The SNARE Motif Is Essential for the Formation of Syntaxin Clusters in the Plasma Membrane (original) (raw)
Abstract
In the plasma membrane, syntaxin 1 and syntaxin 4 clusters define sites at which secretory granules and caveolae fuse, respectively. It is widely believed that lipid phases are mandatory for cluster formation, as cluster integrity depends on cholesterol. Here we report that the native lipid environment is not sufficient for correct syntaxin 1 clustering and that additional cytoplasmic protein-protein interactions, primarily involving the SNARE motif, are required. Apparently no specific cofactors are needed because i), clusters form equally well in nonneuronal cells, and ii), as revealed by nanoscale subdiffraction resolution provided by STED microscopy, the number of clusters directly depends on the syntaxin 1 concentration. For syntaxin 4 clustering the N-terminal domain and the linker region are also dispensable. Moreover, clustering is specific because in both cluster types syntaxins mutually exclude one another at endogenous levels. We suggest that the SNARE motifs of syntaxin 1 and 4 mediate specific syntaxin clustering by homooligomerization, thereby spatially separating sites for different biological activities. Thus, syntaxin clustering represents a mechanism of membrane patterning that is based on protein-protein interactions.
INTRODUCTION
The plasma membrane is a crowded place where numerous biological activities occur simultaneously. For fast and efficient processing, it could be envisaged that required factors are enriched in specialized ‘reaction centers’. It is, therefore, not surprising that lateral protein inhomogeneities have been well documented by fairly different experimental approaches. For instance, tracking of membrane proteins revealed that most do not enjoy continuous, unrestricted lateral diffusion, with certain proteins being transiently confined to small domains (for review, see Kusumi et al. (1)). Other groups have visualized membrane proteins by immunofluorescence and have seen characteristic patterns or even discrete domains. Furthermore, biochemical experiments indirectly suggest the existence of microdomains. Detergent solubilization experiments led to the discovery of detergent-resistant membranes (DRMs, also called membrane rafts) enriched in cholesterol, sphingomyelin, and special proteins (2). The raft hypothesis postulates that DRMs in live cells are stabilized by cholesterol and sphingomyelin and reflect microdomains into which certain proteins are preferentially accumulated. This idea has stimulated the interest in membrane patterning enormously and strengthened the common view that lipids are essential for microdomain formation. Nowadays rafts are suggested to be involved in apoptosis, cell adhesion, cell migration, synaptic transmission, membrane trafficking, cytoskeletal organization, and pathogen entry (for review see, e.g., Brown and London (3) and Munro (4)). However, this does not necessarily mean that lipids alone are sufficient for membrane patterning; protein-protein interactions could also play an important role in this process. From a conceptual point of view, the multitude of proteins and biological processes embedded in the plasma membrane evidently require highly specific segregation mechanisms that could at least partly be achieved by protein-protein interactions. In this scenario, lipids would provide a basic pattern of lipid phases into which certain proteins are preferentially inserted at the start of membrane patterning, with protein-protein interactions eventually refining this process.
Investigating the plasmalemmal distribution of the SNAREs (soluble _N_-ethylmaleimide-sensitive factor attachment protein receptors) syntaxin 1A and syntaxin 4 we found evidence for such a model. SNAREs are a superfamily of small, mostly membrane-bound proteins sharing a homologous sequence of 60–70 amino acids, the SNARE motif (5). In the case of syntaxins 1–4, this motif is anchored to the plasma membrane by a C-terminal transmembrane region (TMR) and attached to a large N-terminal domain via a linker region.
Specific sets of SNAREs drive intracellular membrane fusion steps (6,7). In exocytosis, membranes merge during complex formation between SNAREs associated with the plasma membrane and the corresponding vesicle. For instance regulated vesicle fusion is mediated by the plasma membrane associated SNAREs syntaxin 1A and SNAP-25 and the vesicle associated SNARE synaptobrevin 2, whereas in constitutive exocytosis syntaxin 4 and SNAP-23 (both plasma membrane associated) and cellubrevin (vesicle associated) are involved. In recent years, the organization of plasmalemmal SNAREs has been the subject of several studies. Microscopic analysis of membrane lawns (8–10) and cells (11–13) documented that they are concentrated in microdomain like structures, often called clusters. Moreover, syntaxin 1 and syntaxin 4 clusters have been shown to define docking and fusion sites for secretory vesicles and caveolae, respectively (9,10,12). In microscopic studies, varying degrees of SNARE distribution changes have been observed after cholesterol depletion, ranging from moderate (9,10) to complete disintegration of SNARE domains (8,9), indicating an important role of lipids for SNARE domain integrity. Biochemical experiments based on DRMs isolation documented that cholesterol depletion disturbs SNARE microdomains (14) and led to the suggestion that SNAREs are enriched in membrane rafts (15). However, some SNAREs do not cofloat with raft markers when stringent solubilization conditions are applied (9,16). Nonetheless, it has been established beyond question that the integrity of SNARE domains depends on cholesterol.
Here we report that lipids alone are not sufficient for correct syntaxin clustering but that additional protein-protein interactions are also required. We found that syntaxin clustering in the native membrane is mediated by specific homooligomerization involving the SNARE motif. Hence, by means of syntaxin clustering, cells are able not only to define but also to spatially separate sites with different functions.
MATERIALS AND METHODS
Cell culture and transfection
PC12 cells (clone 251; (17)) and BHK cells were maintained, propagated, and transfected essentially as described (9) apart from the following modifications. For single and cotransfection experiments, 20–40 _μ_g of the corresponding plasmids were used per cuvette. Experiments with PC12 cells were carried out ∼48 h posttransfection. BHK cells were grown in medium containing 1% fetal calf serum and used ∼24 h posttransfection.
Antibodies
Monoclonal antibodies were used for the detection of syntaxin 1 (HPC-1) (18) and the myc tag (CRL-1729 ATCC). For detection of syntaxin 4 an affinity purified rabbit polyclonal antibody was applied (9). As secondary antibodies we used Cy3-coupled goat-anti-mouse and Cy5-coupled goat-anti-rabbit (both from Dianova, Hamburg, Germany). For STED experiments, sheep-anti-mouse immunoglobulins G (catalogue No. 515-005-003, Dianova) were labeled with Atto532 (provided by K. H. Drexhage, Dept. of Chemistry, University of Siegen, Germany).
Plasmids
Plasmids for transient overexpression were produced by standard molecular biological methods. The encoded fusion proteins were epitope tagged with a N-terminal c-myc (M_EQKLISEEDL_NS), and/or the C-terminus was linked by 12 amino acids (LVPRARDPPVAT) to a variant of enhanced green fluorescent protein (EGFP). The single amino acid substitution A206K, previously shown to prevent dimerization of fluorescent proteins (19), was introduced, resulting in monomeric EGFP (mGFP). pBob5.1 (20) was used as the vector backbone for all constructs encoding c-myc tagged proteins. The plasmids carrying the coding sequences of fusion proteins without N-terminal tag are based on the vector pEGFP-N1 (Clontech, Mountain View, CA) (GenBank accession No. U55762). Using the rat sequence of syntaxin 1A and the corrected rat sequence for syntaxin 4 (as described (21)) as references, the coding sequences have been verified by sequencing for all constructs. The constructs used for transient overexpressions coded for the following tagged proteins: Sx1A-green fluorescent protein (GFP) [Sx1A-(1-288) + mGFP]; Sx1A, SNARE motif-TMR-GFP [Sx1A-(1–28 + 183–288) + mGFP]; Sx1A, TMR-GFP [Sx1A-(1–28 + 259–288) + mGFP]; Sx1AmutTMR-GFP [Sx1A-(1–288 carrying the mutations M267A, C271A, and I279A) + mGFP]; Sx4-GFP [Sx4-(1–298) + mGFP]; Sx4, SNARE motif-TMR-GFP [Sx4-(1–37 + 191–298) + mGFP]; Sx4, TMR-GFP [Sx4-(1–37 + 267–298) + mGFP]; myc-Sx1A [myc-tag + Sx1A-(2–288)]; myc-Sx1Aopen [myc-tag + Sx1A-(2–288 carrying the mutations L165A and E166A)]; myc-Sx4 [myc-tag + Sx4-(2–298)]; myc-Sx1A-GFP [myc-tag + Sx1A-(2–288) + mGFP].
Immunofluorescence
Membrane sheets were prepared as previously described (22), except that for onstage sonication a different sonifier was used (Sonifier B12, Branson Ultrasonics, Danbury, CT). In brief, cells were grown on poly-L-lysine-coated coverslips and disrupted by a 100 ms ultrasound treatment in ice cold sonication buffer (20 mM Hepes, pH 7.2, 120 mM potassium glutamate, 20 mM potassium acetate, and 10 mM EGTA). Freshly prepared membrane sheets were fixed for 90–120 min at room temperature in 4% paraformaldehyde in phosphate-buffered saline (PBS) (137 mM NaCl, 2.7 mM KCl, and 8.1 mM Na2HPO4, pH 7.3) and immunostained using standard protocols, essentially as described (9). For Fig. 1, E_–_F, and syntaxin 4 stainings, several steps were performed with high salt PBS (the NaCl concentration was elevated to 500 mM) containing 3% bovine serum albumin. STED microscopy was carried out on HPC-1/sheep-anti-mouse-Atto532 stained coverslips mounted in Mowiol (6 g Glycerol AR (No. 4094, Merck, Darmstadt, Germany), 2.4 g Mowiol 4-88 (Hoechst, Franfort, Germany), 6 ml water, 12 ml 200 mM Tris, pH 7.2 buffer).
FIGURE 1.

Overexpression of syntaxin 1A in BHK and PC12 cells. (A_–_C) Syntaxin 1A-GFP clusters in BHK cells lacking endogenous syntaxin 1. A brightly fluorescent cell was selected and disrupted by ultrasound treatment on the microscope stage. Immediately after rupture, an image was taken in the GFP channel (B; for magnified view see C). To rule out the possibility of areas devoid of fluorescence being holes in the plasma membrane, membrane integrity was documented by staining phospholipids with TMA-DPH (A). (D_–_F) Overexpression of syntaxin 1A-GFP in PC12 cells. Membrane sheets were fixed and immunostained with an antibody visualizing endogenous and overexpressed syntaxin 1 (E; for different scaling see F). As judged from the GFP fluorescence (D), the left membrane sheet contained almost no overexpressed syntaxin 1A-GFP, therefore immunostained clusters arise largely from endogenous syntaxin 1 (E). A highly elevated syntaxin 1 level, as documented for the right membrane sheet, results in a more diffuse appearance (F).
In double immunostaining and coclustering experiments, 0.2 _μ_m Tetraspek beads (Molecular Probes, Eugene, OR) were added and allowed to adsorb to the glass coverslip before imaging in PBS containing 1-(4-trimethyl-ammoniumphenyl)-6-phenyl-1,3,5-hexatriene (TMA-DPH, Molecular Probes). The Tetraspek beads acted as a spatial reference to check the automated correction for the lateral shifts that frequently occur upon filter changes, whereas TMA-DPH visualizes phospholipid membranes and allows assessment of membrane integrity.
Fluorescence microscopy
Membrane sheets were analyzed using a Zeiss Axiovert 100 TV fluorescence microscope with a 100× 1.4 numerical aperture plan apochromat oil objective (Zeiss, Göttingen, Germnay). Illumination was provided by a XBO 75 xenon lamp. For imaging, we used a back-illuminated frame transfer charge-coupled device camera (Princeton Instruments, Princeton, NJ) with a magnifying lens (2.5× Optovar, Zeiss) to avoid spatial undersampling by large pixels. The focal position was controlled using a low voltage piezo translator device and a linear variable transformer displacement sensor/controller (Physik Instrumente, Waldbronn, Germany). Appropriate filter sets were applied for fluorescence excitation and detection. For the images shown in Fig. 1 and for the coclustering experiments the following channels were recorded: TMA-DPH (excitation bandpass (BP) 360/30–50, beamsplitter (BS) 395–420, and emission longpass (LP) 420 or BP 460/50), GFP (excitation BP 480/40, BS LP 505, and emission BP 527/30), Cy3 (excitation BP 565/30, BS LP 595, and emission BP 645/75). For double immunolabeling experiments, the following filter sets were used for TMA-DPH (excitation BP 350/50, BS 395, and emission LP 420), Cy3 (excitation BP 525/30, BS LP 550, and emission BP 575/30) and Cy5 (excitation BP 620/60, BS LP 660, and emission BP 700/75). Images were acquired with Metamorph 5.1 (Universal Imaging, West Chester, PA).
STED microscopy
Stimulated emission depletion (STED) microscopy (23–25) was carried out with a home-built setup in which fluorescence excitation was performed with a pulsed laser diode emitting 100 ps pulses at 470 nm (Picoquant, Berlin, Germany). STED was performed using an optical parametric oscillator (OPO) by the company APE (Berlin, Germany) that was pumped by a mode-locked Ti:Sapphire laser (MaiTai, Spectra Physics, Mountain View, CA) operating at 80 MHz. The excitation diode was triggered by the OPO pulses. STED on the dye Atto532 was accomplished at a central wavelength of 615 nm. The initial duration of the STED pulses of 200 fs was stretched to 200 ps to reduce photobleaching (26). The conversion of the STED beam into a doughnut mode was accomplished by means of a spatial light modulator (Hamamatsu, Hamamatsu City, Japan) delivering a (0–2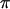 ) helical phase ramp. The excitation and the STED beams were coupled onto an oil immersion lens (HCX PL APO, 100×, Leica Microsystems, Mannheim, Germany) with 1.4 numerical aperture, by means of dichroic mirrors. The average power of the excitation and the STED beams at the sample was 1.9 _μ_W and 18 mW, respectively. The fluorescence was collected by the same lens and directed onto a counting avalanche photodiode. The photodiode featured an opening diameter of 71% of the backprojected Airy disk at the detector plane. The image was obtained by scanning the sample with a piezo stage featuring a positioning accuracy <10 nm.
) helical phase ramp. The excitation and the STED beams were coupled onto an oil immersion lens (HCX PL APO, 100×, Leica Microsystems, Mannheim, Germany) with 1.4 numerical aperture, by means of dichroic mirrors. The average power of the excitation and the STED beams at the sample was 1.9 _μ_W and 18 mW, respectively. The fluorescence was collected by the same lens and directed onto a counting avalanche photodiode. The photodiode featured an opening diameter of 71% of the backprojected Airy disk at the detector plane. The image was obtained by scanning the sample with a piezo stage featuring a positioning accuracy <10 nm.
The point spread function was experimentally determined by measuring the size of fluorescent point sources. For this purpose glass-adsorbed primary antibodies stained by Atto532-labeled secondary antibodies mounted in Mowiol were imaged. Intensity profiles of 426 single spots were fitted by a Lorentz function resulting in an average full width at half-maximum (FWHM) of 72 nm. For comparison, we also determined the FWHM in the confocal mode. Due to the lower resolution, not all spots analyzed in the STED mode were separated in the confocal image. Therefore only 50 spots were fitted by a Gaussian function, resulting in an average FWHM of 192 nm.
Analyzing syntaxin 1 cluster density and expression level
To determine the number of syntaxin 1A microdomains per _μ_m2, 2.4 _μ_m × 2.4 _μ_m regions from the center of the STED images were fast Fourier transform filtered in frequency space using blur (10%) and high pass (30%) options in Metamorph 4.1.7 (Universal Imaging Corporation). The central 0.81 _μ_m2 areas of the processed images were autoscaled and printed. On these printouts the number of clusters was counted by three referees independently, and the averaged number per _μ_m2 was plotted versus the average fluorescence intensity within the respective regions. The result of 80 membrane sheets yielded Fig. 2 C. For presentation, images showing membrane sheets in Fig. 2 were fast Fourier transform filtered applying the blur (30%) option of Metamorph 4.1.7 and scaled accordingly to enhance spotty image features.
FIGURE 2.

STED microscopy reveals a correlation between syntaxin 1 cluster density and expression level. (A and B, upper left) Experimentally determined point spread functions of confocal and STED microscopy. Glass-adsorbed primary antibodies visualized by fluorescently labeled secondary antibodies served as fluorescent point sources. For illustration, signals obtained for several primary antibodies were overlaid (12 for the confocal and 29 for the STED image) and line scans placed through the center of fluorescence. The determined FWHM (see Methods for details) are marked by arrows and approximate the resolution provided by the corresponding imaging technique. For the signal detected by STED microscopy, a FWHM of 72 nm was measured, representing a 7.1-fold reduction in focal area compared to confocal microscopy (FWHM 192 nm). (A and B, upper right) Confocal and STED micrographs from membrane sheets generated from PC12 cells transfected with myc syntaxin 1A. Both endogenous and overexpressed syntaxin 1 were visualized using an antibody recognizing the N-terminal domain of syntaxin 1. The processed images (see Methods for details) show several membrane sheets with varying expression levels ranging from low to high (as indicated in A). (A and B, lower panels) Magnified views from right upper images scaled to visualize individual spots. (C) From STED images as shown in B, we determined the number of fluorescent spots in a defined area and plotted them against the image intensity of the corresponding image (for details see Methods).
Correlation analysis
To quantitate the degree of similarity between images obtained in two different channels, the Pearson correlation coefficient was calculated for the corresponding pair of pictures, yielding an objective measure for the degree of colocalization of the visualized molecules. This value can range form −1 to 1 and reflects the degree of linear relationship between two variables (in this case the pixel intensities at corresponding pixel locations in the two channels).
A custom designed MATLAB 7.0.1.24704 (The MathWorks, Natick, MA) routine was applied. The two images were first automatically aligned and a region of interest (ROI) was defined in the green channel using a freehand tool. When placing the ROI on the membrane sheet, edges and obvious staining artifacts were avoided. ROIs were on average 22 _μ_m2 and 40 _μ_m2 in size for the coclustering and the double immunostaining experiments, respectively.
The Pearson correlation coefficient r was calculated within the ROI for the green and red image (i indicates individual pixel locations and av the average pixel intensity) according to r = Σi(greeni − greenav) × (redi − redav)/{Σi(greeni − greenav)2 × Σi(redi − redav)2}1/2 (for method, see also Manders et al. (27)).
In the coclustering experiments membrane sheets of transiently overexpressing cells were analyzed. To estimate the degree of overexpression the fluorescence intensity was calculated subtracting the local background measured in an area outside the membrane sheet from the mean fluorescence intensity within the ROI analyzed. Overexpressing membrane sheet with a background corrected GFP fluorescence of 200–1500 counts (4 s image) and netto immunostaining signal of 500–2500 counts (1 s image) were included in the analysis.
For each independent experiment, the correlation coefficients obtained from individual membrane sheets were averaged. Experiments yielding <3 sheets were excluded from the overall analysis, resulting in an average of 6.5 membrane sheets per independent experiment.
Colocalization analysis
To determine the colocalization of syntaxin 4 with syntaxin 1 microdomains based on morphological criteria, we used a procedure similar to one previously described (28). After aligning the two images as described for the correlation analysis, 20–21 circles were superimposed on bright fluorescent spots in the syntaxin 4 channel and transferred to identical image locations in the syntaxin 1 channel.
If the fluorescence intensity maximum in the syntaxin 1 channel was located in the same quadrant of the circle and the morphology of the signal resembled that of the syntaxin 4 cluster; the circle was rated as positive (colocalized), if not as negative (not colocalized). Clusters for which a clear assignment was not possible were considered as neutral and excluded from further analysis. To be able to correct for accidental background colocalization, due to the spot density, the circles were also transferred to a mirror image of the syntaxin 1 channel. Corrections were made to ensure that circles on the mirrored image were also placed on the membrane sheet. The assignment as positive, negative, or neutral was carried out as described above.
From five to seven membrane sheets were analyzed for each of three independent experiments. On average 1.20 (6.2%) syntaxin 4 clusters were rated as colocalized with syntaxin 1 microdomains, 18.11 (93.8%) as not colocalized, and 0.73 as neutral (not taken into account when determining the percentages). On the mirrored images an average of 0.96 circles (5.3%) were assigned as positive, 17.45 (94.7%) as negative, and 1.64 as neutral.
Background correction was performed as described (28) according to the following formula: real colocalization = (measured colocalization − background colocalization)/(1 − background colocalization/100), yielding a real colocalization of 0.9% ± 1.5% (n = 3 independent experiments, value given as mean ± SE).
RESULTS
Overexpression of syntaxin 1A in BHK and PC12 cells
To analyze the spatial distribution of syntaxin within the plasma membrane, we used plasma membrane sheets (29). In brief, cells grown on glass coverslips were disrupted by a short ultrasound pulse, which removed the upper part of cells leaving behind intact, two-dimensional plasma membrane sheets. These plasma membrane sheets are ideally suited for fluorescence microscopic examination. Using this preparation, we previously found syntaxin 1 to be concentrated in discrete clusters within the plasma membrane of neuroendocrine PC12 cells (9) (see also Fig. 4, middle panel). Since syntaxin 1 is expressed exclusively in neuronal tissues (30), we asked if syntaxin 1 clusters would also form in the fibroblast cell line BHK (baby hamster kidney) devoid from syntaxin 1, as confirmed by Western blotting (J. J. Sieber and T. Lang, unpublished data). Upon overexpression syntaxin 1A-GFP readily formed clusters (Fig. 1, B and C), demonstrating that neuronal cofactors are not mandatory for clustering.
FIGURE 4.

Syntaxin 4 and syntaxin 1 clusters are strictly separated. Double immunostaining for syntaxin 4 (left, red) and syntaxin 1 (middle, green). The lower panel shows magnified views of the corresponding images in the upper panel. Circles were superimposed onto bright fluorescent spots in the syntaxin 4 channel and transferred to identical image locations in the syntaxin 1 channel and the overlay (right). The colocalization of syntaxin 4 and syntaxin 1 was assessed by two independent approaches. Based on morphological criteria, we first determined the fraction of syntaxin 4 clusters colocalizing with syntaxin 1 clusters (for details see Methods) and found no colocalization (0.9% ± 1.5%; n = 3 independent experiments, values are given as mean ± SE). Second, the correlation coefficient of the two channels was calculated to be −0.01 ± 0.01 (n = 3 independent experiments, values are given as mean ± SE; for details see Methods).
However, this does not rule out the possibility that clustering depends on a more ubiquitously expressed protein. In this case, syntaxin should become more uniformly distributed when elevated to levels largely exceeding endogenous syntaxin, due to depletion of the putative cofactor. Upon strong overexpression in PC12 cells, the syntaxin patterning indeed appeared less discrete (Fig. 1 F; for untransfected cells see Fig. 4). However, it needs to be considered that in untransfected PC12 cells certain clusters are separated by a distance of only some hundred nanometers and, therefore, are hardly resolvable by conventional light microscopy. Hence, the question arises if the change in syntaxin pattern shown in Fig. 1 F is caused by syntaxin molecules unable to cluster or by increased syntaxin cluster density, yielding a more uniform appearance due to diffraction-limited resolution. To clarify this issue, we turned to STED microscopy, a diffraction-unlimited far-field microscopy technique (23) that provides nanoscale optical resolution (24,25,31).
Correlation of cluster density and syntaxin expression
To clarify if increasing syntaxin concentration generates either more clusters or a uniformly distributed syntaxin pool, membrane sheets with highly variable expression levels of syntaxin were analyzed at nanoscale optical resolution. To this end, myc-tagged syntaxin 1A was overexpressed in PC12 cells. Membrane sheets were generated and immunostained for endogenous and overexpressed syntaxin 1. For analysis, a microscope setup was used that simultaneously acquires images both in the confocal and the STED mode, featuring focal spot diameters of 192 and 72 nm, respectively (Fig. 2). In the confocal images, membrane sheets with highly variable syntaxin levels could be distinguished due to their staining intensities and were occasionally present in the same field of view (Fig. 2 A, upper right). When image features like spotty structures were enhanced by corresponding scaling (Fig. 2 A, lower panel), no relation between syntaxin distribution and expression level could be observed due to the limited resolution of confocal imaging. This was different in the STED mode. The (192/72)2 = 7.1-fold reduction in focal area achieved over confocal imaging revealed that the brighter the image the more clusters were present (Fig. 2 B, lower panel). A correlation became apparent when cluster density was plotted against image intensity (Fig. 2 C). Even when syntaxin levels were increased four- to fivefold over the endogenous level (taken to be the intensity of stainings on membrane sheets from untransfected cells; J. J. Sieber, K. I. Willig, S. W. Hell, and T. Lang, unpublished data), we did not observe a uniform syntaxin distribution, structures different from clusters, or clusters becoming obviously larger. It should be noted that upon highest overexpression the clusters become so dense that even the resolution of the STED microscope attained in this setup becomes a limiting factor. Nevertheless, although syntaxin 1 is already very abundant in the membrane it can be increased dramatically with all syntaxin 1 still appearing in clusters. This implies that no additional cofactors, apart from perhaps lipids, are essential for the clustering process. In summary, the overexpression studies presented suggest that syntaxin clustering does not depend on cofactors exclusively expressed in neuronal cells. Moreover, it appears that for syntaxin clustering no additional factors at all are limiting and that upon overexpression cluster number rather than size increases. So the nanoscale resolution provided by STED microscopy has proven to be powerful for studying plasmalemmal microdomains.
Correct clustering of syntaxins primarily requires the SNARE motif
The results so far have documented that cluster formation is an intrinsic property of syntaxin 1A. To test if protein-protein interactions are involved and to identify the responsible domain, we simultaneously overexpressed syntaxin 1A constructs carrying either a myc or a GFP tag, enabling us to discriminate the two corresponding syntaxin populations. Simultaneous overexpression of full-length syntaxin variants containing either tag resulted in high, but not perfect, colocalization of the differently visualized constructs (Fig. 3, A and B, upper panel). A similar result was obtained when a double-tagged syntaxin carrying both the myc epitope and the GFP on the N- and the C-terminus, respectively, was expressed. This shows that the minor, albeit noticeable, differences between the two images are probably due to imperfect epitope accessibility. To obtain an objective measure for the similarity of the two molecule distributions, the Pearson correlation coefficient was calculated for the two corresponding images. A correlation coefficient of 1 indicates perfect (pixel by pixel) colocalization, whereas a value of 0 shows that there is no correlation between the signals of the two channels.
FIGURE 3.

SNARE motif is mandatory for coclustering. (A) An immunostained membrane sheet generated from a PC12 cell cooverexpressing myc-tagged syntaxin 1A (left, red channel) and syntaxin 1A-GFP (middle, green channel). The right image represents a color overlay of both channels. A fluorescent bead (arrow) acts as a spatial reference, for cross-checking the automated alignment. (B) Magnified views from A (upper panel) and an experiment in which a syntaxin 1A-GFP construct (Sx1A, TMR-GFP) lacking the N-terminal domain, the linker region, and the SNARE motif was used (lower panel) instead of the full-length syntaxin. Arrows indicating some of the bright spots in the red channel were transferred to identical pixel locations in the green channel and the overlay. (C) Correlation analysis of experiments like those illustrated in A and B. To obtain an objective measure for the similarity between the red and the green channel, the Pearson correlation coefficient was calculated (see Methods for details). The images acquired for the myc- or GFP-tagged full-length syntaxin were correlated with the ones obtained for the illustrated syntaxin/syntaxin variants carrying the alternate tag (see Methods for details). Constructs analyzed from left to right: full-length syntaxin 1A (Sx1A-GFP), a construct lacking the N-terminal domain, linker region, and SNARE motif (Sx1A, TMR-GFP), syntaxin 1A lacking the N-terminal domain and linker region (Sx1A, SNARE motif-TMR-GFP), a mutant carrying three mutations abolishing TMR oligomerization (Sx1AmutTMR-GFP), and a construct with mutations preventing the closed conformation of syntaxin 1A (myc Sx1Aopen). For each construct, 4–5 independent experiments were performed. Values are given as mean ± SE.
Two-channel visualization of double-tagged syntaxin 1A yielded a correlation coefficient of 0.63 (n = 3 independent experiments), providing a reference for the maximal value obtainable with these tags. For myc syntaxin 1A and syntaxin 1A-GFP we obtained the lower value of 0.42 (Fig. 3 C), possibly because the two differently labeled syntaxins are not always present at a 1:1 stochiometry, mix also with the unlabeled endogenous syntaxin, and perfect clustering is not necessarily achieved by the biological system. In any case, this value is the reference for maximal coclustering in this experiment, since both constructs have the full, identical information required for clustering and therefore should appear in the same clusters. A construct lacking the N-terminal domain, the linker region, and the SNARE motif of syntaxin 1A (Syx1A, TMR-GFP) still showed a nonuniform distribution within the plasma membrane, but its ability to cocluster with the full-length syntaxin dropped dramatically as documented by a correlation coefficient of 0.08 (Fig. 3, B and C). In contrast, wild-type levels of coclustering with full-length syntaxin were obtained with variants that lacked the N-terminal domain and the linker region but maintained the SNARE motif, carried point mutations in the TMR that were previously shown to prevent self-oligomerization of this domain (32) or contained mutations in the linker region resulting in a permanently open conformation (33) (Fig. 3 C).
These data suggest that the SNARE motif is primarily responsible for the protein-protein interactions leading to correct clustering. A role of the N-terminal domain, either via homophilic interactions or by forming “bridges” between adjacent SNARE motifs, can be ruled out. Similarly, the TMR plays no role in syntaxin cluster formation, although it is capable of forming ‘cluster-like’ structures on its own.
Syntaxin 1 and 4 form distinct clusters
We then turned to syntaxin 4, a close relative of syntaxin 1 (65% aa similarity, (34)), which also has been reported to form clusters (9,10). First we tested if syntaxin 1 and 4 are organized in the same, or in distinct, clusters. Membrane sheets were double immunostained for the corresponding syntaxins and colocalization was examined by correlation analysis and by a method based on morphological criteria (28). No colocalization could be detected by either method (Fig. 4). Being strictly segregated, syntaxin 1 and 4 clusters reflect an intrinsic specificity of syntaxins to form homoclusters.
Next we asked if the N-terminal domain and the linker region are also dispensable for syntaxin 4 clustering. The maximal value in these coclustering experiments was given by the correlation of myc syntaxin 4 with syntaxin 4-GFP. No difference was observed for the according deletion construct when compared to this reference (Fig. 5 B). We could not test the effect of additionally deleting the SNARE motif, as all constructs made with varying linker regions between the TMR and GFP were not successfully sorted to the plasma membrane. We further asked if the clustering mechanism is also capable of separating syntaxin 1 and 4 when both are overexpressed. As shown in Fig. 5 B, coclustering of myc syntaxin 4 with syntaxin 1A-GFP is diminished when compared to syntaxin 4-GFP. However, probably due to an increase of unspecific interactions, the segregation of syntaxin 1 and 4 is weakened upon overexpression. Similar observations were made when myc syntaxin 1A was coclustered with syntaxin 4-GFP (J. J. Sieber and T. Lang, unpublished data).
FIGURE 5.

Coclustering experiment with syntaxin 4. (A) Representative images showing a magnified view of a membrane sheet generated from a PC12 cell overexpressing myc syntaxin 4 and syntaxin 4 GFP. Arrows pointing to bright spots in the red channel (left) were transferred to identical pixel locations in the green channel (middle) and the overlay (right). (B) Correlation analysis. From images like those illustrated in A, the correlation coefficient between the red and the green channel was determined (for details, see Methods). GFP-tagged constructs were tested for correlation with myc syntaxin 4. From left to right: full-length syntaxin 4 (Sx4-GFP), full-length syntaxin 1A (Sx1A-GFP), and a syntaxin 4 construct lacking the N-terminal domain and the linker region (Sx4, SNARE motif-TMR-GFP). For each construct, 4–6 independent experiments were performed. Values are given as mean ± SE.
The results document that, under physiological conditions, syntaxin 1 and 4 are strictly separated. Furthermore, the results for syntaxin 4 corroborate that the N-terminal domain is dispensable for syntaxin clustering.
DISCUSSION
The SNARE motif is essential for syntaxin clustering
In this study we have shown that syntaxins in the native plasma membrane form clusters by specific interactions requiring the SNARE motif. This is plausible considering what is known about the biochemistry of syntaxin 1A. In solution, the SNARE motifs of syntaxin 1A self-oligomerize at concentrations above 2 _μ_M, and within the homooligomers helices are aligned in parallel as shown by site-directed spin labeling (35). Also, the full cytoplasmatic domain is capable of forming oligomers, a feature abolished upon deleting part of the SNARE motif (36). This implies that the SNARE motif plays the essential role also in oligomerization of the whole cytoplasmic domain and that the N-terminal domain does not interfere with this process. Compared to in vitro experiments, in the plasma membrane this reaction should be even accelerated because syntaxins cannot rotate and translate in all directions, increasing the probability of effective collisions between syntaxin molecules. In summary, our model of syntaxin clustering is well in agreement with data obtained from in vitro studies.
Interestingly, the TMR is capable of forming separate ‘cluster-like’ structures on its own. At first sight, this casts into doubt the finding that all clusters observed by STED microscopy are mediated exclusively by SNARE motif interactions because, in the absence of cofactors, overexpressed syntaxin 1A could perhaps form clusters via TMR interactions. However, overexpressed myc syntaxin 1A should then cocluster with TMR-GFP. As this is not the case, our conclusion that no cofactors are required for syntaxin clustering remains solid.
That the TMR alone forms clusters is not unexpected, as recent findings have shown that the integrity of syntaxin 1 and syntaxin 4 clusters depends on cholesterol (9,10,12,14). Hence, the clustering of the TMR alone is most likely due to the affinity of the TMR for certain lipids and/or to TMR oligomerization (32). However, for complete and correct clustering cytoplasmic SNARE motif interactions are required. It cannot be ruled out that individual oligomers formed by cytoplasmic interactions are further cross-linked by TMR-mediated interactions or vice versa. Most likely a combination of both mechanisms leads to the concentration of dozens to some hundred syntaxin molecules within one syntaxin cluster, a number in line with our preliminary results (J. J. Sieber and T. Lang, unpublished data).
The physiological role of SNARE clustering
Hetero-SNARE complex formation drives intracellular membrane fusion (6,37), but the biological function of homooligomerization is so far unknown. It has been suggested that several SNARE complexes have to cooperate to mediate a fusion event (38), and syntaxin oligomers could provide the local high concentration required. Further, syntaxin oligomers may represent low stability storage forms, as has been suggested for the homotetrameric coiled-coil structure of the N-terminal domain of SNAP-23 (39). The notion that a hypothetical tetramer formed by four syntaxins aligned in parallel is destabilized (40) implies that syntaxin 1A could be released from for example tetramers, without energy consumption, in contrast to its release from stable heterotetrameric SNARE complexes (41).
The observation that syntaxin 1 and 4 form different clusters documents the specificity of the oligomerization, which, according to our model, lays the ground for the spatial separation of the different biological processes associated with both syntaxins. This appears to be in general the case, as in a recent study also syntaxin 3 and 4 have been described to be concentrated in separate clusters in the plasma membrane of epithelial cells before establishment of cell polarity (42). In both cases, syntaxin clusters could represent nucleation sites, at which other factors are recruited, leading to the formation of more complex, but locally restricted, protein networks. This idea is supported by the observations that syntaxin 1 clusters define sites for regulated exocytosis in both PC12 cells (9) and _β_-cells (12) and that fusion of caveolae occurs at syntaxin 4 clusters (10).
In summary, we propose that self-oligomerization of syntaxins, apart from possibly regulating SNARE activity, is also an important mechanism that, in combination with lipid phase partitioning of proteins, lays the ground for membrane compartmentalization. The attractiveness of this proposal is that it would enable cells to separate sites of different biological activities. The future will show if membrane patterning evolving from a combination of intact lipid infrastructure and specific protein-protein interactions is a general principle widely found in cell biology.
Acknowledgments
The authors thank Drs. Reinhard Jahn and Matthew Holt for helpful suggestions on the manuscript and Dr. Dagmar Schütz for the gift of plasmids.
T.L. was supported by a grant from the Deutsche Forschungsgemeinschaft (LA127212-1).
Rainer Heintzmann's present address is Randall Division of Cell and Molecular Biophysics, King's College London, New Hunt's House, Guy's Campus, London SE1 1UL, UK.
References
- 1.Kusumi, A., C. Nakada, K. Ritchie, K. Murase, K. Suzuki, H. Murakoshi, R. S. Kasai, J. Kondo, and T. Fujiwara. 2005. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: high-speed single-molecule tracking of membrane molecules. Annu. Rev. Biophys. Biomol. Struct. 34:351–378. [DOI] [PubMed] [Google Scholar]
- 2.Brown, D. A., and J. K. Rose. 1992. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell. 68:533–544. [DOI] [PubMed] [Google Scholar]
- 3.Brown, D. A., and E. London. 1998. Functions of lipid rafts in biological membranes. Annu. Rev. Cell Dev. Biol. 14:111–136. [DOI] [PubMed] [Google Scholar]
- 4.Munro, S. 2003. Lipid rafts: elusive or illusive? Cell. 115:377–388. [DOI] [PubMed] [Google Scholar]
- 5.Weimbs, T., S. H. Low, S. J. Chapin, K. E. Mostov, P. Bucher, and K. Hofmann. 1997. A conserved domain is present in different families of vesicular fusion proteins: a new superfamily. Proc. Natl. Acad. Sci. USA. 94:3046–3051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hong, W. 2005. SNAREs and traffic. Biochim. Biophys. Acta. 1744:120–144. [DOI] [PubMed] [Google Scholar]
- 7.Brunger, A. T. 2006. Structure and function of SNARE and SNARE-interacting proteins. Q. Rev. Biophys. 38:1–47. [DOI] [PubMed] [Google Scholar]
- 8.Chamberlain, L. H., and G. W. Gould. 2002. The vesicle- and target-SNARE proteins that mediate Glut4 vesicle fusion are localized in detergent-insoluble lipid rafts present on distinct intracellular membranes. J. Biol. Chem. 277:49750–49754. [DOI] [PubMed] [Google Scholar]
- 9.Lang, T., D. Bruns, D. Wenzel, D. Riedel, P. Holroyd, C. Thiele, and R. Jahn. 2001. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 20:2202–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Predescu, S. A., D. N. Predescu, K. Shimizu, I. K. Klein, and A. B. Malik. 2005. Cholesterol-dependent syntaxin-4 and SNAP-23 clustering regulates caveolae fusion with the endothelial plasma membrane. J. Biol. Chem. 280:37130–37138. [DOI] [PubMed] [Google Scholar]
- 11.Aoyagi, K., T. Sugaya, M. Umeda, S. Yamamoto, S. Terakawa, and M. Takahashi. 2005. The activation of exocytotic sites by the formation of phosphatidylinositol 4,5-bisphosphate microdomains at syntaxin clusters. J. Biol. Chem. 280:17346–17352. [DOI] [PubMed] [Google Scholar]
- 12.Ohara-Imaizumi, M., C. Nishiwaki, T. Kikuta, K. Kumakura, Y. Nakamichi, and S. Nagamatsu. 2004. Site of docking and fusion of insulin secretory granules in live MIN6 beta cells analyzed by TAT-conjugated anti-syntaxin 1 antibody and total internal reflection fluorescence microscopy. J. Biol. Chem. 279:8403–8408. [DOI] [PubMed] [Google Scholar]
- 13.Rickman, C., F. A. Meunier, T. Binz, and B. Davletov. 2004. High affinity interaction of syntaxin and SNAP-25 on the plasma membrane is abolished by botulinum toxin E. J. Biol. Chem. 279:644–651. [DOI] [PubMed] [Google Scholar]
- 14.Chamberlain, L. H., R. D. Burgoyne, and G. W. Gould. 2001. SNARE proteins are highly enriched in lipid rafts in PC12 cells: implications for the spatial control of exocytosis. Proc. Natl. Acad. Sci. USA. 98:5619–5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salaun, C., D. J. James, and L. H. Chamberlain. 2004. Lipid rafts and the regulation of exocytosis. Traffic. 5:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hering, H., C. C. Lin, and M. Sheng. 2003. Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J. Neurosci. 23:3262–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Heumann, R., V. Kachel, and H. Thoenen. 1983. Relationship between NGF-mediated volume increase and “priming effect” in fast and slow reacting clones of PC12 pheochromocytoma cells. Role of cAMP. Exp. Cell Res. 145:179–190. [DOI] [PubMed] [Google Scholar]
- 18.Barnstable, C. J., R. Hofstein, and K. Akagawa. 1985. A marker of early amacrine cell development in rat retina. Brain Res. 352:286–290. [DOI] [PubMed] [Google Scholar]
- 19.Zacharias, D. A., J. D. Violin, A. C. Newton, and R. Y. Tsien. 2002. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science. 296:913–916. [DOI] [PubMed] [Google Scholar]
- 20.Schutz, D., F. Zilly, T. Lang, R. Jahn, and D. Bruns. 2005. A dual function for Munc-18 in exocytosis of PC12 cells. Eur. J. Neurosci. 21:2419–2432. [DOI] [PubMed] [Google Scholar]
- 21.Pabst, S., J. W. Hazzard, W. Antonin, T. C. Sudhof, R. Jahn, J. Rizo, and D. Fasshauer. 2000. Selective interaction of complexin with the neuronal SNARE complex. Determination of the binding regions. J. Biol. Chem. 275:19808–19818. [DOI] [PubMed] [Google Scholar]
- 22.Avery, J., D. J. Ellis, T. Lang, P. Holroyd, D. Riedel, R. M. Henderson, J. M. Edwardson, and R. Jahn. 2000. A cell-free system for regulated exocytosis in PC12 cells. J. Cell Biol. 148:317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hell, S. W., and J. Wichmann. 1994. Breaking the diffraction resolution limit by stimulated emission: stimulated emission depletion microscopy. Opt. Lett. 19:780–782. [DOI] [PubMed] [Google Scholar]
- 24.Klar, T. A., S. Jakobs, M. Dyba, A. Egner, and S. W. Hell. 2000. Fluorescence microscopy with diffraction resolution limit broken by stimulated emission. Proc. Natl. Acad. Sci. USA. 97:8206–8210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Westphal, V., and S. W. Hell. 2005. Nanoscale resolution in the focal plane of an optical microscope. Phys. Rev. Lett. 94:143903. [DOI] [PubMed] [Google Scholar]
- 26.Dyba, M., and S. W. Hell. 2003. Photostability of a fluorescent marker under pulsed excited-state depletion through stimulated emission. Appl. Opt. 42:5123–5129. [DOI] [PubMed] [Google Scholar]
- 27.Manders, E. M., J. Stap, G. J. Brakenhoff, R. van Driel, and J. A. Aten. 1992. Dynamics of three-dimensional replication patterns during the S-phase, analysed by double labelling of DNA and confocal microscopy. J. Cell Sci. 103:857–862. [DOI] [PubMed] [Google Scholar]
- 28.Lang, T., M. Margittai, H. Holzler, and R. Jahn. 2002. SNAREs in native plasma membranes are active and readily form core complexes with endogenous and exogenous SNAREs. J. Cell Biol. 158:751–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lang, T. 2003. Imaging SNAREs at work in ‘unroofed’ cells-approaches that may be of general interest for functional studies on membrane proteins. Biochem. Soc. Trans. 31:861–864. [DOI] [PubMed] [Google Scholar]
- 30.Inoue, A., and K. Akagawa. 1993. Neuron specific expression of a membrane protein, HPC-1: tissue distribution, and cellular and subcellular localization of immunoreactivity and mRNA. Brain Res. Mol. Brain Res. 19:121–128. [DOI] [PubMed] [Google Scholar]
- 31.Hell, S. W. 2003. Toward fluorescence nanoscopy. Nat. Biotechnol. 21:1347–1355. [DOI] [PubMed] [Google Scholar]
- 32.Laage, R., J. Rohde, B. Brosig, and D. Langosch. 2000. A conserved membrane-spanning amino acid motif drives homomeric and supports heteromeric assembly of presynaptic SNARE proteins. J. Biol. Chem. 275:17481–17487. [DOI] [PubMed] [Google Scholar]
- 33.Dulubova, I., S. Sugita, S. Hill, M. Hosaka, I. Fernandez, T. C. Sudhof, and J. Rizo. 1999. A conformational switch in syntaxin during exocytosis: role of munc18. EMBO J. 18:4372–4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bennett, M. K., J. E. Garcia-Arraras, L. A. Elferink, K. Peterson, A. M. Fleming, C. D. Hazuka, and R. H. Scheller. 1993. The syntaxin family of vesicular transport receptors. Cell. 74:863–873. [DOI] [PubMed] [Google Scholar]
- 35.Margittai, M., D. Fasshauer, S. Pabst, R. Jahn, and R. Langen. 2001. Homo- and heterooligomeric SNARE complexes studied by site-directed spin labeling. J. Biol. Chem. 276:13169–13177. [DOI] [PubMed] [Google Scholar]
- 36.Lerman, J. C., J. Robblee, R. Fairman, and F. M. Hughson. 2000. Structural analysis of the neuronal SNARE protein syntaxin-1A. Biochemistry. 39:8470–8479. [DOI] [PubMed] [Google Scholar]
- 37.Jahn, R., and P. I. Hanson. 1998. Membrane fusion. SNAREs line up in new environment. Nature. 393:14–15. [DOI] [PubMed] [Google Scholar]
- 38.Montecucco, C., G. Schiavo, and S. Pantano. 2005. SNARE complexes and neuroexocytosis: how many, how close? Trends Biochem. Sci. 30:367–372. [DOI] [PubMed] [Google Scholar]
- 39.Freedman, S. J., H. K. Song, Y. Xu, Z. Y. Sun, and M. J. Eck. 2003. Homotetrameric structure of the SNAP-23 N-terminal coiled-coil domain. J. Biol. Chem. 278:13462–13467. [DOI] [PubMed] [Google Scholar]
- 40.Misura, K. M., R. H. Scheller, and W. I. Weis. 2001. Self-association of the H3 region of syntaxin 1A. Implications for intermediates in SNARE complex assembly. J. Biol. Chem. 276:13273–13282. [DOI] [PubMed] [Google Scholar]
- 41.Sollner, T., M. K. Bennett, S. W. Whiteheart, R. H. Scheller, and J. E. Rothman. 1993. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 75:409–418. [DOI] [PubMed] [Google Scholar]
- 42.Low, S. H., A. Vasanji, J. Nanduri, M. He, N. Sharma, M. Koo, J. Drazba, and T. Weimbs. 2006. Syntaxins 3 and 4 are concentrated in separate clusters on the plasma membrane prior to the establishment of cell polarity. Mol. Biol. Cell. 17:977–989. [DOI] [PMC free article] [PubMed] [Google Scholar]