Spinal G-Protein-Gated Potassium Channels Contribute in a Dose-Dependent Manner to the Analgesic Effect of μ- and δ- But Not κ-Opioids (original) (raw)
Abstract
Opioids can evoke analgesia by inhibiting neuronal targets in either the brain or spinal cord, and multiple presynaptic and postsynaptic inhibitory mechanisms have been implicated. The relative significance of presynaptic and postsynaptic inhibition to opioid analgesia is essentially unknown, as are the identities and relevant locations of effectors mediating opioid actions. Here, we examined the distribution of G-protein-gated potassium (GIRK) channels in the mouse spinal cord and measured their contribution to the analgesia evoked by spinal administration of opioid receptor-selective agonists. We found that the GIRK channel subunits GIRK1 and GIRK2 were concentrated in the outer layer of the substantia gelatinosa of the dorsal horn. GIRK1 and GIRK2 were found almost exclusively in postsynaptic membranes of putative excitatory synapses, and a significant degree of overlap with the μ-opioid receptor was observed. Although most GIRK subunit labeling was perisynaptic or extrasynaptic, GIRK2 was found occasionally within the synaptic specialization. Genetic ablation or pharmacologic inhibition of spinal GIRK channels selectively blunted the analgesic effect of high but not lower doses of the μ-opioid receptor-selective agonist [d-Ala(2),_N_-Me-Phe(4),Gly(5)-ol]-enkephalin. Dose-dependent contributions of GIRK channels to the analgesic effects of the δ-opioid receptor-selective agonists Tyr-d-Ala-Phe-Glu-Val-Val-Gly amide and [d-Pen(2,5)]-enkephalin were also observed. In contrast, the analgesic effect of the κ agonist (trans)-3,4-dichloro-_N_-methyl-_N_-[2-(1-pyrrolidinyl)-cyclohexyl] benzeneacetamide methanesulfonate hydrate was preserved despite the absence of GIRK channels. We conclude that the activation of postsynaptic GIRK1 and/or GIRK2-containing channels in the spinal cord dorsal horn represents a powerful, albeit relatively insensitive, means by which intrathecal μ- and δ-selective opioid agonists evoke analgesia.
Keywords: knock-out, opioid, tail-flick, Kir3, pain, nociception
Introduction
Opioids modulate nociception by stimulating G-protein-coupled receptors (Kieffer and Gaveriaux-Ruff, 2002). μ-, δ-, and κ-opioid receptors (MORs, DORs, and KORs, respectively) are found spinally and supraspinally, and stimulation of each can elicit analgesia (Yaksh, 1997). Multiple presynaptic and postsynaptic mechanisms have been implicated in opioid analgesia, including voltage-gated Ca2+ channel inhibition, voltage-gated K+ channel activation, and G-protein-gated inwardly rectifying K+ (GIRK) channel activation (Yoshimura and North, 1983; Chang et al., 1989; Vaughan et al., 1997; Yaksh, 1997). The relative significance of presynaptic and postsynaptic inhibition to opioid analgesia is essentially unknown.
GIRK channels are homotetrameric and heterotetrameric complexes of GIRK/Kir3 subunits that contribute to the inhibitory actions of opioids and other neurotransmitters (Krapivinsky et al., 1995a,b; Luscher et al., 1997; Slesinger et al., 1997; Torrecilla et al., 2002; Beckstead et al., 2004; Cruz et al., 2004). Four GIRK subunits (GIRK1-GIRK4) have been identified, and all but GIRK4 are found throughout the CNS (Karschin et al., 1996). GIRK1-GIRK3 are expressed in supraspinal targets of opioids, including the locus ceruleus (LC), periaqueductal gray, and ventral tegmental area (Karschin et al., 1996). GIRK1 and GIRK2 are enriched in the spinal cord dorsal horn, a key spinal target of opioids (Marker et al., 2004).
Several lines of evidence argue that GIRK channels contribute to opioid analgesia. First, the coupling of GIRK channels to MORs, DORs, and KORs has been demonstrated in vitro (Henry et al., 1995; Ikeda et al., 1995; Kobayashi et al., 1996). Second, MOR-, DOR-, and KOR-activated K+ currents exhibiting GIRK-like characteristics have been studied in many neuron populations, including the substantia gelatinosa (North, 1989; Grudt and Williams, 1993; Schneider et al., 1998). Third, opioid inhibition was absent in LC neurons from GIRK knock-out mice (Torrecilla et al., 2002). Finally, mice with Girk gene mutations displayed blunted responses to systemic administration of the MOR-preferring agonist morphine (Ikeda et al., 2000; Marker et al., 2002; Mitrovic et al., 2003). Because systemic morphine modulates spinal and supraspinal targets with synergistic consequences (Yeung and Rudy, 1980), these studies offered little mechanistic insight into the role of GIRK channels in opioid analgesia.
We recently exploited the spatial resolution of intrathecal injections to characterize the contribution of local GIRK channels to spinal morphine analgesia. We found that the analgesic effect of a high nontoxic dose of intrathecal morphine was selectively blunted in mouse models of GIRK channel ablation (Marker et al., 2004). Because morphine at high concentrations can activate DORs and KORs (Takemori et al., 1986), the dose-dependent contribution of GIRK channels to spinal morphine analgesia could reflect preferential coupling of GIRK channels to DORs and/or KORs. In this study, we measured the analgesic effects of selective opioid agonists in pharmacologic and genetic models of GIRK channel ablation. We also characterized the distributions of GIRK subunits and MORs in the dorsal horn. Our data show that spinal GIRK channels represent a powerful, although insensitive, class of postsynaptic effector used by spinal MORs and DORs to evoke analgesia.
Materials and Methods
Drugs. [d-Ala(2),_N_-Me-Phe(4),Gly(5)-ol]-enkephalin (DAMGO), Tyr-d-Ala-Phe-Glu-Val-Val-Gly amide (deltorphin II), [d-Pen(2,5)]-enkephalin (DPDPE), and (trans)-3,4-dichloro-_N_-methyl-_N_-[2-(1-pyrrolidinyl)-cyclohexyl] benzeneacetamide methanesulfonate hydrate (U50488) were purchased from Sigma (St. Louis, MO). rTertiapin-Q was obtained from Alomone Labs (Jerusalem, Israel). All drugs were dissolved in 0.9% saline in high-concentration stocks, divided into single-use aliquots, and stored at -20°C.
Behavioral subjects. All animal use was reviewed and approved by the Institutional Animal Care and Use Committee of the University of Minnesota. Efforts were made to minimize the pain and discomfort of the animals throughout the study. Mice were housed on a 12 h light/dark cycle, with food and water available ad libitum. The generation of GIRK1 knock-out and GIRK2 knock-out mice was described previously (Signorini et al., 1997; Bettahi et al., 2002). The GIRK null mutations were backcrossed through at least nine rounds against the C57BL/6 mouse strain before initiating this study. Genotypes were determined by PCR of crude DNA samples from tail biopsies taken at weaning (3 weeks of age). Both male and female GIRK knock-out mice and wild-type controls were evaluated in the tail-flick test. For tertiapin studies, male wild-type C57BL/6 mice were purchased from The Jackson Laboratory (Bar Harbor, ME). The generation of MOR knock-out mice was described previously (Roy et al., 1998). Breeding pairs consisting of MOR knock-out or strain-matched (C57/129) wild-type mice were established to generate litters of MOR knock-out mice and wild-type controls for behavioral testing. All subjects were tested between 6 and 7 weeks of age and evaluated only once in the tail-flick test.
Tail-flick testing. Thermal nociception was assessed using the warm-water immersion tail-flick assay, as described previously (Marker et al., 2004). Briefly, tails were immersed in a sequential manner in water baths set at 49, 52.5, and 55°C and wiped dry between dips. After baseline latencies were determined for each animal, the mice were given intrathecal injections of saline, drug, or a drug/tertiapin combination, as described previously (Hylden and Wilcox, 1980). Postdrug latencies were determined 10 min after injection. Cutoff values were set at 20, 12, and 10 s, respectively, for the 49, 52.5, and 55°C bath exposures to prevent tissue damage. The investigator was blind to drug status and genotype throughout the experiment. Water bath temperatures were measured daily using a Traceable RTD platinum thermometer (Control Company, Friendswood, TX) and adjusted as necessary to ensure consistency throughout the study. The highest doses of DAMGO, deltorphin II, DPDPE, and U50488 administered were chosen on the basis of pilot studies, in which the intent was to identify the drug dose that produced the maximum analgesic effect without causing toxicity or motor impairment. The optimal dose of intrathecal tertiapin (30 pmol) was determined previously (Marker et al., 2004).
Immunohistochemistry. Conditions for immunohistochemical analysis were as described previously (Marker et al., 2004). Briefly, adult mice were anesthetized and subjected to transcardial perfusion with a 4% paraformaldehyde-based solution. Spinal cords were extracted, and 10 μm sections were made by cryostat. All antibodies were diluted in PBS containing 0.1% Triton X-100. Primary antibodies were applied to the section and incubated overnight at 4°C. The next morning, sections were washed with PBS (three times for 15 min each) and incubated for 1 h at room temperature with the appropriate donkey secondary antibodies conjugated to cyanine 2 (Cy2) (1:200), Cy3 (1:500), or Cy5 (1:100) for immunofluorescence detection (Jackson ImmunoResearch, West Grove, PA). Sections were then washed with PBS, dehydrated, cleared, and coverslipped using dimeric paraxylene (Fluka, Ronkonkoma, NY). Anti-GIRK1 (Chemicon, Temecula, CA; Alomone Labs) and anti-GIRK2 (Alomone Labs) antibodies were used at dilutions of 1:200. The anti-MOR (Chemicon) antibody was used at a dilution of 1:500. The anticalcitonin gene-related peptide (CGRP) (US Biological, Swampscott, MA) antibody was used at a dilution of 1:200, whereas the anti-PKCγ (Fitzgerald Industries International, Concord, MA) antibody was used at a dilution of 1:25. Images were collected using a Bio-Rad (Hercules, CA) MRC 1024 confocal laser head on an upright microscope and an Olympus (Tokyo, Japan) AX70 camera with Bio-Rad LaserSharp software version 3.0. Adobe Photoshop version 6.0 (Adobe Systems, San Jose, CA) was used to add color to the images.
Immunoelectron microscopy. Procedures for the isolation of spinal cord tissue from adult wild-type, GIRK1 knock-out, and GIRK2 knock-out mice for use in immunoelectron microscopy experiments were similar to those described above and previously (Marker et al., 2004), with one noteworthy modification. The 4% paraformaldehyde solution was supplemented with 0.05% glutaraldehyde immediately before perfusion. After the perfusion, spinal cord tissue was extracted, rinsed thoroughly in 0.1 m phosphate buffer (PB), and embedded in 4% agarose before sectioning.
The preembedding immunogold method was described previously (Lujan et al., 1996). Briefly, free-floating sections were incubated in 10% normal goat serum (NGS) diluted in Tris-buffered saline (TBS) for 1 h at room temperature. Sections were then incubated for 48 h at 4°C with primary antibody at final concentrations of 1-2 μg/ml each, diluted in TBS/1% NGS. When a single primary antibody was used, it was visualized by the silver-intensified immunogold reaction. For colabeling experiments, MOR immunoreactivity was visualized by the immunoperoxidase reaction, and GIRK subunit immunoreactivity was revealed with the silver-intensified immunogold reaction. Sections were incubated for 3 h at room temperature with goat anti-rabbit and/or anti-guinea pig Fab fragments coupled to 1.4 nm gold (Nanoprobes, Stony Brook, NY), or biotinylated goat anti-rabbit and/or anti-guinea pig Fab fragments (Vector Laboratories, Burlingame, CA). All secondary antibodies were diluted 1:100 in TBS/1% NGS. Sections were then washed with TBS and double-distilled water, followed by silver enhancement of the gold particles with an HQ Silver kit (Nanoprobes) for 8-10 min. Sections were then incubated for 4 h with the ABC complex (Vector Laboratories) and washed in TBS. Peroxidase was visualized with DAB (0.05% in TBS, pH 7.4) using 0.01% H2O2 as a substrate for 5-10 min. The sections were washed in PBS and postfixed with OsO4 (1% in 0.1 m PB), followed by block staining with uranyl acetate, dehydration in graded series of ethanol solutions, and flat embedding on glass slides in Durcupan resin (Fluka, Buchs, Switzerland). Regions of interest were cut at 70-90 nm using a Reichert Ultracut E ultramicrotome (Leica, Vienna, Austria). Ultrathin sections were mounted on 200 mesh nickel grids, and counterstaining was performed on drops of 1% aqueous uranyl acetate, followed by Reynolds's lead citrate. Unless otherwise stated, electron microscopic samples were obtained from three different mouse spinal cords, and three blocks of each animal were cut for electron microscopy.
For postembedding immunogold experiments, blocks from three spinal cords, prepared as described above, were washed in 0.1 m PB for 2 h. Slices of the spinal cord were cut at 500 μm, incubated in 1 m sucrose/PBS solution overnight, slammed onto copper blocks cooled in liquid nitrogen, and embedded in Lowicryl HM 20 (TAAB Laboratories, Aldermaston, UK) after freeze substitution with methanol. Ultrathin sections (70-90 nm) from three Lowicryl-embedded blocks were incubated for 45 min on coated nickel grids with drops of blocking solution consisting of 2% albumin in 0.05 m TBS, 0.9% NaCl, and 0.03% Triton X-100. The grids were transferred to solutions of GIRK1, GIRK2, or MOR antibodies at concentrations of 10 μg/ml in blocking solution for overnight incubation at room temperature. After being washed in TBS, grids were incubated for 2 h in drops of a mixture of goat anti-rabbit IgG conjugated to 10 nm colloidal gold particles and goat anti-guinea pig IgG conjugated to 20 nm colloidal gold particles (Nanoprobes), each diluted 1:80 in a 0.05 m TBS solution containing 2% normal human serum and 0.5% polyethylene glycol. Grids were then washed in TBS for 30 min and counterstained for electron microscopy with saturated aqueous uranyl acetate and lead citrate.
Ultrastructural analyses were performed with a Jeol (Peabody, MA) 1010 electron microscope. Electron photomicrographs were captured with a CCD camera (MegaView III; Soft Imaging System, Münster, Germany). Digitized electron images were modified for brightness and contrast with Adobe Photoshop version 7.0. Several lines of evidence argue that the observed staining patterns were specific. First, when primary antibody was omitted or replaced with 5% (v/v) normal serum of the species of the primary antibody, no selective labeling was observed. Second, when sections were incubated with both gold-labeled and biotinylated secondary antibodies, followed by the ABC complex and peroxidase reaction without silver intensification, an amorphous horseradish peroxidase (HRP) end product was observed, but no metal particles were detected. Third, the use of silver intensification without the HRP reaction led to the appearance of silver grains, but only infrequent small patches of HRP end product randomly distributed about the section. Finally, the lack of staining for GIRK1, GIRK2, and MORs was observed in tissue processed in parallel from appropriate knock-out mice.
Data analysis. Behavioral data are presented as mean ± SEM. For experiments involving knock-out mice and controls, drug-induced analgesia is presented as the percentage of maximum possible effect [%MPE = ([drug latency - baseline latency]/[cutoff latency - baseline latency]) × 100]. For experiments involving the coinjection of opioid agonists and tertiapin, %MPE values for the control and tertiapin-treated mice are calculated as above but then normalized to account for the acute hyperalgesic effect of tertiapin. The normalization was done to permit direct comparison of drug effects between the two groups of mice and is described by the following equation: %nMPE = %MPE - %MPE0, where %MPE0 is the %MPE value calculated for the control and tertiapin groups after the injection of saline or 30 pmol of tertiapin, respectively. For tertiapin studies, %nMPE values for tertiapin-treated and control subjects (same opioid dose) were analyzed with an unpaired t test. For knock-out studies, baseline and %MPE measures were analyzed at each temperature by one-way ANOVA. Our statistical analysis revealed no significant contribution of gender to behavioral performance in the studies involving knock-out mice, and, as such, data from males and females were combined to increase the study power. All post hoc analysis was performed using Tukey's honestly significant difference test. Differences were considered to be significant if p < 0.05.
Results
Distribution of GIRK subunits in the spinal cord dorsal horn
Previously, we demonstrated that the G-protein-gated K+ channel subunits GIRK1 and GIRK2 were expressed in the superficial layers of the dorsal horn (Marker et al., 2004). Within this region of the spinal cord, GIRK1 staining (Fig. 1_A_) tends to be more uniformly distributed, whereas GIRK2 staining (Fig. 1_B_) is more punctate and threadlike in appearance. For both GIRK subunits, staining is most prominent in a thin band near the marginal layer (lamina I). To precisely characterize the laminar distribution of GIRK subunits in the spinal cord, sections were costained with GIRK subunit-specific antibodies and the well characterized laminar markers CGRP (lamina I) and PKCγ (the inner region of the substantia gelatinosa) (Malmberg et al., 1997; Tie-Jun et al., 2001). The area of concentrated GIRK subunit staining corresponds to the outer layer of the substantia gelatinosa (lamina IIo) (Fig. 1_C-F_), a region consisting of a heterogeneous population of excitatory and inhibitory interneurons receiving and integrating most C-fiber and some Aδ-fiber nociceptive input (Millan, 1999, 2002).
Figure 1.
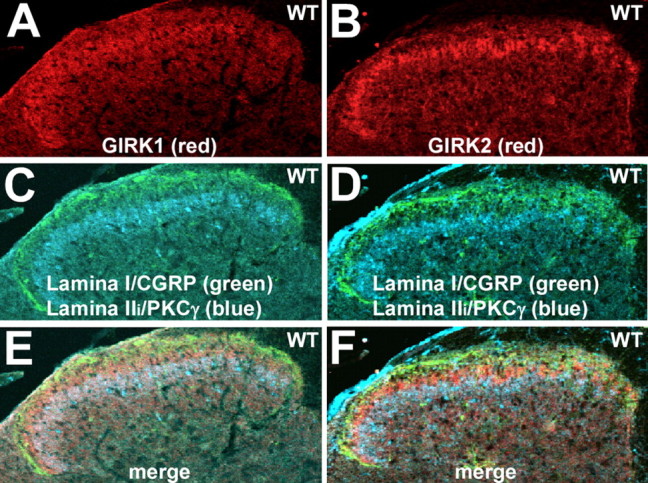
Laminar expression of GIRK subunits in the spinal cord dorsal horn. A, B, Expression of GIRK1 (A) and GIRK2 (B) in the dorsal horn of the spinal cord, shown in serial sections (10 μm) taken from the lumbar enlargement of a wild-type (WT) mouse. Note the concentrated staining for both GIRK1 and GIRK2 in the superficial layer of the dorsal horn. Sections from wild-type mice were processed on the same slide with sections from GIRK1 knock-out and GIRK2 knock-out mice to verify the specificity of GIRK antibody staining (data not shown). The images are representative of data from three different mice. C, D, The same sections were costained with antibodies for the lamina I marker CGRP (green) and PKCγ (blue), a marker for the inner aspect of lamina II (Lamina IIi). E, F, Overlays of the GIRK subunit, CGRP, and PKCγ staining patterns in each section. The region of most concentrated staining for GIRK1 and GIRK2 was found in lamina IIo.
To characterize the subcellular distribution of GIRK1 and GIRK2 subunits in lamina IIo of the mouse spinal cord, we used immunoelectron microscopy and both preembedding and postembedding immunogold techniques (Fig. 2). Results from multiple control experiments validated the observed immunoreactivity (see Materials and Methods), including the complete absence of GIRK1 and GIRK2 immunolabeling in sections taken from respective GIRK knock-out mice (data not shown). In sections from wild-type mice, GIRK1 immunoreactivity was found exclusively at postsynaptic sites (267 of 267 immunoparticles examined). GIRK1 immunoparticles were observed extrasynaptically in the plasma membrane of dendritic shafts of presumptive interneurons (Fig. 2_A-C,E_), as well as perisynaptically at putative excitatory (asymmetrical) synapses, which were easily discerned on the basis of a prominent postsynaptic specialization (Fig. 2_D,E_). We did not detect GIRK1 in the main body of the synaptic specialization (Fig. 2_E_). GIRK2 immunoreactivity was found primarily (209 of 223 immunoparticles examined) at postsynaptic sites in lamina IIo. Specifically, GIRK2 immunoparticles were found extrasynaptically (Fig. 2_F,H_) and perisynaptically at asymmetrical synapses (Fig. 2_G_) in the plasma membrane of dendritic shafts. The remaining 14 immunoparticles were found in the extrasynaptic plasma membrane of axon terminals that established asymmetrical synapses with dendritic shafts (Fig. 2_I_). Interestingly, GIRK2 immunoparticles were sometimes (35 of 153 particles examined) observed within the synaptic specialization of asymmetrical synapses (Fig. 2_J,K_).
Figure 2.
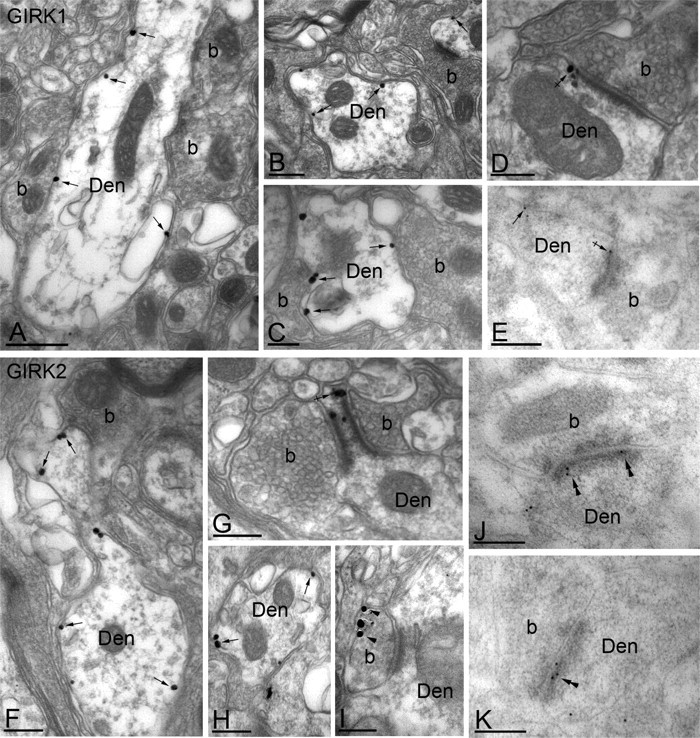
Subcellular distribution of GIRK channels in the spinal cord dorsal horn. A-K, Electron micrographs showing the subcellular distribution of GIRK1 (A-E) and GIRK2 (F-K) subunits in the dorsal horn (lamina IIo) of the mouse spinal cord. Den, Dendritic shafts; b, axon bouton. A-C, Using a preembedding immunogold technique, GIRK1 immunoreactivity was observed along the extrasynaptic plasma membrane (arrows) of dendritic shafts, postsynaptic to excitatory axon terminals. D, GIRK1 immunoparticles were also observed at the edge of asymmetrical synapses (crossed arrow), postsynaptic to excitatory axon terminals. E, Using a postembedding immunogold technique, GIRK1 immunoreactivity was detected at perisynaptic (crossed arrow) and extrasynaptic (arrow) locations but never within the postsynaptic specialization. F-H, Using the preembedding immunogold technique, GIRK2 immunoreactivity was localized to the extrasynaptic plasma membrane (arrows) of dendritic shafts establishing asymmetrical synapses with axon boutons and was associated with asymmetrical synapses (e.g., crossed arrow in G). I, To a much lesser extent, GIRK2 immunoreactivity was observed in the terminal portion (arrowheads) of axon boutons establishing asymmetrical synapses with dendritic shafts. J, K, Using the postembedding immunogold method, GIRK2 immunoparticles were found within the synaptic specialization of asymmetrical synapses (double arrowheads). Scale bars: A, 0.5 μm; B-K, 0.2 μm.
The contribution of GIRK channels to MOR analgesia
The observed localization pattern for GIRK channel subunits is consistent with their involvement in MOR signaling and analgesia, an association further supported by the impact of GIRK subunit ablation and tertiapin treatment on the analgesic effect of morphine (Marker et al., 2004). However, the unusual dose dependence of the GIRK channel contribution to intrathecal morphine analgesia could be explained by preferential coupling of GIRK channels to DORs and KORs stimulated by only relatively high intrathecal morphine doses. To test this possibility, we challenged MOR knock-out mice and wild-type controls with saline and two intrathecal morphine doses shown previously to evoke a modest GIRK-independent (0.3 nmol) and a robust GIRK-dependent (3 nmol) analgesia (Marker et al., 2004). Morphine failed to evoke an analgesic response at either dose in MOR knock-out mice (Fig. 3_A_). The lack of analgesic effect of the higher morphine dose in MOR knock-out mice indirectly supports the notion that GIRK channels contribute to MOR signaling and analgesia.
Figure 3.

Analgesia evoked by MOR agonists in the absence of GIRK channels or MORs. The analgesic effect of intrathecal DAMGO and morphine was measured using the tail-flick test. Comparable data were obtained at three different bath temperatures (49, 52.5, and 55°C), and, as such, only the 52.5°C data are presented here. A, The analgesic effect (%MPE) of intrathecal morphine (0, 0.3, and 3 nmol) was measured in wild-type (black bars; n = 18 per dose) and MOR knock-out (KO; gray bars; n = 5-6 per dose) mice. *p < 0.05 versus MOR KO (same dose); +p < 0.05 versus 0 nmol (same genotype); ++p < 0.01 versus 0 and 0.3 nmol (same genotype). B, The analgesic effect (%nMPE) of intrathecal DAMGO (0, 10, 30, 100, and 200 pmol) was measured in wild-type male C57BL/6 mice in the absence (control; white circles) or presence (black circles) of 30 pmol of tertiapin. Group sizes ranged from 10 to 16 mice per drug condition. *p < 0.05 versus tertiapin-treated mice (same dose). C, The analgesic effect (%MPE) of intrathecal DAMGO (0, 30, and 200 pmol) was measured in wild-type (white bars; n = 19-21 mice per dose), GIRK1 knock-out (black bars; n = 13-14 mice per dose), and GIRK2 knock-out (gray bars; n = 11-13 mice per dose) mice. *p < 0.05 versus GIRK knock-out mice (same dose). Error bars represent SEM.
To more directly probe for a contribution of GIRK channels to MOR signaling in the mouse spinal cord, we challenged wild-type male C57BL/6 mice with the MOR-selective agonist DAMGO (0, 10, 30, 100, and 200 pmol), which was coadministered via intrathecal injection with either saline (control) or 30 pmol of tertiapin. Although tertiapin blocks recombinant GIRK and Kir1.1 channels with similar potencies, GIRK channels are the primary targets of intrathecal tertiapin (Marker et al., 2004). Nociception and analgesia were assessed in naive mice using the warm-water (49, 52.5, and 55°C) immersion tail-flick test. Lower doses of DAMGO (10, 30, and 100 pmol) were equally effective at evoking analgesia in tertiapin-treated and control mice (Fig. 3_B_). The analgesic effect of 200 pmol of DAMGO, however, was significantly smaller in tertiapin-treated mice than in control animals. Similar results were obtained when DAMGO was administered intrathecally to wild-type C57BL/6 and congenic GIRK1 knockout and GIRK2 knock-out mice (Fig. 3_C_). Specifically, an intermediate dose of DAMGO (30 pmol) induced comparable analgesia in GIRK knock-out and wild-type mice, whereas 200 pmol of DAMGO produced a significantly smaller effect in GIRK knock-out mice than in wild-type mice.
MOR and GIRK subunit colocalization in the spinal cord dorsal horn
To ascertain whether the behavioral observations made with morphine and DAMGO reflect a direct functional coupling between GIRK channels and MORs, we costained sections from wild-type and GIRK knock-out mice with anti-GIRK and anti-MOR antibodies (Fig. 4). The most intense MOR staining was found in the most superficial aspect of the dorsal horn (Fig. 4_C,D_), likely reflecting the presence of MORs in lamina I. More diffuse MOR immunoreactivity was also observed in lamina IIo. Indeed, significant overlap in the MOR and GIRK1 (data not shown) and GIRK2 expression patterns was observed in lamina IIo (Fig. 4_E_). No striking differences in the MOR expression pattern or level were observed in sections from GIRK1 knock-out mice (data not shown) and GIRK2 knock-out mice (Fig. 4_F_).
Figure 4.

Overlap between GIRK subunits and MORs in the dorsal horn. Sections (10 μm) from the lumbar enlargement of wild-type (WT; A, C, E) and GIRK2 knock-out (G2KO; B, D, F) mice were stained with antibodies directed against GIRK2 (A, B; red) and MORs (C, D; green), with the overlays presented in E and F. The data presented are representative of staining patterns observed in sections from three different mice. Comparable images were obtained with GIRK1 antibodies as well with tissue from GIRK1 knock-out mice (data not shown).
Consistent with previous studies (Atweh and Kuhar, 1977; Besse et al., 1990; Arvidsson et al., 1995), immunoelectron microscopy revealed both presynaptic and postsynaptic distributions for MORs in lamina IIo of the mouse spinal cord. Of 256 MOR immunoparticles examined, 144 (56%) were found at postsynaptic sites, whereas 121 (44%) were found at presynaptic sites (Fig. 5). MOR immunoreactivity was observed extrasynaptically (Fig. 5_A,B,D_) in dendritic shafts and perisynaptically at asymmetrical synapses (Fig. 5_A,D_). MOR immunoreactivity was also found extrasynaptically in the plasma membrane of axons or axon terminals establishing asymmetrical synapses with the dendritic shafts of spinal cord neurons (Fig. 5_C,D_). In colabeling experiments, we found extensive colocalization of MOR and GIRK2 immunoreactivities in dendritic shafts (Fig. 5_E,F_) and in asymmetrical synapses (Fig. 5_G_). Colocalization was also observed in the small number of presynaptic axon terminals exhibiting GIRK2 immunoparticles (Fig. 5_H_). We also found evidence of MOR and GIRK1 colocalization in dendritic shafts (Fig. 5_I,J_). Thus, the behavioral and immunolocalization approaches together argue that GIRK channels in lamina IIo of the mouse spinal cord dorsal horn are proximal effectors in spinal MOR signaling and analgesia.
Figure 5.
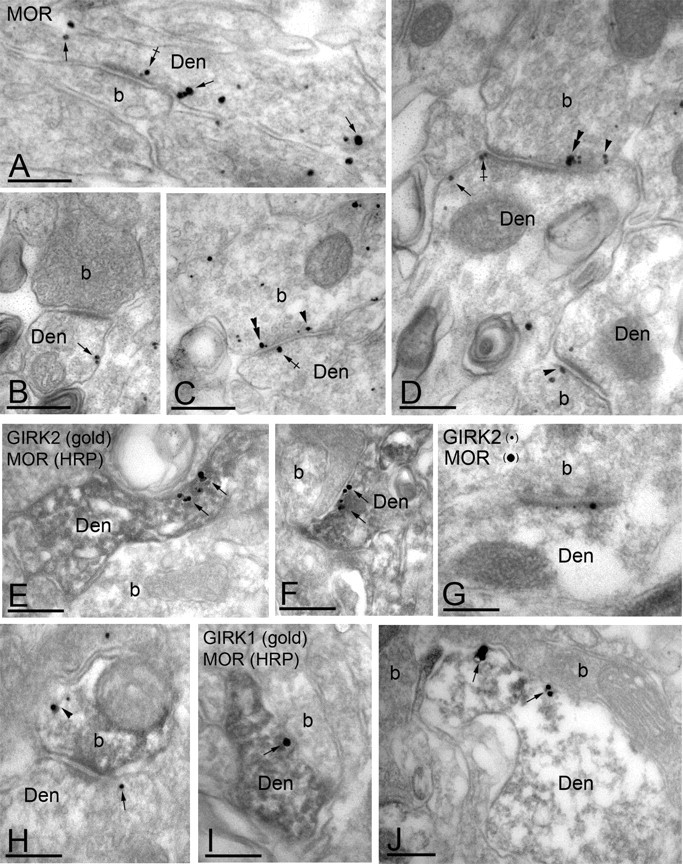
Subcellular distribution of MORs and GIRK channels in the dorsal horn. Electron micrographs showing MOR and GIRK subunit labeling in the dorsal horn (lamina IIo). Den, Dendritic shaft; b, axon bouton. A-D, MOR immunoparticles were found in the extrasynaptic (arrows) and perisynaptic plasma membrane (crossed arrows) of dendritic shafts establishing asymmetrical synapses with presynaptic boutons. MOR immunoreactivity was also observed in axon boutons establishing asymmetrical synapses with dendritic shafts, either along the extrasynaptic plasma membrane (arrowhead) or within active zones (double arrowheads). E, F, Postsynaptic colocalization of MORs and GIRK2 using a preembedding immunogold method. The HRP reaction product represents MOR immunoreactivity, and it filled dendritic shafts with GIRK2 immunoparticles organized along the extrasynaptic plasma membrane (arrows). G, Using a postembedding immunogold method, GIRK2 immunoparticles (10 nm) and MOR immunoparticles (20 nm) were found postsynaptically within the same asymmetrical synapse. H, Presynaptic colocalization of MORs and GIRK2 using a preembedding immunogold method. The HRP reaction product (MOR immunoreactivity) filled the axon terminal, whereas GIRK2 immunoparticles were found along the extrasynaptic plasma membrane (arrowhead). Note the postsynaptic GIRK2 immunoparticle (arrow). I, J, Postsynaptic colocalization of MORs and GIRK1 using a preembedding immunogold method. The HRP reaction product (MOR immunoreactivity) filled dendritic shafts, whereas GIRK1 immunoparticles were found along the extrasynaptic plasma membrane (arrows). Scale bars, 0.2 μm.
The contribution of spinal GIRK channels to DOR and KOR analgesia
To examine whether spinal GIRK channels also contribute the analgesia triggered by stimulation of spinal DORs and KORs, we measured the effects of intrathecal DOR- and KOR-selective agonists in tertiapin-treated wild-type mice. The δ1/DOR-selective agonist DPDPE (1, 3, 10, and 30 pmol) evoked a robust dose-dependent analgesia in wild-type mice at all three temperatures (Fig. 6_A_). When coadministered with 30 pmol of tertiapin, the effect of the 1 and 3 nmol doses of DPDPE was completely preserved, whereas the analgesia produced by 10 and 30 pmol of DPDPE was significantly smaller than that observed for saline-treated control mice. Qualitatively similar effects were documented for tertiapin-treated wild-type mice challenged with the δ2/DOR-selective agonist deltorphin II (Fig. 6_B_). Finally, the effect of the highest dose of deltorphin II (10 nmol) was smaller in GIRK1 knock-out and GIRK2 knock-out mice, whereas the analgesic effect of an intermediate dose (1 nmol) was preserved in these mice (Fig. 6_C_).
Figure 6.

Analgesia evoked by DOR agonists in the absence of GIRK channels. A, B, The analgesic effects of intrathecal DPDPE (0, 1, 3, 10, and 30 nmol) or deltorphin II (0, 0.3, 1, 3, and 10 pmol) were measured with the tail-flick test in wild-type male C57BL/6 mice in the absence (control; white circles) or presence (black circles) of 30 pmol of tertiapin. Comparable data were obtained at three different bath temperatures (49, 52.5, and 55°C), and, as such, only the 52.5°C data are presented here. Group sizes ranged from 10 to 16 mice per drug condition. *p < 0.05 versus tertiapin-treated mice (same dose). C, The analgesic effect of intrathecal deltorphin II (0, 30, and 200 pmol) was measured in wild-type (white bars; n = 11-17 per dose), GIRK1 knock-out (KO; black bars; n = 8-9 per dose), and GIRK2 knock-out (gray bars; n = 5-8 per dose) mice. *p < 0.05 versus GIRK knock-out mice. Error bars represent SEM.
The KOR-selective agonist U50488 evoked a dose-dependent analgesia in wild-type mice (Fig. 7_A_). Unlike our experience with MOR- and DOR-selective agonists, however, the analgesic effect of U50488 was unaffected by 30 pmol of tertiapin. Furthermore, equivalent analgesic responses to U50488 were recorded in wild-type, GIRK1 knock-out, and GIRK2 knock-out mice (Fig. 7_B_). Thus, these experiments suggest that spinal GIRK channels contribute in a dose-dependent manner to the analgesia evoked by DOR, but not KOR, activation.
Figure 7.

Analgesia evoked by a KOR agonist in the absence of GIRK channels. The analgesic effect of intrathecal U50488 was measured using the tail-flick test. Comparable data were obtained at three different bath temperatures (49, 52.5, and 55°C), and, as such, only the 52.5°C data are presented here. A, The analgesic effect of intrathecal U50488 (0, 3, 10, 30, and 60 μg) was measured in wild-type male C57BL/6 mice in the absence (saline; white circles) or presence (tertiapin; black circles) of 30 pmol of tertiapin. Group sizes ranged from 9 to 16 mice per dose. B, The analgesic effect of intrathecal U50488 (0, 10, and 60 μg) was measured in wild-type (white bars; n = 15 per dose), GIRK1 knock-out (KO; black bars; n = 6-13 per dose), and GIRK2 knock-out (gray bars; n = 5-7 per dose) mice. Error bars represent SEM.
Discussion
In this study, we demonstrated that GIRK channel subunits are concentrated in lamina IIo of the spinal cord dorsal horn and that they exhibited an almost exclusive postsynaptic distribution, overlapping with MORs at asymmetrical (excitatory) synapses. Consistent with a direct contribution of GIRK channels to MOR analgesia, we observed that the analgesic effect of a high dose of the MOR-selective agonist DAMGO was selectively blunted in tertiapin-treated wild-type mice and in GIRK knock-out mice. We observed a similar contribution of GIRK channels to the analgesic effect of the DOR-selective agonists DPDPE and deltorphin II. Somewhat surprisingly, we failed to detect a contribution of GIRK channels to the analgesic effect of the KOR-selective agonist U50488. Thus, our data support the contention that the dose dependence of GIRK channel contributions to intrathecal morphine analgesia does not reflect a preferential coupling of GIRK channels to DORs and KORs stimulated by high intrathecal morphine doses. Furthermore, our data suggest that GIRK channel activation and the resultant hyperpolarization of lamina IIo neurons is a powerful, albeit insensitive, postsynaptic mechanism used by MORs, and perhaps DORs, to evoke spinal analgesia.
GIRK channels are widely regarded as postsynaptic effectors in the CNS, primarily because of the clear dissociation of the postsynaptic and presynaptic inhibitory effects of multiple neurotransmitters in hippocampal pyramidal neurons from GIRK2 knock-out mice (Luscher et al., 1997). Here, we offer direct evidence that GIRK channels containing GIRK1 and/or GIRK2 are found almost exclusively at postsynaptic sites, clustering in and around asymmetrical synapses. The presence of GIRK2, but not GIRK1, within the postsynaptic specialization supports the notion that subpopulations of GIRK channels can exist within the same cell and, furthermore, that channel subunit composition determines its precise subcellular distribution. Although the molecular determinants of the subcellular distribution of GIRK channels remain to be elucidated, membrane microdomains or direct channel/receptor physical interactions may play roles. Indeed, GIRK1 was identified recently in lipid rafts isolated from brain tissue (Delling et al., 2002), and GIRK subunits were coimmunoprecipitated with D2 dopamine receptors and adenylyl cyclase from brain extracts (Lavine et al., 2002). In addition, GIRK2 and GIRK3 subunits may interact with scaffolding proteins such as PSD-95 (postsynaptic density-95). Such interactions, however, have not been demonstrated in vivo (Inanobe et al., 1999; Nehring et al., 2000).
Although our data reveal a strong direct functional linkage between GIRK channels and MORs in the mouse spinal cord, experiments with DPDPE and deltorphin II argue for a role of GIRK channels in DOR analgesia as well. Previous studies have shown that the analgesic effects of intrathecal DPDPE (δ1/DOR) and deltorphin II (δ2/DOR) were absent in DOR knockout mice, underscoring the significance of DORs to the analgesic effect of both agonists (Zhu et al., 1999). A significant contribution of MORs, however, to the analgesic effect of intracerebroventricular and intrathecal DPDPE has been observed (Hosohata et al., 2000). Although MOR knock-out mice exhibited normal analgesic responses to deltorphin II, the doseresponse relationship for DPDPE analgesia was shifted to the right by nearly one order of magnitude (Hosohata et al., 2000). Thus, we cannot rule out a significant contribution of MORs to the analgesic effect of DPDPE observed in our study.
The lack of contribution of spinal GIRK channels to the analgesic effect of intrathecal U50488 was unexpected. When KORs were coexpressed with GIRK subunits in Xenopus oocytes, U50488 evoked robust GIRK currents (Henry et al., 1995; Ikeda et al., 1995, 2003; Kobayashi et al., 1996). In addition, U50488 stimulated a K+ conductance in ∼15% of lamina II neurons in rat spinal cord (Eckert et al., 2001; Eckert and Light, 2002). It is possible that the K+ currents evoked by U50488 in rat lamina II neurons were not GIRK related, because KOR stimulation has been linked to other K+ channel classes, including a dendrotoxin-sensitive voltage-gated K+ current (Simmons and Chavkin, 1996). Alternatively, significant differences between rats and mice may exist with respect to the effectors relevant to spinal KOR signaling. It is also possible that significant KOR/GIRK coupling may exist, but does not occur to a sufficient extent in nociceptive and antinociceptive pathways triggered in our study.
The dose-dependent contribution of spinal GIRK channels to MOR and DOR analgesia could be explained in several ways. First, a relative insensitivity to G-protein stimulation may be an intrinsic property of GIRK channels. There may, for instance, be a higher cooperativity inherent in the Gβγ-dependent, GIRK channel activation process compared with other G-protein-dependent effector modulation schemes. Indeed, multiple Gβγ/GIRK subunit binding events are apparently required to evoke the maximal increase in GIRK channel open probability (Nemec et al., 1999; Sadja et al., 2002, 2003). Support for this line of reasoning comes from experiments involving the cardiac G-protein-gated potassium channel _I_KACh (GIRK1/GIRK4 heterotetramer), which represents the dominant but relatively insensitive component of the vagally mediated decrease in heart rate (Yamada, 2002). In atrial myocytes, the cAMP/PKA-dependent modulation of the L-type voltage-gated calcium channel and the “pacemaker” current (_I_f) occur with ACh concentrations too low to stimulate _I_KACh (DiFrancesco, 1995).
In the context of this study, neuroanatomical explanations for the dose-dependent contribution of spinal GIRK channels to MOR and DOR analgesia seem tenable. The tail-flick response to thermal stimuli in the 49-55°C range likely reflects significant contributions of both Aδ- and C-fibers (Yeomans et al., 1996; Millan, 1999, 2002). Evidence from rat studies suggests that C-fibers are more potently inhibited by intrathecal DPDPE than Aδ-fibers (Le Bars et al., 1976; Yeomans and Proudfit, 1996) and that the mechanisms underlying the inhibition of C- and Aδ-fibers by intrathecal DAMGO are distinct. The latter observation likely reflects, at least in part, the differential distribution of MORs in C-fiber compared with Aδ-fiber information relays. Indeed, MOR expression in intrinsic dorsal horn neurons impacts both C-fiber and Aδ- fiber signaling, but MORs are also found on presynaptic terminals of C-fibers, but not Aδ- fibers (Millan, 1999, 2002). Thus, it is possible that the inhibition of C-fiber neurotransmission explains the analgesic effect of low doses of MOR- and DOR-selective agonists, whereas the robust analgesia seen with higher doses might reflect the combined influence of both C-fiber and Aδ-fiber inhibition. One argument against this explanation is that GIRK channels are most prominently located in lamina IIo, the primary central target of C-fibers, whereas Aδ-fibers terminate primarily in lamina I. Nevertheless, additional experiments that directly measure opioid effects on either C-fibers or Aδ-fibers will be useful for clarifying the role of spinal GIRK channels in intrathecal opioid analgesia.
An explanation for the observed contribution of GIRK channels to opioid analgesia that draws on aspects of the two mentioned previously is that presynaptic, non-GIRK targets of opioid agonists may be more potently modulated than postsynaptic GIRK channels. Indeed, considerable evidence argues that MOR- and DOR-selective agents inhibit excitatory (glutamatergic) neurotransmitter release by C-fibers in a more potent manner than how they inhibit intrinsic dorsal horn neurons. Specifically, DAMGO, DPDPE, and deltorphin II reduced the amplitude of evoked EPSC/Ps in lamina II neurons at smaller concentrations than those required to blunt the postsynaptic response to AMPA (Glaum et al., 1994; Kohno et al., 1999). Similarly, DAMGO and the endogenous MOR-selective opioid peptides endomorphin I and II suppressed evoked glutamatergic EPSCs at smaller concentrations than those required to evoke significant outward hyperpolarizing inwardly rectifying K+ currents in lamina II neurons (Wu et al., 2003). Using internalization of MORs in lamina II neurons as a marker of postsynaptic MOR-dependent signaling, Trafton et al. (2000) demonstrated that postsynaptic inhibition likely contributes to the analgesia produced by intrathecal DAMGO but not to the analgesic effects of endogenous opioid peptides released under normal or inflammatory conditions in response to noxious stimuli. In the context of this body of literature, our data suggest that, although opioids potently modulate presynaptic neurotransmitter release and perhaps some non-GIRK postsynaptic targets yielding a modest degree of analgesia, the maximum analgesic effect of MOR-selective agonists results from the relatively insensitive GIRK-dependent blunting of excitatory afferent input to lamina IIo neurons.
In summary, we demonstrated that spinal GIRK channels underlie a dominant component of the analgesia evoked by spinal MOR activation. We also present evidence that spinal GIRK channels contribute in a dose-dependent manner to the analgesic effect of intrathecal DOR-selective, but not KOR-selective, agonists. Altogether, our study suggests that the effect of opioids on presynaptic and postsynaptic targets in the dorsal horn can be dissected and measured independently at the behavioral level. Future experiments will be directed at measuring the individual sensitivities of downstream effectors to opioid inhibition and at exploring whether the separable nature of GIRK-dependent and GIRK-independent processes can be exploited for clinical benefit.
Footnotes
This work was supported by National Institutes of Health Grants MH61933 (K.W.), DA00564 and DA01583 (H.H.L.), P50 DA011806 (K.W. and H.H.L.), and T32 DA07234 (C.L.M.). We thank Dr. Christian Lüscher for reading this manuscript and offering suggestions and Drs. Carolyn Fairbanks and George Wilcox for helpful discussions and input related to study design and data interpretation.
Correspondence should be addressed to Kevin Wickman, Department of Pharmacology, University of Minnesota, 6-120 Jackson Hall, 321 Church Street Southeast, Minneapolis, MN 55455. E-mail: wickm002@tc.umn.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/253551-09$15.00/0
References
- Arvidsson U, Dado R, Riedl M, Lee J, Law P, Loh H, Elde R, Wessendorf M (1995) Distribution and targeting of a μ-opioid receptor (MOR1) in brain and spinal cord. J Neurosci 15: 3328-3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atweh S, Kuhar M (1977) Autoradiographic localization of opiate receptors in rat brain I. Spinal cord and lower medulla. Brain Res 124: 53-67. [DOI] [PubMed] [Google Scholar]
- Beckstead MJ, Grandy DK, Wickman K, Williams JT (2004) Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron 42: 939-946. [DOI] [PubMed] [Google Scholar]
- Besse D, Lombard M, Zajac J, Roques B, Besson J (1990) Pre- and postsynaptic distribution of mu, delta, and kappa opioid receptors in the superficial layers of the cervical dorsal horn of the rat spinal cord. Brain Res 521: 15-22. [DOI] [PubMed] [Google Scholar]
- Bettahi I, Marker CL, Roman MI, Wickman K (2002) Contribution of the Kir3.1 subunit to the muscarinic-gated atrial potassium channel IKAChJ Biol Chem 277: 48282-48288. [DOI] [PubMed] [Google Scholar]
- Chang H, Berde C, Holz G, Steward G, Kream R (1989) Sufentanil, morphine, met-enkephalin, and k-agonist (U-50,488h) inhibit substance P release from primary sensory neurons: a model for presynaptic spinal opioid actions. Anesthesiology 70: 672-677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz HG, Ivanova T, Lunn ML, Stoffel M, Slesinger PA, Luscher C (2004) Bi-directional effects of GABA(B) receptor agonists on the mesolimbic dopamine system. Nat Neurosci 7: 153-159. [DOI] [PubMed] [Google Scholar]
- Delling M, Wischmeyer E, Dityatev A, Sytnyk V, Veh RW, Karschin A, Schachner M (2002) The neural cell adhesion molecule regulates cell-surface delivery of G-protein-activated inwardly rectifying potassium channels via lipid rafts. J Neurosci 22: 7154-7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco D (1995) The onset and autonomic regulation of cardiac pacemaker activity: relevance of the f current. Cardiovasc Res 29: 449-456. [PubMed] [Google Scholar]
- Eckert III WA, Light AR (2002) Hyperpolarization of substantia gelatinosa neurons evoked by mu-, kappa-, delta 1-, and delta 2-selective opioids. J Pain 3: 115-125. [DOI] [PubMed] [Google Scholar]
- Eckert III WA, Willcockson HH, Light AR (2001) Interference of biocytin with opioid-evoked hyperpolarization and membrane properties of rat spinal substantia gelatinosa neurons. Neurosci Lett 297: 117-120. [DOI] [PubMed] [Google Scholar]
- Glaum S, Miller R, Hammond D (1994) Inhibitory actions of delta 1-, delta 2-, and μ-opioid receptor agonists on excitatory transmission in lamina II neurons of adult rat spinal cord. J Neurosci 14: 4965-4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grudt T, Williams J (1993) Kappa-opioid receptors also increase potassium conductance. Proc Natl Acad Sci USA 90: 11429-11432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry DJ, Grandy DK, Lester HA, Davidson N, Chavkin C (1995) Kappaopioid receptors couple to inwardly rectifying potassium channels when coexpressed by Xenopus oocytes. Mol Pharmacol 47: 551-557. [PubMed] [Google Scholar]
- Hosohata Y, Vanderah TW, Burkey TH, Ossipov MH, Kovelowski CJ, Sora I, Uhl GR, Zhang X, Rice KC, Roeske WR, Hruby VJ, Yamamura HI, Lai J, Porreca F (2000) Delta-opioid receptor agonists produce antinociception and [35S]GTPγS binding in mu receptor knockout mice. Eur J Pharmacol 388: 241-248. [DOI] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL (1980) Intrathecal morphine in mice: a new technique. Eur J Pharmacol 67: 313-316. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Kobayashi T, Ichikawa T, Usui H, Kumanishi T (1995) Functional couplings of the delta- and the kappa-opioid receptors with the G-protein-activated K+ channel. Biochem Biophys Res Commun 208: 302-308. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Kobayashi T, Kumanishi T, Niki H, Yano R (2000) Involvement of G-protein-activated inwardly rectifying K+ (GIRK) channels in opioid-induced analgesia. Neurosci Res 38: 113-116. [DOI] [PubMed] [Google Scholar]
- Ikeda K, Yoshii M, Sora I, Kobayashi T (2003) Opioid receptor coupling to GIRK channels. In vitro studies using a Xenopus oocyte expression system and in vivo studies on weaver mutant mice. Methods Mol Med 84: 53-64. [DOI] [PubMed] [Google Scholar]
- Inanobe A, Yoshimoto Y, Horio Y, Morishige KI, Hibino H, Matsumoto S, Tokunaga Y, Maeda T, Hata Y, Takai Y, Kurachi Y (1999) Characterization of G-protein-gated K+ channels composed of Kir3.2 subunits in dopaminergic neurons of the substantia nigra. J Neurosci 19: 1006-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karschin C, Dissmann E, Stuhmer W, Karschin A (1996) IRK(1-3) and GIRK(1-4) inwardly rectifying K+ channel mRNAs are differentially expressed in the adult rat brain. J Neurosci 16: 3559-3570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer BL, Gaveriaux-Ruff C (2002) Exploring the opioid system by gene knockout. Prog Neurobiol 66: 285-306. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Ikeda K, Ichikawa T, Togashi S, Kumanishi T (1996) Effects of sigma ligands on the cloned mu-, delta- and kappa-opioid receptors coexpressed with G-protein-activated K+ (GIRK) channel in Xenopus oocytes. Br J Pharmacol 119: 73-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Kumamoto E, Higashi H, Shimoji K, Yoshimura M (1999) Actions of opioids on excitatory and inhibitory transmission in substantia gelatinosa of adult rat spinal cord. J Physiol (Lond) 518: 803-813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapivinsky G, Gordon EA, Wickman K, Velimirovic B, Krapivinsky L, Clapham DE (1995a) The G-protein-gated atrial K+ channel IKACh is a heteromultimer of two inwardly rectifying K+ channel proteins. Nature 374: 135-141. [DOI] [PubMed] [Google Scholar]
- Krapivinsky G, Krapivinsky L, Velimirovic B, Wickman K, Navarro B, Clapham DE (1995b) The cardiac inward rectifier K+ channel subunit, CIR, does not comprise the ATP-sensitive K+ channel, IKATPJ Biol Chem 270: 28777-28779. [DOI] [PubMed] [Google Scholar]
- Lavine N, Ethier N, Oak JN, Pei L, Liu F, Trieu P, Rebois RV, Bouvier M, Hebert TE, Van Tol HH (2002) G protein-coupled receptors form stable complexes with inwardly rectifying potassium channels and adenylyl cyclase. J Biol Chem 23: 23. [DOI] [PubMed] [Google Scholar]
- Le Bars D, Guilbaud G, Jurna I, Besson JM (1976) Differential effects of morphine on responses of dorsal horn lamina V type cells elicited by A and C fibre stimulation in the spinal cat. Brain Res 115: 518-524. [DOI] [PubMed] [Google Scholar]
- Lujan R, Nusser Z, Roberts JD, Shigemoto R, Somogyi P (1996) Perisynaptic location of metabotropic glutamate receptors mGluR1 and mGluR5 on dendrites and dendritic spines in the rat hippocampus. Eur J Neurosci 8: 1488-1500. [DOI] [PubMed] [Google Scholar]
- Luscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA (1997) G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron 19: 687-695. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Chen C, Tonegawa S, Basbaum AI (1997) Preserved acute pain and reduced neuropathic pain in mice lacking PKCgamma. Science 278: 279-283. [DOI] [PubMed] [Google Scholar]
- Marker CL, Cintora SC, Roman MI, Stoffel M, Wickman K (2002) Hyperalgesia and blunted morphine analgesia in G protein-gated potassium channel subunit knockout mice. NeuroReport 13: 2509-2513. [DOI] [PubMed] [Google Scholar]
- Marker CL, Stoffel M, Wickman K (2004) Spinal G-protein-gated K+ channels formed by GIRK1 and GIRK2 subunits modulate thermal nociception and contribute to morphine analgesia. J Neurosci 24: 2806-2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ (1999) The induction of pain: an integrative review. Prog Neurobiol 57: 1-164. [DOI] [PubMed] [Google Scholar]
- Millan MJ (2002) Descending control of pain. Prog Neurobiol 66: 355-474. [DOI] [PubMed] [Google Scholar]
- Mitrovic I, Margeta-Mitrovic M, Bader S, Stoffel M, Jan LY, Basbaum AI (2003) Contribution of GIRK2-mediated postsynaptic signaling to opiate and alpha 2-adrenergic analgesia and analgesic sex differences. Proc Natl Acad Sci USA 100: 271-276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nehring RB, Wischmeyer E, Doring F, Veh RW, Sheng M, Karschin A (2000) Neuronal inwardly rectifying K+ channels differentially couple to PDZ proteins of the PSD-95/SAP90 family. J Neurosci 20: 156-162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemec J, Wickman K, Clapham DE (1999) Gbetagamma binding increases the open time of IKACh: kinetic evidence for multiple Gbetagamma binding sites. Biophys J 76: 246-252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North A (1989) Drug receptors and the inhibition of nerve cells. Br J Pharmacol 98: 13-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Barke R, Loh H (1998) Mu-opioid receptor-knockout mice: role of mu-opioid receptor in morphine mediated immune functions. Brain Res Mol Brain Res 61: 190-194. [DOI] [PubMed] [Google Scholar]
- Sadja R, Alagem N, Reuveny E (2002) Graded contribution of the Gβγ binding domains to GIRK channel activation. Proc Natl Acad Sci USA 99: 10783-10788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadja R, Alagem N, Reuveny E (2003) Gating of GIRK channels: details of an intricate, membrane-delimited signaling complex. Neuron 39: 9-12. [DOI] [PubMed] [Google Scholar]
- Schneider SP, Eckert III WA, Light AR (1998) Opioid-activated postsynaptic, inward rectifying potassium currents in whole cell recordings in substantia gelatinosa neurons. J Neurophysiol 80: 2954-2962. [DOI] [PubMed] [Google Scholar]
- Signorini S, Liao YJ, Duncan SA, Jan LY, Stoffel M (1997) Normal cerebellar development but susceptibility to seizures in mice lacking G protein-coupled, inwardly rectifying K+ channel GIRK2. Proc Natl Acad Sci USA 94: 923-927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons M, Chavkin C (1996) Kappa-opioid receptor activation of a dendrotoxin-sensitive potassium channel mediates presynaptic inhibition of mossy fiber neurotransmitter release. Mol Pharmacol 50: 80-85. [PubMed] [Google Scholar]
- Slesinger P, Stoffel M, Jan Y, Jan L (1997) Defective γ-aminobutyric acid type B receptor-activated inwardly rectifying K+ currents in cerebellar granule cells isolated from weaver and Girk2 null mutant mice. Proc Natl Acad Sci USA 94: 12210-12217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemori A, Ikeda M, Portoghese P (1986) The mu, kappa, and delta properties of various opioid agonists. Eur J Pharmacol 123: 357-361. [DOI] [PubMed] [Google Scholar]
- Tie-Jun SS, Xu Z, Hokfelt T (2001) The expression of calcitonin generelated peptide in dorsal horn neurons of the mouse lumbar spinal cord. NeuroReport 12: 739-743. [DOI] [PubMed] [Google Scholar]
- Torrecilla M, Marker CL, Cintora SC, Stoffel M, Williams JT, Wickman K (2002) G-protein-gated potassium channels containing Kir3.2 and Kir3.3 subunits mediate the acute inhibitory effects of opioids on locus ceruleus neurons. J Neurosci 22: 4328-4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trafton J, Abbadie C, Marek K, Basbaum A (2000) Postsynaptic signaling via the mu-opioid receptor: responses of dorsal horn neurons to exogenous opioids and noxious stimulation. J Neurosci 20: 8578-8584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Connor MA, Christie MJ (1997) How opioids inhibit GABA-mediated neurotransmission. Nature 390: 611-614. [DOI] [PubMed] [Google Scholar]
- Wu SY, Ohtubo Y, Brailoiu GC, Dun NJ (2003) Effects of endomorphin on substantia gelatinosa neurons in rat spinal cord slices. Br J Pharmacol 140: 1088-1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL (1997) Pharmacology and mechanisms of opioid analgesic activity. Acta Anaesthesiol Scand 41: 94-111. [DOI] [PubMed] [Google Scholar]
- Yamada M (2002) The role of muscarinic K+ channels in the negative chronotropic effect of a muscarinic agonist. J Pharmacol Exp Ther 300: 681-687. [DOI] [PubMed] [Google Scholar]
- Yeomans DC, Proudfit HK (1996) Nociceptive responses to high and low rates of noxious cutaneous heating are mediated by different nociceptors in the rat: electrophysiological evidence. Pain 68: 141-150. [DOI] [PubMed] [Google Scholar]
- Yeomans DC, Pirec V, Proudfit HK (1996) Nociceptive responses to high and low rates of noxious cutaneous heating are mediated by different nociceptors in the rat: behavioral evidence. Pain 68: 133-140. [DOI] [PubMed] [Google Scholar]
- Yeung J, Rudy T (1980) Multiplicative interaction between narcotic agonisms expressed at spinal and supraspinal sites of antinociceptive action as revealed by concurrent intrathecal and intracerebroventricular injections of morphine. J Pharmacol Exp Ther 215: 633-642. [PubMed] [Google Scholar]
- Yoshimura M, North R (1983) Substantia gelatinosa neurones hyperpolarized in vitro by enkephalin. Nature 305: 529-530. [DOI] [PubMed] [Google Scholar]
- Zhu Y, King M, Schuller A, Nitsche J, Reidl M, Elde R, Unterwald E, Pasternak G, Pintar J (1999) Retention of supraspinal delta-like analgesia and loss of morphine tolerance in delta opioid receptor knockout mice. Neuron 24: 243-252. [DOI] [PubMed] [Google Scholar]