HIC1 attenuates Wnt signaling by recruitment of TCF-4 and β-catenin to the nuclear bodies (original) (raw)
Abstract
The hypermethylated in cancer 1 (HIC1) gene is epigenetically inactivated in cancer, and in addition, the haploinsufficiency of HIC1 is linked to the development of human Miller–Dieker syndrome. HIC1 encodes a zinc-finger transcription factor that acts as a transcriptional repressor. Additionally, the HIC1 protein oligomerizes via the N-terminal BTB/POZ domain and forms discrete nuclear structures known as HIC1 bodies. Here, we provide evidence that HIC1 antagonizes the TCF/β-catenin-mediated transcription in Wnt-stimulated cells. This appears to be due to the ability of HIC1 to associate with TCF-4 and to recruit TCF-4 and β-catenin to the HIC1 bodies. As a result of the recruitment, both proteins are prevented from association with the TCF-binding elements of the Wnt-responsive genes. These data indicate that the intracellular amounts of HIC1 protein can modulate the level of the transcriptional stimulation of the genes regulated by canonical Wnt/β-catenin signaling.
Keywords: β-catenin, nuclear HIC1 bodies, TCF-4, Wnt signaling
Introduction
The Wnt signaling pathway plays essential roles in different developmental processes, including cell determination, stem cell survival and organogenesis. In addition, mutational activation of this pathway is implicated in deregulated cell growth and cancerogenesis (reviewed in Logan and Nusse, 2004; Reya and Clevers, 2005). The stabilization of β-catenin is central to the canonical Wnt pathway. In the absence of Wnt signals, β-catenin is phoshorylated by a complex of proteins, including adenomatous polyposis coli (APC), glycogen synthase kinase-3β and axin. Phosphorylation of β-catenin results in its ubiquitylation and degradation by the proteasome. Wnt factors inhibit the APC complex. The result of such inhibition is the stabilization of β-catenin, which accumulates in the cell and translocates into the nucleus where it associates with transcription factors of the TCF/LEF family. TCF/LEF proteins function as nuclear effectors of the Wnt signaling pathway. The DNA-binding specificity of these polypeptides is defined by the HMG box, an 80 amino-acid domain whose primary sequence is virtually identical in all TCF/LEF family members (reviewed in Clevers and van de Wetering 1997). TCF/LEF factors possess only a limited ability to activate transcription. They act as ‘connectors' linking other polypeptides to a distinct set of promoters. β-Catenin contains a strong transcription activation domain, thus its interaction with TCFs results in transcription of the Wnt-responsive genes. Many of these genes execute Wnt-mediated cell specification during development or regulate cell proliferation (for more detailed information, refer to the Wnt homepage http://www.stanford.edu/~rnusse/wntwindow.html). In contrast, mammalian TCFs also bind to TLE/Groucho corepressors and, in the absence of the Wnt signal, repress the transcription of TCF-specific promoters (Roose et al, 1998). Additionally, two TCF/LEF family members, TCF-3 and TCF-4, associate with C-terminal binding proteins (CtBPs) (Brannon et al, 1999; Valenta et al, 2003).
The CtBP proteins bind to a short sequence motif PLDLS conserved among the E1A proteins of all human and primate adenoviruses. Different variants of this motif are also present in many other CtBP-interacting partners that function mainly as sequence-specific DNA-binding transcription factors.
In this study, we focused on one of the CtBP-associating proteins, hypermethylated in cancer 1 (HIC1), which redirects CtBP to a specific set of nuclear dot-like structures called HIC1 bodies (Deltour et al, 2002). The HIC1 gene was identified as a candidate tumor suppressor gene frequently epigenetically silenced or deleted in different types of solid tumors (Herman and Baylin, 2003). HIC1 encodes a zinc-finger transcription factor that belongs to a group of proteins known as the BTB/POZ family (Broad-Complex, Tramtrack, Bric à brac/poxvirus, and zinc finger) (reviewed in Albagli et al, 1995). A 714 amino-acid human HIC1 polypeptide contains the N-terminal BTB/POZ domain involved in dimerization and in protein–protein interactions. The C-terminal region interacts with a specific DNA sequence; the GLDLSKK motif responsible for the interaction with the CtBP proteins is located in the central part. Gene inactivation experiments in mice recently confirmed that HIC1 is a genuine tumor suppressor. Heterozygous Hic1+/− mice develop malignant spontaneous tumors after a year of life (Chen et al, 2003, 2004). These tumors show dense methylation of the remaining wild-type Hic1 allele promoter accompanied by a complete absence of Hic1 expression in the cancer tissue. HIC1 gene resides within a 350 kb region on chromosome 17p13.3, deleted in most patients with Miller–Dieker syndrome (MDS) (Dobyns and Truwit, 1995). This links (in addition to tumorigenesis) the haploinsufficiency of HIC1 to the development of MDS.
In the present study, we show specific binding between HIC1 and a principal Wnt signaling pathway component, TCF-4. We further demonstrate that overexpression of HIC1 suppresses the TCF-mediated transcription, and vice versa, the inactivation of endogenous HIC1 by RNA interference (RNAi) increases the basal expression of the Axin2 gene and elevates the transcriptional response of this Wnt signaling target to Wnt stimulation. A deletion mutant of HIC1 lacking the oligomerization BTB/POZ domain can neither form the nuclear bodies, nor antagonize Wnt signaling, nor interact with TCF-4 in vivo. This clearly indicates that the HIC1 inhibitory function depends on the ability to form nuclear bodies and to recruit TCF-4 into these structures. Interestingly, β-catenin is also relocated by HIC1, but this sequestration seems to be indirect and mediated via its interaction with TCF-4. In addition, we provide evidence that CtBP1 increases the efficiency of recruitment of the TCF-4 into the HIC1 bodies and further strengthens the suppressive effect of the HIC1 protein on Wnt signaling. Finally, using chromatin immunoprecipitation (ChIP), we show that as a consequence of the relocation into the HIC1 speckles, TCF-4 and β-catenin are prevented from binding to the promoters of the TCF-responsive genes.
Results
HIC1 sequesters TCF-4 into nuclear bodies
Exogenous CtBP1 is distributed in the cytoplasm and in the nucleus in a mostly diffused pattern. Only a fraction of nuclear CtBP1 is localized into distinct structures, described previously as CtBP bodies (Sewalt et al, 1999). Cotransfection of the full-length HIC1 construct results in complete relocation of CtBP1 into the nuclear HIC1 bodies (Figure 1B). This relocation is dependent on the direct interaction between HIC1 and CtBP1, as the HIC1-ΔCtBP polypeptide lacking the CtBP-interacting motif loses the translocative properties of the wild-type HIC1 protein. The HIC1 mutant with a deletion encompassing the N-terminal oligomerization BTB/POZ domain still preserved the ability to ‘pull' CtBP1 into the nucleus. However, the localization of both proteins was diffuse and no formation of nuclear punctuated structures was observed.
Figure 1.
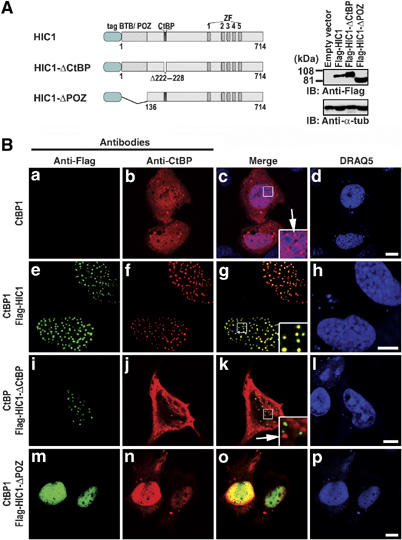
HIC1 targets CtBP into the nuclear bodies. (A) A schematic representation of the human HIC1 constructs used in this study. tag, Flag or EGFP tag; BTB/POZ, the BTB/POZ domain; CtBP, CtBP-binding site; ZF, five C2H2 Krüppel-like zinc fingers. Right, Western blots of total cell extracts after transfection with the Flag-HIC1 constructs, probed with anti-Flag or with anti-α-tubulin as internal control. (B) Confocal microscopy images of CtBP(−/−) cells trasfected with constructs indicated on the left and subsequently stained with mouse anti-Flag and rabbit anti-CtBP antibody. The DRAQ5 nuclear stain was gained in the blue channel. Mutant HIC1-ΔCtBP polypeptide lacking the CtBP-interacting motif displays the punctuated expression of the wild-type HIC1 protein (compare (e) and (i)) but does not influence the distribution of CtBP (compare (b) and (j)). As seen in (j, k), a fraction of nuclear CtBP is still localized in the CtBP bodies (arrows in insets (c, k)), which evidently differ from the HIC1 bodies. Bar, 10 μm.
Previously, we have shown an interaction between CtBP1 and TCF-4 in yeast and in vitro (Valenta et al, 2003). However, we and others in following studies were unable to detect the association of these proteins in mammalian cells. We took advantage of the clear nuclear targeting of CtBP1 by HIC1 and visualized TCF-4 and CtBP1 using confocal microscopy.
In CtBP-positive COS-7 cells, TCF-4 was efficiently sequestered into the HIC1 bodies (Figure 2B). Triple staining and the overlay of the images showed that in these bodies TCF-4, HIC1 and CtBP1 co-localized. CtBP interacts with both, TCF-4 and HIC1 proteins, thus we supposed that the TCF-4 recruitment was mediated by CtBP. As expected, mutated TCF-4 (TCF-4mutCtBP), which is unable to bind CtBP, was not sequestered into the HIC1 bodies even in the CtBP1 background (Figure 2C). Surprisingly, in CtBP(−/−) cells (these cells were derived from _CtBP1_−/−_CtBP2_−/− embryos (Hildebrand and Soriano, 2002)), wild-type HIC1 and TCF-4 still partially colocalized in the nuclear dots. We examined next, whether variants of HIC1 differ in their ability to concentrate TCF-4 into the HIC1 bodies in CtBP1(+) cells. Whereas wild-type proteins perfectly colocalized in nuclear dots, the HIC1-ΔCtBP mutant displayed only limited capability to sequester nuclear TCF-4.
Figure 2.
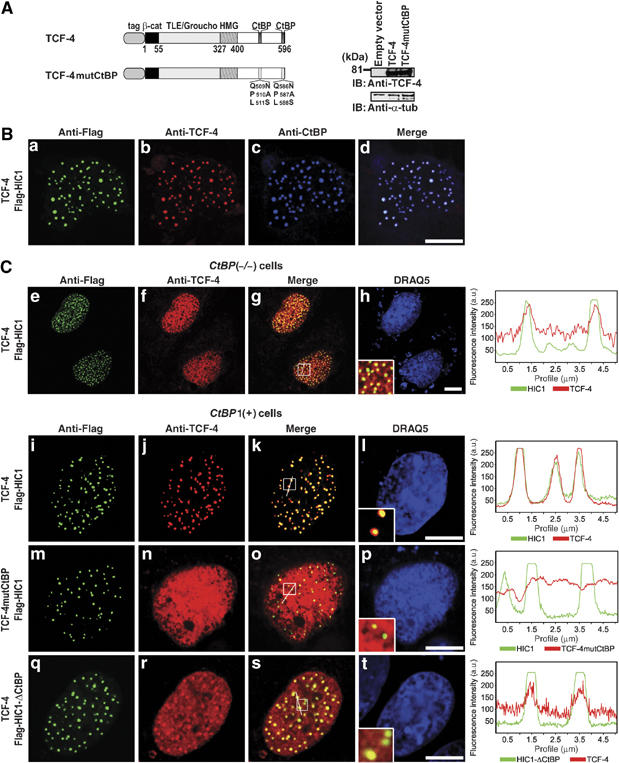
TCF-4 and HIC1 form nuclear protein complexes in mammalian cells. (A) TCF-4 expression constructs. The TCF-4mutCtBP polypeptide has a triple amino-acid substitution of each CtBP-binding motif as indicated. tag, myc or EGFP tag; β-cat, β-catenin interaction domain; TLE/Groucho, TLE/Groucho binding domain; CtBP, CtBP binding sites; HMG, DNA-binding domain. Right, Western blots of total cell extracts after transfection with the indicated TCF-4 constructs, probed with anti-TCF-4 or with anti-α-tubulin. (B) CtBP, HIC1 and TCF-4 colocalize in COS-7 cells. (C) Simultaneous interaction between CtBP, TCF-4 and HIC1 is essential for the efficient nuclear sequestration of TCF-4 into the HIC1 bodies. Confocal microscopy images of CtBP(−/−) and CtBP1(+) cells transfected with the indicated constructs (left) and stained with anti-Flag and anti-TCF-4 antibody. The right panel shows the overlap of fluorescence intensity peaks along profiles as indicated in the merged micrographs. The nuclear sequestration of TCF-4 by HIC1 is less efficient in CtBP(−/−) than in CtBP(+) cells (compare (e, f, g, h) to (i, j, k, l)). The formation of the TCF-4/HIC1 bodies in CtBP(+) cells strictly depends on the presence of the intact CtBP-binding sites in TCF-4 (m, n, o, p). Notice only a partial colocalization of TCF-4 and HIC1-ΔCtBP (q, r, s, t). Bar, 10 μm.
Taken together, these results suggested that TCF-4 and HIC1 could form nuclear aggregates even in the absence of CtBP; nevertheless, CtBP mediates more efficient recruitment of TCF-4 into the HIC1 bodies.
TCF-4 binds directly to HIC1
The existence of HIC1/TCF-4 complexes in mammalian cells was evidenced using co-immunoprecipitation of HIC1 with endogenous TCF-4. By using anti-TCF-4 antibody, a robust coisolation of TCF-4 with wild-type HIC1, and with HIC1-ΔCtBP, was obtained from lysates of human 293 cells; the truncated HIC1-ΔPOZ protein did not co-immunoprecipitate with TCF-4 (Figure 3A). TCF-4 was also isolated from the Flag-tagged HIC1 using anti-Flag or anti-HIC1 antibodies. Furthermore, we immunoprecipitated the endogenous HIC1/TCF-4 complexes from whole-cell lysates prepared from mouse embryos on day 12.5 p.c. We also performed coimmunoprecipitation experiments in mouse CtBP(−/−) and CtBP1(+) cells, and we detected association of EGFP-tagged HIC1 and TCF-4 in both _CtBP_-negative and _CtBP1_-positive cells. The coimmunoprecipitation was specific for HIC1, because a parallel assay did not show any binding of a control, EGFP-nls protein to TCF-4 (Figure 3B, only results from CtBP(−/−) cells are shown). These results implied a direct interaction between TCF-4 and HIC1.
Figure 3.
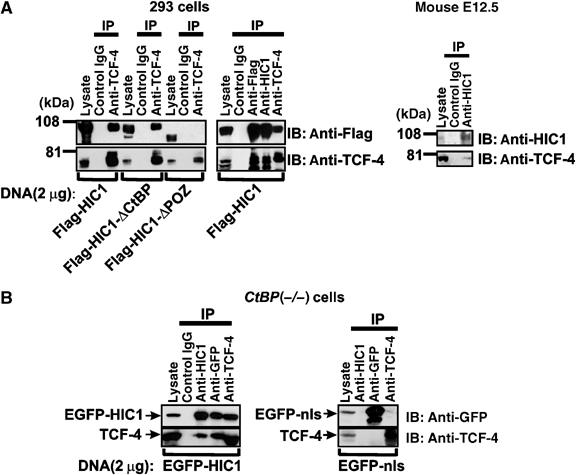
Association between TCF-4 and HIC1 in vivo. (A) Left, coimmunoprecipitations between endogenous TCF-4 and various exogenous HIC1 proteins in human 293 cells. Right, coimmunoprecipitations of endogenous HIC1 and TCF-4 in cells derived from mouse embryos on day 12.5 p.c. IP, immunoprecipitation; IB, immunoblotting; in lanes denoted ‘lysate' five percent of the total sample were loaded. (B) TCF-4 associates with HIC1 in CtBP(−/−) cells. Coimmunoprecipitation of endogenous TCF-4 with EGFP-HIC1 is specific for HIC1 as evidenced by no detectable interaction between TCF-4 and EGFP-nls in a control experiment (right).
The direct binding between TCF-4 and HIC1 was studied in vitro by pull-down assays between bacterially expressed GST-tagged TCF-4 and in vitro translated HIC1. GST-TCF-4 associated only with in vitro translated full-length HIC1 and not with the N-terminally truncated HIC1-ΔPOZ (Figure 4B). As was shown by others, the deletion of the BTB/POZ domain involved in the homo- and heteromeric interactions often prevents the association of HIC1 with other partners, although this domain is not directly included in the protein–protein interaction (Deltour et al, 2002). Due to extensive degradation of GST-HIC1 in bacterial lysates, we could not test the binding of the full-length HIC1 immobilized on the glutathione-Sepharose beads to in vitro translated TCF-4. Nevertheless, we prepared GST-fusion proteins containing partly overlapping N-terminal, internal, and C-terminal HIC1 fragments (Figure 4A), and used these more stable proteins in pull-down assays. As shown in Figure 4B, all three GST-HIC proteins interacted with full-length TCF-4; however, the strongest interacting domain was localized in the internal and C-terminal region of HIC1. Importantly, TCF-4mutCtBP binds to GST-HIC1 proteins equally well as the wild-type protein. Two nonoverlapping TCF-4 fragments were used to delineate domains involved in binding to HIC1. Whereas the C-terminus of TCF-4 interacted strongly with the GST-linked C-terminal part and the HIC1 internal fragments, a substantially less avid interaction was observed between the TCF-4 C-terminus and the GST-HIC1 N-terminus. Finally, the N-terminal part of TCF-4 did not show any affinity to HIC1. TCF-4 and HIC1 bind specific DNA sequences and their interaction domains were mapped in part to the DNA-binding regions of these proteins. To exclude the possibility that the association between TCF-4 and HIC1 is indirect and might be mediated by a DNA bridge from contaminating DNA, we performed a GST-pull-down with translated HIC1-mutZF3 protein containing a single amino acid exchange in the third zinc finger (Figure 4A). This mutation abolishes the DNA binding of the mutated protein to its recognition motif (Pinte et al, 2004). We also treated translated TCF-4 and glutathione-Sepharose bound GST-HIC1 with DNase I prior to the pull-down. As shown in Figure 4C, the HIC1-ZF3 associates with GST-TCF-4 with a comparable avidity to the wild-type protein and, moreover, the DNase I treatment even slightly improved in vitro binding of TCF-4 and HIC1.
Figure 4.
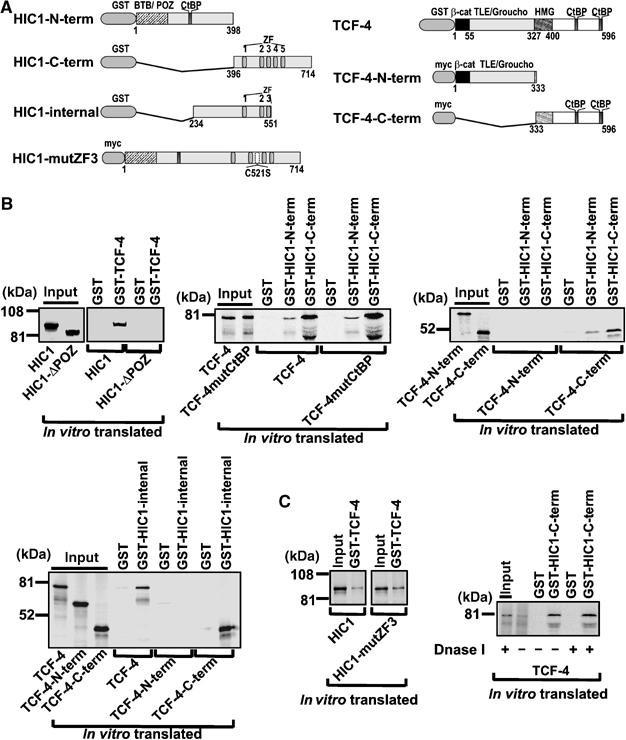
In vitro interaction of TCF-4 and HIC1. (A) Structures of HIC1 and TCF-4 proteins used in the in vitro pull-down assays (see also the diagrams of the additional HIC1 and TCF-4 constructs depicted in Figures 1A and 2A). (B) Pull-down assays between bacterially expressed GST-fusion and in vitro translated proteins, as indicated. Ten percent of the total reactions were loaded in lanes denoted ‘input'. (C) Right, TCF-4/HIC1 interaction is resistant to DNase I treatment; left, the intact DNA-binding domain of HIC1 is not essential for the interaction with TCF-4.
Altogether, the data reported here indicated that HIC1 interacts directly with TCF-4 in a complex multidomain mode of interaction. These data also implied that this interaction is not dependent on the presence of the intact CtBP-binding sites in TCF-4.
HIC1 inhibits TCF/_β_-catenin-driven transcription
To examine whether HIC1-mediated sequestration of TCF-4 affects the Wnt-dependent transcription, pTOPFLASH was cotransfected with each of the HIC1 constructs into 293 cells, the cells were subsequently stimulated by the Wnt ligand and the levels of the TCF-mediated transcription were determined. In 293 cells, Wnt3a-containing medium induced robust 25-five fold activation of the Tcf reporter pTOPFLASH as compared to the control medium (Figure 5). The cotransfection of wild-type HIC1 resulted in a substantial decrease of the pTOPFLASH activity, the HIC1-ΔCtBP was less than half as effective as wild-type HIC1, and finally, the HIC1-ΔPOZ mutant appeared to be completely inefficient in the downregulation of the pTOPFLASH-driven transcription. We did not observe any effect whatsoever on the pTOPFLASH reporter in the nonstimulated cells. Nevertheless, HIC1 repressed the Wnt-induced transcription of a luciferase reporter containing a 5 kb promoter region of the well-established Wnt target Axin2 gene (Figure 5) (Jho et al, 2002). Conversely, the HIC1 overexpression did not disturb the transcription from the negative-control reporter pFOPFLASH, or from the synthetic reporter G1E1B-Luc activated by Gal4-DBD-VP16 fusion protein (data not shown).
Figure 5.
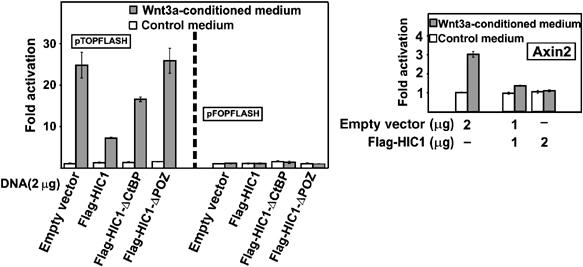
HIC1 represses the Wnt-stimulated transcription. Reporter gene assay with the Wnt-responsive promoters. 293 cells were cotransfected with the reporters and the HIC1 constructs as indicated and stimulated for 24 h with Wnt3a-conditioned or control medium. Luciferase (firefly) activities were corrected for the efficiency of transfection using the internal control Renilla luciferase expression plasmid. The reporter activity in unstimulated mock-transfected cells was arbitrarily set to 1. The histograms represent mean values of triplicate experiments and SDs (standard deviations) are shown by error bars.
HIC1 contains five Krüppel-like C2H2 zinc fingers in its C-terminal part. Recently, Pinte et al (2004) investigated the DNA binding properties of the isolated zinc finger domain and defined a specific DNA motif recognized by HIC1. Full-length HIC1 binds probes with a single recognition site poorly; however, the wild-type protein interacts cooperatively with complex probes containing multiple HIC1-specific sequences. In contrast, the N-terminally truncated HIC1-ΔPOZ, lacking the BTB/POZ domain, interacts preferentially with a probe containing a single HIC1-binding site. We performed an electrophoretic mobility-shift assay (EMSA) with in vitro translated TCF-4 and HIC1. HIC1-ΔPOZ, and with a lower efficiency, full-length HIC1, bound to a simple HIC1-specific probe, but these proteins did not interact directly with probes containing single or multiple TCF recognition motifs (Supplementary Figure 1).
The inhibitory effect of HIC1 on the transcriptional activation of pTOPFLASH was also found in DLD-1 cells, that is, cells with a mutant APC gene. The lack of functional APC protein in these adenocarcinoma cells results in the accumulation of β-catenin and in constitutive activation of the TCF-dependent target genes (van de Wetering et al, 2002; Rosin-Arbesfeld et al, 2003). By retroviral transduction, we generated DLD/HIC1 cells containing the HIC1-EGFP regulated by AP21967, a synthetic dimerizer (Ariad). We examined the levels of the TCF/β-catenin-dependent transcription simultaneously in three independent DLD/HIC1 cell lines at three different levels of HIC1-EGFP expression (dimerizer concentrations: 0, 0.25 and 25 nM). The result of a representative experiment is shown in Figure 6A. At maximum induction when HIC1 was produced in amounts comparable to the physiological levels of endogenous HIC1 in primary human WI38 cells (Figure 9C), the pTOPFLASH activity decreased to approximately 40% when compared to the DLD/HIC1 cells growing without the inducer (Figure 6A). Transcription from the negative control reporter pFOPFLASH did not change during the experiment. Since DLD-1 cells express high amounts of both TCF-4 and β-catenin (Korinek et al, 1997), we asked first whether HIC1 can function by decreasing the intracellular levels of β-catenin and/or TCF-4. Using anti-TCF-4 and β-catenin antibodies, we performed immunoblotting of cell lysates prepared from DLD/HIC1 growing with dimerizer for 5 days or without HIC1 induction. The analysis revealed that HIC1 overexpression does not reduce the overall levels of the endogenous TCF-4 and β-catenin proteins in the cells (Figure 6A). However, HIC1 sequestered endogenous β-catenin and TCF-4 into the nuclear HIC1 bodies (Figure 6B and Supplementary Figure 2). Using anti-GFP antibody, we coimmunoprecipitated β-catenin, HIC1 and TCF-4 in one complex from 293 cells (Figure 6C). As we did not observe a direct interaction between β-catenin and HIC1 in vitro using pull-down assays (Supplementary Figure 3), we concluded that β-catenin associates with HIC1 indirectly, possibly by binding to TCF-4.
Figure 6.
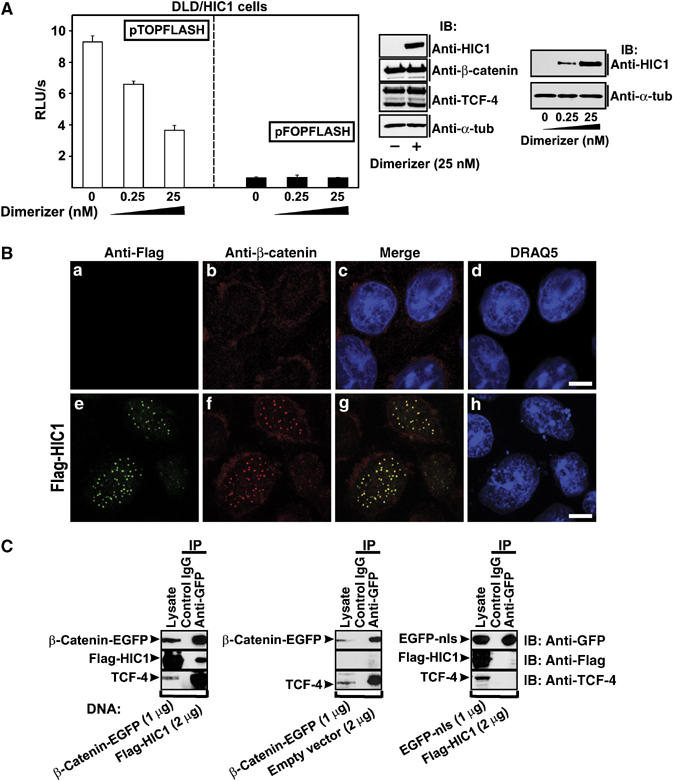
HIC1 represses TCF/β-catenin signaling in DLD-1 adenocarcinoma cells. (A) Right, transgenic DLD/HIC1 cells growing at higher concentrations of a synthetic compound AP21967 (dimerizer) contain increasing amounts of the HIC1 protein as evidenced by Western blots of total cell extracts probed with anti-HIC1 antibody. Middle, HIC1 expression does not influence the protein levels of TCF-4 and β-catenin. Left, the constitutive activity of the TCF-dependent reporter pTOPFLASH is suppressed by increasing amounts of the HIC1 protein. Average luciferase light units per second (RLU/s) corrected for the efficiency of transfection determined as the luciferase/Renilla ratio from five experiments are given (right, pFOPFLASH values). (B) Colocalization of HIC1 with endogenous β-catenin. Confocal micrographs of DLD-1 cells transfected with the full-length Flag-HIC1 construct stained with anti-Flag and anti-β-catenin antibody. Bar, 10 μm. (C) HIC1 expression does not disrupt the binding between TCF-4 and β-catenin. Coimmunoprecipitation of endogenous TCF-4 with ectopically expressed β-catenin is not affected by co-expression of HIC1 (compare left and middle panel). The coimmunoprecipitation is specific for β-catenin as indicated by a control experiment using the EGFP-nls instead of β-catenin-EGFP fusion protein (right).
Figure 9.
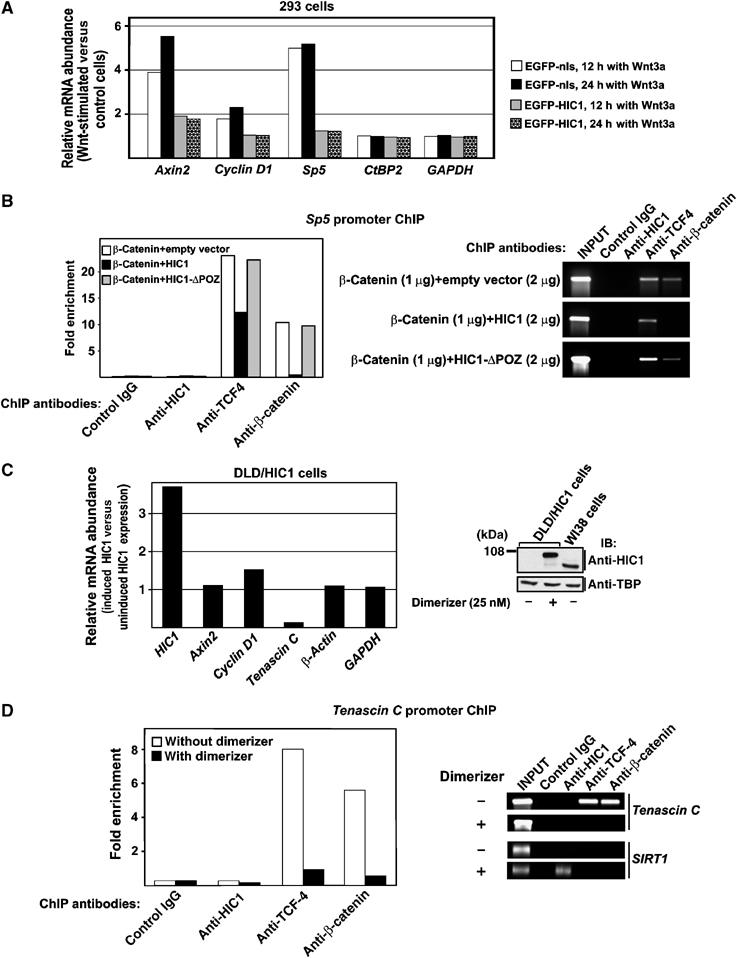
HIC1 sequesters TCF and β-catenin from the TCF-dependent promoters. (A) HIC1 blocks transcriptional activation of the Wnt signaling target genes. Results of qRT–PCR analyses performed with cDNA generated from 293 cells transfected with the indicated constructs upon 12- or 24-h stimulation with Wnt3a. Four to six PCR reactions were performed for each primer set. The relative abundance of the indicated mRNA in Wnt3a-stimulated versus control cells was derived from the average CT values after normalizing to the levels of β_-actin_ cDNA. (B) HIC1 is not associated with the Sp5 promoter but sequesters TCF-4 and β-catenin from the TCF-specific DNA element of this promoter. ChIP analysis of chromatin isolated from 293 cells transfected with the indicated constructs. The diagram at the left represents real-time PCR values obtained with primers spanning the respective DNA element, normalized to the inputs. The image on the right depicts relevant PCR products after 29 cycles of amplification. (C) HIC1-EGFP blocks transcription of the Tenascin C promoter in DLD-1 cells. Left, results of qRT–PCR analyses performed with cDNA prepared from DLD/HIC1 cells growing in the presence of the dimerizer (25 nM; HIC1 induction) or without induction. Right, Western blot analysis of nuclear extracts isolated from DLD/HIC1 and WI38 cells. (D) HIC1 sequesters TCF-4 and β-catenin from the Tenascin C promoter. Left, ChIP analysis of chromatin isolated from DLD/HIC1 cells prior to and upon HIC1-EGFP induction. The diagram at the left represents real-time PCR values obtained with primers spanning the proximal TCF-binding element in the Tenascin C promoter, normalized to the inputs. The image on the right depicts relevant PCR products after 29 cycles of amplification. Right bottom, although HIC1-EGFP is not associated with the Tenascin C promoter, it binds its recognition element in the SIRT1 promoter.
In summary, these data indicated that HIC1 specifically represses TCF-mediated transcription. Intriguingly, the repression is dependent on the recruitment of the TCF-4 to the HIC1 bodies rather than on the direct interaction of HIC1 with the promoters of the repressed genes.
HIC1 regulates Axin2 transcription
The HIC1 gene is silenced by promoter methylation in most of the tumor-derived cell lines tested so far; nevertheless, we detected HIC1 mRNA expression in human medulloblastoma DAOY cells, human primary fibroblast WI38 cells and also in mouse embryo STO cells. Using anti-HIC1 antibody, we visualized the nuclear HIC1 bodies in all these cells (see higher magnification insets in Figures 7B and 8A; staining of STO cells is not shown). Treatment of DAOY cells with 5-aza-2′-deoxycytidine (5-aza-2′-dCyt), that is, with an agent blocking DNA methylation, resulted in two-fold increase in the levels of HIC1 mRNA (Figure 7A). This increased HIC1 expression was also detected at the protein level and the 5-aza-2′-dCyt-treated cells contained a higher amount of larger HIC1 bodies than untreated cells (Figure 7B, insets). We further tested the effect of HIC1 knockdown on Wnt signaling. We found that in WI38 cells the HIC1 mRNA level was reduced to 20% upon transfection with HIC1 short inhibitory RNAs (siRNAs), compared to an irrelevant control (a mixture of anti-GFP and anti-luciferase siRNAs) (Figure 8A). The effective downregulation of HIC1 in the transfected cells was confirmed by Western blotting and confocal microscopy (Figure 8A, inset). Recently, it was shown by Chen et al (2005) that HIC1 binds the SIRT1 promoter and directly represses its transcription. Thus, as expected, upon HIC1 knockdown we observed increased levels of the SIRT1 mRNA; we also noted a 60% increase of basal transcription of Axin2, the Wnt signaling pathway target gene (Figure 8A). Interestingly, upon Wnt stimulation, the HIC1 siRNAs remarkably (almost two-fold) elevated only the transcriptional response of the Axin2 promoter, while the expression of the SIRT1 and two housekeeping genes remained unchanged (Figure 8B). Such robust activation of Axin2 was quite astonishing as the HIC1 siRNAs treatment already increased the levels of the Axin2 mRNA in unstimulated cells. These data, obtained with physiological amounts of HIC1 and with the endogenous Wnt signaling target, supported our observations about HIC1 antagonizing Wnt signaling.
Figure 7.
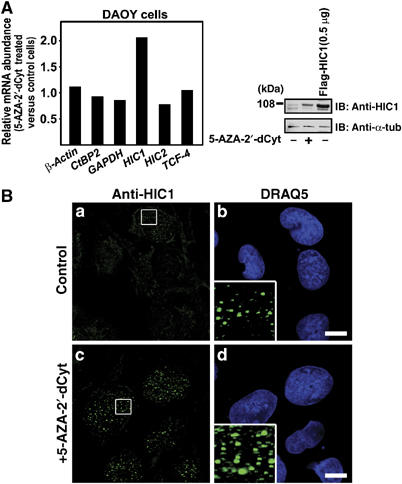
The nuclear HIC1 bodies in DAOY cells. (A) Increased expression of HIC1 mRNA and protein in the cell line DAOY after treatment with 1 μM 5-aza-2′-deoxycytidine for 6 days. The expression was analyzed by qRT–PCR (left) or by Western blotting using anti-HIC1 antibody. In lane 3, a lysate from 293 cells transfected with 0.5 μg of the Flag-HIC1 construct was loaded. (B) Confocal micrographs of DAOY cells treated with 5-aza-2′-deoxycytidine (c, d) or with a vehicle (a, b) stained with the affinity purified anti-HIC1 antibody. Bar, 10 μm.
Figure 8.
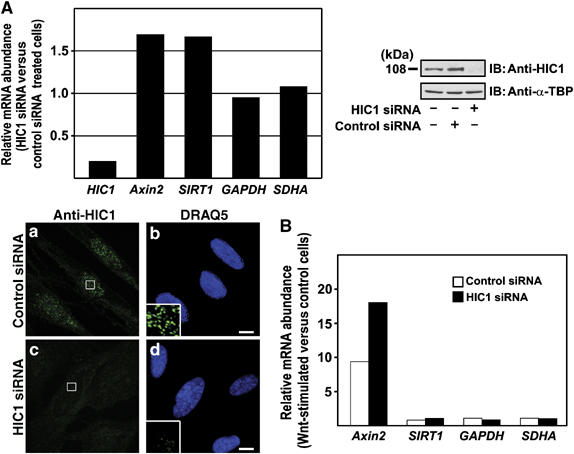
HIC1 knockdown increases the TCF-mediated transcription. (A) Human primary cells WI38 were transfected with HIC1 siRNAs or control siRNAs and the changes in the levels of HIC1 mRNA or protein were tested 24 h post-transfection. Left, results of the qRT–PCR analysis. The relative abundance of the given mRNA in HIC1 siRNA versus control siRNA-transfected cells was derived from the average CT values of four independent experiments after normalizing to the levels of β_-actin_ cDNA. Right, Western blots of nuclear extracts prepared from the indicated cells. Bottom, confocal micrographs of WI38 cells transfected with the indicated siRNAs and stained with the affinity purified anti-HIC1 antibody. Bar, 10 μm. (B) The activity of the Wnt-dependent promoter of the Axin2 gene is increased by HIC1 knockdown. Results of qRT–PCR analysis performed with cDNA generated from WI38 cells transfected with the indicated siRNAs upon 24-h stimulation with Wnt3a. Six PCR reactions were done for each primer set. The relative abundance of the indicated mRNA in Wnt3a-stimulated versus control cells was derived from the average CT values after normalizing to the levels of β_-actin_ cDNA.
HIC1 diverts TCF-4 and _β_-catenin from the Wnt-responsive promoters
The mechanism of the HIC1 action on the endogenous TCF-responsive promoters was first studied by ectopic expression of full-length HIC1 in 293 cells. At the mRNA level, HIC1 overexpression partly blocked the increase in abundance of three Wnt signaling responsive genes Axin2, Sp5 and Cyclin D1 (Shtutman et al, 1999; Leung et al, 2002; Weidinger et al, 2005) observed in control cells upon 12- or 24-h stimulation with Wnt3a (Figure 9A). The inhibition appeared to be incomplete because about 50% transfection efficiency was routinely achieved for the 293 cells (data not shown). Thus, although HIC1 obviously inhibited the Axin2, Sp5 or CyclinD1 stimulation in the transfected cells, the overall amounts of the corresponding mRNAs were moderately elevated in Wnt3a-treated cell cultures. The mRNA abundance of GAPDH and CtBP2, that is genes with no direct relationship to Wnt signaling, did not change in the experiment.
To clarify the mechanisms of the HIC1-mediated repression, ChIP was performed on a cluster of TCF-binding sites in the Sp5 promoter (Supplementary Figure 4) (Takahashi et al, 2005). The ChIP assay showed that HIC1 did not associate with the Sp5 promoter; furthermore, the binding of endogenous TCF-4 to this promoter was decreased by wild-type HIC1 overexpression and not by HIC1-ΔPOZ in both stimulated and nonstimulated 293 cells (Figure 9B, only data for Wnt3a-induced cells are shown). We then co-transfected HIC1 and β-catenin, and performed ChIP with an antibody directed against this Wnt effector protein. The results unambiguously showed the presence of β-catenin on the Sp5 promoter in control cells (transfected with an empty vector or with the HIC1-ΔPOZ mutant which is deficient in TCF binding). In contrast, wild-type HIC1 completely eliminated association of exogenous β-catenin with the TCF-responsive element of the Sp5 gene (Figure 9B).
In DLD-1 cells, we did not detect any Sp5 mRNA and, moreover, expression of Axin2 and Cyclin D1 seemed to be independent of the TCF/β-catenin as the transient transfection of a dominant-negative form of TCF-4 (ΔN-TCF-4) did not reduce transcription of these Wnt signaling target genes. However, ΔN-TCF-4 inhibited production of Tenascin C, a recently identified TCF/β-catenin target gene active in colon carcinoma cells growing at the invasive front of the tumors (data not shown) (Beiter et al, 2005). For the ChIP assay we used DLD/HIC1 cells with regulated expression of the HIC1-EGFP transgene. Predictably, HIC1 expression did not inhibit the activity of the in DLD-1 cells TCF-independent Axin2 and Cyclin D1 promoters (Figure 9C). This was not caused by a dysfunction of the HIC1-EGFP fusion protein as HIC1-EGFP efficiently attenuated pTOPFLASH transcription (Figure 6A) and the ectopically expressed HIC1-EGFP construct displayed the same activity as wild-type HIC1 in suppressing. Nevertheless, HIC1 efficiently blocked transcription from a promoter of the Tenascin C gene (Figure 9C). ChIP analysis of the proximal TCF-dependent DNA element in the Tenascin C promoter (Supplementary Figure 4) revealed a clear binding of TCF-4 and β-catenin in parental DLD-1 cells. In DLD/HIC1, upon induction with the dimerizer HIC1 significantly decreased the association of both TCF-4 and β-catenin with the Tenascin C promoter, but no interaction of this promoter, with HIC1 was observed (Figure 9D). Simultaneously, the functionality of HIC1-EGFP to bind its cognate DNA motif was confirmed by ChIP of the HIC1-recognition element in the SIRT1 promoter (Figure 9D). These data indicate that as a result of the recruitment to the HIC1 bodies, TCF-4 and β-catenin are prevented from associating with TCF target genes.
Discussion
Since the canonical Wnt/β-catenin signaling alone is unlikely to regulate multiple developmental programs initiated by the Wnt ligands, we searched for additional proteins that could modulate the function of the TCF/LEF factors. In the present study, we identified tumor suppressor HIC1 as a new nuclear modulator of the Wnt signaling pathway.
Nuclear sequestration of TCF-4 into the HIC1 bodies
HIC1 encodes a zinc-finger transcription factor that acts as a transcriptional repressor. Additionally, the HIC1 protein binds CtBP and via its N-terminal BTB/POZ domain forms nuclear aggregates known as HIC1 bodies. Here, we present convincing data based on confocal microscopy showing the colocalization of the Wnt signaling effector protein, TCF-4, HIC1 and CtBP1 in these nuclear bodies. We further show that HIC1 directly associates with TCF-4 in vitro and in vivo. However, the efficient sequestration of TCF-4 into the HIC1 bodies depends on the presence of the CtBP in the TCF-4/HIC1 complex. Strikingly, in CtBP-positive cells, wild-type HIC1 and the TCF-4mutCtBP protein (this variant lacks the CtBP-binding motifs but is still able to interact with HIC1 in vitro (Figure 4A and B)) do not display an overlapping localization (Figure 2C). We speculate that CtBP induces a specific spatial arrangement of the HIC1 bodies. Such arrangement possibly favors the recruitment of TCF-4 via the interaction with CtBP. This conclusion is supported by a different morphology of the HIC1 aggregates, which in the presence of CtBP show more compact appearance than the HIC1/TCF-4 bodies without CtBP (Figure 2C). The absence of HIC1 and TCF-4mutCtBP colocalization argues against the obvious objection that overexpressed proteins aggregate and sequester other proteins that they do not normally interact with.
Using in vitro pull-down assays, we mapped the main regions of the interaction to the C-terminal parts of HIC1 and TCF-4. Surprisingly, whereas GST-HIC C-terminal and GST-HIC internal fragments interacted with in vitro translated TCF-4, the N-terminally truncated HIC-ΔPOZ variant failed to interact with full-length GST-TCF-4. We assume that the anchoring of the HIC1 protein fragments to GST-beads possibly prevents the incorrect folding induced by the deletion of the structurally essential BTB/POZ domain.
Many nuclear factors involved in pre-mRNA splicing, regulation of transcription, apoptosis or cell cycle progression are localized in distinct structures called speckles or nuclear bodies (reviewed in Lamond and Spector, 2003). Using confocal microscopy we visualized HIC1 bodies at endogenous expression levels in the nuclei of three different cell types. Is the physical sequestration of the transcription factors main function of the HIC1 bodies or are there some other physiological roles for these structures? What is the proportion between the ‘free' HIC1 protein (i.e. HIC1 associated with promoters or other factors) and HIC1 aggregated in the bodies? More experimental work needs to be carried out to answer these questions.
HIC1 represses TCF-mediated transcription
The results presented here show that full-length HIC1 substantially reduced the levels of TCF-mediated transcription of two different TCF/β-catenin-regulated reporters (Figure 5), and furthermore, several selected endogenous Wnt signaling target genes were also affected by HIC1 expression (Figures 8 and 9). Importantly, HIC1 knockdown in normal cells enhanced the levels of the transcriptional stimulation induced by the Wnt3a ligand (Figure 8B). Thus, HIC1 specifically repressed transcription dependent on TCF and this repression occurred at physiological levels of the HIC1 protein. Interestingly, we observed that the activator role of Wnt signaling was dependent on the particular cellular background. For example, the stimulation by Wnt3a activated transcription of Sp5, Axin2 and Cyclin D1 in 293; in primary fibroblast WI38 cells, only robust transactivation of the Axin2 gene was detected, while the expression levels of Sp5 and Cyclin D1 remained under detection limits. In colon carcinoma DLD-1 cells, that is, in cells with constitutive active Wnt signaling, ectopic expression of a dominant-negative (blocking) form of TCF-4 inhibited expression of Tenascin C but not Axin2 and Cyclin D1 mRNA. This clearly indicates that gene expression is in general regulated by inputs from various cellular pathways that integrate in the regulatory regions of a particular gene. Nevertheless, HIC1 regulated the transcriptional response of all tested genes showing the reactivity to the Wnt signal in the given cell type.
Mechanisms of HIC1-mediated inhibition
It was well documented that Kaiso, a member of the BTB/POZ protein family, interacts with sequence-specific elements in several Wnt target genes (Park et al, 2005). In Xenopus, Kaiso and TCF act in concert on the siamois promoter. The Kaiso's general role is not completely understood, but it is likely to also include a direct binding and the recruitment of corepressors such as N-CoR to a subset of Wnt targets. Recently, Chen et al (2005) showed that HIC1 forms a transcriptional repression complex with SIRT1 deacetylase. This complex directly binds and represses transcription from the promoter of the SIRT1 deacetylase gene.
ChIP analysis revealed that HIC1 does not bind directly or indirectly (i.e. via TCF-4) the regulatory elements in the TCF-responsive genes. In contrast, HIC1 partly reduces the occupancy of the promoter of the Sp5 gene by endogenous TCF-4. This partial sequestration was expected as only a fraction of the cells expressed exogenous HIC1. In addition, HIC1 eliminated binding of ectopically expressed β-catenin to the Sp5 promoter.
ChIP analysis of the TCF-binding element in the Tenascin C promoter in DLD/HIC1 cells showed a substantial decrease in association of TCF-4 and β-catenin upon HIC1 induction. Taken together, these data indicate that HIC1-mediated sequestration prevents TCF-4 from binding its target promoter. Although β-catenin targeting to the HIC1 bodies seems to be indirect and mediated via its interaction with TCF-4, we cannot completely exclude participation of an unknown factor involved in relocation of β-catenin into the HIC1 bodies (Figure 10). These results imply that HIC1-mediated sequestration can uncouple the TCF/β-catenin-regulated promoters from various inputs related to Wnt signaling, but still may leave such promoters responsive to other regulatory signals.
Figure 10.
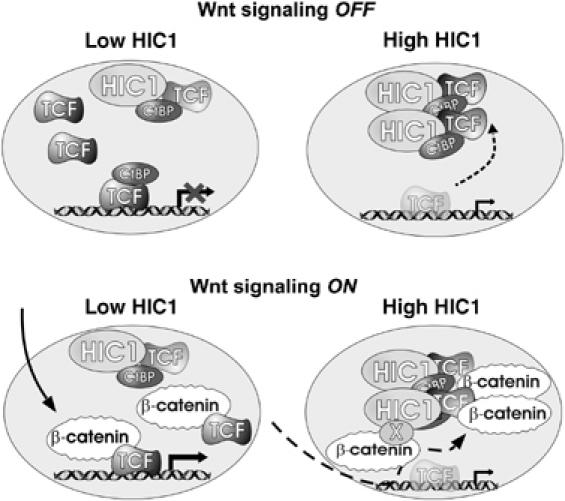
A model for HIC1 suppression of the transcriptional response induced by Wnts. The regulation of a complex promoter integrating inputs from the Wnt and other signaling pathways is depicted. At low levels of HIC1 the activity of the promoter depends mainly on the Wnt signaling components. High levels of HIC1 uncouple the promoter from Wnt signaling. X depicts a hypothetical factor mediating besides TCF-4 the interaction between β-catenin and HIC1.
Materials and methods
Plasmids and RNAi
All constructs were made by standard molecular biology techniques. Triple amino-acid substitutions (Q509 to N509, P510 to A510, L511 to S511, Q586 to N586, P587 to A587, L588 to S588) were introduced into each of the CtBP-binding sites in TCF-4mutCtBP by using a site-directed mutagenesis kit (Stratagene). For gene knockdowns HIC1 siRNAs were purchased from Ambion. See the Supplementary data for more detailed description of the plasmids used in this study.
Cell culture and transfections
Cell lines were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Hyclone) and antibiotics. All cell lines were regularly checked for the presence of mycoplasma. Transfections were performed using the Lipofectamine reagent (Invitrogen) as described by the manufacturer. DLD/HIC1 cells were produced using ARGENT Regulated Transcription Retrovirus Kit (Ariad). CtBP(−/−) cells derived from the CtBP1−/−CtBP2−/− embryos were kindly provided by J Hildebrand.
Production of Wnt3a-conditioned medium
Mouse Wnt3a-producing L cells (L Wnt-3A; ATCC No. CRL-2647) were cultured in complete DMEM supplemented with G418 (0.5 mg per ml; Alexis). Control and Wnt3a-conditioned medium was prepared according to the protocol provided by the supplier.
Luciferase assays
Reporter gene assays were performed as described previously (Valenta et al, 2003). To assay TCF-mediated transcription, firefly luciferase pTOPFLASH and pFOPFLASH (Korinek et al, 1997) and the Axin2 promoter reporter constructs (Jho et al, 2002) (a gift from F Costantini) were used. The G1-E1B-Luc reporter and the Gal4-DBD-VP16 construct were kindly provided by C Svensson.
EMSA
Full-length TCF-4, full-length human HIC1 and HIC1-ΔPOZ protein were produced in vitro using the Quick TNT Coupled Reticulocyte System (Promega). The assay was performed as reported previously (Valenta et al, 2003).
GST interaction assays and DNase I treatment
GST-TCF-4, GST-β-catenin, GST-HIC1-N-term (aa 1–398), GST-HIC1-C-term (aa 396–714) and GST-HIC1-internal (aa 234–551) fusion proteins were expressed in the BL21 (DE3) strain of Escherichia coli using the pET-42b vector (Novagen). Relevant proteins were produced in vitro using the Quick TNT Coupled Reticulocyte System (Promega). The detailed protocol for the GST pull-downs was described previously (Valenta et al, 2003). DNase I treatment of GST-bound or in vitro translated proteins was performed in 1 × DNase I buffer (Invitrogen), for 30 min at RT with 0.2 U of DNase I (Invitrogen) per 1 μl of the reaction mixture.
Antibodies
Antisera to TCF-4, HIC1, CtBP1, β-catenin and EGFP were produced by immunization of rabbits with bacterially expressed proteins; mouse mAbs to TCF-4 and HIC1 were prepared using standard techniques from splenocytes of mice immunized with a bacterially produced TCF-4 fragment (aa 31–333) and HIC1 fragment (aa 230–404) respectively. The following commercially available mouse monoclonal antibodies were used: anti-β-catenin (Santa Cruz Biotechnology), anti-dephospho-β-catenin (Alexis), anti-CtBP (Santa Cruz Biotechnology), anti-GFP (BD Clontech), anti-Myc 9E10 (Roche Molecular Biochemicals), anti-Flag M2 (Sigma), anti-Flag (Exbio Praha).
Immunofluorescent microscopy
Cells grown on coverslips were fixed 24 h after transfection in cold methanol (−20°C, 5 min) and then briefly in acetone (−20°C). Fluorochromes were ALEXA 488, 594, and 680 dyes (dilution 1:500; Invitrogen), Cy5 dye (1:500, Amersham Pharmacia Biotech). The samples were mounted in MOWIOL (Calbiochem) containing nuclear staining dye DRAQ 5 (1:750; Alexis). Immunofluorescences were visualized using a confocal laser scanning microscope (TCS SP; Leica). All images were scanned separately in the ‘sequential scanning mode' for the green, red and blue channels using a × 100/1.40 oil-immersion objective. The ratio of colocalization was quantified by measuring the overlap in the fluorescence intensities of corresponding channels along selected profiles using Leica confocal software. Image files were processed with Adobe Photoshop.
RNA purification and real-time qRT–PCR
Standard procedures were used for RNA purification and reverse transcription. The primers used are listed in Supplementary Table S1 (Supplementary data). The cycling was performed in an Mxp3000 instrument (Stratagene).
ChIP
293 cells stimulated with Wnt3a-conditioned or control medium, DLD-1 and DLD/HIC1 cells were subjected to the ChIP assays according to Kirmizis et al (2004). See the Supplementary data for the primer sequences.
Supplementary Material
Supplementary Information
Acknowledgments
We. thank J Hildebrand, O Machon, F Costantini, D Leprince, C Svensson, F Gage and ARIAD Pharmaceuticals, Inc. for sharing reagents; and L Andera, L Cermak and S Takacova for critically reading the manuscript. This work was supported by the Grant Agency of the Czech Republic (204/04/0532), the project Center of Molecular and Cellular Immunology (1M6837805001), and in part by the institutional grant (AV0Z50520514).
References
- Albagli O, Dhordain P, Deweindt C, Lecocq G, Leprince D (1995) The BTB/POZ domain: a new protein–protein interaction motif common to DNA- and actin-binding proteins. Cell Growth Differ 6: 1193–1198 [PubMed] [Google Scholar]
- Beiter K, Hiendlmeyer E, Brabletz T, Hlubek F, Haynl A, Knoll C, Kirchner T, Jung A (2005) Beta-Catenin regulates the expression of tenascin-C in human colorectal tumors. Oncogene 24: 8200–8204 [DOI] [PubMed] [Google Scholar]
- Brannon M, Brown JD, Bates R, Kimelman D, Moon RT (1999) XCtBP is a XTcf-3 co-repressor with roles throughout Xenopus development. Development 126: 3159–3170 [DOI] [PubMed] [Google Scholar]
- Chen W, Cooper TK, Zahnow CA, Overholtzer M, Zhao Z, Ladanyi M, Karp JE, Gokgoz N, Wunder JS, Andrulis IL, Levine AJ, Mankowski JL, Baylin SB (2004) Epigenetic and genetic loss of Hic1 function accentuates the role of p53 in tumorigenesis. Cancer Cell 6: 387–398 [DOI] [PubMed] [Google Scholar]
- Chen WY, Wang DH, Yen RC, Luo J, Gu W, Baylin SB (2005) Tumor suppressor HIC1 directly regulates SIRT1 to modulate p53-dependent DNA-damage responses. Cell 123: 437–448 [DOI] [PubMed] [Google Scholar]
- Chen WY, Zeng X, Carter MG, Morrell CN, Chiu Yen RW, Esteller M, Watkins DN, Herman JG, Mankowski JL, Baylin SB (2003) Heterozygous disruption of Hic1 predisposes mice to a gender-dependent spectrum of malignant tumors. Nat Genet 33: 197–202 [DOI] [PubMed] [Google Scholar]
- Clevers H, van de Wetering M (1997) TCF/LEF factor earn their wings. Trends Genet 13: 485–489 [DOI] [PubMed] [Google Scholar]
- Deltour S, Pinte S, Guerardel C, Wasylyk B, Leprince D (2002) The human candidate tumor suppressor gene HIC1 recruits CtBP through a degenerate GLDLSKK motif. Mol Cell Biol 22: 4890–4901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobyns WB, Truwit CL (1995) Lissencephaly and other malformations of cortical development: 1995 update. Neuropediatrics 26: 132–147 [DOI] [PubMed] [Google Scholar]
- Herman JG, Baylin SB (2003) Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med 349: 2042–2054 [DOI] [PubMed] [Google Scholar]
- Hildebrand JD, Soriano P (2002) Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol Cell Biol 22: 5296–5307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jho EH, Zhang T, Domon C, Joo CK, Freund JN, Costantini F (2002) Wnt/beta-catenin/Tcf signaling induces the transcription of Axin2, a negative regulator of the signaling pathway. Mol Cell Biol 22: 1172–1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirmizis A, Bartley SM, Kuzmichev A, Margueron R, Reinberg D, Green R, Farnham PJ (2004) Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev 18: 1592–1605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW, Vogelstein B, Clevers H (1997) Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC−/− colon carcinoma. Science 275: 1784–1787 [DOI] [PubMed] [Google Scholar]
- Lamond AI, Spector DL (2003) Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol 4: 605–612 [DOI] [PubMed] [Google Scholar]
- Leung JY, Kolligs FT, Wu R, Zhai Y, Kuick R, Hanash S, Cho KR, Fearon ER (2002) Activation of AXIN2 expression by beta-catenin-T cell factor. A feedback repressor pathway regulating Wnt signaling. J Biol Chem 277: 21657–21665 [DOI] [PubMed] [Google Scholar]
- Logan CY, Nusse R (2004) The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol 20: 781–810 [DOI] [PubMed] [Google Scholar]
- Park JI, Kim SW, Lyons JP, Ji H, Nguyen TT, Cho K, Barton MC, Deroo T, Vleminckx K, McCrea PD (2005) Kaiso/p120-catenin and TCF/beta-catenin complexes coordinately regulate canonical Wnt gene targets. Dev Cell 8: 843–854 [DOI] [PubMed] [Google Scholar]
- Pinte S, Stankovic-Valentin N, Deltour S, Rood BR, Guerardel C, Leprince D (2004) The tumor suppressor gene HIC1 (hypermethylated in cancer 1) is a sequence-specific transcriptional repressor: definition of its consensus binding sequence and analysis of its DNA binding and repressive properties. J Biol Chem 279: 38313–38324 [DOI] [PubMed] [Google Scholar]
- Reya T, Clevers H (2005) Wnt signalling in stem cells and cancer. Nature 434: 843–850 [DOI] [PubMed] [Google Scholar]
- Roose J, Molenaar M, Peterson J, Hurenkamp J, Brantjes H, Moerer P, van de Wetering M, Destree O, Clevers H (1998) The Xenopus Wnt effector XTcf-3 interacts with Groucho-related transcriptional repressors. Nature 395: 608–612 [DOI] [PubMed] [Google Scholar]
- Rosin-Arbesfeld R, Cliffe A, Brabletz T, Bienz M (2003) Nuclear export of the APC tumour suppressor controls beta-catenin function in transcription. EMBO J 22: 1101–1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sewalt RG, Gunster MJ, van der Vlag J, Satijn DP, Otte AP (1999) C-Terminal binding protein is a transcriptional repressor that interacts with a specific class of vertebrate Polycomb proteins. Mol Cell Biol 19: 777–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R, Ben-Ze'ev A (1999) The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci USA 96: 5522–5527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi M, Nakamura Y, Obama K, Furukawa Y (2005) Identification of SP5 as a downstream gene of the beta-catenin/Tcf pathway and its enhanced expression in human colon cancer. Int J Oncol 27: 1483–1487 [PubMed] [Google Scholar]
- Valenta T, Lukas J, Korinek V (2003) HMG box transcription factor TCF-4's interaction with CtBP1 controls the expression of the Wnt target Axin2/Conductin in human embryonic kidney cells. Nucleic Acids Res 31: 2369–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering M, Sancho E, Verweij C, de Lau W, Oving I, Hurlstone A, van der Horn K, Batlle E, Coudreuse D, Haramis AP, Tjon-Pon-Fong M, Moerer P, van den Born M, Soete G, Pals S, Eilers M, Medema R, Clevers H (2002) The beta-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 111: 241–250 [DOI] [PubMed] [Google Scholar]
- Weidinger G, Thorpe CJ, Wuennenberg-Stapleton K, Ngai J, Moon RT (2005) The Sp1-related transcription factors sp5 and sp5-like act downstream of Wnt/beta-catenin signaling in mesoderm and neuroectoderm patterning. Curr Biol 15: 489–500 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information