The phosphoinositide-3-kinase inhibitor PX-866 overcomes resistance to the EGFR inhibitor gefitinib in A-549 human non small cell lung cancer xenografts (original) (raw)
. Author manuscript; available in PMC: 2006 Sep 1.
Abstract
Epidermal growth factor receptor (EGFR) inhibitors such as gefitinib show antitumor activity in a subset of non small cell lung (nscl) cancer patients having mutated EGFR. Recent work shows that phosphatidylinositol-3-kinase (PtdIns-3-kinase) is coupled to the EGFR only in nscl cancer cell lines expressing ErbB-3 and that EGFR inhibitors do not inhibit PtdIns-3-kinase signaling in these cells. The central role PtdIns-3-kinase plays in cell survival suggests that a PtdIns-3-kinase inhibitor offers a strategy to increase the antitumor activity of EGFR inhibitors in resistant nscl tumors that do not express ErbB-3. We show that PX-866, a Ptdins-3-kinase inhibitor with selectivity for p110α, potentiates the antitumor activity of gefitinib against even large A-549 nscl xenografts giving complete tumor growth control in the early stages of treatment. A-549 xenograft phospho-Akt was inhibited by PX-866 but not by gefitinib. A major toxicity of PX-866 adminsitration was hyperglycemia with decreased glucose tolerance, which was reversed upon cessation of treatment. The decreased glucose tolerance caused by PX-866 was insensitive to the AMPK inhibitor metformin but reversed by insulin, and by the PPARγ activator pioglitazone. Prolonged PX-866 administration also caused increased neutrophil counts. Thus, PX-866, by inhibiting PtdIns-3-kinase signaling may have clinical utility in increasing the response to EGFR inhibitors such as gefitinib in patients with nscl cancer, and possibly in other cancers, who do not respond to EGFR inhibition.
Keywords: PX-866, gefitinib, PtdIns-3-kinase, EGFR, blood glucose
INTRODUCTION
Increased cell survival is a fundamental characteristic of cancer cells and limits the effectiveness of cancer therapy (1). An important mechanism for increased cell survival in many cancers is mediated by the phosphatidylinositol-3-kinase (PtdIns-3-kinase)/Akt (protein kinase B) signaling pathway that is activated by receptor and oncogenic protein tyrosine kinases (2). Eight mammalian PtdIns-3-kinases are divided into 3 main classes; Class I PtdIns-3-kinases phosphorylate membrane PtdIns to give PtdIns(3,4,5)P3 which recruits the cytoplasmic serine/threonine kinase Akt by binding to its pleckstrin homology (PH) domain. Membrane-associated Akt is activated by Ser473 phosphorylation by membrane-associated phosphoinositide dependent kinase-1 (PDK1) (3) and Thr308 phosphorylation by a second incompletely characterized PDK2 (4). Activated Akt detaches from the plasma membrane and moves to the cytoplasm and the nucleus, where it phosphorylates a battery of targets to prevent the expression of death genes, and induces cell survival (5). PtdIns- 3-kinase activity is increased in human small cell lung cancer, ovarian, head and neck, urinary tract, colon and cervical cancers (6–8). The tumor suppressor protein PTEN (phosphatase and tensin homologue deleted on chromosome ten), a dual specificity tyrosine-threonine/PtdIns-3 phosphatase, prevents the accumulation of PtdIns(3,4,5)P3 and attenuates PtdIns-3-kinase signaling (9). PTEN is mutated or deleted in a variety of human cancers including advanced prostate, endometrial, renal, glial, melanoma, and small cell lung cancers (10).
The protein kinase family has more that 800 human members (11) among which receptor protein tyrosine kinases are frequently targets for cancer therapy. They include the epidermal growth factor receptor (EGFR, ErbB-1, HER1), that when activated by ligand binding to its extracellular domain, homo or heterodimerizes with any of 3 other family members, ErbB-2 (HER2), ErbB-3 (HER3) and ErbB-4 (HER4), leading to autophosphorylation of cytoplasmic C-terminal tyrosine residues. These phosphorylations recruit signal transducers leading to activation of signaling pathways that include the Ras-MEK-MAPK pathway, the STAT pathway and the PtdIns-3-kinase/Akt survival pathway. The EGFR is amplified or overexpressed in a wide range of human cancers where it is thought to play an important role in tumor progression (12). In non small cell (nsc) lung cancer EGFR expression is correlated with decreased patient survival (13). A number of small molecule inhibitors of the EGFR kinase as well as EGFR monoclonal antibodies are under development or approved for clinical use. Gefitinib (ZD 1839, IressaTM) is a small molecule EGFR inhibitor that when administered to patients with relapsed nsc lung cancer has shown a response rate of 10 to 20% and stabilized the disease in another 20 to 30% of patients (14). However, the addition of gefitinib to chemotherapy in untreated patients with nsc lung cancer had no effect on overall survival, time to progression, or response rate (15). A majority, but not all, nsc lung cancer patients responding to single agent gefitinib contain somatic mutations of unknown functional significance in the EGFR tyrosine kinase domain(16). However, there are also nsc lung cancer patients that do not have mutated EGFR receptors who may derive benefit from gefitinib and other EGFR inhibitors. Furthermore, even though activating mutations of the EGFR are very rare in human colorectal cancer and absent in glioblastoma (17) some of these tumors may be responsive to EGFR inhibitors (18). A recent study has shown that gefitinib inhibits cell growth and downregulates PtdIns-3-kinase signaling only in nsc lung cancer cell lines with ErbB-3 expression (19). This is because PtdIns-3-kinase couples to ErbB-3 leading to PtdIns-3-kinase/Akt signaling activation only in nsc lung cancer cell lines with either wild type or mutant EGFR receptor, and ErbB-3. Gefitinib is able to block the association of PtdIns-3-kinase with ErbB-3, thus, preventing PtdIns-3-kinase/Akt activation in these cell lines. The central role PtdIns-3-kinase plays in determining the response to gefitinib suggests that an inhibitor of PtdIns-3-kinase may provide a strategy to increase the antitumor activity of gefitinib in resistant nsc lung cancer tumors that do not express ErbB-3. PX-866 is a novel inhibitor of PtdIns-3-kinase that is currently in advanced preclinical development as an antitumor agent (20). We used the A-549 human nsc lung cancer cell line that does not express ErbB-3 and is resistant to gefitinib (19). We found that in A-549 tumor xenografts gefitinib did not inhibit PtdIns-3-kinase/Akt signaling and the administration of PX-866 either intravenously (iv) or orally (po) markedly potentiated the antitumor activity of gefitinib. We also report on the toxicity of long term administration of PX-866, showing that it increases blood glucose associated with a decrease in insulin sensitivity.
MATERIALS AND METHODS
Compounds
PX-866 (acetic acid (1_S_,4_E_,10_R_,11_R_,13_S_,14_R_)-[4-diallylaminomethylene-6-hydroxy-1-methoxymethyl-10,13-dimethyl-3,7,17-trioxo-1,3,4,7,10,11,12,13,14,15,16,17-dodecahydro-2-oxa-cyclopenta[a]phenanthren-11-yl ester) was synthesized as previously described (21) . For iv administration to mice PX-866 was dissolved at 10 mg/ml in 5% ethanol in 0.9% NaCl, and for po administration at 5 mg/ml in 5% ethanol in water. Gefitinib was obtained from Astra Zeneca (Macclesfield, UK) and suspended at 7.5 mg/ml in 0.1% Tween 20 in water for po administration.. Rabbit purified anti-phosphoSer473-Akt antibody, anti-Akt antibody and anti-phospho EGF-receptor antibody were obtained from Cell Signaling Technology (Beverly, MA). Bovine recombinant p110α /p85α PtdIns-3-kinase was obtained from Jena Bioscience (Jena, Germany) and human recombinant p110β/p85α, p120γ and p110δ /p85α from Upstate (Charlottesville, VA). Metformin hydrochloride was obtained from Spectrum Chemical (Gardena,CA), pioglitazone hydrochloride and recombinant human insulin from Sigma Chemical (St. Louis).
Cells
A-549 non small cell lung cancer cells were obtained from the American Tissue Type Collection (Rockville, MD). The cells were grown in humidified 95% air, 5% CO2 at 37°C in Dulbecco=s modified Eagle=s medium (DMEM) supplemented with 10% fetal bovine serum (fbs). All cell lines were tested to be mycoplasma free using a PCR ELISA kit (Roche Diagnostics Inc., Indianapolis, IN).
Measurement of PtdIns-3-kinase
The ability of PX-866 to inhibit recombinant bovine p110α /p85α and recombinant human p110β/p85α, p120γ and p110δ/p85α was measured by the [32 P]γ-ATP dependent phosphorylation of PtdIns as described by Stirdivant et al (22). Inhibition of cellular PtdIns-3-kinase was measured as the ratio of phosphoSer473 -Akt to total Akt measured by Western blotting, as previously described (20).
Antitumor Studies
Approximately 107 A-549 nsc lung cancer cells in log cell growth were injected subcutaneously in 0.2ml phosphate buffered saline into the flanks of severe combined immunodeficient (scid) mice. When the tumors reached 100 or 600 mm3 the mice were stratified into groups of 8 animals having approximately equal mean tumor volumes and drug administration was started. Dosing was every other day with gefitinib at 75 mg/kg po; PX-866 at 4, 9 or 12 mg/kg iv; PX-866 at 1, 2.5 and 3 mg/kg po, or PX-866 administered 4 hr before gefitinib. Animals were weighed weekly and tumor diameters were measured twice weekly at right angles (d short and d long) with electronic calipers and tumor volumes calculated by the formula volume = (dshort)2 x (dlong) _ 2 (23). When the tumor reached 2,000 mm3 or more, or became necrotic the animals were euthanized.
Pharmacodynamic Studies
107 A-549 nsc lung cancer cells were injected subcutaneously into the flanks of male scid mice and allowed to grow to approximately 300 mm3. Mice were administered PX-866 12 mg/kg iv, 3 mg/kg po and gefitinib 75g/kg po, every other day for 5 days. Tumors were removed 24 hr after the last dose and immediately frozen in liquid N2. For assay, the tumors were homogenized in 50mM HEPES buffer, pH 7.5, 50mM NaCl, 1% Nonidet P40 and 0.25 % sodium deoxycholate and Western blotting performed using anti- phosphoSer473-Akt and anti-Akt antibodies. Tumor Akt activity was expressed as the ratio of phospho-Ser473-Akt to total Akt.
Toxicity Studies
Male scid mice were administered PX-866 at 10 mg/kg iv, or 3 and 1.5 mg/kg po, every other day for 14 doses. C57Bl/6 mice were administered PX-866 at 3 mg/kg po every other day for 15 doses. The mice were killed 24 hr after the last dose and changes in body weight, blood lymphocyte, neutrophil, red blood cell, platelet counts, serum glucose, aspartate amino transferase (AST), and amino alanine transferase (ALT) were measured.
Glucose Tolerance Studies
Female C5781/6 mice were fasted overnight and administered a single dose of D(+) glucose (1 mg/kg) as a 0.1 g/ml solution po. Blood was collected at 0, 10, 20, 30, 60, 90, 120 and 180 min and plasma glucose measured using a blood glucose kit (Sigma Chemical Co., St Louis, MO) to obtain a plasma glucose area under the curve (AUC 0-180 min). Mice were administered PX-866 10 mg/kg po as a single dose and glucose administered 4 hours later, or 3 mg/kg PX-866 po every other day for 20 doses and glucose administered 24 hours and 8 days after the last dose. Metformin was administered at 250 mg/kg po daily for 5 days (24) and 10 mg/kg pioglitazone ip daily for 7 days (25) before the glucose administration. Human recombinant insulin was administered at 0.075 μg/kg ip (26) at the same time as glucose administration.
Bone Marrow Colony Formation
After sacrifice, mouse bone marrow was extracted from each femur and red blood cells lysed with 0.2% hypotonic NaCl followed by the addition of a 1.6% hypertonic NaCl. Approximately 20,000 cells were plated in 1ml of Methocult™ GF M3434 (Stemcell Technologies Inc, Vancouver, BC, Canada) containing 1% methylcellulose in Iscove’s Minimum Essential Media, 15% fbs, 1% bovine serum albumin, 10 μg/ml recombinant human insulin, 200 μg/ml human transferrin, 10 mM β-mercaptoethanol, 2mM L-glutamine, 50 ng/ml rm stem cell factor, 10 ng/ml recombinant mouse interleukin-3, 10 ng/ml recombinant human interleukin-6, and 3 units/ml recombinant erythropoietin. Cells were plated in triplicate and grown at 37° C and 5% CO2 in a humid environment for 14 days before scoring. Colonies (> 40 cells/colony) or clusters (3-40 cells) were scored and growth of colony-forming unit granulocyte, erythroid, macrophage, megakaryocyte (CFU-GEMM; burst-forming units-erythroid (BFU-E), colony-forming units-granulocyte macrophage (CFU-GM), assessed using standard criteria (27). Qualitative observations were made on background levels of single cells.
RESULTS
PtdIns-3-kinase inhibition
The ability of PX-866 to inhibit recombinant PtdIns-3-kinases compared to the activity of wortmannin is shown in Table 1. PX-866 is a more potent inhibitor of p110α (20 fold) and p120γ (10 fold) than wortmannin, has a similar similar potency for p110δ and, unlike wortmannin, is a poor inhbitor of p110β.
Table 1. Inhibition of PtdIns-3-kinases by PX-866 and wortmannin.
Recombinant PtdIns-3-kinase were assayed for activity as described in the text.
| PtdIns-3-kinase | PX-866 IC 50 (nm) | wortmannin IC 50 (nm) |
|---|---|---|
| p110α /p85α | 0.1 | 2.0 |
| p110β /p85α | > 300 | 0.6 |
| p120γ | 1.0 | 10.0 |
| p110δ /p85α | 2.9 | 3.9 |
Cell culture studies
PX-866 inhibited phospho-Akt in A-549 human breast cancer cells in media containing 10% fbs with an IC50 of 25 nM. Gefitinib only inhibited phospho-Akt in cells that were serum starved for 24 hr and then stimulated with EGF 25 ng/ml but not in media with 10% fbs. This suggests that the PtdIns-3-kinase pathway is stimulated by growth factors in serum, in addition to EGF. Cell growth inhibition studies confirmed previous reports (19) that A-549 cells are resistant to growth inhibition by gefitinib, with an IC50 of 1.1 μM. PX-866 at concentrations up to 100 nM did not enhance the growth inhibition by gefitinib.
In vivo antitumor studies
Administration of gefitinib at 75 mg/kg po every other day to mice with 100 mm3 A-549 human nsc lung cancer xenografts inhibited xenograft growth with a T/C of 51 % at the end of the dosing period (Figure 1A). We have previously reported that PX-866 is approximately 4 times more potent as an antitumor agent when given po than given iv, and doses were adjusted accordingly (Table 2). When administered alone to mice with 100 mm3 A-549 tumor xenografts, PX-866 inhibited tumor growth with T/Cs of 31% at 9 mg/kg iv and 41% at 2.5 mg/kg po. Preliminary studies showed that PX-866 in combination with gefitinib on an alternating day schedule was more active when administered 4 hr before rather than 24 hr after gefitinib (data not shown). When PX-866 was administered 4 hr before gefitinib the combination gave T/Cs of 22 % at 9 mg/kg PX-866 iv and 18 % at 2.5 mg/kg PX-866 po. Tumor growth was held stationary for the first half of the treatment period with PX-866 and then began to slowly increase towards the end of the period (Figure 1A). Increased combination antitumor activity was also seen with very large 600 mm3 A-4549 tumor xenografts (Figure 1B).
Figure 1. Potentiation of the antitumor activity of gefitnib by PX-866.
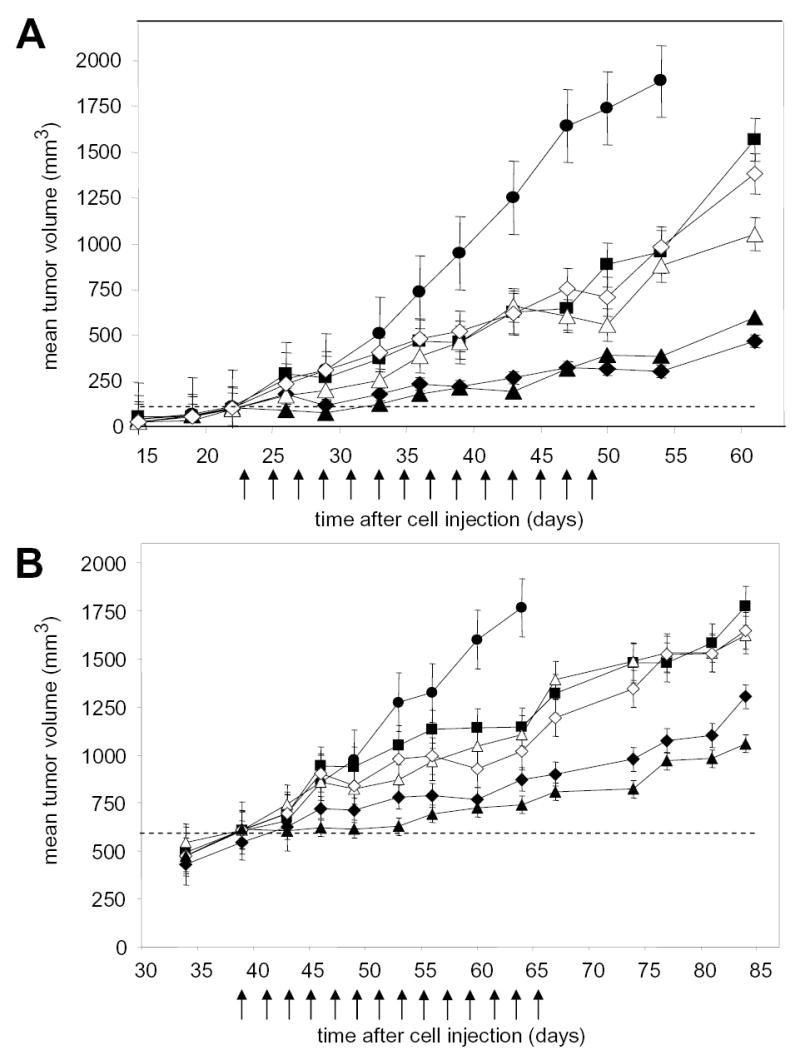
Female scid mice were implanted subcutaneously with 107 A-549 human non small cell lung cancer cells. A, Tumors were 100 mm3 on day 22 (shown by dashed line) when dosing was begun every other day for 15 doses shown by the arrows with () vehicle alone; (_) gefitinib 75 mg/kg po; (Δ) PX-866 9 mg/kg iv; (_) PX-866 2.5 mg/kg po; (_) PX-866 9 mg/kg iv 4 hr before gefitinib 75 mg/kg po; (_) PX-866 2.5 mg/kg po 4 hr before gefitinib 75 mg/kg po. Values are the mean of 8 mice per group and bars are SE. B. Tumors were 600 mm3 on day 39 (shown by dashed line) when dosing was begun every other day for 14 doses shown by the arrows with (_ ) vehicle alone; (_) gefitinib 75 mg/kg po; (Δ) PX-866 12 mg/kg iv; (_) PX-866 4 mg/kg po; (_) PX-866 12 mg/kg iv 4 hr before gefitinib 75 mg/kg po; (_) PX-866 4 mg/kg po 4 hr before gefitinib 75 mg/kg po. Values are the mean of 8 mice per group and bars are SE.
Table 2. Antitumor Activity of PX-866 in combination with gefitinib.
Female scid mice were implanted subcutaneously in the flank with 107 A-549 human nsc lung cancer cells. Tumors were allowed to grow to a mean volume of 100 mm3 before drug treatment was stared every other day for 14 doses. Antitumor activity is expressed as the % volume of the treated tumor/ control tumor (T/C %) at the end of the dosing period. There were 8 mice in each group and all differences are p < 0.01.
| Treatment and route | dose mg/kg | schedule | Tumo r T/C % | PX-866 4 hr before gefitinib Tumor T/C % |
|---|---|---|---|---|
| gefitinib po | 75 | QOD x 14 | 50.8 | - |
| PX-866 iv | 4 | QOD x 14 | 65.3 | 20.5 |
| PX-866 iv | 9 | QOD x 14 | 31.5 | 22.3 |
| PX-866 po | 1 | QOD x 14 | 54.8 | 40.8 |
| PX-866 po | 2.5 | QOD x 14 | 40.8 | 18.1 |
Inhibition of tumor EGFR and PtdIns-3-kinase signaling
Administration of gefitnib 75 mg/kg po to mice with A-549 tumor xenografts every other day for 5 days inhibited tumor phospho-EGFR by 43% but had no significant effect upon tumor phospho-Akt (Figure 2). PX-866 12 mg/kg iv or 3 mg/kg po, every other day for 5 days, had no significant effect upon tumor phospho-EGFR but inhibited tumor phospho-Akt by 51% and 48 %, respectively. The combination of gefitinib and PX-866 inhibited both tumor phospho-EGFR and tumor phospho-Akt. Thus, in A-549 tumor xenografts the EGFR and PtdIns-3-kinase pathways appear to function independently and to be selectively inhibited by gefitinib and PX-866, respectively.
Figure 2.
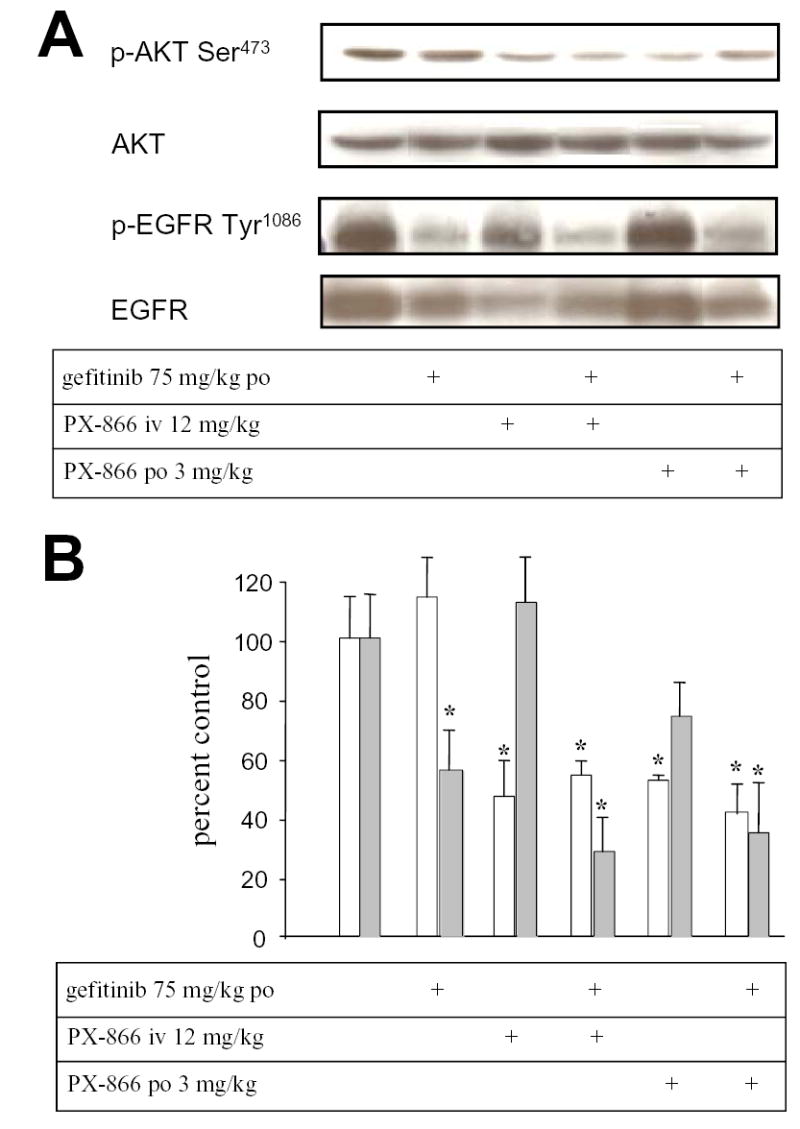
Inhibition of EGFR and phospho-Akt in A-549 non small cell lung cancer xenografts by gefitinib and PX-866.. Female scid mice with 300 mm3 A-549 non small cell lung cancer xenografts were administered gefitinib 75 mg/kg po, PX-866 12 mg/kg iv or PX-866 3 mg/kg po, alone or with the PX-866 4 hr before the gefitinib, daily every other day for 5 days. 24 hr after the last dose tumors were removed for measurement of phospho-Akt/total Akt shown by the open bars or phospho-EGFR shown by the shaded bars. Values are the mean of 3 mice and bars are SE. * p < 0.05 compared to non treated control.
Toxicity of long term PX-866 administration
The toxicity of long term administration of PX-866 to scid mice is summarized in Table 3. There was a decreased gain in body weight over the 4 weeks of treatment with PX-866 at 10 mg/kg iv and 3 mg/kg po, to 83% and 28% of the control weight gain, respectively (p < 0.05). There was no change in the plasma liver enzymes ALT and AST but a significant increase in plasma glucose caused by **PX-866** at 1.5 mg/kg and 3 mg/kg po of 113 % and 142%, respectively (p < 0.05). The increase in blood glucose with **PX-866** at 10 mg/kg iv was 62% but was not significant (p > 0.05). There was a significant increase in white blood cell counts following oral administration of PX-866, due primarily to increased blood neutrophil counts. All of the changes in body weight, plasma glucose and blood cell counts had returned to normal by 9 days after treatment stopped. The decrease in body weight and an increase in blood glucose were confirmed in two additional studies using scid mice, but the increase in blood cell counts was less pronounced in these studies (data not shown).
Table 3. Toxicity of long term PX-866 administration.
PX-866 was administered either iv or po as 14 doses every other day to male scid mice. Twenty four hr after the last dose blood was collected for serum chemistry and differential blood counts. Values are the mean of 4 mice per group ± SE.
| Treatment Group | ALT U/l | AST U/l | glucose mg/dl | WBC K/μ l | NE K/μ l | LY K/μ l | MO K/μ l | RBC M/μ l | Hb g/dl | Plt K/μ l | Weight change g |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Control | 52.6 ± 13.6 | 142.9 ± 46.6 | 46.9 ± 5.1 | 8.9 ± 1.0 | 6.9 ± 0.8 | 1.1 ± 0.2 | 0.8 ± 0.1 | 11.0 ± 0.3 | 15.4 ± 0.2 | 1427 ± 60 | 4.7 ± 0.3 |
| PX-866 10 mg/Kg | 35.5 ± 11.7 | 105.2 ±19.2 | 76.2 ± 3.6 | 14.6 ± 4.2 | 14.0 ± 2.7 | 1.8 ± 0.5 | 1.1 ± 0.2 | 10.6 ± 0.0 | 14.4 ± 0.1 | 1390 ± 43 | 3.9 ± 0.2* |
| PX-866 3 mg/Kg | 47.6 ±16.8 | 152.0 ± 47.2 | 113.5 ± 23.4* | 67.8 ± 19.7* | 53.6 ± 10.7** | 5.2 ±3.9 | 9.2 ±3.6 | 10.4 ± 0.3 | 14.7 ± 0.4 | 1665 ± 227 | 1.3 ± 0.5** |
| PX-866 1.5 mg/Kg po | 65.6 ±27.5 | 140.5 ± 35.2 | 100.1 ±10.9** | 16.6 ± 2.4* | 12.5 ± 1.9* | 3.1 ± 0.6* | 1.8 ± 0.2* | 1030 ± 0.3 | 14.6 ± 0.3 | 1221 ± 18* | 3.9 ± 0.6 |
PX-866 and glucose tolerance
In order to gain further insight into the mechanism for the increase in plasma glucose by PX-866, studies were conducted on insulin levels and on glucose tolerance following an oral dose of 1 g glucose/kg to fasted C57Bl/6 mice (Figure 3). Administration of PX-866 as a single dose of 10 mg/kg po caused an increase in plasma insulin levels for up to 5 hr. PX-866 also deceased glucose tolerance in the mice leading to an increase in plasma glucose, particularly at time points after 1 hr after glucose administration where where plasma glucose was decreasing in non treated mice but increasing in the PX-866 treated mice. The results expressed as AUC 0-180 min for all the glucose tolerance studies are shown in Table 4. Treatment with insulin at high doses overcame the increase in plasma glucose caused by PX-866 and significantly decreased the glucose AUC 0-180 min in both control and PX-866 treated mice. The antihyperglycemic drug metformin had no effect upon the increase in blood glucose by PX-866, but the hypoglycemic thiazolidinedione drug pioglitazone almost completely blocked the increase (Figure 3 and Table 4). Long term treatment with PX-866 at 9 mg/kg iv every other day for 15 doses gave an increase in non-fasting glucose levels (± S.E., n = 4) from 133.7 ± 16 mg/dl in control mice to 269.4 ± 27.8 mg/dl (p < 0.05) in the PX-866 treated mice. The treatment also gave an increase in plasma glucose AUC 0-180 min 24 hr after the last dose of PX-866, but this had recovered to control values 8 days after the last dose (Table 4). Pioglitazone significantly decreased the glucose AUC 0-120 min 24 hr after the last dose of long term PX-866 treatment to a value not significantly different to control (Table 4).
Figure 3. Effect of PX-866 on plasma insulin and glucose tolerance.
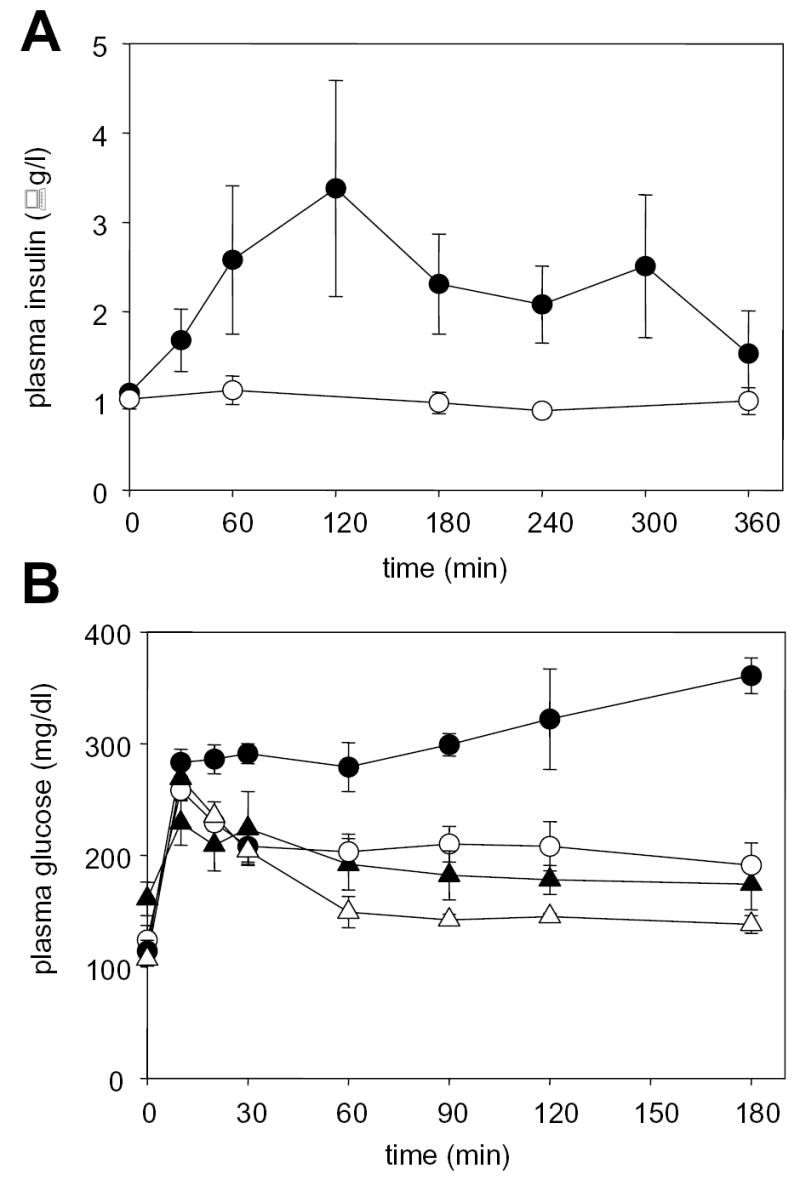
A, Plasma insulin in female C57Bl/6 mice fasted for 16 hr. (○) vehicle control and (•) mice administered PX-866 10 mg/kg po. Values are the mean of 3 mice and bars are SE. B, Plasma glucose in female C57Bl/6 mice fasted for 16 hr and administered a dose of glucose of 1 g/kg po. (○) vehicle control; (•) administered PX-866 10 mg/kg 4 hr previously; (_) treated daily for 7 days with pioglitazone 10 mg/kg ip; and (Δ) treated daily for 7 days with pioglitazone 10 mg/kg ip and administered PX-866 10 mg/kg 4 hr previously. Values are the mean of 4 mice and bars are SE.
Table 4. Effects of PX-866 on glucose tolerance in mice.
Female C5781/6 mice were fasted overnight and administered D(+) glucose 1 mg/kg as a 0.1 g/ml solution po. Blood was collected at various time up to 180 min and plasma glucose measured to obtain a plasma glucose area under the curve (AUC 0-180 min). Mice were administered PX-866 10 mg/kg po and glucose administered 4 hours later, or PX-866 3 mg/kg po every other day for 20 doses and glucose administered 24 hours or 8 days after the last dose. Metformin was administered at 250 mg/kg daily for 5 days and pioglitazone 10 mg/kg ip daily for 7 days before glucose administration. Human recombinant insulin was administered at 0.075 μ/kg ip at the same time as glucose. Values are the mean ± SE of 4 mice per group.
| Treatment | AUC 0-180 min (mg.min.ml−1 ) | AUC 0-180 min (mg.min.ml−1 ) |
|---|---|---|
| PX-866 10 mg/kg po | ||
| No drug | 369 ± 25 | 533 ± 17 a |
| insulin 0.075 μ /kg ip | 64 ± 25 a | 64 ± 5 |
| metformin 250 mg/kg ip QD x 5 | 367 ± 46 | 537 ± 4 b |
| pioglitazone 10 mg/kg ip QD x 7 | 274 ± 3 a | 340 ± 39 c |
| PX-866 3 mg/kg po QOD x 20 | ||
| 520 ± 14 a | ||
| 8 day recovery | 343 ± 30 | |
| pioglitazone 10 mg/kg ip QD x 7 | 405 ± 26 d |
PX-866 and increased neutrophils
When PX-866 was administered to C57Bl/6 mice at 3 mg/kg po every other day for 15 doses there was a significant increase in neutrophil counts (± S.E., n = 4) from 1.2 ± 0.3 K/μl in control mice to 3.7 ± 1.8 K/μl in PX-866 treated mice (p < 0.05), but with no significant change in any other blood elements. Bone marrow colony forming units showed no significant change in erythroid lineage CFU-GEMM, BFU-E or CFU-E and a small but significant decrease in myeloid CFU-GM (± S.E., n = 4) from 388 ± 52 colonies per 60,000 bone marrow cells plated in control mice to 168 ± 59 colonies ( p < 0.05) in the PX-866 treated mice. At the same time there was an increase in the numbers of individual white cells in the cultures from PX-866 treated mice suggestive of altered cell adhesion.
DISCUSSION
Sensitivity of nsc lung cancer cell lines to growth inhibition by gefitinib is associated with inhibition of EGF-stimulated EGFR autophosphorylation, down regulation of cell surface EGFR, ERK1/2 down regulation and inhibition of PtdIns-3-kinase/Akt signaling (28). The PtdIns-3-kinase/Akt pathway is a critical pathway for cancer cell survival (29, 30). In a study by Ono et al (28) gefitinib inhibited EGF-induced PtdIns-3-kinase/Akt signaling, as measured by phospho-Akt levels, in nearly all nsc lung cancer cell lines, however, only a few lines (3/11) showed inhibition of phospho-Akt under serum stimulated growth conditions. These results suggest that in many nsc lung cancer cell lines factors other than EGF are responsible for the activation of PtdIns-3-kinase/Akt signaling. Tumor cells with this phenotype may show limited responsiveness to the cytostatic and/or cytotoxic activities of EGFR inhbitors. Engelman et al (19) have recently reported that ErbB-3 couples EGFR signaling to the activation of Ptdins-3-kinase/Akt, and that gefitinib inhibits phospho-Akt and cell growth only in nsc lung cancer cell lines expressing EGFR, either wild type or mutant, and ErbB-3. However, forced ErbB-3 expression did not render nsc lung cancer cells sensitive to gefitinib suggesting that pathways other than EGFR must activate the Ptdins-3-kinase/Akt signaling in ErbB-3 deficient cells. Other members of the ErbB receptor family may also couple with ErbB-3 to activate PtdIns-3-kinase and promote the cancer phenotype (19, 31). We reasoned that inhibiting PtdIns-3-kinase could offer a rational strategy to potentiate the antitumor activity of gefitinib in gefitinib resistant nsc lung cancer cell lines.
For our studies we chose the A-549 nsc lung cancer cell line that is among the most resistant of nsc lung cancer lines to gefitinib (19, 28) and does not express ErbB-3 (19). PTEN deficiency can also render cells resistant to gefitinib growth inhibition presumably through constitutive activation of Ptdins-3-kinase/Akt signaling (32). However, genetic abnormalities of PTEN are relatively rare in lung cancer (33, 34) and A-549 cells, as do most nsc lung cancer cell lines, has wild type PTEN and non-constitutively activated levels of phospho-Akt (35). To inhibit PtdIns-3-kinase we used PX-866 that has been shown to downregulate tumor phospho-Akt and to exhibit antitumor activity in a number of human tumor xenograft models when administered either intravenously or orally (20).
We found that PX-866 administered either iv or po inhibited the growth of A-549 nsc lung tumor xenografts in scid mice as effectively as gefitinib. Both agents were most active when administered long term giving tumor T/Cs around 50%. However, when PX-866 was administered together with gefitinib, A-549 tumor growth was held stationary during the first part of the treatment and increased only slightly during the later part of treatment. This was seen with both 100 mm3 tumors and with large advanced 600 mm3 tumors. Gefitinib failed to inhibit phospho-Akt in A-549 tumor xenografts. In the A-549 cell culture studies gefitinib also did not inhibit phospho-Akt cells under serum stimulated growth conditions and was only inhibitory in EGF-stimulated, serum deprived A-549 cells. In contrast, PX-866 inhibited phospho-Akt of A-549 cells under serum simulated growth conditions, and in A-549 human tumor xenografts. Gefitinib inhibited phospho-EGFR in the A-549 human tumor xenografts and PX-866 did not. Thus, PX-866 potentiated the antitumor activity of gefitinib against even very large A-549 tumor xenografts giving complete tumor growth control in the early stages of treatment. The inhibition of tumor growth was associated with inhibition of Ptdins-3-kinase/Akt signaling by PX-866 and was not observed with gefitinib alone.
A previous study has reported LY294002, a relatively toxic and non-specific PtdIns-3-kinase inhibitor with limited potential for clinical development (36), administered ip potentiates the antitumor activity of gefitinib against small, 6 to 100 mm3, U87:ΔEGFR human glioma cell xenografts that coexpress wild type and mutant tumor derived activated EGFR (37). In this study neither gefitinib nor LY294002 showed antitumor activity alone.
The major toxicity of prolonged administration of PX-866 was hyperglycemia and decreased glucose tolerance that reversed when drug administration was stopped. Insulin signals are relayed predominantly by the PtdIns-3-kinase isoform p110β, but also by p110α (38, 39) while growth signals are relayed by PtdIns-3-kinase p110α (40). PX-866 is a more potent PtdIns-3-kinase p110α inhibitor than wortmannin but, unlike wortmannin, PX-866 is a poor inhbitor of inhibitor of PtdIns-3-kinase p110β. Acute administration of PX-866 to mice decreased glucose tolerance at the same time that plasma insulin levels were increased suggesting a decrease in sensitivity to insulin. This is similar to the phenotype of mice deficient in the Akt2 isoform that includes marked hyperglycemia, hyperinsulinemia and an impaired ability of insulin to lower blood glucose (41, 42). In the present study a high dose of insulin was able to overcame the increase in blood glucose caused by PX-866. Metformin, a widely used drug for the treatment of hyperglycemia of type 2 diabetes, lowers blood glucose by stimulating AMP-activated protein kinase (AMPK) downstream of PtdIns-3-kinase to increase fatty acid oxidation and to decrease triglyceride synthesis, hepatic glucose production and glucose utilization (43, 44). AMPK mediates the stimulation of glucose uptake through translocation of the glucose transporter 4 (GLUT-4) to the plasma membrane (45). It has been suggested that an AMPK activator such as metformin might enhance tumor cell survival if used with agents such as PtdIns-3-kinase or Akt inhibitors that impair glucose utilization (46). We found that metformin had no effect on the decreased glucose tolerance caused by by PX-866. Itshould be noted that a parallel pathway mediated by the recruitment of the Cbl proto-oncogene to the activated insulin receptor also increases glucose uptake by insulin (47).
In contrast to metformin, the thiazolidinedione hyperglycemic drug pioglitazone reversed the inhibitory effects of both acute and chronic PX-866 administration on glucose tolerance. Thiazolidenediones sensitize the body to the metabolic effects of insulin by acting as ligands for the peroxisome proliferator-activated receptor-γ (PPARγ) transcription factor that is present at high levels in adipose tissue (48). PPARγ also induces differentiation of tumor cells and PPARγ activation by pioglitazone has been reported to inhibit the growth of A-549 nsc lung tumor xenograft in scid mice (49). While all the details of insulin signaling through PtdIns-3-kinase and the effects of glucose lowering drugs such as metformin and pioglitazone remain to be elucidated, it appears that hypergylcemia caused by PtdIns-3-kinase inhibition by PX-866 is responsive to insulin and pioglitazone, which could be important for the clinical use of PX-866. The selectivity of PX-866 as an inhibitor of p110γ relative to p110β, unlike wortmannin which inhibits both p110α and p110β, may also explain the more pronounced growth inhbitory effects of PX-86, and the ability of insulin and pioglitazone to reverse **PX-866-**induced hyperglycia.
The other pharmacological effect of PX-866 administration was an increase in circulating neutrophils at the same time there is a a decrease in bone marrow CFU-GM colony formation The decrease in CFU-GM induced by PX-866 is consistent with the decreased sensitivity to granulocyte macrophage-colony stimulating factor (GM-CSF) observed in bone marrow derived macrophages of p85α −/− knockout mice (50). The increase in circulating neutrophils by PX-866 may reflect increased mobilization of progenitor cells into the peripheral circulation, perhaps associated with the decreased cell adhesion as seen in the p85α −/− knockout mice (50).
In summary, we have shown that the Ptdins-3-kinase inhibitor PX-866 which shows selectivity for p110α compared to p110β potentiates the antitumor activity of the EGFR inhibitor gefitinib against even large A-549 nsc lung cancer xenografts, with complete tumor growth control in the early stages of treatment. This therapeutic effect of PX-866 was associated with inhibition of tumor Akt phosphorylayion which was not seen with gefitinib alone. The major toxicity of chronic PX-866 was a target-related hyperglycemia with a reversible decrease in glucose tolerance due to decreased sensitivity to insulin. The decreased glucose tolerance was insensitive to the AMPK inhibitor metformin but was reversed by insulin and the PPARγ activator pioglitazone. Long term PX-866 also caused increased neutrophils counts, apparently due to vascular mobilization. Thus, PX-866 by inhibiting PtdIns-3-kinase/Akt signaling, may have clinical utility in increasing the response to EGFR inhibitors such as gefitinib in patients with nsc lung cancer who do not respond to therapy with EGFR inhibitors.
Acknowledgments
Supported by NIH grants CA52995 and CA90821
References
- 1.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 2.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2000;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 3.Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PRJ, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates activates protein kinase Bα. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- 4.Hill MM, Feng J, Hemmings BA. Identification of a plasma membrane Raft-associated PKB Ser473 kinase activity that is distinct from ILK and PDK1. Curr Biol. 2002;12:1251–1255. doi: 10.1016/s0960-9822(02)00973-9. [DOI] [PubMed] [Google Scholar]
- 5.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signaling. 2002;14:381–395. doi: 10.1016/s0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 6.Moore SM, Rintoul RC, Walker TR, Chilvers ER, Haslett C, Sethi T. The presence of a constitutively active phosphoinositide 3-kinase in small cell lung cancer cells mediates anchorage-independent proliferation via a protein kinase B p70s6k-dependent pathway. Cancer Res. 1998;58:5239–5247. [PubMed] [Google Scholar]
- 7.Shayesteh L, Lu Y, Kuo WL, Baldocchi R, Godfrey T, Collins C, Pinkel D, Powell B, Mills GB, Gray JW. PIK3Cα is implicated as an oncogene in ovarian cancer. Nature Genetics. 1999;21:99–102. doi: 10.1038/5042. [DOI] [PubMed] [Google Scholar]
- 8.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, Willson KV, Markowitz S, Kinzler KW, Vogelstein B, Velculescu VE. High frequency of mutations of the PI3KCα gene in human cancer. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 9.Maehama T, Dixon JE. The tumor suppressor PTEN/MMAC1 dephosphorylates the lipid second messenger phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 10.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 1999;96:4240–4245. doi: 10.1073/pnas.96.8.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Manning G, Whyte D, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- 12.Arteaga CL. Epidermal growth factor receptor dependence in human tumors: more than just expression? Oncologist. 2002;7:31–39. doi: 10.1634/theoncologist.7-suppl_4-31. [DOI] [PubMed] [Google Scholar]
- 13.Brabender J, Danenberg KD, Metzger R. Epidermal growth factor receptor and HER2-neu mRNA expression in non-small cell lung cancer is correlated with survival. Clin Cancer Res. 2001;7:1850–1855. [PubMed] [Google Scholar]
- 14.Fukuoka M, Yano S, Giaccone G, Tamura T, Nakagawa K, Douillard JY, Nishiwaki Y, Vansteenkiste J, Kudoh S, Rischin D. Multi-institutional randomized phase II trial of gefitinib for previously treated patients with advanced non-small-cell lung cancer. J Clin Oncol. 2003;21:2237–2246. doi: 10.1200/JCO.2003.10.038. [DOI] [PubMed] [Google Scholar]
- 15.Giaccone G, Herbst RS, Manegold C. Gefitinib in combination with gemcitabine and cisplatin in advanced non-small-cell lung cancer: a phase III trial-INTACT. J Clin Oncol. 2004;22:784. doi: 10.1200/JCO.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 16.Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 17.Barber TD, Vogelstein B, Kinzler KW, Velculescu VE. Somatic mutations of EGFR in colorectal cancers and glioblastomas. N Engl J Med. 2004;351:2883. doi: 10.1056/NEJM200412303512724. [DOI] [PubMed] [Google Scholar]
- 18.Redlinger M, Kramer A, Flaherty K, Sun W, Halter D, O'Dwyer P. A phase II trial of gefitinib in combination with 5-FU/LV/irinotecan in patients with colorectal cancer. J Clin Oncol. 2004;22:3767. [Google Scholar]
- 19.Engleman JA, Jänne PA, Mermel C, Pearlberg J, Mukohara T, Fleet C, Cichowski K, Johnson BE, Cantley LC. ErbB-3 mediates phosphoinositide 3-kinase activity in gefitinib-sensitive non-small cell lung cancer cell lines. Proc Natl Acad Sci USA. 2005;102:3788–3793. doi: 10.1073/pnas.0409773102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ihle NT, Williams R, Chow S, Chew W, Berggren MI, Paine-Murrieta G, Minion DJ, Halter RJ, Wipf P, Abraham R, Kirkpatrick DL, Powis G. Molecular pharmacology and antitumor activity of PX-866, a novel inhibitor of phosphoinositide-3-kinase signaling. Mol Cancer Ther. 2004;3:1–10. [PubMed] [Google Scholar]
- 21.Wipf P, Minion DJ, Halter RJ, Berggren MI, Ho CB, Chiang GG, Kirkpatrick L, Abraham R, Powis G. Synthesis, biological evaluation of synthetic viridins derived from C(20)-Heteroalkylation of the steroidal PI-3-kinase inhibitor wortmannin. Org Biomol Chem. 2004;2:1911–1920. doi: 10.1039/b405431h. [DOI] [PubMed] [Google Scholar]
- 22.Stirdivant SM, Ahern J, Conroy RR, Stanley FB, Ledder LM, Oliff A, Heimbrook DC. Cloning mutagenesis of the p110a subunit of human phosphoinositide 3'-hydroxykinase. Biorganic Medl Chem. 1997;5:65–74. doi: 10.1016/s0968-0896(96)00196-4. [DOI] [PubMed] [Google Scholar]
- 23.Paine GD, Taylor CW, Curtis RA, Lopez MHA, Dorr RT, Roe D, Funk CY, Thompson F, Hersh EM. Human tumor models in the severe combined immune deficient scid mouse. Cancer Chemother Pharmacol. 1997;40:209–214. doi: 10.1007/s002800050648. [DOI] [PubMed] [Google Scholar]
- 24.Thomas CR, Turner SL, Jefferson WH, Bailey CJ. Prevention of dexamethasone-induced insulin resistance by metformin. Biochem Pharmacol. 1998;56:1145–1150. doi: 10.1016/s0006-2952(98)00151-8. [DOI] [PubMed] [Google Scholar]
- 25.Tanimoto M, Fan Q, Gohda T, Shike T, Makita Y, Tomino Y. Effect of pioglitazone on the early stage of type 2 diabetic nephropathy in KK/Ta mice. Metabolism. 2004;53:1473–1479. doi: 10.1016/j.metabol.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 26.Miyake K, Ogawa W, Matsumot M, Nakamura T, Sakaue H, Kasuga M. Hyperinsulinemia, glucose intolerance, and dyslipidemia induced by acute inhibition of phosphoinositide 3-kinase signaling in the liver. J Clin Invest. 2002;110:1483–1491. doi: 10.1172/JCI15880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Coutinho L.H., Gilleece M.H., De Wynter E.A., Will A. and Testa N.G. Clonal and long term cultures using human bone marrow. In: N. G. Testa (ed.), Haematology: A Practical Approach,1993. p. 75. Oxford University: Oxford, UK.
- 28.Ono M, Akira H, Kometani T, Miyagawa M, Ueda S, Kinoshita H, Fujii T, Kuwano M. Sensitivity to gefitinib (Iressa, ZD1839) in non-small cell lung cancer cell lines correlates with dependence on the epidermal growth factor (EGF) receptor/extracellular signal-regulated kinase 1/2 and EGF receptor/Akt pathway for proliferation. Mol Cancer Ther. 2004;3:465–472. [PubMed] [Google Scholar]
- 29.Toker A, Cantley LC. Signalling through the lipid products of phosphoinositide-3-OH kinase. Nature. 1997;387:673–676. doi: 10.1038/42648. [DOI] [PubMed] [Google Scholar]
- 30.Lin J, Adam RM, Santiestevan E, Freeman MR. The phosphatidylinositol 3'-kinase pathway is a dominant growth factor-activated cell survival pathway in LNCaP human prostate carcinoma cells. Cancer Res. 1999;59:2891–2897. [PubMed] [Google Scholar]
- 31.Holbro T, Beerli RR, Maurer F, Koziczak M, Barbas CF, Hynes NE. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc Natl Acad Sci USA. 2003;100:8933–8938. doi: 10.1073/pnas.1537685100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bianco R, Shin I, Ritter CA, Yakes FM, Basso A, Rosen N, Tsurutani J, Dennis PA, Mills GB, Arteaga CL. Loss of PTEN/MMACI/TEP in EGF receptor-expressing tumor cells counteracts the antitumor action of EGFR tyrosine kinase inhibitors. Oncogene. 2003;22:2812–2822. doi: 10.1038/sj.onc.1206388. [DOI] [PubMed] [Google Scholar]
- 33.Forgacs E, Biesterveld EJ, Sekido Y, Fong K, Muneer S, Wistuba II, Milchgrub S, Brezinschek R, Virmani A, Gazdar AF, Minna JD. Mutation analysis of the PTEN/MMAC1 gene in lung cancer. Oncogene. 1998;17:1557–1565. doi: 10.1038/sj.onc.1202070. [DOI] [PubMed] [Google Scholar]
- 34.Soria J, Lee H, Lee JI, Wang L, Issa J, Kemp BL, Liu DD, Kurie JM, Mao L, Khuri FR. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin Cancer Res. 2002;8:1178–1184. [PubMed] [Google Scholar]
- 35.Brognard J, Clark AS, Ni Y, Dennis PA. Akt/protein kinase B is constitutively active in non-small cell lung cancer cells and promotes cellular survival and resistance to chemotherapy and radiation. Cancer Re. 2001;61:3986–3997. [PubMed] [Google Scholar]
- 36.Stein RC. Prospects for phosphoinositide 3-kinase inhibition as a cancer treatment. Endocrine Rel Cancer. 2001;8:237–248. doi: 10.1677/erc.0.0080237. [DOI] [PubMed] [Google Scholar]
- 37.Fan W, Musa Specht K, Zhang C, Goldenberg DD, Shokat KM, Weiss WA. Combinatorial efficacy achieved through two-point blockade within a signaling pathway-a chemical genetic approach. Cancer Res. 2003;63:8930–8938. [PubMed] [Google Scholar]
- 38.Roche S, Downward J, Raynal P, Courtneidge SA. A function for phosphatidylinositol 3-kinase β (p85α -p110β) in fibroblasts during mitogenesis: requirement for inslulin- and lysophosphatidic acid-mediated signal transduction. Mol Cell Biol. 1998;18:7119–7129. doi: 10.1128/mcb.18.12.7119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hooshmand-Rad R, Hajkova L, Klint P, Karlsson R, Vanhaesebroeck B, Claesson-Welsh L, Heldin CH. The PI 3-kinase isoforms p110(alpha) and p110(beta) have differential roles in PDGF- and insulin-mediated signaling. J Cell Science. 2000;113:207–214. doi: 10.1242/jcs.113.2.207. [DOI] [PubMed] [Google Scholar]
- 40.Roche S, Koegl M, Courtneidge SA. The phosphatidylinositol 3-kinase alpha is required for DNA synthesis induced by some, but not all, growth factors. Proc Natl Acad Sci USA. 1994;91:9185–9189. doi: 10.1073/pnas.91.19.9185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho H, Mu J, Lim JK, Thorvaldsen JL, Chu Q, Crenshaw EB, Kaestner KH, Bartolomei MS, Shulman GI, Birnbaum MJ. Insulin resistance and a diabetes mellitus-like syndrome in mice lacking the protein kinase Akt2 (PKBβ) Science. 2001;292:1728–1731. doi: 10.1126/science.292.5522.1728. [DOI] [PubMed] [Google Scholar]
- 42.Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks JR, McNeish JD, Coleman KG. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKBβ. J Clin Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebbler T, Fujii N, Musi N, Hirshman MF, Goodyear LJ, Moller DE. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. doi: 10.1172/JCI13505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zou M, Kirkpatrick SS, Davis BJ, Nelson JS, Wiles WG, Schlattner U, Neumann D, Brownlee M, Freeman MB, Goldman MH. Activation of the AMP-activated protein kinase by the anti-diabetic drug metformin in vivo - role of mitochondrial reactive nitrogen species. J Biol Chem. 2005;279:43940–43951. doi: 10.1074/jbc.M404421200. [DOI] [PubMed] [Google Scholar]
- 45.Holmes BF, Kurth-Kraczek EJ, Winder WW. Chronic activation of 5'-AMP-activated protein kinase increases GLUT-4, hexokinase, and glycogen in muscle. J Appl Physiol. 1999;87:1990–1995. doi: 10.1152/jappl.1999.87.5.1990. [DOI] [PubMed] [Google Scholar]
- 46.Buzzai M, Bauer DE, Jones RG, DeBerardinis RJ, Hatzivassiliou G, Elstrom RL, Thompson CB. The glucose dependence of Akt-transformed cells can be reversed by pharmacologic activation of fatty acid β-oxidation. Oncogene. 2005:1–9. doi: 10.1038/sj.onc.1208622. [DOI] [PubMed] [Google Scholar]
- 47.Baumann CA, Ribon V, Kanzaki M. CAP defines a second signalling pathway required for insulin-stimulated glucose transport. Nature. 2000;407:202–207. doi: 10.1038/35025089. [DOI] [PubMed] [Google Scholar]
- 48.Cheng AYY, Fantus IG. Oral antihyperglycemic therapy for type 2 diabetes mellitus. Can Med Assoc J. 2005;172:213–226. doi: 10.1503/cmaj.1031414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Keshamouni VG, Reddy RC, Arenberg DA, Joel B, Thannickal VJ, Kalemkerian GP, Standiford TJ. Peroxisome proliferator-activated receptor-g activation inhibits tumor progression in non-small-cell lung cancer. Oncogene. 2004;23:100–108. doi: 10.1038/sj.onc.1206885. [DOI] [PubMed] [Google Scholar]
- 50.Munugalavadla V., Borneo J., Ingram D.A. and Kapur R. p85α subunit of class 1A PI-3 kinase is crucial for macrophage growth and migration, Blood, 2005.March 15 [epub ahead of print]. [DOI] [PMC free article] [PubMed]