Role of Dok-1 and Dok-2 in Myeloid Homeostasis and Suppression of Leukemia (original) (raw)
Abstract
Dok-1 and Dok-2 are closely related rasGAP-associated docking proteins expressed preferentially in hematopoietic cells. Although they are phosphorylated upon activation of many protein tyrosine kinases (PTKs), including those coupled with cytokine receptors and oncogenic PTKs like Bcr-Abl, their physiological roles are largely unidentified. Here, we generated mice lacking Dok-1 and/or Dok-2, which included the double-deficient mice succumbed to myeloproliferative disease resembling human chronic myelogenous leukemia (CML) and chronic myelomonocytic leukemia. The double-deficient mice displayed medullary and extramedullary hyperplasia of granulocyte/macrophage progenitors with leukemic potential, and their myeloid cells showed hyperproliferation and hypo-apoptosis upon treatment and deprivation of cytokines, respectively. Consistently, the mutant myeloid cells showed enhanced Erk and Akt activation upon cytokine stimulation. Moreover, loss of Dok-1 and/or Dok-2 induced blastic transformation of chronic phase CML-like disease in mice carrying the bcr-abl gene, a cause of CML. These findings demonstrate that Dok-1 and Dok-2 are key negative regulators of cytokine responses and are essential for myeloid homeostasis and suppression of leukemia.
Keywords: hematopoiesis, granulocyte, macrophage, cytokine, bcr-abl
Introduction
Dok-1 was first identified as a major substrate of oncogenic protein tyrosine kinases (1, 2). Among the mammalian Dok family, Dok-1, Dok-2, and Dok-3 are preferentially expressed in hematopoietic cells and share structural similarities characterized by NH2-terminal PH and PTB domains, followed by COOH-terminal SH2 target motifs (3–7). However, Dok-3 appears relatively distant and does not bind p120 rasGAP unlike Dok-1 and Dok-2 (6, 7). More distant Dok family members, Dok-4 and Dok-5, are virtually absent in hematopoietic cells. A variety of growth factors and cytokines induce tyrosine phosphorylation of Dok-1 and Dok-2, providing multiple docking sites for SH2-containing proteins such as Nck, SHP-1, SHIP-1, and p120 rasGAP, an inhibitor of Ras (4, 8–10). Experiments with Dok-1-deficient (Dok-1−/−) mice demonstrated an indispensable role in the negative regulation of Ras and Erk downstream of protein tyrosine kinases (11–13). Although enhanced Ras signaling often leads to cell proliferative disease or malignancy (14), Dok-1−/− mice did not display overt defects in hematopoiesis, suggesting overlap in functions of Dok-1 and its closest family, Dok-2. Although ectopic expression of Dok-2 negatively regulates Erk and T cell development (5, 15), the physiological function of Dok-2 has yet to be revealed. To understand the role of Dok-1 and Dok-2 in vivo, we have generated a null mutation in the mouse dok-2 gene and have crossed Dok-2−/− mice with Dok-1−/− mice.
Materials and Methods
Mice.
Dok-2−/− mice were generated by homologous recombination (see Fig. S1 and Supplemental Materials and Methods below) and crossed with Dok-1−/− mice to generate Dok-1/Dok-2 double-deficient (dKO) animals. Although initial experiments had been performed with the mixed background mice of 129/SvJ and C57BL/6, mice were crossed over eight times to the C57BL/6 background for detailed experiments (see Figs. 2, D–I, and 3). The bcr-abl transgenic line was generated elsewhere (16). Mice were maintained under specific pathogen-free conditions. Experiments and animal care were performed according to institutional guidelines.
Figure 3.

Cell autonomous progression of myeloproliferative disease in the absence of Dok-1 and Dok-2. Hematopoietic cells in lethally irradiated wild-type (Ly5.1 or Ly5.2 allotype) or dKO mice (Ly5.2) were reconstituted with bone marrow cells from 8-wk-old wild-type (Ly5.1 or Ly5.2) or dKO (Ly5.2) mice. Flow cytometry on peripheral blood cells of each recipient was performed at 9 mo after transplantation. The donor-derived myeloid cell compartment is circled in forward scatter (FSC) versus side scatter (SSC) parameters (top). Gr-1 and Mac-1 expression on donor-derived cells in peripheral blood of each recipient was also examined (bottom). Mice are with the C57BL/6 genetic background.
Histology and Immunohistochemistry.
Tissue sections were stained with hematoxylin and eosin. Cytospin preparations and blood smears were stained with Wright and Giemsa solutions (Sigma-Aldrich). For immunohistochemistry, tissue sections were stained with antibodies to Mac-1 (BD Biosciences) or to F4/80 (Serotec). Isotype-matched immunoglobulins were used as controls.
Flow Cytometry.
Mononuclear cells were stained with the following FITC-, PE-, or biotin-conjugated primary antibodies: anti–Gr-1, Mac-1, CD4, CD8, CD45.1/Ly5.1, and CD45.2/Ly5.2 (BD Biosciences). Streptavidin–quantum red conjugate (Sigma-Aldrich) was used for biotin-coupled antibody. Antibody-stained cells were analyzed with FACSCalibur (BD Biosciences).
Colony Formation Assay.
2–2.5 × 104 bone marrow cells or 105 spleen cells were plated in 1.2% αMEM-based methylcellulose medium containing 30% FCS and 1% BSA in the absence or presence of a combination of 100 ng/ml mSCF (PeproTech), 10 ng/ml mIL-3, 10 ng/ml hG-CSF, 2 U/ml hEpo (Kirin Brewery), 100 ng/ml hIL-6 (Tosoh), and 100 U/ml mIL-7 (Toray) in 35-mm plates. The number of colonies over 50 cells was scored as described previously (17).
Bone Marrow–derived Mast Cells (BMMCs) and Bone Marrow–derived Macrophages (BMMφs).
Bone marrow cells from 8-wk-old mice were cultured in RPMI 1640 medium containing 10% FCS and 10 ng/ml mIL-3 (PeproTech) for the first 2 wk, and then 10 ng/ml mSCF was added. More than 95% of the nonadherent cells were confirmed as FcɛRI+ c-Kit+ Sca-1+ after 4 wk of culture, and the BMMCs were used up to an additional 4 wk. Bone marrow cells from 8-wk-old mice were cultured in RPMI 1640 medium containing 10% FCS and 10 ng/ml mM-CSF (PeproTech) for 7 d to establish adherent BMMφs.
Proliferation and Apoptosis Assays.
For proliferation assay, BMMCs were seeded at 105 cells/well in 96-well plates, cultured for 48 h in RPMI 1640 medium containing 10% FCS and mSCF or mIL-3 (PeproTech), and then pulse labeled for 8 h with 0.2 μCi [3H]thymidine/well. BMMφs were plated at 104 cells/well in 96-well plates, cultured in RPMI 1640 medium containing 10% FCS and mM-CSF or mGM-CSF (PeproTech), and then viable cells were stained with MTT (Sigma-Aldrich). Bone marrow cells were seeded at 5 × 104 cells/well in 96-well plates, cultured in RPMI 1640 medium containing 10% FCS in the presence or absence of mSCF or hG-CSF, and then stained with MTT. For apoptosis assay, BMMCs were seeded at 106 cells/well in 24-well plates, cultured in RPMI 1640 medium containing 10% FCS, mSCF, and mIL-3, and then subjected to flow cytometry with an annexin V-FITC apoptosis detection kit (BD Biosciences).
Immunoblotting.
BMMφs were solubilized in the SDS-loading buffer. Proteins in cell lysates were separated by SDS-PAGE, transferred to PVDF membrane, and visualized with the following antibodies: anti-Erk1/Erk2 (Santa Cruz Biotechnology, Inc.), phospho-Erk (p-Erk; New England Biolabs, Inc.), and phospho-Akt (p-Akt) or Akt (Cell Signaling).
Transplantation.
2 × 106 bone marrow cells from the donor were injected into the tail vein of lethally irradiated (9.5 Gy) recipients at 2–4 mo of age. The donor and recipient cells were distinguished by the expression of Ly5.1 and Ly5.2.
Online Supplemental Material.
Fig. S1 shows a generation of _dok-2_–null mice and dok-1 and dok-2 expression in hematopoietic cells. Table S1 shows accumulation of immature myeloid cells and neutrophils/monocytes in dKO spleen and peripheral blood, respectively. Fig. S2 and Table S2 show down-regulation of Dok-1 and Dok-2 in human leukemia cell lines. Tables S3 and S4 show the pathological examination of bcr-abl transgenic mice lacking Dok-1 and/or Dok-2. Fig. S3 shows clonal evolution of immature T cell blasts developed in Bcr-Abl mice lacking Dok-1 and/or Dok-2. Methods regarding these experiments are described in Supplemental Materials and Methods. Figs. S1–S3, Tables S1–S4, and Supplemental Materials and Methods are available at http://www.jem.org/cgi/content/full/jem.20041247/DC1.
Results and Discussion
Dok-1/Dok-2 Double-deficient Mice Succumbed to Myeloproliferative Disease.
Dok-2−/− mice were generated by homologous recombination and crossed with Dok-1−/− mice to generate Dok-1/Dok-2 double-deficient (dKO) animals (Fig. S1, available at http://www.jem.org/cgi/content/full/jem.20041247/DC1). These mice were viable, fertile, and born at the Mendelian frequency. 7 of 13 dKO mice, but not the others, developed myeloproliferative disease at ∼1 yr of age, accompanied by an enlargement of the spleen where immature and mature granulocytes/monocytes, erythroblasts, and atypical myeloid cells were accumulated (Fig. 1, A and B, and Table S1, available at http://www.jem.org/cgi/content/full/jem.20041247/DC1). These atypical cells resemble the myelomonocytic cells in Gfi-1–deficient mice (18). dKO bone marrow or spleen cells showed an increased ratio of immature granulocytic and/or monocytic precursors (Gr-1−/low Mac-1+; Fig. 1 C). Pathological studies of dKO mice also revealed extramedullary hematopoiesis in the spleen, hyperplasia of megakaryocytes and myeloid progenitors at various stages in the bone marrow, and pronounced cellular infiltrations of granulocytes and lymphocytes in several organs (Fig. 1 D and not depicted). The leukocyte number in the dKO peripheral blood was much higher than that of the other mice (Fig. 1 E), and the dKO leukocytes showed enlarged neutrophil (Gr-1high Mac-1+) and monocyte (Gr-1−/low Mac-1+) populations (Fig. 1, C and E, and Table S1). Interestingly, half of the dKO mice with myeloproliferative disease developed histiocytic sarcoma of macrophage origin (Fig. 1 F). These results indicate that the dKO mice developed myeloproliferative disease resembling human chronic myelogenous leukemia (CML) and chronic myelomonocytic leukemia (CMMoL) as well as macrophage tumor. However, it is of note that these dKO mice required several months of latency to develop CML-like disease and that 6 of 13 dKO mice did not succumb to it until ∼1 yr of age, suggesting an involvement of additional genetic changes.
Figure 1.

dKO mice develop a myeloproliferative disease resembling human CML and CMMoL. (A) Massive enlargement of the spleen in dKO mice compared with wild-type controls at 13 mo of age (top). Spleen weights of wild-type (n = 25), Dok-1−/− (n = 10), Dok-2−/− (n = 17), and dKO (n = 12) mice measured at 13–15 mo of age (bottom). The mean value with the SE is demonstrated. (B) Cytospin specimens of splenocytes from dKO mice at 15 mo of age are characteristic of myeloproliferative disease with marked myeloid hyperplasia comprised of granulocytes/monocytes in various stages of differentiation, including atypical myeloid cells (arrowheads). (C) Flow cytometry for Mac-1 and Gr-1 on bone marrow, spleen, or peripheral blood cells from mice at 15 mo of age. (D) Mononuclear cell infiltration into perivascular areas in dKO kidney and liver. Higher magnification of each boxed area (bottom) shows the infiltrating neutrophils (arrowheads). (E) Leukocytes (left) or erythrocytes (middle) in peripheral blood of wild-type (n = 22), Dok-1−/− (n = 6), Dok-2−/− (n = 7), or dKO (n = 12) mice at 13–15 mo of age were enumerated, and mean values with each SE are demonstrated. Differential cell counts were determined (right) from peripheral blood smears of wild-type (○) or dKO (•) mice and are demonstrated with each mean value. Ne, neutrophil; Mo, monocyte; Eo, eosinophil; Ly, lymphocyte. (F) Gross appearance of enlarged dKO kidneys bearing histiocytic sarcoma or wild-type controls for each mouse at 15 mo of age (left). Histological sections of dKO kidney were stained for macrophage markers Mac-1 or F4/80 (right). Positively stained tumor cells surround unstained cells in the glomerulus (G) and uriniferous tubules (U). Statistical significance compared with wild-type was determined with the Student's t test. *, P < 0.01.
As dKO mice developed hyperplasia of myeloid precursors, we evaluated the clonogenic potential of the common myeloid progenitor (CFU-GEMM), granulocyte/macrophage lineage-restricted progenitor (CFU-GM), or B cell progenitor (CFU-preB). Numbers of CFU-preB in the bone marrow were not significantly different among the mice tested at 4 mo of age, and the number of CFU-GEMM in the bone marrow was slightly increased in dKO mice (Fig. 2, A and B). The number of CFU-GM in the bone marrow of Dok-1−/−, Dok-2−/−, and dKO mice was increased as compared with the number in wild-type mice. However, the number of CFU-GM in the spleen of dKO mice was dramatically increased, whereas the number of CFU-GEMM in the spleen varied only slightly (Fig. 2 C). These results indicate that the neutrophilic and monocytic hyperplasia observed in dKO mice, at least in part, is due to hyperplasia of CFU-GM in the bone marrow and subsequent extramedullary myelopoiesis in the spleen. Given that dKO mice showed hyperplasia of erythroblasts and megakaryocytes, Dok-1 and Dok-2 may also have an inhibitory function in the generation of their precursor cells.
Figure 2.
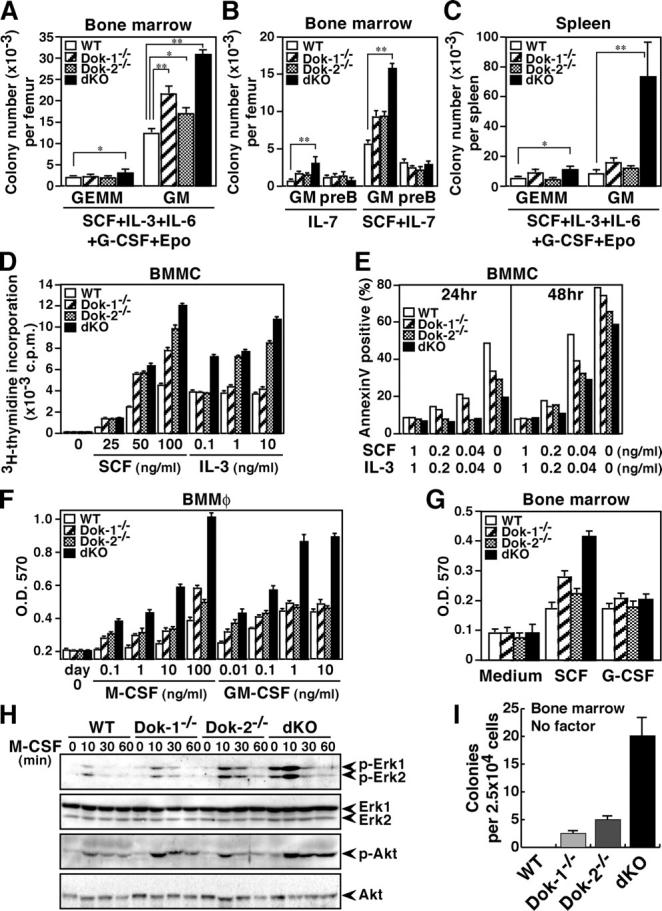
Hyperplasia of granulocyte/macrophage progenitors in dKO mice and hyperactivation of their myeloid cells upon stimulation with cytokines. (A–C) Colony formation assays for CFU-GEMM (GEMM), CFU-GM (GM), or CFU-preB (preB) were performed with bone marrow or spleen cells of the mice at 4 mo of age in the presence of the indicated cytokine(s). Statistical significance compared with wild-type was determined with the Student's t test. *, P < 0.01; **, P < 0.001. (D and E) Proliferation and apoptosis of BMMCs were evaluated based on [3H]thymidine incorporation and annexin V binding, respectively, in the presence or absence of the indicated cytokine(s). Apoptotic rates were determined after the indicated culture period. (F) The proliferation of BMMφs was evaluated with the MTT assay after 5 d of culture in the presence of the indicated cytokine. (G) The proliferation of bone marrow cells from mice at 8 wk of age was evaluated with the MTT assay after 4 d of culture in the absence or presence (10 ng/ml) of the indicated cytokine. (A–G) Mean values and SEs are from triplicate experiments. (H) Activated Erk (p-Erk1/2), Akt (p-Akt), total Erk1/2, or Akt was examined with immunoblotting upon M-CSF treatment (10 ng/ml) of BMMφs. The position of each protein and period of stimulation are indicated. (I) Colony formation assay was performed with bone marrow cells of mice at 8 wk of age in the absence of cytokine. Mean values and SEs are from triplicate experiments.
Myeloid Cells Lacking Dok-1 and Dok-2 Are Hyperresponsive to Cytokines.
To elucidate how the myeloproliferative disease developed in dKO mice, we examined growth or survival responses of BMMCs or BMMφs from wild-type, Dok-1−/−, Dok-2−/−, or dKO mice at 8 wk of age upon stimulation with cytokines involved in myelopoiesis. Note that these mutant mice appeared normal in hematopoiesis at this age. All mutant BMMCs showed a greater proliferative response than the wild-type cells upon SCF stimulation, whereas only dKO BMMCs were hyperproliferative to the lower dose of IL-3 (Fig. 2 D). Moreover, all mutant BMMCs displayed attenuated apoptosis induced by a reduction or deprivation of SCF and IL-3 (Fig. 2 E). In any case, dKO BMMCs showed the best proliferative or antiapoptotic response to the cytokines. Although Dok-1−/− or Dok-2−/− BMMφs displayed a significant increase of proliferation upon M-CSF treatment or the lower dose of GM-CSF treatment, dKO BMMφs always showed the greatest response, especially to the higher dose of each cytokine (Fig. 2 F). Finally, we examined the growth response upon G-CSF stimulation of primary bone marrow cells, as neither BMMCs nor BMMφs react to this cytokine, and found that bone marrow cells lacking Dok-1 and/or Dok-2 show normal growth to G-CSF, but elevated growth to SCF (Fig. 2 G). Together, our findings indicate that Dok-1 and Dok-2 work cooperatively in the negative regulation of these cytokine signaling except G-CSF. As we confirmed their expression in premature and mature myeloid cells (Fig. S1), this cooperative function likely explains why dKO mice, but not Dok-1−/− or Dok-2−/− mice, displayed aberrant myelopoiesis, macrophage tumors, and myeloid leukemia. However, of note is that these cytokines can induce CFU-GEMM as well as CFU-GM in vitro, suggesting that Dok-1 and Dok-2 have limited inhibitory functions in CFU-GEMM and/or that these cytokines have limited effects to it in vivo.
M-CSF stimulation of monocytic cells triggers Ras/Erk and PI-3K/Akt pathways facilitating cellular proliferation and survival, and it also induces tyrosine phosphorylation of Dok-1 and Dok-2 (8, 9). Thus, we evaluated M-CSF–induced Erk and Akt activation in Dok-1−/−, Dok-2−/−, or dKO BMMφs (Fig. 2 H). Although cells lacking Dok-1 or Dok-2 showed enhanced activation of both Erk and Akt, a more vigorous activation was obvious in the dKO macrophages, indicating that Dok-1 and Dok-2 cooperatively suppress Erk and Akt upon cytokine stimulation. Further, the dKO macrophages showed cytokine-independent activation of Erk. Therefore, we addressed the cytokine-independent clonogenicity of bone marrow cells from 8-wk-old mice and found that dKO bone marrow cells formed ∼20 colonies of >100 cells and Dok-1−/− or Dok-2−/− cells formed a few colonies of <100 cells, whereas wild-type cells formed no colony (Fig. 2 I). These results indicate that dKO progenitors gained enhanced growth capability resulting in their cytokine-independent hyper-clonogenicity.
Myeloproliferative Disease in dKO Mice Is Cell Autonomous.
To test whether myeloproliferative disease in dKO mice is cell autonomous, bone marrow cells from 8-wk-old Ly5.2+ dKO mice were transferred into irradiated Ly5.1+ wild-type mice. 9 mo after the transplantation, 7 of 10 recipients reconstituted with dKO cells showed hyperplasia of circulating neutrophils (Gr-1high Mac-1+; Fig. 3); however, none of the recipients reconstituted with Ly5.2+ or Ly5.1+ wild-type cells showed such abnormality, regardless of the dok-1 and dok-2 mutations. Although the transmitted abnormality is weaker than that seen in dKO mice with mixed genetic background (Fig. 1 C), it is probably because the transplantation was performed with C57BL/6 background, which is relatively resistant to _bcr-abl_–induced leukemogenesis (19). Together, these results indicate that the myeloproliferative disease in dKO mice is cell autonomous.
Dok-1 and Dok-2 Suppress Blastic Transformation of CML-like Disease.
Human CML is caused by Bcr-Abl, which phosphorylates Dok-1 and Dok-2 in hematopoietic stem/progenitor cells of patients with CML (1, 3). As the disease progresses, lethal blastic transformation into acute leukemia or lymphoma, termed blast crisis, occurs. Recently, the dok-2 gene was identified as 1 of the 21 most down-regulated genes in the bone marrow cells of patients with CML at blast crisis among 5,315 genes analyzed (20). In addition, we found that four of eight leukemia cell lines established from such patients express undetectable or marginal levels of Dok-1 and/or Dok-2, suggesting their negative roles in the blastic transformation (Fig. S2 and Table S2). However, it was difficult to examine the role of these adaptors in the blastic transformation because Bcr-Abl–induced CML-like disease in mice usually displayed no chronic phase (12, 19). Exceptionally, a tec promoter-p210_bcr-abl_transgenic line (Bcr-Abl mice, hereafter) routinely developed chronic phase CML-like disease at ∼6–8 mo after birth, and the chronic phase lasted at least until 1 yr of age (16). Therefore, we prepared Bcr-Abl mice lacking Dok-1 and/or Dok-2 to elucidate the roles of Dok-1 and Dok-2 in the clinical progression of CML (Table S3). 5 of the 30 Bcr-Abl mice lacking Dok-1 (TKH), Dok-2 (THK), or both (TKK) died at 4–5 mo after birth, and most of the others died at 6–9 mo after birth, whereas very few of the controls died (Fig. 4 A). Although loss of either adaptor induced early lethality of Bcr-Abl mice, this could be due to haploinsufficiency of each gene to replace the other's function because these Bcr-Abl mice lacking Dok-1 or Dok-2 had only one allele of wild-type dok-2 or dok-1 gene, respectively. Alternatively, Dok-1 and Dok-2 functions may not be mutually replaceable in this respect. Pathological examination of dead or moribund mice revealed that some of the THK, TKH, and TKK mice developed a severe enlargement of the thymus and lymph nodes with a massive infiltration of lymphoblasts at 4–5 mo of age (Fig. 4, B and C, and Table S4). The infiltrated tumor cells were composed of CD4/CD8 double-positive immature T lymphoblasts, which also comprised a large part of the peripheral blood cells, indicating severe blastic transformation (Fig. 4 D and Table S4). Southern blot analysis further revealed that these tumors carried rearrangements in the T cell receptor γ locus, indicating that the blast cells were clonal in origin (Fig. S3). At 7–8 mo of age, many of the THK, TKH, and TKK mice, but not the controls, were moribund or dead. They showed a massive infiltration of lymphoblastoid cells in the gastrointestinal tracts in addition to aggravated CML-like symptoms (Fig. 4 E and Table S4). The infiltrates showed a monotonous morphology and the pattern was diffuse in many parts, accompanied with several foci of lymphoepithelial lesions, indicating blastic transformation into gastrointestinal lymphoma. Together, these results demonstrate that Dok-1 and Dok-2 are essential to suppress the blastic transformation of the Bcr-Abl–induced CML-like disease. However, it is of note that in case of patients with CML, blast crisis mostly results in myeloid or B cell leukemia/lymphoma, usually not in the T cell variety. That Bcr-Abl mice carrying a p53 mutation also developed T cell lymphoma suggests involvement of genetic background (21).
Figure 4.
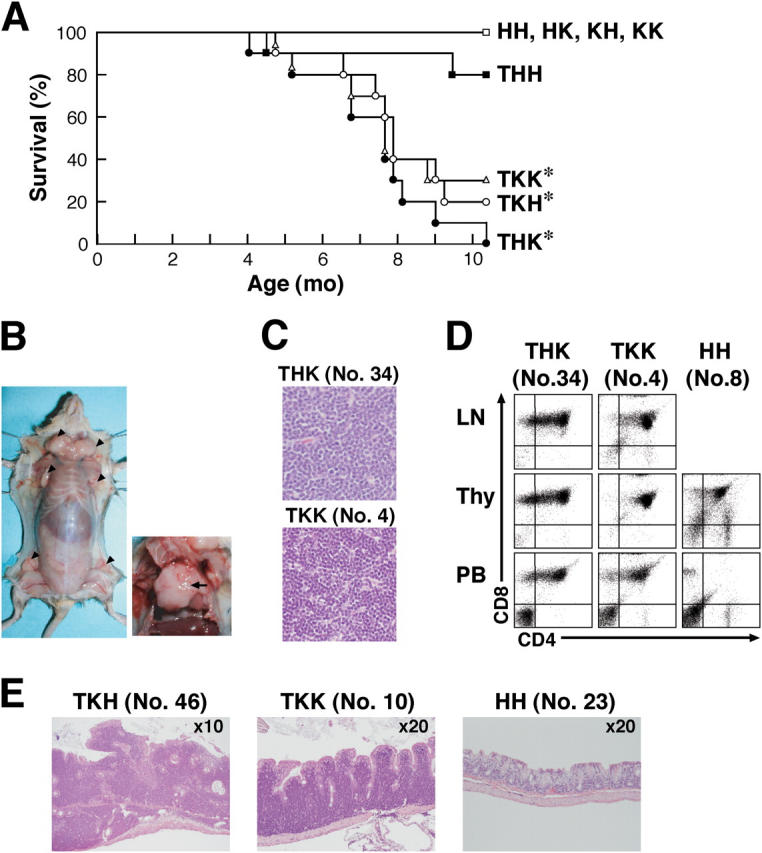
Dok-1 and Dok-2 suppress the blastic transformation of the Bcr-Abl–induced CML-like disease. (A) Survival curves for Bcr-Abl mice lacking Dok-1 and/or Dok-2 and the nontransgenic controls. HH, HK, KH, and KK stand for mice with the dok-1 +/−:dok-2 +/−, dok-1 +/−: dok-2 −/−, dok-1 −/−: dok-2 +/−, and dok-1 −/−: dok-2 −/− loci, respectively, and T stands for mice carrying the bcr-abl transgene. For each genotype, 10 mice were monitored. Log rank statistical analysis was performed (*, P < 0.03, compared with the THH mice). (B) Tumors that developed in the lymph nodes (left, arrowheads) and thymus (right, arrow) of a TKK mouse (No. 4). (C) Lymphoblasts in tumors developed in lymph nodes of mice. (D) Surface expression of CD4 and CD8 on mononuclear cells in lymph nodes (LN), thymus (Thy), or peripheral blood (PB) of mice at 4 mo of age examined with flow cytometry. The HH mice were analyzed as an age-matched control. (E) Lymphoblasts in gastrointestinal lymphomas developed in large intestines of mice at 7 mo of age. The HH mouse was analyzed as an age-matched control.
Here, we reported that Dok-1 and Dok-2 inhibit cytokine-mediated proliferative and antiapoptotic signaling in myeloid cells and oppose myeloid leukemogenesis and blastic transformation of CML-like disease in mice. That loss of Dok-1 and/or Dok-2 induced the blastic transformation of Bcr-Abl–induced CML-like disease into lymphoma suggests their roles in hematopoietic stem cells, where the tec gene promoter, which controls the bcr-abl transgene, is preferentially active (16). Moreover, as dKO mice developed myeloproliferative disease in the absence of Bcr-Abl, Dok-1 and Dok-2 may oppose a wide range of myeloid leukemogenesis in humans. Consistently, undetectable or marginal levels of their expression was observed in about half of the leukemic cell lines established from patients with myeloid leukemia, regardless of whether it is CML or not (Fig. S2 and Table S2). Further investigation of the tumor suppressive function of Dok-1 and Dok-2 in human malignancies, especially myeloid leukemias including CML and CMMoL, might lead to an understanding of the molecular mechanisms of such diseases and contribute to designing effective therapies against them.
Acknowledgments
We thank Y. Matsuo and K. Orita for leukemia cell lines; H. Honda for TCR probe; R. Horai, K. Nakamura, N. Suzuki, and H. Suzuki for help in generating dok-2+/− mice; T. Usami, T. Tsubata, R. Miura, and C. Kai for animal care; S. Tronick for antibodies; and K. Tani, Y. Morishita, S. Mori, M. Kurokawa, T. Suzuki, S. Ogawa, and T. Akiyama for discussions.
This work was supported by Grants-in-Aid for Scientific research from the Ministry of Education, Culture, Sports, Science and Technology and by grants from the Naito, Kato, Uehara, Sagawa, Japan Leukemia Research, the Tokyo Biochemical Research, and the Yamanouchi Foundations.
The authors have no conflicting financial interests.
T. Yasuda and M. Shirakata contributed equally to this work.
References
- 1.Carpino, N., D. Wisniewski, A. Strife, D. Marshak, R. Kobayashi, B. Stillman, and B. Clarkson. 1997. p62dok: a constitutively tyrosine-phosphorylated, GAP-associated protein in chronic myelogenous leukemia progenitor cells. Cell. 88:197–204. [DOI] [PubMed] [Google Scholar]
- 2.Yamanashi, Y., and D. Baltimore. 1997. Identification of the Abl- and rasGAP-associated 62 kDa protein as a docking protein, Dok. Cell. 88:205–211. [DOI] [PubMed] [Google Scholar]
- 3.Di Cristofano, A., N. Carpino, N. Dunant, G. Friedland, R. Kobayashi, A. Strife, D. Wisniewski, B. Clarkson, P.P. Pandolfi, and M.D. Resh. 1998. Molecular cloning and characterization of p56dok-2defines a new family of RasGAP-binding proteins. J. Biol. Chem. 273:4827–4830. [DOI] [PubMed] [Google Scholar]
- 4.Jones, N., and D.J. Dumont. 1998. The Tek/Tie2 receptor signals through a novel Dok-related docking protein, Dok-R. Oncogene. 17:1097–1108. [DOI] [PubMed] [Google Scholar]
- 5.Nelms, K., A.L. Snow, J. Hu-Li, and W.E. Paul. 1998. FRIP, a hematopoietic cell-specific rasGAP-interacting protein phosphorylated in response to cytokine stimulation. Immunity. 9:13–24. [DOI] [PubMed] [Google Scholar]
- 6.Cong, F., B. Yuan, and S.P. Goff. 1999. Characterization of a novel member of the DOK family that binds and modulates Abl signaling. Mol. Cell. Biol. 19:8314–8325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lemay, S., D. Davidson, S. Latour, and A. Veillette. 2000. Dok-3, a novel adaptor molecule involved in the negative regulation of immunoreceptor signaling. Mol. Cell. Biol. 20:2743–2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Berg, K.L., K.A. Siminovitch, and E.R. Stanley. 1999. SHP-1 regulation of p62DOK tyrosine phosphorylation in macrophages. J. Biol. Chem. 274:35855–35865. [DOI] [PubMed] [Google Scholar]
- 9.Suzu, S., M. Tanaka-Douzono, K. Nomaguchi, M. Yamada, H. Hayasawa, F. Kimura, and K. Motoyoshi. 2000. p56dok-2as a cytokine-inducible inhibitor of cell proliferation and signal transduction. EMBO J. 19:5114–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tamir, I., J.C. Stolpa, C.D. Helgason, K. Nakamura, P. Bruhns, M. Daeron, and J.C. Cambier. 2000. The RasGAP-binding protein p62dok is a mediator of inhibitory FcγRIIB signals in B cells. Immunity. 12:347–358. [DOI] [PubMed] [Google Scholar]
- 11.Yamanashi, Y., T. Tamura, T. Kanamori, H. Yamane, H. Nariuchi, T. Yamamoto, and D. Baltimore. 2000. Role of the rasGAP-associated docking protein p62dokin negative regulation of B cell receptor-mediated signaling. Genes Dev. 14:11–16. [PMC free article] [PubMed] [Google Scholar]
- 12.Di Cristofano, A., M. Niki, M. Zhao, F.G. Karnell, B. Clarkson, W.S. Pear, L.V. Aelst, and P.P. Pandolfi. 2001. p62dok, a negative regulator of Ras and mitogen-activated protein kinase (MAPK) activity, opposes leukemogenesis by p210bcr-abl. J. Exp. Med. 194:275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhao, M., A.A.P. Schmitz, Y. Qin, A.D. Cristofano, P.P. Pandolfi, and L.V. Aelst. 2001. Phosphoinositide 3-kinase–dependent membrane recruitment of p62dok is essential for its negative effect on mitogen-activated protein (MAP) kinase activation. J. Exp. Med. 194:265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rodenhuis, S. 1992. ras and human tumors. Semin. Cancer Biol. 3:241–247. [PubMed] [Google Scholar]
- 15.Gugasyan, R., C. Quilici, S.T.T. I, D. Grail, A.M. Verhagen, A. Roberts, T. Kitamura, A.R. Dunn, and P. Lock. 2002. Dok-related protein negatively regulates T cell development via its RasGTPase-activating protein and Nck docking sites. J. Cell Biol. 158:115–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Honda, H., H. Oda, T. Suzuki, T. Takahashi, O.N. Witte, K. Ozawa, T. Ishikawa, Y. Yazaki, and H. Hirai. 1998. Development of acute lymphoblastic leukemia and myeloproliferative disorder in transgenic mice expressing p210bcr/abl: a novel transgenic model for human Ph1-positive leukemias. Blood. 91:2067–2075. [PubMed] [Google Scholar]
- 17.Yang, F.-C., S. Watanabe, K. Tsuji, M. Xu, A. Kaneko, Y. Ebihara, and T. Nakahata. 1998. Human granulocyte colony-stimulating factor (G-CSF) stimulates the in vitro and in vivo development but not commitment of primitive multipotential progenitors from transgenic mice expressing the human G-CSF receptor. Blood. 92:4632–4640. [PubMed] [Google Scholar]
- 18.Hock, H., M.J. Hamblen, H.M. Rooke, D. Traver, R.T. Bronson, S. Cameron, and S.H. Orkin. 2003. Intrinsic requirement for zinc finger transcription factor Gfi-1 in neutrophil differentiation. Immunity. 18:109–120. [DOI] [PubMed] [Google Scholar]
- 19.Wong, S., and O.N. Witte. 2001. Modeling Philadelphia chromosome positive leukemias. Oncogene. 20:5644–5659. [DOI] [PubMed] [Google Scholar]
- 20.Nowicki, M.O., P. Pawlowski, T. Fischer, G. Hess, T. Pawlowski, and T. Skorski. 2003. Chronic myelogenous leukemia molecular signature. Oncogene. 22:3952–3963. [DOI] [PubMed] [Google Scholar]
- 21.Honda, H., T. Ushijima, K. Wakazono, H. Oda, Y. Tanaka, S. Aizawa, T. Ishikawa, Y. Yazaki, and H. Hirai. 2000. Acquired loss of p53 induces blastic transformation in p210bcr/abl-expressing hematopoietic cells: a transgenic study for blast crisis of human CML. Blood. 95:1144–1150. [PubMed] [Google Scholar]