Thrombospondin-1 up-regulates expression of cell adhesion molecules and promotes monocyte binding to endothelium (original) (raw)
. Author manuscript; available in PMC: 2006 Sep 18.
Published in final edited form as: FASEB J. 2005 Apr 15;19(9):1158–1160. doi: 10.1096/fj.04-3310fje
Abstract
Expression of cell adhesion molecules (CAM) responsible for leukocyte-endothelium interactions plays a crucial role in inflammation and atherogenesis. Up-regulation of vascular CAM-1 (VCAM-1), intracellular CAM-1 (ICAM-1), and E-selectin expression promotes monocyte recruitment to sites of injury and is considered to be a critical step in atherosclerotic plaque development. Factors that trigger this initial response are not well understood. As platelet activation not only promotes thrombosis but also early stages of atherogenesis, we considered the role of thrombospondin-1 (TSP-1), a matricellular protein released in abundance from activated platelets and accumulated in sites of vascular injury, as a regulator of CAM expression. TSP-1 induced expression of VCAM-1 and ICAM-1 on endothelium of various origins, which in turn, resulted in a significant increase of monocyte attachment. This effect could be mimicked by a peptide derived from the C-terminal domain of TSP-1 and known to interact with CD47 on the cell surface. The essential role of CD47 in the cellular responses to TSP-1 was demonstrated further using inhibitory antibodies and knockdown of CD47 with small interfering RNA. Furthermore, we demonstrated that secretion of endogenous TSP-1 and its interaction with CD47 on the cell surface mediates endothelial response to the major proinflammatory agent, tumor necrosis factor α (TNF-α). Taken together, this study identifies a novel mechanism regulating CAM expression and subsequent monocyte binding to endothelium, which might influence the development of anti-atherosclerosis therapeutic strategies.
Keywords: integrin-associated protein, cardiovascular disease, TNF-α
Expression of cell adhesion molecules (CAM) is one of the initial responses of activated endothelium during injury or atherosclerotic plaque formation. CAM mediate monocyte and leukocyte adhesion and transmigration into the subendothelial space (1, 2). In larger blood vessels, these processes are critical for foam cell formation and subsequent development of atherosclerotic lesions. In the microvasculature, these are important for leukocyte recruitment and inflammatory response (3). Both processes require up-regulation of members of the selectin family, such as P- and E-selectin, and members of the immunoglobulin (Ig) superfamily, most notably, ICAM-1 and VCAM-1. The importance of these particular CAM is underscored by the consequences of null mutations in mice. Deficiency of ICAM-1 in mice on ApoE null background appears to limit the development of atherosclerotic lesions (4). In animals lacking domain 4 of VCAM-1, the area of early atherosclerotic lesions is reduced significantly compared with control littermates (5). ICAM-1 and VCAM-1 expression is known to be activated by a number of agents including free radicals and cytokines.
TNF-α is often used as a representative inducer of CAM expression in endothelial cells (EC; refs. 6, 7). Originally discovered as an anti-tumor agent (8), TNF-α serves as a key mediator of immune and inflammatory response. In its primary target, EC, TNF-α induces shape and motility changes and increases EC monolayer permeability and secretion of a number of additional cytokines (9). Among numerous activities assigned to TNF-α, stimulation of CAM (ICAM-1, VCAM-1, and E-selectin) expression on EC and subsequent leukocyte infiltration is one of its most important functions.
As an important role of CAM in a variety of pathological conditions, including atherogenesis, the mechanisms of their regulation is a subject of extensive investigation. Despite the significant advances in atherosclerosis research, molecular events that trigger the development of vascular lesions are not well defined.
Recently, it was shown that activation of circulating platelets may significantly influence the initial stage of atherosclerotic lesion development (10). Platelet activation is known to result in a release of considerable numbers of factors, which in turn, might stimulate early vascular responses, i.e., expression of CAM and recruitment of macrophages. TSP-1, one of the major proteins released from activated platelets (11), seems to be an appropriate candidate to serve as a “proatherogenic” factor. Moreover, a postnatal pattern of TSP-1 expression links it to the development of a number of pathological conditions. TSP-1 expression in tissues of healthy adult individuals is significantly lower compared with embryos (12, 13). However, it is increased extensively in response to injury (13). TSP-1 is secreted by a variety of cells including platelets, EC, macrophages, fibroblasts, and smooth muscle cells (13). Elevated TSP-1 expression was observed during wound healing (14), after balloon catheter injury in rats (15), and in EC, glial, neuronal, and macrophage cells after ischemia (16). It is important that TSP-1 is overexpressed in aorta and carotid artery of diabetic rats (17) as well as in many other pathological conditions (13). Immunohistochemical studies showed up-regulation of TSP-1 in the atherosclerotic plaques compared with normal blood vessels (18). Specifically, TSP-1 expression was limited to primary hypocellular atherosclerotic plaques (19). Recent genetic studies also linked TSP-1 to coronary artery disease, in particular, to the development of premature heart attack. TSP-1 gene mutation (Asn700Ser) results in a significantly increased risk of myocardial infarction in persons homozygous for this mutation (20, 21).
Thus, there is compelling evidence linking genetics of TSP-1 as well as its expression pattern to atherosclerotic lesion development. It is not clear at present if elevated expression of TSP-1 is a cause or result of atherosclerosis development.
At the cellular level, TSP-1 plays an active role in tissue remodeling (12, 13). TSP-1 inhibits vascular EC proliferation (22, 23) and morphogenesis (24). Additionally, TSP-1 has anti-inflammatory activities, as evidenced by multiorgan inflammation in TSP-1 null mice (25, 26) and by its suppression of cytokine release (27). Although multiple lines of evidence suggest a critical role of TSP-1 in responses associated with vascular injury, its direct function is not understood. To elucidate the direct role of TSP-1 in early events of atherogenesis, we assessed its effect on the expression of CAM by endothelium. In this report, we provide evidence that TSP-1 is able to stimulate ICAM-1, VCAM-1, and E-selectin expression and consequently, monocyte adhesion to EC. As TSP-1 is the multidomain homotrimer, we elucidated which domain of TSP-1 is involved in stimulation of CAM expression. The complex TSP-1 structure supports a diversity of functions. Each domain of the TSP-1 molecule exhibits distinct and sometimes opposite biological activities through interactions with specific receptors. For example, the TSP-1 CSVTCG peptide from the type-1 domain binds to CD36, and this interaction leads to apoptosis of EC (28, 29). The C-terminal fragment of TSP-1, when recognized by integrin-associated protein (IAP), inhibits homotypic aggregation of monocytes (29). TSP-1-IAP interactions seem to be of particular importance, as these proteins are up-regulated simultaneously during vascular injury (30). The Arg-Gly-Asp motif in the TSP-1 sequence has been identified as a binding site for αIIbβ3 and αvβ3 integrins (31). Several studies suggest a cooperation between integrin and IAP in TSP-1 recognition (32).
In this report, we show that the IAP binding peptide, derived from the TSP-1 sequence, mimics cellular response caused by the full-length TSP-1 molecule, suggesting that CAM expression is mediated by TSP-1-IAP interaction. The blockade of TSP-1-IAP binding by antibodies or by down-regulation of IAP using small interfering RNA (siRNA) completely abolished expression of CAM in response to TSP-1. It is interesting that TSP-1-IAP interactions appear to be involved in CAM expression stimulated by TNF-α, as siRNA against IAP, similar to blocking TSP-1 interaction with IAP, significantly inhibited TNF-α-stimulated CAM expression. We also show that TNF-α at low concentration stimulates TSP-1 and IAP expression. Taken together, our data suggest that the TSP-1-IAP complex is an important regulatory element of CAM expression on EC.
MATERIALS AND METHODS
Protein and peptide purification
TSP-1 was purified from platelets (33) or expressed as a recombinant protein TSP-1 (rTSP-1) in a baculovirus expression system as described (34). Endotoxin was removed during TSP-1 preparation using Detoxi-Gel from Pierce (Rockford, IL, USA). The endotoxin level was assessed by a quantative chromogenic Limulus amoebocyte lysate (LAL) test QCL-1000 (BioWhittaker, Walkersville, MD, USA). The N-terminal part of TSP-1 (NoC, amino acids 1–365) was kindly provided by Dr. Deane Mosher (University of Wisconsin, Madison, WI, USA). All proteins were >95% pure, as judged by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) at reducing (50 mM dithiothreitol, Pierce) and nonreducing conditions and staining with Coomassie brilliant blue. The CD36 binding peptide, CSVTCG, was derived from the TSP-1 sequence (amino acids 429–434), and the IAP binding peptide, RFYVVMWK (VV; amino acids 1116–1123 of TSP-1), and the control peptide, RFYGGMWK (GG), were synthesized and purified using high-pressure liquid chromatography. The functions of these peptides have been reported previously by others (35, 36). Peptide purity was assessed by mass spectroscopy.
Expression of CAM
The effect of TSP-1 on CAM expression was demonstrated using three types of EC: human microvascular EC line (HMVEC), human aortic EC (HAEC), and human umbilical vein EC (HUVEC). HMVEC (HMEC-1's, a gift from Dr. Fransico J. Candal, Center for Disease Control, Atlanta, GA, USA) were cultured in MCDB131 media supplemented with 10% fetal calf serum (FCS), 10 ng/ml epidermal growth factor (EGF), and 1 μg/ml hydrocortisone as described previously (37). HUVEC were obtained from cords collected through Birthing Services Department at the Cleveland Clinic Foundation (CCF) and the Prenatal Clinical Research Center at the Cleveland Metrohealth Hospital (Cleveland, OH, USA). Dr. Donald W. Jacobsen (CCF) provided HAEC. HUVEC and HAEC were cultured as described (38). To assess the expression of E-selectin, ICAM-1, or VCAM-1, EC were maintained in 0.5% FCS in the presence of various concentrations of TSP-1, peptides, or TNF-α for 4 (for E-selectin) and 17 (for VCAM-1 and ICAM-1) h (39). Cells treated with the same volume of phosphate-buffered saline (PBS) were used as negative controls. After trypsinization, cells were washed with Hanks' balanced salt solution, supplemented with 25 mM HEPES, 1 mM MgCl2, and 1 mM CaCl2, and incubated with primary antibodies: rabbit antibodies against ICAM-1 αICAMDER803 (20 μg/ml); mouse monoclonal antibodies (mAb) against VCAM-1 CBL206 (10 μg/ml, Cymbus Biotech, Chandlers Ford, UK), or anti-E-selectin antibodies MAB2150 (10 μg/ml, Chemicon, Temecula, CA, USA) in the presence of 0.2% bovine serum albumin for 1 h on ice. Cells were then incubated with secondary Alexa–Fluor-labeled antibodies (Molecular Probes, Eugene, OR, USA). Results were analyzed by flow cytometry. The CAM expression in the presence of different agents was represented as the increase in the means of fluorescence intensity (MFI) value over control (nonstimulated cells).
Cell lysates were subjected to Western blotting using rabbit polyclonal antibodies against ICAM-1. β-actin, detected by AC-15 mAb (Sigma Chemical Co., St. Louis, MO, USA), was used as a loading control. Intensities of the band were quantified using the ScanImage program.
Mouse mAb against TSP-1, Ab-3 (4–10 μg/ml, Lab Vision, Fremont, CA, USA), were used to block TSP-1 interaction with IAP.
Immunocytohistochemistry
HAEC were grown on coverslips and preincubated with 3 μg/ml TSP-1, 10 μg/ml TNF-α, or PBS in 0.5% fetal bovine serum for 17 h. Staining of formaldehyde-fixed cells was performed as described previously (40) using anti-ICAM-1 mAb sc-107 (2 μg/ml, Santa Cruz Biotechnology, Santa Cruz, CA, USA). Slides were coated with mounting medium containing 4′-6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA, USA) to stain the nuclei and were then covered and analyzed with a Leica microscope. Controls containing only the secondary antibody were processed as above, omitting the primary antibody, to determine nonspecific binding. ImagePro software was used to compare the square and intensity of color (green) staining on selected areas (arbitrary values). For statistical analysis, the areas with identical number of cells were chosen. The average values are presented in the text.
Monocyte binding
HMVEC were cultured in 24-well plates in MCDB131 media supplemented with 10% FCS, 10 ng/ml EGF, and 1 μg/ml hydrocortisone as described previously (37). Once a confluent monolayer was formed, cells were treated for 17 h with TSP-1 or TSP-1-derived peptides in Dulbecco's modified Eagle's medium containing 1% FCS. The endothelial monolayer was then rinsed three times with serum-free media, and 51Cr-labeled THP-1 monocytes were added at 3.75 × 105 cells per well. After a 45-min incubation at 37°C, the unbound monocytes were removed by repeated washing, and bound cells were quantified by measuring the remaining radioactivity.
Construction of siRNA-expressing plasmids
The human IAP cDNA sequence was obtained from Gene Bank (NM 001777). Two 64 nucleotide-long primers with a 21-nucleotide homology to this cDNA were designed (siRNA-IAP-_Bam_HI-direct, 5′gatccggatggataagagtgatgctgtcttcctgtcaacagcatcactcttatccatctttttg, and siRNA-IAP-_Eco_RI-reverse, 5′aattcaaaaagatggataagagtgatgctgttgacaggaagacagcatcactcttatccatccg). Following primer annealing, the resulting double-stranded DNA fragment with _Bam_HI and _Eco_RI cohesive ends was cloned into a lentiviral pLSLG plasmid (a generous gift from Dr. Peter Chumakov, CCF). The resulting pLSLG-IAP-siRNA plasmid expressed an anti-IAP-siRNA as a stem-loop RNA under H1 promoter control. Anti-luciferase (luc)-siRNA was used as a control for unspecific siRNA effects in target cells. The luciferase sequence was obtained from pSP-luc+, a luciferase cassette vector sequence (U47122), and cloned into the pLSLG plasmid using the above procedure and the following primers: siRNA-luc-_Bam_HI-direct, 5′gatccgcacttacgctgagtacttcgacttcctgtcatcgaagtactcagcgtaagtgtttttg, and siRNA-luc-_Eco_RI-reverse, 5′aattcaaaaacacttacgctgagtacttcgatgacaggaagtcgaagtactcagcgtaagtgcg.
Lentivirus packaging and EC infection procedure
For lentiviral stock preparation, 293T packaging cells (41) were transfected with pLSLG-IAP-siRNA or pLSLG-luciferase-siRNA plasmid DNA and two helper plasmids (pCMVΔR8.2 and pVSVG coding for lentivirus packaging proteins) using Lipofectamin-Plus (Invitrogen, Carlsbad, CA, USA). Thirty-six hours after transfection, media containing packaged lentivirus were collected, passed through 0.45 μ filters, and added to target EC cells for at least 12 h. Polycation hexadimethrine bromide (polybrene) was obtained from Sigma (Deisenhofen, Germany) and added to lentiviral stocks at a concentration of 4 μg/ml to improve infection efficiency. IAP expression was assessed using fluorescein-activated cell sorter (FACS) analysis and Western blot with B6H12 mAb (BD Pharmingen, Palo Alto, CA, USA).
Luciferase reporter assay
HUVEC were seeded in 12-well plates (80% confluence). Six hours later, part of the wells were infected with virus expressing inhibitor of nuclear factor-κB (NF-κB)-I-κB or green fluorescent protein (GFP; control). Eighteen hours later, cells were transfected with a luciferase reporter plasmid detecting NF-κB activity (pNF-κB-Luc) (Stratagene, La Jolla, CA, USA) at a final concentration of 1 μg/ml using lipofectine reagent. Simian virus 40 promoter-driven β-galactosidase reporter vector (0.1 μg/ml) was used as a transfection control. Following transfection, the cells were allowed to recover overnight and were then treated with TNF-α (R&D Systems, Minneapolis, MN, USA) at a concentration of 10 ng/ml in growth medium for 6 h. Cells were then washed twice in PBS and harvested by scraping in 100 μl lysis buffer (Promega, Madison, WI, USA). Cell debris was pelleted by centrifugation, and the supernatant was assayed for luciferase and β-galactosidase activity. Luciferase activity was determined by combining 30 μl cell extracts with 100 μl luciferase reagent (Promega) and measuring luminescence using a microplate luminometer. β-Galactosidase activity was determined using the Galacto-Light Plus assay kit from Tropix (Bedford, MD, USA). Transfections were carried out in triplicate, and the data were corrected for transfection efficiency using β-galactosidase activity.
Isolation of mouse aortic EC
TNF receptor (TNFR)I null and TNFRII null mice (C57BL/6 background) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). EC from the thoracic aorta were isolated by a technique described previously (42). EC authenticity was verified by staining for von Willebrand factor and endothelial-specific gene expression profiling by real-time polymerase chain reaction.
RESULTS
In view of TSP-1 importance in atherothrombosis, studies were designed to examine whether TSP-1 affects the expression of CAM and monocyte adhesion to endothelium. For this purpose, TSP-1 purified from platelets as well as a recombinant TSP-1 expressed in a baculovirus system were used (Fig. 1A). Both protein preparations were more than 95% pure, as determined by SDS-PAGE (Fig. 1Aa). The identity of purified proteins was verified by Western blot analysis using TSPB-7 mAb (Sigma Chemical Co.) (Fig. 1Ab). To analyze an effect of TSP-1 on EC, various concentrations of purified protein were added to EC of different origin, including HUVEC, HAEC, and microvascular HMVEC, and CAM expression was assessed by FACS analysis. The peak of cellular response to TNF-α, which is known to up-regulate major CAM on the various EC, was observed 17–18 h after stimulation, which is in agreement with previous observations (43). MFI for ICAM-1 on HUVEC were 230, 584, 692, 1278, and 1115 at 2, 4, 8, 17, and 24 h of stimulation with TNF-α. Likewise, maximum expression of VCAM-1 and ICAM-1 was also observed 17 h after the addition of TSP-1 (Fig. 1B).
Figure 1.
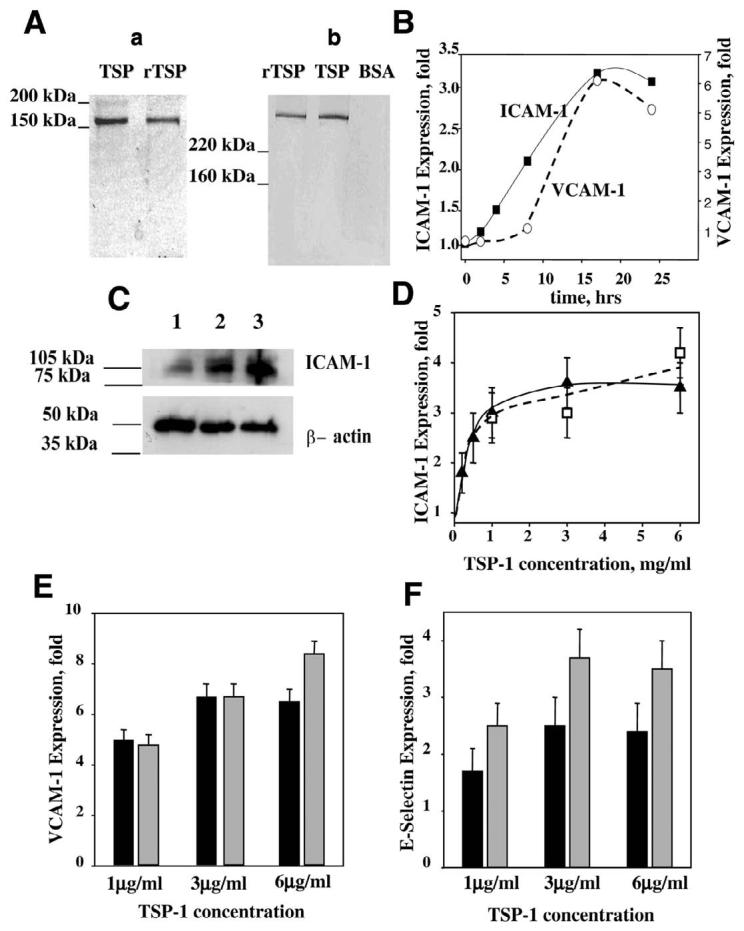
A) SDS-PAGE analysis of TSP-1 purified from platelets and recombinant TSP-1 expressed in the baculovirus system: Aa) Coomassie staining (reducing conditions, 50 mM DTT); Ab) Western blot (nonreducing conditions). Expression of CAM on HUVEC. B) Time-dependence of ICAM-1 (■) and VCAM-1 (○) surface expression on TSP-1-treated cells. TSP-1 concentration was 3 μg/ml. C) Western blot shows the level of ICAM-1 in control-untreated HUVEC (lane 1) or in HUVEC after 17 h of treatment with rTSP-1 (lane 2) or TNF-α (lane 3). The cell lysate was subjected to Western blot using rabbit polyclonal antibodies against ICAM-1. β-actin was used as a loading control. Effect of various concentrations of TSP-1 on ICAM-1 (D), VCAM-1 (E), and E-selectin (F) expression on HUVEC. Cells were incubated in the presence of recombinant TSP-1 (D, open symbols; E and F, shaded bars) or TSP-1 purified from platelets (D, closed symbols; E and F, solid bars) for 4 or 17 h to assess the expression of E-selectin or ICAM-1 and VCAM-1, respectively. The surface expression of CAM was analyzed by flow cytometry, as described in Materials and Methods. The increases in MFI over control (PBS buffer) are shown (_P_≤0.008). The data are means ± sd of three to seven separate experiments.
Expression of CAM by HUVEC in response to a number of proinflammatory stimuli, such as TNF-α, interleukin (IL)-1α, and IL-4 (43-45), has been characterized extensively in the literature, and hence, we first assessed the effect of TSP-1 treatment on these cells. Expression of ICAM-1 in HUVEC was evaluated by Western blotting analysis (Fig. 1C). Treatment with rTSP-1 significantly increased ICAM-1 content in EC lysate (Fig. 1C, lane 2). As a result of rTSP-1 stimulation, the density of the ICAM-1 band (normalized to β-actin) increased 3.8-fold compared with nonstimulated cells. TNF-α, which served as a positive control, induced a 5.2-fold increase in ICAM-1 expression. Surface expression of CAM monitored by FACS also was notably increased by TSP-1. Platelet-derived, as well as recombinant TSP-1, induced surface expression of ICAM-1, VCAM-1, and E-selectin on HUVEC in a concentration-dependent manner shown in Fig. 1D-F, respectively). For all three CAM monitored, maximal expression was observed at a concentration of 3 μg/ml platelet TSP-1. Maximum increases of MFI were 3.5-, 6.7-, and 2.4-fold over control for ICAM-1 (Fig. 1D), VCAM-1 (Fig. 1E), and E-selectin (Fig. 1F), respectively. rTSP-1 also had a marked effect on the expression of CAM, as it induced 4.8-, 8.2-, and 3.7-fold increases in an expression of ICAM-1, VCAM-1, and E-selectin, respectively.
The increase in CAM expression was not a result of contamination of TSP-1 with endotoxin, as observed effects were not influenced by polymixin treatment of TSP (data are not shown). Moreover, by a quantitative, chromogenic LAL test, the amount of endotoxin in TSP-1 (or peptides used subsequently) solutions was similar or lower than that in control samples (buffer or control peptides), and this amount of endotoxin did not stimulate CAM expression in HUVEC.
To demonstrate an effect of TSP-1 on EC of a different vascular origin, HAEC were used. As shown in Fig. 2A, this cell type was also responsive to TSP-1 treatment. A significant increase in ICAM-1 expression on HAEC was observed when monolayer cells were treated with rTSP-1 (Fig. 2A). The stimulatory effect of TSP-1 on HAEC was comparable with that of TNF-α. As analyzed by ImagePro software, the average intensity of ICAM-1 staining in rTSP-1-treated cells was 56 compared with 74 in TNF-α-treated cells and 27 in control. When quantified by FACS, rTSP-1 increased expression of ICAM-1 on HAEC threefold compared with control, and TNF-α induced an approximate sixfold increase (Fig. 2B).
Figure 2.

Expression of CAM on HAEC and HMVEC. Cells were incubated for 17 h in the presence of 3 μg/ml TSP-1 or rTSP-1, 10 ng/ml TNF-α, or PBS buffer as a negative control. A) Immunostaining of formaldehyde-fixed HAEC using anti-ICAM-1 mAb as described in Materials and Methods. B, C) Expression of ICAM-1 on HAEC and VCAM-1 on HMVEC, respectively. The data were analyzed by flow cytometry. The increases in MFI over control (PBS) are shown (_P_≤0.01).
In view of the fact that microvascular EC regulate their CAM differently than EC from large vessels (46), the effect of TSP-1 on microvascular HMVEC was examined. A significant increase in CAM expression in the presence of TSP-1 was observed on HMVEC as assessed by FACS analysis. The expression of VCAM-1 in the presence of TSP-1 increased 2.5-fold over control. Under the same conditions, TNF-α produced a 3.4-fold increase of VCAM-1 over control (Fig. 2C). TSP-1 and TNF-α were also effective stimulators of ICAM-1 in HMVEC with an expression increase of 3.8- and 9.1-fold over control, respectively (Fig. 3A).
Figure 3.
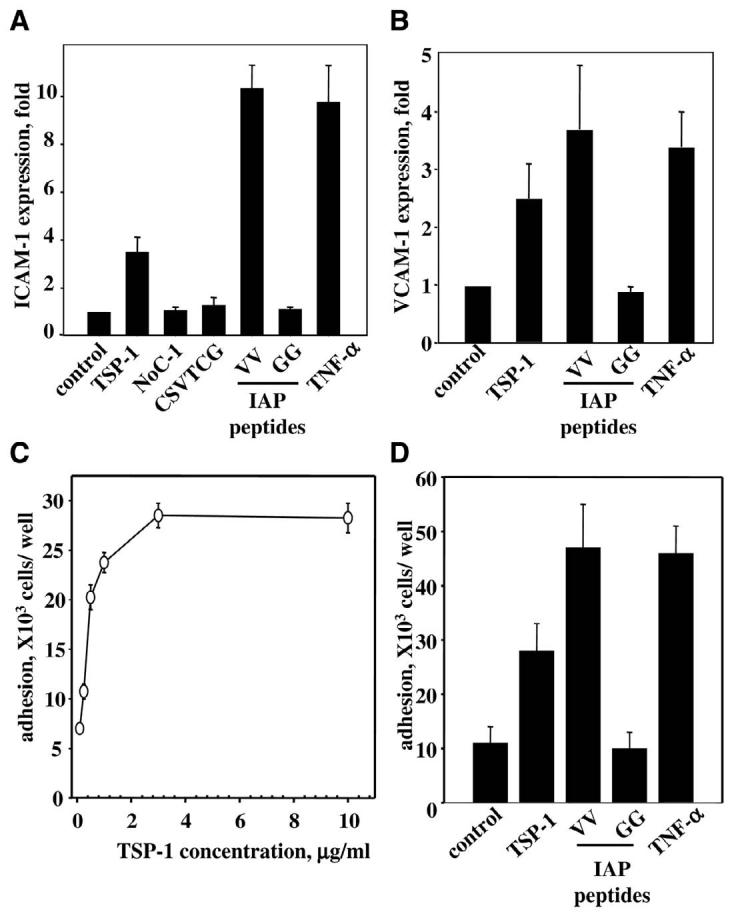
Expression of CAM and monocyte adhesion to HMVEC. A, B) Expression of ICAM-1 (A) and VCAM-1 (B) on HMVEC. Cells were incubated for 17 h in the presence of 10 ng/ml TNF-α, 3 μg/ml TSP-1, 3 μg/ml N-terminal fragment of TSP-1 (NoC-1; amino acids 1–356 of TSP-1), or 50 μM TSP-1-derived peptides: CD-36 binding peptide CSVTCG, IAP binding peptide VV (RFYVVMWK), and control peptide GG (RFYGGMWK). The expression of CAM was analyzed by flow cytometry. The increases in MFI over control (PBS) are shown (_P_≤0.02). The data are means ± SD of three to five separate experiments. C, D) Adhesion of monocytes to endothelium. HMVEC grown in 24-well plates were treated with TSP-1 for 17 h or remained untreated. THP-1 cells were added to HMVEC at 3.75 × 105 cells per well. After 45 min incubation at 37°C, the unbound monocytes were removed by repeated washing, and bound cells were quantified by measuring radioactivity remaining in wells. Data are normalized on background level. C) Effect of various concentrations of TSP-1 on monocyte adhesion. The data are means ± SD of three separate experiments. D) Effect of TSP-1 and TSP-1-derived peptides on adhesion of THP-1 cells. The concentration of TSP-1 as well as NoC-1 fragment of TSP-1 (amino acids 1–356) was 3 μg/ml; IAP binding peptide VV (RFYVVMWK), control peptide GG (RFYGGMWK), and CD-36 binding peptide (CSVTCG), 75 μM; TNF-α, 10 ng/ml. The data are means ± SD of three separate experiments.
In contrast, TSP-1 treatment did not affect the expression of the well-described TSP-1 receptor, CD36, as well as the most prominent integrin on EC, α5β1: MFI for CD36 were 87 ± 10 and 83 ± 12 and for α5β1 were 1300 ± 50 and 1290 ± 40 for cells treated with and without TSP-1, respectively. Thus, TSP-1 appears to specifically affect the expression of CAM associated with leukocyte recruitment in inflammation and atherosclerosis development.
As TSP-1 is a multidomain protein, next, we sought to determine what part of TSP-1 is responsible for up-regulation of CAM. Accordingly, we tested a series of peptides and fragments derived from TSP-1, which are known to interact with different TSP-1 receptors (Fig. 3). Under the conditions of analysis, the CD36 binding peptide (CSVTCG) from the TSP-1 sequence and the N-terminal fragment of TSP-1 (NoC-1, residues 1–356) were ineffective in stimulating ICAM-1 expression on HMVEC. However, the RFYVVMWK (VV) peptide, derived from the C-terminal domain of TSP-1, up-regulated ICAM-1 by 5.4-fold, which was comparable with the effect of TNF-α. At the same concentration, the control peptide RFYGGMWK (GG) did not have any effect on the expression of ICAM-1 (Fig. 3A). The RFYVVMWK peptide is well described in literature as a binding site for IAP. The IAP binding peptide, but not the control peptide, was also a potent stimulator of VCAM-1 expression on HMVEC: MFI for the RFYVVMWK-treated HMVEC increased 3.6-fold over control compared with 3.4-fold over control in the presence of TNF-α (Fig. 3B). These data indicate a potential role of IAP in CAM expression stimulated by TSP-1. Considering the important role of CAM in interactions of blood cells with endothelium, we examined whether treatment of EC for 17 h with TSP-1 promotes monocyte adhesion. The amount of 51Cr-labeled THP-1 monocytes adherent to HMVEC after 45 min of incubation was assessed by measuring the amount of bound radioactivity after washing as described in Materials and Methods. Preincubation of HMVEC with TSP-1 resulted in increased cell adhesion to HMVEC. As with CAM expression, TSP-1 stimulated monocyte adhesion to endothelium in a concentration-dependent manner, and a saturation level was achieved at the concentration of 3 μg/ml TSP-1 (Fig. 3C). The number of monocytes bound to HMVEC treated with TSP-1 was 2.8-fold higher compared with untreated cells. Similar to results regarding CAM expression, treatment of HMVEC with the IAP-specific peptide RFYVVMWK resulted in a dramatic increase of monocyte adhesion (4.6-fold over control), whereas the control RFYGGMWK peptide did not have any effect (Fig. 3D). Thus, TSP-1 appears to induce monocyte adhesion to EC via its interaction with IAP.
To further elucidate the role of IAP in TSP-1-induced CAM expression, we took advantage of siRNA technology, which permits specific knockdown of a target gene (47). We introduced siRNA specific to IAP (siIAP) to HUVEC and HMVEC. To distinguish the unspecific effect of lentiviruses and the effect of siRNA, we used anti-luciferase siRNA (siLuc) as a control. pLSLG-IAP-siRNA and pLSLG-luciferase-siRNA plasmids contained a GFP as an infection marker to identify the subpopulation of infected cells by FACS analysis (Fig. 4A). Infection efficiency was more than 80% for both siRNAs, as determined by GFP expression. Based on FACS analysis, infection with siIAP resulted in more than a twofold decrease of surface IAP expression in HMVEC: MFI was 88 for siIAP-infected cells compared with 211 for noninfected cells and 185 for siLuc-infected HMVEC. Using Western blot analysis of cell lysates, more than a fourfold decrease in IAP protein was observed as a result of infection with siIAP as compared with noninfected and siLuc-infected HMVECs (Fig. 4B). Similar data were obtained for HUVEC (data not shown). Therefore, siIAP was an efficient tool to down-regulate IAP expression in EC. Upon siIAP infection, no significant changes in cell morphology and behavior were observed in HMVEC or HUVEC, with the exception of a slightly decreased proliferation rate in siIAP-infected cells, as the average time required to double the cell number in the culture was 28 h for siIAP-infected HMVEC (HMVEC-siIAP) compared with 19 h for uninfected cells and 21 h for HMVEC infected with siLuc (HMVEC-siLuc).
Figure 4.

The infection of EC by lentivirus-expressing siRNA. A) Micrographs of uninfected HMVEC and HMVEC infected with lentiviruses expressing the GFP gene along with siIAP or siLuc under visible and ultraviolet light. B) IAP expression in HMVEC. Cell lysates were subjected to Western blot using B6H12 mAb against IAP: (lane 1) cells infected with siIAP; (lane 2) noninfected cells; (lane 3) cells infected with siLuc. β-actin was used as a loading control.
siIAP completely inhibited the stimulating effect of TSP-1 on CAM expression in HMVEC (Fig. 5A) and HUVEC (Fig. 5B). In contrast, TSP-1 did stimulate ICAM-1 expression up to a 1.8-fold increase in control HMVEC-siLuc (Fig. 5A) and up to 1.75 in control HUVEC-siLuc (Fig. 5B). Moreover, antibody Ab-3, which is known to specifically block TSP-1 binding to IAP but not antibody Ab-1, which blocks TSP-1-CD36 binding, also provided significant (more than 80%) inhibition of TSP-1-induced CAM expression (data not shown). Taken together, these results clearly demonstrate a key role of IAP as a mediator of TSP-1-induced CAM expression.
Figure 5.
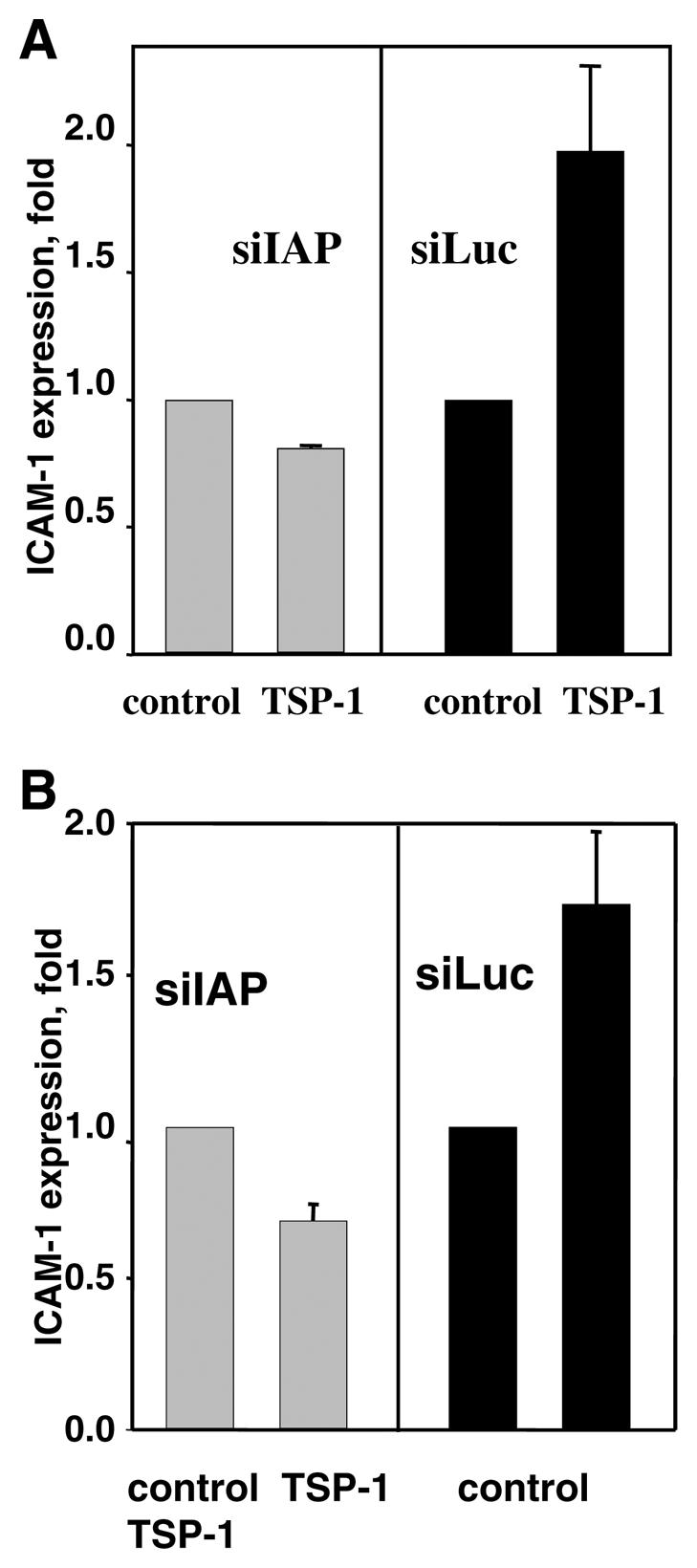

Expression of CAM on EC infected with siIAP or siLuc. A, B) The expression of ICAM-1 on HMVEC (A) or HUVEC (B) in the presence of 3 μg/ml TSP-1 (17 h of treatment). C, D) The expression of VCAM-1 on HUVEC (C) and ICAM-1 on HMVEC (D) in the presence of 10 ng/ml TNF-α (17 h of treatment). E) Effect of Ab-3 TSP-1-blocking antibodies or normal IgG on the expression of ICAM-1 on HUVEC in the presence of 2, 5, and 10 ng/ml TNF-α. Antibody concentration was 4 μg/ml. Data were analyzed by flow cytometry: The increases in MFI over control (PBS) are shown (_P_≤0.01). The data are means ± sd of five separate experiments.
EC response to TNF-α was also impaired as a result of IAP knockdown. For example, VCAM-1 expression on HUVEC-siIAP in the presence of 10 ng/ml TNF-α was decreased twofold compared with control HUVEC-siLuc (Fig. 5C). Similar results were obtained using HMVEC: Up-regulation of expression of ICAM-1 by TNF-α was diminished in HMVEC-siIAP compared with HMVEC-siLuc (1.4-fold increase vs. 4.8-fold increase over control; Fig. 5D). The increase in TNF-α concentration up to 20 ng/ml partially reduced but not completely impeded the siIAP effect (data not shown). It appears that the TSP-1-IAP interaction may also be involved in TNF-α-stimulated CAM expression. If this is the case, neutralization of TSP-1 should also decrease the TNF-α-induced response. To address this possibility, we used Ab-3 antibodies against TSP-1, which are known to recognize the C-terminal part of TSP-1 and specifically block TSP-1 interaction with IAP. Indeed, as shown by FACS analysis, Ab-3, at concentrations as low as 4 μg/ml, significantly (>80%) inhibited binding of exogenous TSP-1 to the EC surface as well as its effect on CAM expression (data not shown). Similar to the effect of siIAP, Ab-3 mAb significantly diminished CAM expression stimulated by TNF-α (Fig. 5E). At 4 μg/ml, Ab-3 was able to totally block CAM up-regulation by TNF-α at concentrations of 2 or 5 ng/ml. Thus, the blockade of TSP-1-IAP interaction as well as knockdown of IAP on EC resulted in the down-regulation of TNF-α-induced CAM expression. At a concentration of 10 ng/ml TNF-α, the inhibitory effect of Ab-3 was ∼60% (Fig. 5E). Higher concentration of Ab-3 (10 μg/ml) was even more effective and provided 80% inhibition of cellular response to 10 ng/ml TNF-α. However, the complete blockade of ICAM-1 expression induced by 10 ng/ml TNF-α was not achieved using Ab-3 at concentrations up to 20 μg/ml. Taken together, these results demonstrated that a TSP-1-IAP interaction plays an important role in regulation of CAM expression stimulated by low concentrations of TNF-α. Cellular response induced by higher concentrations of TNF-α seems to be controlled by alternative pathways.
NF-κB is known to be a key mediator of CAM expression after stimulation of endothelium with TNF-α (48). To shed light on the signaling pathway induced by TSP-1 and compare this response with that of TNF-α, we sought to assess whether TSP-1 is able to activate NF-κB. Using a NF-κB-dependent promoter construct, we monitored NF-κB activity after EC treatment with TSP-1 (Fig. 6A). We found that TSP-1 induced a threefold increase in promoter activity (normalized to control). In agreement with the literature (49), TNF-α provided a fivefold increase of NF-κB-dependent promoter activity. In both cases, the increased activity was blocked completely by infection of EC with adenovirus encoding I-κB, the canonical suppressor of NF-κB activity, but not with control GFP adenovirus (Fig. 6A).
Figure 6.

A) The effect of rTSP-1 and TNF-α on NF-κB activity in HUVEC (solid bars), HUVEC infected with adenovirus-encoding inhibitor of NF-κB (I-κB; shaded bars), and HUVEC infected with adenovirus-encoding GFP (striped bar). rTSP-1 concentration was 5 μ g/ml; TNF-α concentration was 10 ng/ml. NF-κB activity was analyzed using a luciferase reporter assay. Effect of TNF-α on the expression of IAP and TSP-1 on the EC. B) Western blot shows the level of TSP-1 synthesis in HUVEC after 17 h of treatment with TNF-α. The cell lysate was subjected to Western blot using TSPB-7 mAb against TSP-1. β-actin was used as a loading control. C) Effect of various concentrations of TNF-α on ICAM-1 (solid box) and TSP-1 (shaded circle) expression on HUVEC. The increases in MFI over control (PBS buffer) are shown. The data are means ± sd of three experiments. D) Representative data of FACS analysis. B6H12 mAb were used to assess the surface expression of IAP: shaded spectra, nontreated cells; open spectra, cells treated with 10 ng/ml TNF-α for 17 h.
As little is known about activators of TSP-1 expression in EC, experiments were performed to test whether this protein is up-regulated in TNF-α-activated endothelium. Expression of TSP-1 on HUVEC was evaluated by Western blotting and FACS analysis. TNF-α treatment for 17 h significantly increased TSP-1 content in an EC lysate, as assessed by Western blotting (Fig. 6A). Likewise, surface expression of TSP-1 on EC was notably increased. FACS analysis demonstrated more than a fourfold increase of TSP-1 on HMVEC treated with TNF-α for 17 h compared with untreated control (Fig. 6B). TNF-α induced a bell-shaped effect on TSP-1 expression. Similar to ICAM-1 expression, a maximum of TSP-1 on EC surface was observed at 10 ng/ml TNF-α (Fig. 6C). These findings were further supported by an analysis of TSP-1 expression using a TSP-1 promoter-luciferase construct. In these experiments, TNF-α induced a concentration-dependent increase of TSP-1 transcription in EC (data not shown). In agreement with the previous report (50), IAP expression was also affected by TNF-α. Figure 6D shows more than a twofold increase of surface expression of IAP as assessed by FACS analysis. Thus, it appears that TNF-α up-regulates IAP and TSP-1, and these two proteins are involved in the TNF-α-stimulated expression of CAM.
There are two distinct TNF-α receptors (TNFRI and TNFRII) expressed on EC, which are known to regulate cellular responses to this cytokine differentially (51, 52). Thus, next, we sought to investigate which receptor is critical for up-regulation of TSP-1 in response to TNF-α. To address this issue, we isolated primary EC from mice lacking TNFRI or TNFRII and assessed expression of TSP-1 in response to TNF-α. We have found that stimulation of TNFRI null cells resulted in a fivefold increase of TSP-1 expression as determined by FACS analysis. At the same time, TNFRII null cells did not exhibit a significant increase in TSP-1 expression. This result implicates TNFRII in regulation of TSP-1 expression in EC.
DISCUSSION
The expression of CAM, including ICAM-1 and VCAM-1, is one of the major markers of EC dysfunction that occurs at the early stages of inflammation and atherosclerosis development (4). In this manuscript, we considered a possible role of the extracellular matrix protein, TSP-1, which is known to be present and up-regulated at the sites of vascular injury, in the regulation of CAM expression on endothelium from various origins. The major findings of our studies are as follows: 1) TSP-1 (purified from platelets or recombinant) specifically induces expression of ICAM-1, VCAM-1, and E-selectin in a concentration-dependent manner, reaching a maximum at TSP-1 concentration of 3 μg/ml. At the same time, the expression levels of CD36 and integrin α5β1 remained unchanged. This effect was observed using EC of different origins, including macrovascular EC, such as HUVEC and HAEC, and microvascular EC. 2) The expression of CAM on endothelium was stimulated by the IAP-specific peptide derived from TSP-1 but not by peptides corresponding to other fragments of TSP-1. Disruption of TSP-1-IAP interaction by blocking antibodies or by knockdown of IAP with siRNA completely inhibited TSP-1-stimulated CAM expression. 3) TSP-1 and the IAP-specific peptide derived from TSP-1 dramatically increased monocyte adhesion to endothelium. Thus, we have shown for the first time that TSP-1, via its receptor IAP, targets endothelium and promotes expression of CAM and subsequent attachment of monocytes. 4) The activation of EC by TNF-α, at least in part, depends on TSP-1 expression and its binding to IAP. TNF-α stimulates the expression of TSP-1 and IAP on EC in a concentration-dependent manner. Knockdown of IAP as well as blockade of TSP-1-IAP interaction diminish TNF-α-induced CAM expression. Together, these data suggest that TNF-α acts via a TSP–IAP complex to stimulate CAM expression on EC. An up-regulation of TSP-1 and IAP enhances the stimulatory effect of TNF-α on CAM expression.
TSP-1 is a complex, multi-domain matrix protein with a number of regulatory and structural functions, some of them directly relating to vascular injury and atherosclerosis. Immunohistochemical studies demonstrated that TSP-1 is deposited within the vessel wall at the early stages of atherosclerotic plaque development (18, 19). The blockade of TSP-1 by antibodies accelerates re-endothelization and reduces neointima formation in balloon-injured carotid arteries (15). All these data together show a vital role of TSP-1 in inflammatory processes and in particular, in atherosclerosis development. However, the molecular mechanisms of TSP-1 action in processes associated with vascular injury have not been determined. Our finding that TSP-1 regulates an expression of CAM and monocyte binding to endothelium assigns a new role to TSP-1 and suggests that this matrix protein, which is abundant at the sites of vascular injury and platelet activation, might trigger the initial stages of inflammation and atherogenesis. This finding is in agreement with the recently proposed role of activated platelets as inducers of atherosclerosis in mice (10). As TSP-1 is the major protein released from activated platelets, its effect on CAM expression and monocyte adhesion to endothelium provides a mechanism for an observed stimulatory effect of activated platelets on atherosclerosis development. Stimulation of CAM expression by TSP-1 appears to be selective, as TSP-1 does not affect the expression of other adhesion molecules such as CD-36 and β1 integrins.
The effects of TSP-1 on vascular cells are mediated by interactions with cell-surface receptors. One of the known TSP-1 receptors, CD36, binds TSP-1 and mediates signal transduction in human monocytes (53). This function appears to involve the SVTCG peptide derived from TSP-1, which binds to CD36 with high affinity (54). However, in our experiments, the SVTCG peptide as well as the N-terminal fragment of TSP-1 was not able to stimulate CAM expression. TSP-1 has been reported to interact with another receptor on the cell surface: IAP (CD47; ref. 55). Interaction of TSP-1 with an αVβ3 integrin-IAP complex appears to regulate the flow-induced apoptosis of endothelium (56). It is interesting that vascular expression of IAP is increased simultaneously with TSP-1 after mechanical injury, providing in vivo evidence for potential communication between IAP and TSP-1 (30). The RFYVVMWK peptide, which is highly conserved in all isoforms of the thrombospondin family among all species (57), has been identified as an IAP agonist (32). Results of recent studies by Kvansakul and coauthors (58) demonstrate that the RFYVVMWK sequence appears to be buried and not available for the direct interaction with IAP. These results do not exclude, however, the possibility that a small population of TSP-1 molecules might exist in a conformation with this sequence exposed or may become exposed as a result of conformational changes. Based on studies using IAP-deficient mice, it is clear that the RFYVVMWK peptide is crucial for efficient TSP-1-IAP interaction. For example, aortic smooth muscle cells from wild-type but not from IAP null mice are able to migrate toward this peptide (59). In addition, platelets from IAP-deficient mice fail to aggregate on a fibrinogen-coated surface in response to the RFYVVMWK peptide (60). Taken together, it appears that the RFYVVMWK peptide is specifically recognized by IAP. Our analysis revealed that the RFYVVMWK peptide, but not the control RFYGGMWK peptide (36), is able to promote CAM expression as well as monocyte adhesion, suggesting a possible involvement of IAP.
Using siIAP to decrease IAP expression, we have shown a complete ablation of the effect of TSP-1 on CAM production. It is interesting that infection of EC with siIAP, similar to blocking TSP-1 interaction with IAP, also inhibited TNF-α-stimulated CAM expression. This effect is more prominent at low concentrations of TNF-α. Moreover, similar to TNF-α, TSP-1 treatment of EC activates the NF-κB transcriptional factor, which is known to be an important regulator of CAM expression. Therefore, our results suggest that TSP-1 might affect CAM expression at the transcriptional level.
We have also shown that TNF-α up-regulates TSP-1 expression in EC. These data are not in agreement with previously published results of Morandi and coauthors (61), which show a modest inhibitory effect of TNF-α on TSP-1 expression. Discrepancy in results may be a result of slightly different experimental conditions, i.e., confluence level, which is known to be important for TSP-1 production. In our experiments, we observed a bell-shaped concentration response to TNF-α. It appears that expression of TSP-1 is regulated via TNFRII but not TNFRI. This is in agreement with several reports (62, 63), which emphasize a preferential role for this particular receptor in cell activation by TNF-α. Notably, the expression profile of ICAM-1 follows the TSP-1 pattern, showing a link between these two events. Consistent with the literature, we found that IAP is also overexpressed on the endothelium upon TNF-α treatment (50). Moreover, the blockade of the TSP-1-IAP interaction by a specific antibody or by knockdown of IAP significantly inhibits a response to TNF-α. Taken together, we speculate that the TSP-1-IAP complex is necessary for expression of CAM in response to TNF-α, at least at the low concentrations of this cytokine. This observation provides a previously unrecognized mechanism for the regulation of TNF-α-induced CAM expression. It appears that besides modulation of integrin-mediated processes, IAP has numerous regulatory functions that are independent from the integrins. In addition to control of the EC response to TNF-α, documented in this manuscript, it was demonstrated recently that the TSP-1-IAP interaction enhances proliferation of vascular cells induced by insulin-like growth factor-1 (64). As cytokine expression followed by up-regulation of CAM is a main response to bacterial infection, our findings explain, at least in part, the decreased resistance to infection in IAP-deficient mice (10).
To summarize, our findings suggest that TSP-1 promotes CAM expression and monocyte adhesion to endothelium. IAP is involved in this process as a mediator of TSP-1-enodothelium interaction. The TSP-1-IAP complex is an important part of EC activation by TNF-α. Thus, our results reveal a new role for TSP-1 in the regulation of endothelial functions related to vascular injury and atherosclerosis.
ACKNOWLEDGMENTS
This study was supported by National Institutes of Health (NIH) Grants p50 HL77107 and HL071625. N. V. N. is a recipient of an American Heart Association Fellowship (0325229B). HUVEC were obtained from cords collected through the Birthing Services Department at the Cleveland Clinic Foundation and the Prenatal Clinical Research Center (NIH GCRC Award RR-00080) at the Cleveland Metrohealth Hospital. The authors thank Dr. Deane Mosher for the viral stock of human TSP-1 and NoC-1 recombinant fragment from the TSP-1 molecule and Dr. Eric Brown for 1F7 antibodies. We thank Vicky J. Byers-Ward for her technical help.
REFERENCES
- 1.Wood KM, Cadogan MD, Ramshaw AL, Parums DV. The distribution of adhesion molecules in human atherosclerosis. Histopathology. 1993;22:437–444. doi: 10.1111/j.1365-2559.1993.tb00157.x. [DOI] [PubMed] [Google Scholar]
- 2.Krieglstein CF, Granger DN. Adhesion molecules and their role in vascular disease. Am. J. Hypertens. 2001;14:44S–54S. doi: 10.1016/s0895-7061(01)02069-6. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez-Amaro R, Diaz-Gonzalez F, Sanchez-Madrid F. Adhesion molecules in inflammatory diseases. Drugs. 1998;56:977–988. doi: 10.2165/00003495-199856060-00003. [DOI] [PubMed] [Google Scholar]
- 4.Collins RG, Velji R, Guevara NV, Hicks MJ, Chan L, Beaudet AL. P-Selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E- deficient mice. J. Exp. Med. 2000;191:189–194. doi: 10.1084/jem.191.1.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW, Milstone DS. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Invest. 2001;107:1255–1262. doi: 10.1172/JCI11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lenardo MJ, Baltimore D. NF-κ B: a pleiotropic mediator of inducible and tissue-specific gene control. Cell. 1989;58:227–229. doi: 10.1016/0092-8674(89)90833-7. [DOI] [PubMed] [Google Scholar]
- 7.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc. Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 8.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc. Natl. Acad. Sci. USA. 1975;72:3666–3670. doi: 10.1073/pnas.72.9.3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Madge LA, Pober JS. TNF signaling in vascular endothelial cells. Exp. Mol. Pathol. 2001;70:317–325. doi: 10.1006/exmp.2001.2368. [DOI] [PubMed] [Google Scholar]
- 10.Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, Littman DR, Weber C, Ley K. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat. Med. 2003;9:61–67. doi: 10.1038/nm810. [DOI] [PubMed] [Google Scholar]
- 11.Baenziger NL, Brodie GN, Majerus PW. A thrombin-sensitive protein of human platelet membranes. Proc. Natl. Acad. Sci. USA. 1971;68:240–243. doi: 10.1073/pnas.68.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schellings MW, Pinto YM, Heymans S. Matricellular proteins in the heart: possible role during stress and remodeling. Cardiovasc. Res. 2004;64:24–31. doi: 10.1016/j.cardiores.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 13.Adams JC. Thrombospondins: multifunctional regulators of cell interactions. Annu. Rev. Cell Dev. Biol. 2001;17:25–51. doi: 10.1146/annurev.cellbio.17.1.25. [DOI] [PubMed] [Google Scholar]
- 14.Reed MJ, Puolakkainen P, Lane TF, Dickerson D, Bornstein P, Sage EH. Differential expression of SPARC and thrombospondin 1 in wound repair: immunolocalization and in situ hybridization. J. Histochem. Cytochem. 1993;41:1467–1477. doi: 10.1177/41.10.8245406. [DOI] [PubMed] [Google Scholar]
- 15.Chen D, Asahara T, Krasinski K, Witzenbichler B, Yang J, Magner M, Kearney M, Frazier WA, Isner JM, Andres V. Antibody blockade of thrombospondin accelerates reendothelialization and reduces neointima formation in balloon-injured rat carotid artery. Circulation. 1999;100:849–854. doi: 10.1161/01.cir.100.8.849. [DOI] [PubMed] [Google Scholar]
- 16.Lin TN, Kim GM, Chen JJ, Cheung WM, He YY, Hsu CY. Differential regulation of thrombospondin-1 and thrombospondin-2 after focal cerebral ischemia/reperfusion. Stroke. 2003;34:177–186. doi: 10.1161/01.str.0000047100.84604.ba. [DOI] [PubMed] [Google Scholar]
- 17.Stenina OI, Krukovets I, Wang K, Zhou Z, Forudi F, Penn MS, Topol EJ, Plow EF. Increased expression of thrombospondin-1 in vessel wall of diabetic Zucker rat. Circulation. 2003;107:3209–3215. doi: 10.1161/01.CIR.0000074223.56882.97. [DOI] [PubMed] [Google Scholar]
- 18.van Zanten GH, de Graaf S, Slootweg PJ, Heijnen HF, Connolly TM, de Groot PG, Sixma JJ. Increased platelet deposition on atherosclerotic coronary arteries. J. Clin. Invest. 1994;93:615–632. doi: 10.1172/JCI117014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Riessen R, Kearney M, Lawler J, Isner JM. Immunolocalization of thrombospondin-1 in human atherosclerotic and restenotic arteries. Am. Heart J. 1998;135:357–364. doi: 10.1016/s0002-8703(98)70105-x. [DOI] [PubMed] [Google Scholar]
- 20.Topol EJ, McCarthy J, Gabriel S, Moliterno DJ, Rogers WJ, Newby LK, Freedman M, Metivier J, Cannata R, O'Donnell CJ, Kottke-Marchant K, Murugesan G, Plow EF, Stenina O, Daley GQ. Single nucleotide polymorphisms in multiple novel thrombospondin genes may be associated with familial premature myocardial infarction. Circulation. 2001;104:2641–2644. doi: 10.1161/hc4701.100910. [DOI] [PubMed] [Google Scholar]
- 21.Peyvandi F, Palla R, Ardissino D, Foco L, Bernardinelli L, Bauer K, Pier A, Mannucci M. Vagaries of genetic association studies in complex diseases: thrombospondins and acute myocardial infarction. Blood. 2003;102:1129. doi: 10.1182/blood-2003-05-1389. [DOI] [PubMed] [Google Scholar]
- 22.Bagavandoss P, Wilks JW. Specific inhibition of endothelial cell proliferation by thrombospondin. Biochem. Biophys. Res. Commun. 1990;170:867–872. doi: 10.1016/0006-291x(90)92171-u. [DOI] [PubMed] [Google Scholar]
- 23.Sengupta K, Banerjee S, Saxena NK, Banerjee SK. Thombospondin-1 disrupts estrogen-induced endothelial cell proliferation and migration and its expression is suppressed by estradiol. Mol. Cancer Res. 2004;2:150–158. [PubMed] [Google Scholar]
- 24.Iruela-Arispe ML, Bornstein P, Sage H. Thrombospondin exerts an antiangiogenic effect on cord formation by endothelial cells in vitro. Proc. Natl. Acad. Sci. USA. 1991;88:5026–5030. doi: 10.1073/pnas.88.11.5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lawler J, Sunday M, Thibert V, Duquette M, George EL, Rayburn H, Hynes RO. Thrombospondin-1 is required for normal murine pulmonary homeostasis and its absence causes pneumonia. J. Clin. Invest. 1998;101:982–992. doi: 10.1172/JCI1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agah A, Kyriakides TR, Lawler J, Bornstein P. The lack of thrombospondin-1 (TSP1) dictates the course of wound healing in double-TSP1/TSP2-null mice. Am. J. Pathol. 2002;161:831–839. doi: 10.1016/S0002-9440(10)64243-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doyen V, Rubio M, Braun D, Nakajima T, Abe J, Saito H, Delespesse G, Sarfati M. Thrombospondin 1 is an autocrine negative regulator of human dendritic cell activation. J. Exp. Med. 2003;198:1277–1283. doi: 10.1084/jem.20030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jimenez B, Volpert OV, Crawford SE, Febbraio M, Silverstein RL, Bouck N. Signals leading to apoptosis-dependent inhibition of neovascularization by thrombospondin-1. Nat. Med. 2000;6:41–48. doi: 10.1038/71517. [DOI] [PubMed] [Google Scholar]
- 29.Yamauchi Y, Kuroki M, Imakiire T, Uno K, Abe H, Beppu R, Yamashita Y, Kuroki M, Shirakusa T. Opposite effects of thrombospondin-1 via CD36 and CD47 on homotypic aggregation of monocytic cells. Matrix Biol. 2002;21:441–448. doi: 10.1016/s0945-053x(02)00036-7. [DOI] [PubMed] [Google Scholar]
- 30.Sajid M, Hu Z, Guo H, Li H, Stouffer GA. Vascular expression of integrin-associated protein and thrombospondin increase after mechanical injury. J. Investig. Med. 2001;49:398–406. doi: 10.2310/6650.2001.33784. [DOI] [PubMed] [Google Scholar]
- 31.Lawler J, Weinstein R, Hynes RO. Cell attachment to thrombospondin: the role of ARG-GLY-ASP, calcium, and integrin receptors. J. Cell Biol. 1988;107:2351–2361. doi: 10.1083/jcb.107.6.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gao AG, Lindberg FP, Dimitry JM, Brown EJ, Frazier WA. Thrombospondin modulates α v β 3 function through integrin-associated protein. J. Cell Biol. 1996;135:533–544. doi: 10.1083/jcb.135.2.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aiken ML, Ginsberg MH, Plow EF. Identification of a new class of inducible receptors on platelets. Thrombospondin interacts with platelets via a GPIIb-IIIa-independent mechanism. J. Clin. Invest. 1986;78:1713–1716. doi: 10.1172/JCI112767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schultz-Cherry S, Chen H, Mosher DF, Misenheimer TM, Krutzsch HC, Roberts DD, Murphy-Ullrich JE. Regulation of transforming growth factor-β activation by discrete sequences of thrombospondin 1. J. Biol. Chem. 1995;270:7304–7310. doi: 10.1074/jbc.270.13.7304. [DOI] [PubMed] [Google Scholar]
- 35.Kosfeld MD, Frazier WA. Identification of a new cell adhesion motif in two homologous peptides from the COOH-terminal cell binding domain of human thrombospondin. J. Biol. Chem. 1993;268:8808–8814. [PubMed] [Google Scholar]
- 36.Gao AG, Lindberg FP, Finn MB, Blystone SD, Brown EJ, Frazier WA. Integrin-associated protein is a receptor for the C-terminal domain of thrombospondin. J. Biol. Chem. 1996;271:21–24. doi: 10.1074/jbc.271.1.21. [DOI] [PubMed] [Google Scholar]
- 37.Ades EW, Candal FJ, Swerlick RA, George VG, Summers S, Bosse DC, Lawley TJ. HMEC-1: establishment of an immortalized human microvascular endothelial cell line. J. Invest. Dermatol. 1992;99:683–690. doi: 10.1111/1523-1747.ep12613748. [DOI] [PubMed] [Google Scholar]
- 38.Byzova TV, Goldman CK, Jankau J, Chen J, Cabrera G, Achen MG, Stacker SA, Carnevale KA, Siemionow M, Deitcher SR, DiCorleto PE. Adenovirus encoding vascular endothelial growth factor-D induces tissue-specific vascular patterns in vivo. Blood. 2002;99:4434–4442. doi: 10.1182/blood.v99.12.4434. [DOI] [PubMed] [Google Scholar]
- 39.Unger RE, Krump-Konvalinkova V, Peters K, Kirkpatrick CJ. In vitro expression of the endothelial phenotype: comparative study of primary isolated cells and cell lines, including the novel cell line HPMEC-ST1.6R. Microvasc. Res. 2002;64:384–397. doi: 10.1006/mvre.2002.2434. [DOI] [PubMed] [Google Scholar]
- 40.Collard CD, Agah A, Reenstra W, Buras J, Stahl GL. Endothelial nuclear factor-κB translocation and vascular cell adhesion molecule-1 induction by complement: inhibition with anti-human C5 therapy or cGMP analogues. Arterioscler. Thromb. Vasc. Biol. 1999;19:2623–2629. doi: 10.1161/01.atv.19.11.2623. [DOI] [PubMed] [Google Scholar]
- 41.Kootstra NA, Matsumura R, Verma IM. Efficient production of human FVIII in hemophilic mice using lentiviral vectors. Mol. Ther. 2003;7:623–631. doi: 10.1016/s1525-0016(03)00073-x. [DOI] [PubMed] [Google Scholar]
- 42.Shi W, Haberland ME, Jien ML, Shih DM, Lusis AJ. Endothelial responses to oxidized lipoproteins determine genetic susceptibility to atherosclerosis in mice. Circulation. 2000;102:75–81. doi: 10.1161/01.cir.102.1.75. [DOI] [PubMed] [Google Scholar]
- 43.Swerlick RA, Lee KH, Li LJ, Sepp NT, Caughman SW, Lawley TJ. Regulation of vascular cell adhesion molecule 1 on human dermal microvascular endothelial cells. J. Immunol. 1992;149:698–705. [PubMed] [Google Scholar]
- 44.Zapolska-Downar D, Siennicka A, Kaczmarczyk M, Kolodziej B, Naruszewicz M. Simvastatin modulates TNFα-induced adhesion molecules expression in human endothelial cells. Life Sci. 2004;75:1287–1302. doi: 10.1016/j.lfs.2004.03.005. [DOI] [PubMed] [Google Scholar]
- 45.Mackay F, Loetscher H, Stueber D, Gehr G, Lesslauer W. Tumor necrosis factor α (TNF-α)-induced cell adhesion to human endothelial cells is under dominant control of one TNF receptor type, TNF-R55. J. Exp. Med. 1993;177:1277–1286. doi: 10.1084/jem.177.5.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Swerlick RA, Garcia-Gonzalez E, Kubota Y, Xu YL, Lawley TJ. Studies of the modulation of MHC antigen and cell adhesion molecule expression on human dermal microvascular endothelial cells. J. Invest. Dermatol. 1991;97:190–196. doi: 10.1111/1523-1747.ep12479643. [DOI] [PubMed] [Google Scholar]
- 47.Novina CD, Sharp PA. The RNAi revolution. Nature. 2004;430:161–164. doi: 10.1038/430161a. [DOI] [PubMed] [Google Scholar]
- 48.Collins T, Read MA, Neish AS, Whitley MZ, Thanos D, Maniatis T. Transcriptional regulation of endothelial cell adhesion molecules: NF-κ B and cytokine-inducible enhancers. FASEB J. 1995;9:899–909. [PubMed] [Google Scholar]
- 49.Chiu JJ, Lee PL, Chen CN, Lee CI, Chang SF, Chen LJ, Lien SC, Ko YC, Usami S, Chien S. Shear stress increases ICAM-1 and decreases VCAM-1 and E-selectin expressions induced by tumor necrosis factor-[α] in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2004;24:73–79. doi: 10.1161/01.ATV.0000106321.63667.24. [DOI] [PubMed] [Google Scholar]
- 50.Favaloro EJ. Differential expression of surface antigens on activated endothelium. Immunol. Cell Biol. 1993;71:571–581. doi: 10.1038/icb.1993.63. [DOI] [PubMed] [Google Scholar]
- 51.Baker SJ, Reddy EP. Modulation of life and death by the TNF receptor superfamily. Oncogene. 1998;17:3261–3270. doi: 10.1038/sj.onc.1202568. [DOI] [PubMed] [Google Scholar]
- 52.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 53.Schuepp BJ, Pfister H, Clemetson KJ, Silverstein RL, Jungi TW. CD36-mediated signal transduction in human monocytes by anti-CD36 antibodies but not by anti-thrombospondin antibodies recognizing cell membrane-bound thrombospondin. Biochem. Biophys. Res. Commun. 1991;175:263–270. doi: 10.1016/s0006-291x(05)81229-x. [DOI] [PubMed] [Google Scholar]
- 54.Li WX, Howard RJ, Leung LL. Identification of SVTCG in thrombospondin as the conformation-dependent, high-affinity binding site for its receptor, CD36. J. Biol. Chem. 1993;268:16179–16184. [PubMed] [Google Scholar]
- 55.Brittain JE, Mlinar KJ, Anderson CS, Orringer EP, Parise LV. Integrin-associated protein is an adhesion receptor on sickle red blood cells for immobilized thrombospondin. Blood. 2001;97:2159–2164. doi: 10.1182/blood.v97.7.2159. [DOI] [PubMed] [Google Scholar]
- 56.Freyberg MA, Kaiser D, Graf R, Buttenbender J, Friedl P. Proatherogenic flow conditions initiate endothelial apoptosis via thrombospondin-1 and the integrin-associated protein. Biochem. Biophys. Res. Commun. 2001;286:141–149. doi: 10.1006/bbrc.2001.5314. [DOI] [PubMed] [Google Scholar]
- 57.Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11:130–135. doi: 10.1016/s0962-8924(00)01906-1. [DOI] [PubMed] [Google Scholar]
- 58.Kvansakul M, Adams JC, Hohenester E. Structure of a thrombospondin C-terminal fragment reveals a novel calcium core in the type 3 repeats. EMBO J. 2004;23:1223–1233. doi: 10.1038/sj.emboj.7600166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang XQ, Lindberg FP, Frazier WA. Integrin-associated protein stimulates α2β1-dependent chemotaxis via Gi-mediated inhibition of adenylate cyclase and extracellular-regulated kinases. J. Cell Biol. 1999;147:389–400. doi: 10.1083/jcb.147.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chung J, Wang XQ, Lindberg FP, Frazier WA. Thrombospondin-1 acts via IAP/CD47 to synergize with collagen in α2β1-mediated platelet activation. Blood. 1999;94:642–648. [PubMed] [Google Scholar]
- 61.Morandi V, Cherradi SE, Lambert S, Fauvel-Lafeve F, Legrand YJ, Legrand C. Proinflammatory cytokines (interleukin-1 β and tumor necrosis factor-α) down regulate synthesis and secretion of thrombospondin by human endothelial cells. J. Cell. Physiol. 1994;160:367–377. doi: 10.1002/jcp.1041600218. [DOI] [PubMed] [Google Scholar]
- 62.Laegreid A, Medvedev A, Nonstad U, Bombara MP, Ranges G, Sundan A, Espevik T. Tumor necrosis factor receptor p75 mediates cell-specific activation of nuclear factor κ B and induction of human cytomegalovirus enhancer. J. Biol. Chem. 1994;269:7785–7791. [PubMed] [Google Scholar]
- 63.Rothe M, Sarma V, Dixit VM, Goeddel DV. TRAF2-mediated activation of NF-κ B by TNF receptor 2 and CD40. Science. 1995;269:1424–1427. doi: 10.1126/science.7544915. [DOI] [PubMed] [Google Scholar]
- 64.Maile LA, Clemmons DR. Integrin-associated protein binding domain of thrombospondin-1 enhances insulin-like growth factor-I receptor signaling in vascular smooth muscle cells. Circ. Res. 2003;93:925–931. doi: 10.1161/01.RES.0000101754.33652.B7. [DOI] [PubMed] [Google Scholar]