecond isomerization of rhodopsin. Science. 254:412–415. [DOI] [PubMed] [Google Scholar]
- Shick, G. A., T. M. Cooper, R. A. Holloway, L. P. Murray, and R. R. Birge. 1987. Energy storage in the primary photochemical events of rhodopsin and isorhodopsin. Biochemistry. 26:2556–2562. [DOI] [PubMed] [Google Scholar]
- Stratmann, R. E., G. E. Scuseira, and M. J. Frisch. 1998. An efficient implementation of time-dependent density-functional theory for the calculation of excitation energies of large molecules. J. Chem. Phys. 109:8218–8224. [Google Scholar]
- Sugihara, M., V. Buss, P. Entel, M. Elstner, and T. Frauenheim. 2002a. 11-_cis_-retinal protonated Schiff base: influence of the protein environment on the geometry of the rhodopsin chromophore. Biochemistry. 41:15259–15266. [DOI] [PubMed] [Google Scholar]
- Sugihara, M., P. Entel, and V. Buss. 2002b. A first-principles study of 11-_cis_-retinal: modelling the chromophore-protein interaction in rhodopsin. Phase Trans. 75:11–17. [Google Scholar]
- Svensson, M., S. Humbel, R. Froese, T. Matsubara, S. Sieber, and K. Morokuma. 1996. ONIOM: a multilayered integrated MO + MM method for geometry optimizations and single point energy predictions. A test for Diels-Alder reactions and Pt(Pt)–Bu(3)(2)+ H−2 oxidative addition. J. Phys. Chem. 100:19357–19363. [Google Scholar]
- Tallent, J., E. Hyde, L. Findsen, G. Fox, and R. Birge. 1992. Molecular dynamics of the primary photochemical event in rhodopsin. J. Am. Chem. Soc. 114:1581–1592. [Google Scholar]
- Teller, D., T. Okada, C. Behnke, K. Palczewki, and R. Stenkamp. 2001. Advances in determination of a high-resolution three-dimensional structure of rhodopsin, a model of G-protein-coupled receptors (GPCRs). Biochemistry. 40:7761–7772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreven, T., F. Bernardi, M. Garavelli, M. Olivucci, M. A. Robb, and H. B. Schlegel. 1997. Ab initio photoisomerization dynamics of a simple retinal chromophore model. J. Am. Chem. Soc. 119:12687–12688. [Google Scholar]
- Vreven, T., B. Mennucci, C. daSilva, K. Morokuma, and J. Tomasi. 2001. The ONIOM-PCM method: combining the hybrid molecular orbital method and the polarizable continuum model for solvation. Application to the geometry and properties of a merocyanine in solution. J. Chem. Phys. 115:62–72. [Google Scholar]
- Vreven, T., and K. Morokuma. 2000a. On the application of the IMOMO (integrated molecular orbital plus molecular orbital) method. J. Comp. Chem. 16:1419–1432. [Google Scholar]
- Vreven, T., and K. Morokuma. 2000b. The ONIOM (our own _N_-layered integrated molecular orbital plus molecular mechanics) method for the first singlet excited (_S_1) state photoisomerization path of a retinal protonated Schiff base. J. Chem. Phys. 113:2969–2975. [Google Scholar]
- Vreven, T., and K. Morokuma. 2003. Investigation of the _S_0 → _S_1 excitation in bacteriorhodopsin with the ONIOM(MO:MM) hybrid method. Theor. Chem. Acc. 109:125–132. [Google Scholar]
- Wang, Q., R. Schoenlein, L. Peteanu, R. Mathies, and C. Shank. 1994. Vibrationally coherent photochemistry in the femtosecond primary event of vision. Science. 266:422–424. [DOI] [PubMed] [Google Scholar]
- Warshel, A. 1976. Bicycle-pedal model for first step in vision process. Nature. 260:679–683. [DOI] [PubMed] [Google Scholar]
- Warshel, A., and N. Barboy. 1982. Energy storage and reaction pathways in the first step of the vision process. J. Am. Chem. Soc. 104:1469–1476. [Google Scholar]
- Warshel, A., and Z. Chu. 2001. Nature of the surface crossing process in bacteriorhodopsin: computer simulations of the quantum dynamics of the primary photochemical event. J. Phys. Chem. B. 105:9857–9871. [Google Scholar]
- Warshel, A., and M. Karplus. 1974. Calculation of π –π* excited state conformations and vibronic structure of retinal and related molecules. J. Am. Chem. Soc. 96:5677–5689. [DOI] [PubMed] [Google Scholar]
- Weiss, R., and A. Warshel. 1979. A new view of the dynamics of singlet _cis_-trans photoisomerization. J. Am. Chem. Soc. 101:6131–6133. [Google Scholar]
- Yamada, A., T. Kakitani, S. Yamamoto, and T. Yamato. 2002. A computational study on the stability of the protonated Schiff base of retinal in rhodopsin. Chem. Phys. Lett. 366:670–675. [Google Scholar]
- Yan, E., M. Kazmi, S. De, B. Chang, C. Seibert, E. Marin, R. Mathies, and T. Sakmar. 2002. Function of extracellular loop 2 in rhodopsin: glutamic acid 181 modulates stability and absorption wavelength of metarhodopsin II. Biochemistry. 41:3620–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan, E. C. Y., M. A. Kazmi, Z. Ganim, J. Hou, D. Pan, B. S. W. Chang, T. P. Sakmar, and R. A. Mathies. 2003. Retinal counterion switch in the photoactivation of the G-protein-coupled receptor rhodopsin. Proc. Natl. Acad. Sci. USA. 100:9262–9267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhukovsky, E., and D. Oprian. 1989. Effect of carboxylic-acid side-chains on the absorption maximum of visual pigments. Science. 246:928–930. [DOI] [PubMed] [Google Scholar]