COMMD1 promotes the ubiquitination of NF-κB subunits through a cullin-containing ubiquitin ligase (original) (raw)
Abstract
NF-κB is a pleiotropic transcription factor involved in multiple processes, including inflammation and oncogenesis. We have previously reported that COMMD1 represses κB-dependent transcription by negatively regulating NF-κB–chromatin interactions. Recently, ubiquitination of NF-κB subunits has been similarly implicated in the control of NF-κB recruitment to chromatin. We report here that COMMD1 accelerates the ubiquitination and degradation of NF-κB subunits through its interaction with a multimeric ubiquitin ligase containing Elongins B and C, Cul2 and SOCS1 (ECSSOCS1). COMMD1-deficient cells demonstrate stabilization of RelA, greater nuclear accumulation of RelA after TNF stimulation, de-repression of several κB-responsive genes, and enhanced NF-κB-mediated cellular responses. COMMD1 binds to Cul2 in a stimulus-dependent manner and serves to facilitate substrate binding to the ligase by stabilizing the interaction between SOCS1 and RelA. Our data uncover that ubiquitination and degradation of NF-κB subunits by this COMMD1-containing ubiquitin ligase is a novel and critical mechanism of regulation of NF-κB-mediated transcription.
Keywords: COMMD1, cullin, NF-κB, transcription, ubiquitination
Introduction
NF-κB is a dimeric transcription factor that controls expression of genes involved in multiple processes including immunity, apoptosis, and cell cycle progression (Karin and Lin, 2002). In mammals, NF-κB subunits are encoded by five genes (RELA, RELB, REL, NFKB1, and NFKB2). All subunits share an ∼300 amino-acid region of homology termed the Rel homology domain (RHD) that mediates DNA binding and dimerization. The NFKB1 and NFKB2 genes encode large precursor polypeptides known as p105 and p100, which are cleaved into the mature p50 and p52 subunits, respectively.
NF-κB is ordinarily sequestered in the cytoplasm in a transcriptionally inactive form as a result of interactions with IκB proteins or, in the case of RelB, through its dimerization with p105 or p100, which contain IκB-like domains in their carboxy-termini. In response to a variety of stimuli, transient nuclear translocation of NF-κB dimers takes place, a prerequisite for κB-mediated transcription to occur. The translocation event is triggered by phosphorylation of IκB proteins by the multimeric IκB kinase, which leads to their ubiquitination by the SCFβ-TrCP ubiquitin ligase (Henkel et al, 1993; Chen et al, 1995; Yaron et al, 1998). Ubiquitinated IκB is then quickly degraded allowing nuclear translocation of NF-κB to occur. Once in the nucleus, NF-κB complexes bind to cognate DNA sequences present in an array of promoters resulting in gene expression.
In addition to nuclear translocation, transcriptional activation by NF-κB requires recruitment of co-activators and the displacement of co-repressor complexes. This is mediated through an intricate set of nuclear events that involve post-translational modifications of NF-κB subunits, including phosphorylation and acetylation (Sakurai et al, 1999; Chen et al, 2001; Zhong et al, 2002).
In order to achieve controlled expression of its gene targets, the termination of NF-κB-mediated transcriptional responses is tightly regulated. Nuclear export of NF-κB complexes is a major step in the regulation of this pathway. Through a negative feedback loop, NF-κB induces the resynthesis of IκB proteins, ultimately driving nuclear export of NF-κB and termination of transcription (Hoffmann et al, 2002). Despite the important role that IκB proteins play in regulating the nuclear pool of NF-κB, other factors are likely involved. For example, NF-κB remains largely excluded from the nucleus in cells that are profoundly deficient in IκB proteins, in contrast to the mostly nuclear localization that results from stimulus-dependent degradation of IκB (Tergaonkar et al, 2005), suggesting that other pathways are also involved in the control of nuclear NF-κB levels.
We have previously reported that a factor named MURR1, now referred to as COMMD1, is a ubiquitously expressed inhibitor of NF-κB (Ganesh et al, 2003; Burstein et al, 2005). COMMD1 interferes with HIV-1 replication, a process that is dependent on κB-mediated transcription (Ganesh et al, 2003). More recently, we identified that COMMD1 is the prototype of a family of 10 factors known as COMMD proteins that also function to repress NF-κB (Burstein et al, 2005). The defining characteristic of all family members is the presence of an ∼70 amino-acid region of high homology in their extreme carboxy-termini termed the COMM domain, which serves as an interface for COMMD–COMMD protein interactions (Burstein et al, 2005). COMMD proteins interact with NF-κB subunits but, unlike IκB-α, do not control the translocation of NF-κB from the cytosol to the nucleus (Burstein et al, 2005). Rather, COMMD1 is recruited to κB-responsive promoters and decreases the duration of RelA–chromatin interactions (Burstein et al, 2005). However, the precise mechanism for this effect remained elusive.
Recent work has demonstrated that ubiquitination and proteasomal degradation of nuclear RelA also participate in the termination of NF-κB–chromatin interactions (Saccani et al, 2004), prompting us to investigate a potential role for COMMD1 in RelA ubiquitination. Indeed we demonstrate here that COMMD1 promotes the ubiquitination and degradation of NF-κB subunits. COMMD1 deficiency results in stabilization of NF-κB and increased nuclear accumulation of RelA following TNF stimulation. Furthermore, NF-κB activation in COMMD1-deficient cells results in de-repressed κB-dependent transcription and enhanced release of chemokines. We show that endogenous COMMD1 immune complexes contain E3 ubiquitin ligase activity, as well as components of a Cullin-containing ubiquitin ligase comprised of Elongins B and C, Cul2 and SOCS1 (also known as ECSSOCS1), which has been previously implicated in RelA ubiquitination. Immunoprecipitation of COMMD1 from cells expressing Cul2 or the ECSSOCS1 complex recovered ubiquitin ligase against RelA in vitro. Finally, our data indicate that the interaction of COMMD1 with ECSSOCS1 serves to stabilize SOCS1–RelA interactions, thereby promoting binding of the substrate to the enzyme. Overall, these studies define a novel pathway by which κB-mediated transcription is regulated through COMMD1-mediated ubiquitination of NF-κB subunits.
Results
COMMD1 promotes the ubiquitination of NF-_κ_B subunits
We have previously reported that COMMD1 inhibits κB-mediated transcription by decreasing the duration of RelA–chromatin interactions (Burstein et al, 2005). Recently, ubiquitination and proteasomal degradation of DNA-bound RelA was implicated in the control of RelA–chromatin interactions (Saccani et al, 2004). Therefore, we explored the possibility that ubiquitination of RelA might be a mechanism for the inhibitory effects of COMMD1 on κB-mediated transcription.
To investigate this, we examined the effect of COMMD1 expression on the levels of ubiquitinated NF-κB. First, ubiquitination of ectopically expressed, GST-tagged RelA was evaluated by precipitating RelA and immunoblotting the recovered material for polyubiquitin (Figure 1A). Heavily ubiquitinated RelA was recovered after proteasomal blockade only in cells coexpressing COMMD1. Utilizing a similar approach, we next examined the effect of COMMD1 expression on the ubiquitination of endogenous RelA (Figure 1B). In this case, endogenous RelA was immunoprecipitated and the recovered material immunoblotted for polyubiquitin. Again, expression of COMMD1 resulted in increased recovery of ubiquitinated RelA. To corroborate that the ubiquitinated material recovered in Figure 1A and B was RelA itself, a complementary approach was utilized. Here ubiquitinated proteins were recovered by precipitating ubiquitin (ectopically expressed as a fusion protein containing the His6 affinity tag, His6-Ubi) and detecting the presence of RelA in this fraction by immunoblotting. As shown in Figure 1C, precipitation of cell lysates with Ni+-NTA agarose beads resulted in the recovery of polyubiquitinated proteins only when the cells were transfected to express His6-Ubi (and not from vector-transfected cells). Slight and nonspecific binding of RelA to the beads was detected, but modified RelA consisting of material of increasing molecular weight was only recovered from cells expressing His6-Ubi, indicating that this material is ubiquitinated RelA (Figure 1C, upper panel). As in Figure 1A and B, increased expression of COMMD1 accelerated RelA ubiquitination (Figure 1C, left panel), whereas decreased expression of endogenous COMMD1 after RNA interference (RNAi) diminished the amount of ubiquitinated RelA (Figure 1C, right panel). Importantly, these changes in the recovery of ubiquitinated RelA were not the result of uneven expression of RelA or uneven precipitation of polyubiquitinated proteins across the samples.
Figure 1.
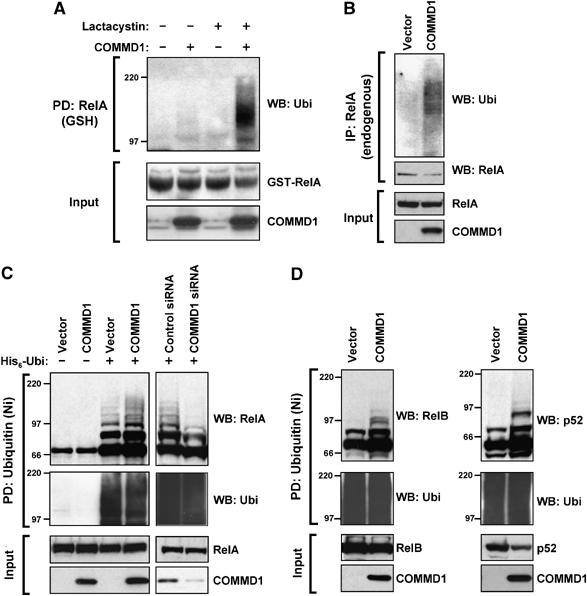
COMMD1 promotes the ubiquitination of NF-κB subunits. (A) Ubiquitination of RelA. HEK 293 cells were transfected with COMMD1 or a vector control, along with GST-RelA. Cells were treated with Lactacystin (10 μM) for 3 h before lysis with Triton X-100 buffer. RelA was precipitated (PD) using glutathione (GSH) beads and the recovered material was immunoblotted for ubiquitin (Ubi). (B) Ubiquitination of endogenous RelA. Cells were transfected with COMMD1 (or vector control) as in (A), and endogenous RelA was immunoprecipitated from cell lysates prepared with Triton X-100 buffer. The recovered material was immunoblotted for ubiquitin. (C) Effects of overexpression and underexpression of COMMD1 on RelA ubiquitination. HEK 293 cells were transfected with His6-ubiquitin (His6-Ubi) or a vector control, along with HA-RelA. These cells were co-transfected with a vector control or COMMD1 (left panel), or with control or COMMD1-specific siRNA (right panel). Ubiquitinated material was precipitated from cell lysates prepared with Triton X-100 buffer, using Ni-NTA agarose beads. The recovered material was immunoblotted for RelA (HA) and ubiquitin. (D) Effects on other NF-κB subunits. HEK 293 cells were transfected as in (C), except that HA-RelB (left panel) or HA-p52 (right panel) was used.
COMMD1 is a member of a larger protein family, which also shares the ability to bind to and inhibit NF-κB activity; therefore, we investigated the possibility of a similar effect by other COMMD family members. Decreased expression of COMMD6, 9, and 10 also resulted in decreased recovery of ubiquitinated RelA (Supplementary Figure S1). Finally, using a similar approach, we observed that COMMD1 expression accelerated the ubiquitination of two other NF-κB subunits, RelB and p52 (Figure 1D). Altogether, these results indicate that COMMD1 promotes the ubiquitination of NF-κB.
COMMD1 affects the stability of RelA
If COMMD1-mediated ubiquitination of NF-κB subunits results in their degradation, we reasoned that COMMD1 should alter the half-life and possibly the steady-state levels of NF-κB subunits in cells. We examined this possibility in U2OS cells with stable RNAi of COMMD1, resulting in nearly undetectable COMMD1 protein expression (Figure 2A) and 94% suppression of the COMMD1 transcript (Figure 2B). In these cells, basal levels of RelA protein were increased (Figure 2A) and were not accompanied by changes in RELA mRNA expression, suggesting a post-transcriptional effect of COMMD1 on RelA (Figure 2B). In addition, similar increases in the steady-state protein levels of RelB, p105, and p100 were observed (Figure 2A) in the absence of transcriptional upregulation of their respective genes (data not shown).
Figure 2.
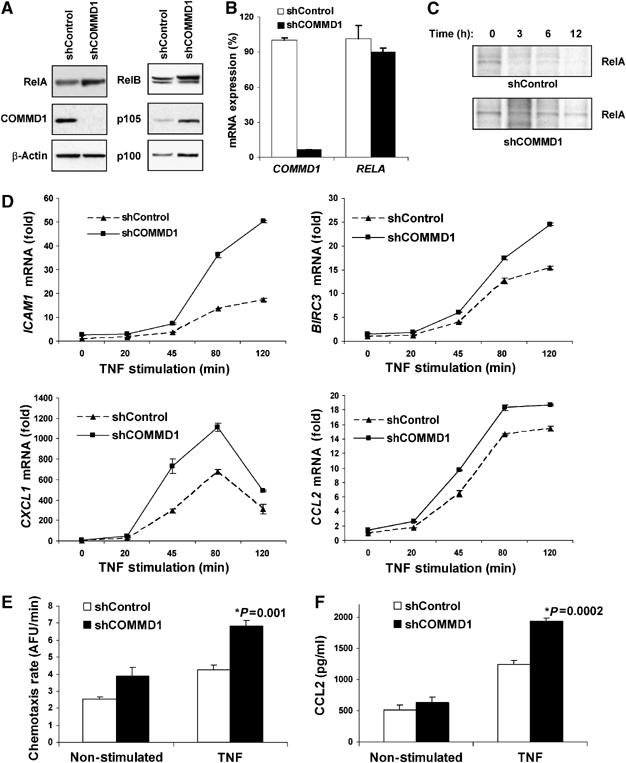
COMMD1 deficiency stabilizes RelA and de-represses endogenous κB-dependent transcription and cellular events. (A) COMMD1 deficiency results in higher basal levels of RelA. U2OS cells were stably infected with lentiviruses targeting a control gene (shControl) or COMMD1 (shCOMMD1). Once stable polyclonal cell lines were established, cell lysates were prepared with Triton X-100 buffer for immunoblotting as indicated. (B) COMMD1 deficiency does not affect mRNA levels of RELA. In the same cells, mRNA was extracted and the levels of COMMD1 and RELA expression were determined by qRT–PCR and expressed as % compared to the control line. (C) The half-life of RelA is prolonged in COMMD1-deficient cells. Cells were metabolically labeled with 35S-labeled methionine and cysteine and cell lysates were prepared at increasing time intervals as indicated. After immunoprecipitation of RelA, the recovered material was resolved by SDS–PAGE and subjected to autoradiography. (D) COMMD1 deficiency results in enhanced transcription of several κB-responsive genes. COMMD1-deficient and control cell lines were stimulated with a 10 min pulse of TNF, rinsed in PBS, and placed in normal media. Extraction of mRNA was performed for qRT–PCR of the indicated gene transcripts. (E) COMMD1-deficient cells produce greater amounts of chemotactic factors. Conditioned media from untreated or TNF-treated cells (12 h) were utilized. The ability of the media to induce chemotaxis was determined by monitoring the migration of fluorescently labeled peripheral mononuclear cells across a membrane barrier (and expressed as a rate of fluorescence change over time). (F) COMMD1-deficient cells secrete greater amounts of CCL2. Levels of CCL2 in the conditioned media (prepared similarly as in (E)) were determined by ELISA.
This suggested an effect of COMMD1 on the protein stability of RelA, and therefore the half-life of the protein was examined. After metabolic labeling with 35S-labeled methionine and cysteine, cell lysates were prepared at different time points and endogenous RelA was immunoprecipitated, resolved by SDS–PAGE, and then detected by autoradiography. Consistent with the previous results, the half-life of RelA was prolonged in COMMD1-deficient cells (Figure 2C). Altogether, this indicated that COMMD1 destabilizes RelA by promoting its ubiquitination and targeting the protein for proteasomal degradation.
COMMD1 controls endogenous _κ_B-mediated transcription and cellular responses
The predicted functional effect of the stabilization of RelA that we observed in COMMD1-deficient cells would be an increase in κB-dependent transcription. Therefore, we examined the inducible mRNA expression of NF-κB-responsive genes following TNF stimulation. Treatment with TNF resulted in time-dependent increases in mRNA expression of known NF-κB target genes such as ICAM1, BIRC3 (encoding c-IAP2), CXCL1, and CCL2 (Figure 2D). In each case, COMMD1-deficient cells demonstrated increased transcription of these genes, consistent with the notion that endogenous levels of COMMD1 serve to restrict κB-mediated transcription. Interestingly, the transcriptional effects observed varied in each case (Figure 2D), and for other NF-κB-regulated genes such as IL8 or TNF, the effects were minimal (Supplementary Figure S2). These data indicate that endogenous levels of COMMD1 serve to repress transcription of a specific subset of NF-κB-inducible genes.
NF-κB-mediated transcription plays a critical role in a number of cellular events, particularly the expression of mediators of the inflammatory response in vivo, including the production of several chemokines. Given the increased rates of NF-κB-mediated transcription observed in COMMD1-deficient cells (Figure 2D), we anticipated that these cells might produce larger amounts of chemokines. To test this possibility, we examined the ability of conditioned media to induce chemotaxis of freshly isolated peripheral mononuclear cells across a membrane barrier. As shown in Figure 2E, conditioned media from COMMD1-deficient cells induced a higher rate of chemotaxis than the control cell line, particularly after TNF stimulation. As predicted, this increased chemotaxis rate correlated with increased secretion of NF-κB-inducible chemokines into the media such as CCL2 (Figure 2F), a chemokine whose mRNA expression was also upregulated in these cells (Figure 2D, fourth panel).
COMMD1 deficiency alters the kinetics of nuclear accumulation of RelA
The data thus far indicated that COMMD1 promotes the ubiquitination of RelA and affects κB-mediated transcription and cellular responses, such as the production of chemokines. Given our prior observations that COMMD1 negatively regulates RelA–chromatin interactions (Burstein et al, 2005), we reasoned that one potential outcome of COMMD1 deficiency would be decreased RelA ubiquitination leading to enhanced accumulation of nuclear RelA. To study this latter possibility, nuclear extracts of control and COMMD1-deficient cells were prepared at various time points following pulsed TNF stimulation. As shown in Figure 3A, COMMD1 deficiency resulted in greater nuclear accumulation of RelA, and the differences observed were not accounted for by the levels of IκB-α as COMMD1-deficient cells had equal or higher levels of IκB-α at every time point (Figure 3B). Interestingly, despite the prolongation in the half-life of RelA (Figure 2C), the increased accumulation of nuclear RelA in these cells was not a sustained effect and was only noticeable during the peak times of 20 and 45 min post-stimulation. This observation correlated with a faster and higher rate of IκB-α protein resynthesis in COMMD1-deficient cells (Figure 3B), explained in part by modest increases in mRNA expression (Supplementary Figure S3). This is consistent with the known negative feedback loop by which NF-κB activates transcription of NFKBIA (the gene encoding IκB-α), an event known to promote nuclear export of NF-κB (Hoffmann et al, 2002).
Figure 3.
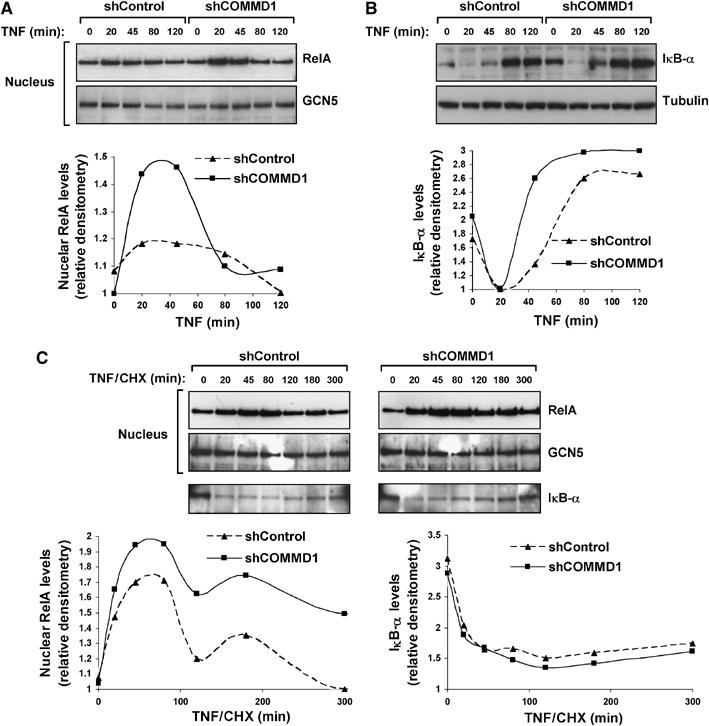
COMMD1 deficiency alters the nuclear accumulation of RelA. (A) COMMD1 deficiency results in greater accumulation of nuclear RelA after TNF treatment. U2OS COMMD1-deficient and control cell lines were stimulated with a 10 min pulse of TNF. At the indicated time point, cells were harvested and nuclear extracts were prepared for Western blot analysis of RelA in the nuclear fraction (and GCN5 as a loading control). Densitometry analysis is also presented. (B) Greater nuclear accumulation of RelA is not due to alterations in IκB-α degradation or resynthesis. U2OS cells were treated in an identical manner as in the experiment performed in (A) and IκB-α degradation was evaluated by immunoblotting and densitometry. (C) COMMD1 deficiency promotes greater nuclear accumulation of RelA independent of IκB-α levels. Similar to (A), U2OS cells were stimulated with a 10 min pulse of TNF, and medium containing CHX (30 μM) was then added. Nuclear extracts were prepared at the indicated time intervals and immunoblotted for RelA. In addition, IκB-α levels were evaluated utilizing the cytosolic fractions (bottom panel). Representative data and densitometry analysis are presented.
In order to examine the effect of COMMD1 on nuclear RelA levels in the absence of IκB-mediated nuclear export, we examined the kinetics of TNF-induced nuclear accumulation of RelA after blocking new protein synthesis with cycloheximide (CHX). The presence of this inhibitor largely abrogated IκB-α resynthesis (Figure 3C compared to B). Under these conditions, nuclear accumulation of RelA following TNF treatment was more sustained and, importantly, remained significantly elevated in COMMD1-deficient cells compared to the control cells (Figure 3C). Altogether, these data indicate that COMMD1-mediated ubiquitination of RelA affects its stability in the nuclear compartment, consistent with the observation that COMMD1 decreases RelA–chromatin interactions.
COMMD1 associates with an endogenous ubiquitin ligase
Although COMMD1 can accelerate the ubiquitination of RelA, it remained to be determined whether this was a direct effect on ubiquitination, or an indirect effect that subsequently facilitates the ubiquitination of RelA. Given that COMMD proteins do not possess any known catalytic activity, we examined first whether COMMD1 could interact with an endogenous ubiquitin ligase. To that end, COMMD1 immunoprecipitates were offered as a potential source for E3 ubiquitin ligase activity to an in vitro ubiquitination reaction containing recombinant ubiquitin and the E1 and E2 enzymes. At the end of the reaction, the presence of E3 ubiquitin ligase activity was determined by the formation of polyubiquitin chains. As can be appreciated in Figure 4A, COMMD1 immunoprecipitates catalyzed the formation of polyubiquitinated material in the presence of ATP. This effect was far stronger than the contaminating activity that co-precipitated with the preimmune serum and indicated that endogenous COMMD1 interacts with a protein complex possessing E3 ubiquitin ligase activity.
Figure 4.
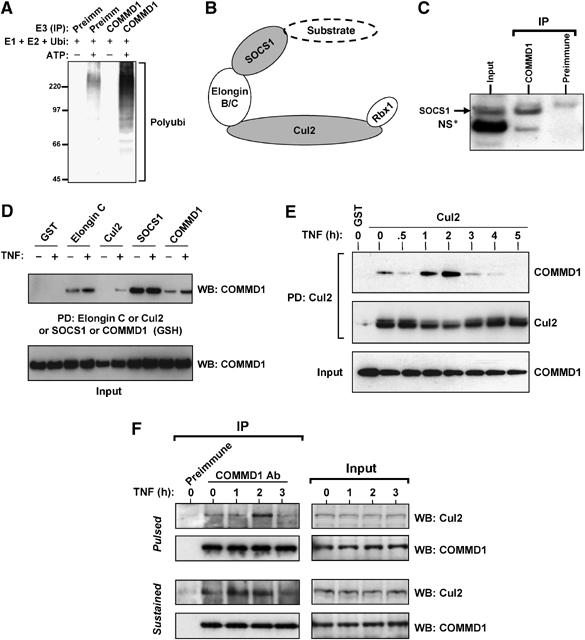
COMMD1 interacts with the Cullin-containing ECSSOCS1 complex. (A) COMMD1 immunoprecipitates possess endogenous E3 ubiquitin ligase activity. An in vitro ubiquitination reaction containing recombinant E1, E2, and ubiquitin was supplemented with immunoprecipitates prepared with COMMD1 antibody or preimmune serum control. The presence of E3 activity was determined by the formation of polyubiquitin chains, as detected by immunoblotting. (B) Schematic representation of the ECSSOCS1 complex. SOCS1 is the substrate binding subunit of the complex. An Elongin B and C complex serves as an adaptor linking SOCS1 to Cul2, which in turn interacts with the RING finger protein Rbx1. (C) Interaction between endogenous COMMD1 and SOCS1. NIH-SR cells were lysed with Triton X-100 buffer and COMMD1 antibodies or a control preimmune serum was used for immunoprecipitation. The recovered material was immunoblotted for SOCS1. (D) COMMD1 interacts with several components of ECSSOCS1. HEK 293 cells were transfected with plasmids expressing Elongin C, Cul2, SOCS1, or COMMD1 in fusion with GST, along with COMMD1-Flag in each case. TNF stimulation consisted of 30 min. After lysis with RIPA buffer, ECS components (or COMMD1-GST) were precipitated with GSH beads and the recovered material was immunoblotted for COMMD1 (Flag). (E) COMMD1–Cul2 interactions are inducible upon TNF stimulation. Similar to the experiment shown in (D), cells were transfected to express GST-Cul2 and COMMD1-Flag. Cells were stimulated with TNF for 10 min and then placed in fresh media. The cells were lysed in RIPA buffer at the indicated time points and Cul2 was precipitated using GSH beads. The recovered material was immunoblotted for COMMD1 (Flag). (F) Endogenous COMMD1 and Cul2 interactions. After TNF stimulation, either as a 10 min pulse or a sustained exposure, endogenous COMMD1 was immunoprecipitated at the indicated time points or the recovered material was immunoblotted for endogenous Cul2.
COMMD1 interacts with the ECSSOCS1 complex, a Cullin-containing ubiquitin ligase
Previous work identified that SOCS1 can induce the ubiquitination and degradation of RelA (Ryo et al, 2003). SOCS1 is a member of a large family of proteins containing the conserved carboxy-terminal SOCS box domain. These factors can form, through their SOCS box, part of multimeric ubiquitin ligases that contain a Cullin protein. These complexes are referred to as ECS and contain Elongins B and C, Cullin 2 or 5, and a SOCS box containing protein (Figure 4B) and are similar in structure to the better known Cul1-containing SCF complexes (Willems et al, 2004). An ECS complex containing SOCS1 (designated as ECSSOCS1) is involved in the ubiquitination of various targets, including JAK2, IRS1, and IRS2 (Rui et al, 2002; Ungureanu et al, 2002).
We have previously reported that COMMD1 interacts with Cul1, a member of the Cullin family that is highly homologous to Cul2 (Ganesh et al, 2003). Given the finding that COMMD1 immune complexes contain E3 ubiquitin ligase activity and that ECSSOCS1 has been implicated in the ubiquitination of RelA, we investigated the possibility that COMMD1 could interact with this complex. Immunoprecipitation of endogenous COMMD1 resulted in the co-precipitation of endogenous SOCS1, consistent with an interaction between these two proteins (Figure 4C). Next, various components of the ECSSOCS1 complex were expressed in Human embryonic kidney (HEK) 293 cells, precipitated from cell lysates, and the recovered material was immunoblotted for COMMD1. As shown in Figure 4D, Elongin C, Cul2, and SOCS1 (but not the control GST protein) co-precipitated with COMMD1. In addition, the interactions of COMMD1 with Elongin C and Cul2 (but not SOCS1) were enhanced by TNF stimulation. Interestingly, the ability of COMMD1 to interact with itself, an event mediated by the conserved COMM domain (Burstein et al, 2005), was also enhanced by TNF stimulation (Figure 4D). In reciprocal experiments, precipitation of COMMD1 resulted in the recovery of components of the ECS complex (including Elongin C, Cul2, SOCS1, and Rbx1) and these interactions were mediated by the COMM domain of COMMD1 (Supplementary Figure S4).
Given the TNF-inducible nature of COMMD1–Cul2 binding, we investigated next the temporal profile of this interaction. Cells expressing Cul2 were stimulated with a pulse of TNF and lysates were prepared at different time points following stimulation. Cul2 was immunoprecipitated and the recovered material was immunoblotted for COMMD1. As before, TNF treatment induced greater recovery of COMMD1 over time, with the peak of the effect taking place 2 h after stimulation (Figure 4E). Similar experiments were performed by immunoprecipitating endogenous COMMD1 and detecting the co-precipitation of endogenous Cul2 after TNF stimulation (Figure 4F). As before, TNF augmented the interaction between COMMD1 and Cul2, and this effect peaked at 2 h, irrespective of whether the stimulation was provided as a pulse or sustained exposure to TNF. Altogether, these data demonstrated a physical interaction between COMMD1 and the ECSSOCS1 complex, which is inducible upon activation of the NF-κB pathway, and suggested the possibility that this might be responsible for COMMD1-directed RelA ubiquitination.
COMMD1-directed ubiquitination is mediated through ECSSOCS1
Given the interaction of COMMD1 with the ECSSOCS1 complex, previously implicated in RelA ubiquitination, we investigated if COMMD1 could be operating through this ligase. To that end, we examined the ability of COMMD1 to induce RelA ubiquitination after RNAi-mediated suppression of either Cul2 or SOCS1. COMMD1 expression induced the accumulation of ubiquitinated RelA in control cells, whereas this event was prevented in cells deficient in either Cul2 or SOCS1 (Figure 5A). This experiment was consistent with the notion that expression of the endogenous ECSSOCS1 complex is required for the effects of COMMD1. Next, we examined the effects of ECSSOCS1 expression on the rates of RelA ubiquitination. Expression of Cul2 and SOCS1 increased the recovery of ubiquitinated RelA and this effect was enhanced by concurrent expression of COMMD1, particularly in Cul2-expressing cells (Figure 5B), and to a lesser extent after proteasomal blockade in SOCS1-expressing cells (data not shown).
Figure 5.
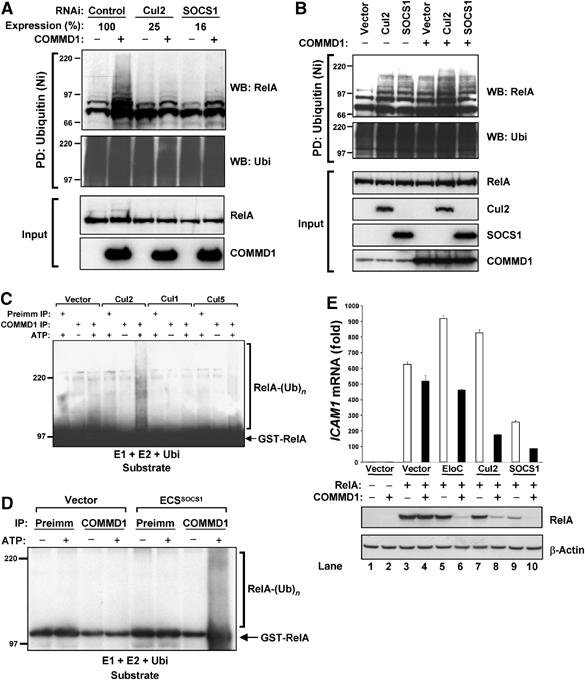
The ECSSOCS1 complex is required for COMMD1-mediated ubiquitination of RelA. (A) COMMD1-mediated ubiquitination of RelA depends on the endogenous ECSSOCS1 complex. HEK 293 cells were transfected with His6-Ubi and RelA as before (Figure 1C), as well as siRNA against Cul2, SOCS1, or a control target. Two days later, mRNA expression levels of Cul2 and SOCS1 were determined by qRT–PCR. In addition, cell lysates were prepared with an 8 M urea buffer and ubiquitinated proteins were precipitated with Ni-NTA beads. The recovered material was immunoblotted for RelA (HA) and ubiquitin. (B) COMMD1 and ECS components have additive effects on RelA ubiquitination. As before, cells were transfected with His6-Ubi and RelA, along with SOCS1, Cul2, and COMMD1, as indicated. Lysates prepared with an 8 M urea buffer were utilized for ubiquitin precipitation; the recovered material was immunoblotted for RelA and ubiquitin. (C, D) In vitro ubiquitination of RelA by COMMD1 immunoprecipitates. An in vitro ubiquitination reaction containing recombinant E1, E2, ubiquitin, and GST-RelA as substrates was provided with immunoprecipitates prepared with COMMD1 antibody or preimmune serum as a source for E3 activity. In (C), the immunoprecipitates were prepared from cells expressing Cul1, Cul2, Cul5, or a vector control. In (D), the immunoprecipitates were prepared from cells expressing ECSSOCS1 subunits (Elongin C, Cul2, SOCS1, and Rbx1) or a vector control. (E) COMMD1 and ECSSOCS1 cooperatively inhibit RelA-mediated ICAM1 expression and promote RelA degradation. HEK 293 cells were transfected with RelA, along with Elongin C (EloC), Cul2, SOCS1, or COMMD1 as indicated. Two days later, ICAM1 mRNA abundance was determined by qRT–PCR and expressed as fold over the vector control sample. In addition, RelA (HA) and β-actin protein levels were concurrently determined by Western blot.
The effect that the expression levels of ECSSOCS1 had on the recovery of ubiquitinated RelA from cells supported the notion that COMMD1-directed ubiquitination is mediated through the ECSSOCS1 complex. Therefore, we set out to test this possibility in an in vitro ubiquitination system. COMMD1 immunoprecipitates were provided as an E3 ubiquitin ligase to a reaction containing recombinant ubiquitin, the E1 and E2 enzymes, and purified GST-RelA as ubiquitination substrate for the reaction. We prepared COMMD1 immunoprecipitates from cells transfected with Cul1, Cul2, Cul5, or the corresponding expression vector as a control. Despite the ability of COMMD1 to interact with all three Cullins (data not shown), only COMMD1 immune complexes recovered from cells expressing Cul2 were capable of catalyzing detectable polyubiquitination of GST-RelA in vitro (Figure 5C). In a similar approach, COMMD1-containing complexes were recovered from cells coexpressing several ECSSOCS1 subunits (Elongin C, Cul2, SOCS1, and Rbx1) or transfected with the corresponding expression vector as a control. Again, the immunoprecipitation of COMMD1 from ECSSOCS1-expressing cells was able to recover an activity that ubiquitinates RelA in vitro (Figure 5D).
COMMD1 and ECSSOCS1 cooperate to inhibit RelA by promoting its degradation
Our data thus far indicated that the interaction of COMMD1 with the ECSSOCS1 complex is required for COMMD1-mediated ubiquitination of RelA in cells and in vitro. Therefore, we reasoned that these factors should have a synergistic negative impact on NF-κB-mediated transcription. We examined this possibility by determining the effects of COMMD1 and ECSSOCS1 components on RelA-mediated ICAM1 expression (Figure 5E). Cells were transfected with RelA and, as expected, this induced strong expression of the endogenous ICAM1 gene (Figure 5E, lane 3). Under the conditions of this experiment, expression of COMMD1, Elongin C, or Cul2 by themselves did not have a substantial inhibitory effect on RelA-mediated transcription of ICAM1 (Figure 5E, lanes 4, 5, and 7). However, when combined with COMMD1, both Elongin C and Cul2 had significant inhibitory activity (Figure 5E, lanes 6 and 8). Similarly, SOCS1-mediated inhibition of ICAM1 expression was further enhanced by COMMD1 (Figure 5E, lanes 9 and 10).
The inhibition of ICAM1 expression correlated directly with the ability of COMMD1 and ECSSOCS1 components to induce reductions in RelA protein levels (Figure 5E, second and third panels). The decreases in protein levels observed were not due to changes in mRNA levels of RelA, and did not occur with a deletion mutant of SOCS1 that cannot interact with the catalytic components of the ubiquitin ligase (data not shown), consistent with a post-transcriptional effect that requires the activity of the ubiquitin ligase. Altogether, the synergistic effects of COMMD1 and ECSSOCS1 on transcription and RelA degradation indicate that these factors cooperatively repress NF-κB.
COMMD1 serves as an adaptor between SOCS1 and Cul2
Next, we sought to understand the role of COMMD1 in the activity of the ECSSOCS1 ubiquitin ligase. We began by examining to which component(s) of the complex COMMD1 binds to directly. To this end, we tested the ability of COMMD1 to bind to deletion mutants of Cul2 and SOCS1 that cannot interact with the rest of the ECSSOCS1 complex (schematic representation of domains involved is shown in Figure 6E). As expected, an amino-terminal deletion of Cul2 (Cul2 ΔN) could not bind to Elongin C; however, COMMD1 binding to this mutant was unaffected (Figure 6A, lanes 2 and 3). This suggested that the carboxy-terminal end of Cul2 is involved in COMMD1 binding and indeed, a carboxy-terminal deletion of Cul2 (Cul2 ΔC) abrogated binding to COMMD1 (Figure 6A, lane 4). Similar studies performed with SOCS1 indicated that a deletion of its carboxy-terminal SOCS box (SOCS1 ΔSB) did not affect COMMD1 binding although as expected it prevented binding to Elongin C (Figure 6A, lanes 5 and 6). In fact, the SH2 domain of SOCS1 was necessary and sufficient for COMMD1 binding (Figure 6A, lanes 7 and 8), even though this region alone is incapable of binding to the ECS complex. Altogether, these studies indicated that COMMD1 can bind independently with two components of the ECSSOCS1, namely Cul2 and SOCS1, resembling the binding characteristics of the adaptor Elongin B/C complex (Figures 4B and 6E).
Figure 6.
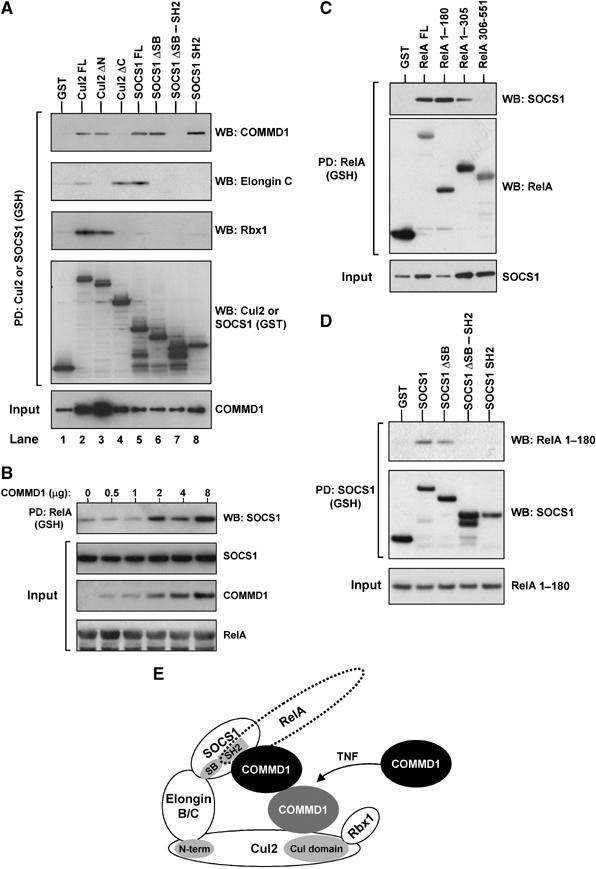
COMMD1 interacts with both Cul2 and SOCS1 and facilitates SOCS1–RelA binding. (A) COMMD1 interacts independently with both SOCS1 and Cul2. Full-length and specific deletion mutants of Cul2 and SOCS1 were expressed in fusion with GST, along with COMMD1-Flag. Cul2 and SOCS1 were precipitated from cell lysates (RIPA buffer) and the recovered material was immunoblotted as indicated. (B) COMMD1 facilitates SOCS1–RelA interactions. HEK 293 cells were transfected with GST-RelA, SOCS1, and increasing amounts of COMMD1 as indicated. Two days after transfection, the cells were lysed in RIPA buffer. RelA was precipitated from the lysate and the recovered material was immunoblotted for SOCS1 (Flag). (C) SOCS1 binds to the amino-terminus of the RHD of RelA. Cells were transfected as in (B), this time utilizing GST-RelA full-length and deletion constructs encompassing the amino acids indicated in the figure. Two days after transfection, cell lysates were prepared with RIPA buffer and RelA was precipitated. The recovered material was immunoblotted for SOCS1 (HA) or RelA (GST). (D) RelA binds to the entire amino-terminus of SOCS1. HEK 293 cells were transfected to express GST-SOCS1 or the deletion mutants indicated, as well as RelA 1–180. Two days after transfection, the cells were lysed in RIPA buffer. SOCS1 was precipitated from the lysate and the recovered material was immunoblotted for RelA 1–180 (HA) or SOCS1 (GST). (E) Schematic representation of COMMD1 interactions with the ECSSOCS1 complex. COMMD1 interacts independently with both SOCS1, at the level of its SH2 domain, and Cul2, at the level of its conserved carboxy-terminus. The interaction between COMMD1 and Cul2 is inducible upon TNF stimulation, and COMMD1 in turn facilitates the binding of RelA to SOCS1.
COMMD1 facilitates the binding of SOCS1 to RelA
SOCS1 is the component of the ECSSOCS1 complex that binds to its substrates, including RelA (Ryo et al, 2003). Interestingly, COMMD1 associates with both SOCS1 and RelA, and accelerates the ubiquitination of RelA, prompting us to investigate if COMMD1 facilitates SOCS1–RelA binding. To this end, we examined the ability of SOCS1 to co-precipitate with RelA under increasing expression of COMMD1. As shown in Figure 6B, precipitation of RelA resulted in co-precipitation of SOCS1, which was enhanced by increased expression of COMMD1 in a dose-dependent manner.
The ability of COMMD1 to interact with both SOCS1 and RelA and the cooperative effect of COMMD1 on the SOCS1–RelA interaction suggested the possibility of a stabilizing protein–protein interaction. Interestingly, SOCS1 co-precipitated with the first 180 amino acids of RelA (Figure 6C), the same amino-terminal motif of the RHD that is also involved in COMMD1 binding (Burstein et al, 2005). Similar to the ability of COMMD1 to enhance SOCS1 binding to full-length RelA, expression of COMMD1 also augmented the binding of SOCS1 to this region of RelA (Supplementary Figure S5).
This raised the possibility that SOCS1 binding to RelA might be indirect and mediated through COMMD1. However, unlike SOCS1 binding to COMMD1 that only required the SH2 domain (Figure 6A), the entire amino-terminal end of SOCS1 was required for SOCS1–RelA interactions (Figure 6D). These data indicate that COMMD1 does not mediate but rather enhances binding of SOCS1 with RelA, altogether suggesting that COMMD1 stabilizes the SOCS1–RelA interaction by cooperatively binding to the same amino-terminal motif in RelA (Figure 6E).
Discussion
In this study, we examined the potential mechanism responsible for COMMD1-mediated repression of NF-κB and its ability to accelerate the dissociation of RelA from chromatin (Burstein et al, 2005). Interestingly, others had reported that ubiquitination of RelA also accelerates its release from chromatin (Saccani et al, 2004), suggesting that ubiquitination of RelA might be responsible for the effects of COMMD1. Indeed, we present evidence that COMMD1 promotes the ubiquitination of NF-κB subunits and our data also indicate that this effect is mediated by the interaction of COMMD1 with ECSSOCS1, a Cullin-containing ubiquitin ligase complex previously implicated in RelA ubiquitination (Ryo et al, 2003). These events are physiologically important because COMMD1 deficiency results in de-repression of several endogenous NF-κB-responsive genes and enhanced κB-mediated cellular responses such as the production of chemokines. Finally, our studies indicated that the role of COMMD1 in this process is to induce greater binding of the substrate, RelA, to the SOCS1 subunit, ultimately promoting its ubiquitination.
SOCS1 has been previously implicated in the control of RelA levels and the data presented here indicate that SOCS1 functions in concert with the ECS complex. More importantly, we present data indicating that the ability of COMMD1 to ubiquitinate RelA is mediated through ECSSOCS1. COMMD1 interacts with components of the ECS complex, as shown at the level of endogenous SOCS1 and Cul2 (Figure 4C and F) and overexpressed ECS components (Figures 4D, E, and 6A and Supplementary Figure S4). Similarly, COMMD1-mediated ubiquitination of RelA in cells is hampered by ECS deficiency (Figure 5A) and accentuated by overexpression of ECS components (Figure 5B). Finally, COMMD1 immune complexes contain ubiquitin ligase activity and can mediate the ubiquitination of RelA in vitro when recovered from cells expressing Cul2 and not other Cullins, or from cells expressing ECS components (Figure 5C and D).
SOCS1 expression is under the control of the JAK/STAT pathway, which stimulates transcription of the SOCS1 gene (Naka et al, 1997) and promotes the stability of the SOCS1 protein through Pim-mediated phosphorylation (Chen et al, 2002). These effects predict that STAT activation should result in negative crosstalk with the NF-κB pathway. In this regard, NF-κB activation results in the expression of various inducers of the STAT pathway (such as IL-6) that can upregulate SOCS1 expression (Zhang et al, 1994), possibly providing an autocrine negative feedback loop. This mechanism could be important during inflammatory responses in vivo, when multiple cytokines are coordinately activated. In addition, our studies demonstrate that the SOCS1–COMMD1 interaction is mediated by the SH2 domain of SOCS1 (Figure 5A), a region that preferentially interacts with tyrosine phosphorylated targets (Ungureanu et al, 2002). This suggests the possibility that activation of tyrosine kinase(s) that might target COMMD1 could enhance this interaction and provide additional crosstalk regulation of the NF-κB pathway.
Our previously reported data suggest that COMMD1-mediated inhibition of NF-κB occurs in the nucleus, a compartment where the protein can be readily identified (Burstein et al, 2005). Although it is largely accepted that the cytoplasmic pool of SOCS1 is responsible for its ability to regulate JAK2 kinase activity (Endo et al, 1997), the presence of a nuclear pool of SOCS1 is also well documented (Vuong et al, 2004). Indeed, our analysis indicates that the main components of the ECSSOCS1 complex, including SOCS1, Elongin C, Cul2, and Rbx1, can be found in nuclear extracts (Supplementary Figure S6).
Another important observation in our studies is that there is dissociation between the duration of transcriptional responses and nuclear RelA accumulation. The mRNA levels for all κB-responsive genes evaluated here continued to accumulate at time points when nuclear levels of RelA were back to baseline (after 45 min; see Figures 2D and 3A). This suggests that after RelA has been effectively exported into the cytosol, other mechanisms are likely involved in the late termination of transcription. In fact, the inducible COMMD1–Cul2 interaction peaks at 2 h (Figure 4E and F), a time point when nuclear export of RelA has been completed, suggesting that the physiologic role of NF-κB ubiquitination is in late transcriptional termination.
We have previously reported that COMMD1 binds to Cul1 (Ganesh et al, 2003), and more detailed examination indicates that COMMD1 can bind to additional Cullins (GN Maine and E Burstein, unpublished observations). This suggests the possibility that COMMD1 might be a component of various ubiquitin ligases targeting proteins other than NF-κB subunits and might ultimately explain functions of COMMD1, such as its role in copper metabolism, that appear unrelated to its effects on NF-κB (van de Sluis et al, 2002; Burstein et al, 2004).
Altogether, this study demonstrates that COMMD1, acting via a Cullin-containing ubiquitin ligase, represses κB-mediated transcription through ubiquitination of NF-κB subunits. COMMD1-mediated regulation of NF-κB through ECSSOCS1 is part of a larger paradigm of transcriptional regulation. Indeed, various Cullin-containing complexes have been shown to play critical roles in the regulation of other transcription factors such as c-Myc (von der Lehr et al, 2003) and HIF1α (Pause et al, 1997). Our data uncover that in addition to the well-known events that control IκB protein stability, regulation of the stability of the NF-κB subunits by this COMMD1-containing ubiquitin ligase is a critical event in the termination of κB-mediated transcription.
Materials and methods
Plasmids and small interfering RNA
Expression vectors for RelB and p52 were generated by PCR amplification of the corresponding coding sequences from plasmid templates kindly provided by Dr Ranjeny Thomas and Dr Colin Duckett, respectively. Expression vectors for Elongin C, Cul2, SOCS1, and Rbx1 were generated by PCR amplification using as templates the following IMAGE cDNA clones: 3923736, 4104375, 5179306/5093311, and 3138751, respectively. Deletion constructs for Cul2 and SOCS1 were similarly generated with the amino-acid boundaries of the encoded mutant proteins being Cul2 Δ_N_=109–745, Cul2 ΔC=1–415, SOCS1 ΔSB=1–157, SOCS1 ΔSB-SH2=1–78, SOCS1 SH2=78–174. Information about the sequences of small interfering RNA (siRNA) oligonucleotides utilized (chloramphenicol acetyl transferase, COMMD1, SOCS1, and Cul2, obtained from Invitrogen) are available upon request. For stable RNAi utilizing lentiviral delivery we introduced a cassette containing the histone 1 promoter and a short hairpin RNA into the FG12 plasmid (kindly provided by Dr David Baltimore). All other plasmids used have been described previously (Burstein et al, 2004, 2005).
Cell culture, transfection, and lentiviral production
HEK 293 cells, HEK 293T cells, U2OS cells, and NIH-SR cells were obtained from ATCC. HEK 293, HEK 293T, and U2OS cells were cultured in DMEM supplemented with 10% FBS and L-glutamine (2 mM). NIH-SR cells were cultured in RPMI supplemented with 10% FBS, L-glutamine (2 mM), sodium pyruvate (1 mM), and glucose (4.5 g/l). A standard calcium phosphate transfection protocol was used to transfect plasmids and siRNA into HEK 293 cells (Burstein et al, 2004). TNF treatments (1000 U/ml; Roche) were for variable amounts of time, as indicated in each experiment. Production of lentiviruses for stable delivery of short hairpin RNA constructs was performed as previously described utilizing plasmids kindly provided by Dr David Baltimore (Lois et al, 2002).
Quantitative RT–PCR
RNA extraction and RT–PCR methods were performed as previously described (Burstein et al, 2005). Oligonucleotides and probes for RELA, ICAM1, BIRC3, CXCL1, CCL2, TNF, and IL8 transcripts were obtained from Applied Biosystems.
Immunoblotting and immunoprecipitation
The compositions of Triton lysis buffer and RIPA buffer have been previously described (Burstein et al, 2005). In some experiments, an 8 M urea buffer (8 M urea, 50 mM Tris, pH 8.0, 300 mM NaCl, 50 mM NaPO4, 0.5% NP-40) was used. All buffers were supplemented with 1 mM sodium orthovanadate and protease inhibitors (Roche). Preparation of cytosolic and nuclear extracts, immunoprecipitations, GSH precipitations, Ni-NTA precipitations, and immunoblotting were performed as described previously (Burstein et al, 2004, 2005). The following antibodies were used: COMMD1 (Burstein et al, 2005; Abnova), Flag (Sigma), HA (Sigma), ubiquitin (Stressgen), RelA (Santa Cruz Biotechnology), RelB (Santa Cruz Biotechnology), p52 (Upstate Biotechnology), Cul2 (Zymed, Upstate Biotechnology, and Abnova), Elongin C (BioLegend), SOCS1 (Zymed, Abcam, Santa Cruz Biotechnology), Rbx1 (LabVision), IκB-α (Upstate Biotechnology), GST (Santa Cruz), β-actin (Sigma), α-tubulin (Molecular Probes), and GCN5 (Santa Cruz).
Metabolic labeling
One day prior, 8 × 105 cells were seeded in a 10cm dish. They were placed in a cysteine- and methionine-deficient medium (with 10% dialyzed FBS) for 30 min, and 500 μCi of 35S-labeled methionine and cysteine (GE Healthcare) was added per plate for 1 h. The media were then replaced with regular growth media supplemented with excess non-radiolabeled methionine (2 mM) and cysteine (2 mM). Denatured lysates were prepared by applying 50 mM Tris, 1% SDS, 5 mM DTT, and boiling the samples for 10 min at 95°C. The buffer was then supplemented with Triton X-100 lysis buffer to decrease the SDS concentration to 0.2%. Samples were pre-cleared with protein G Sepharose beads for 1 h at 4°C and the supernatant obtained after centrifugation was subjected to RelA immunoprecipitation (Santa Cruz Biotechnology, sc-372). The recovered material at the end of immunoprecipitation was resolved by SDS–PAGE and RelA was detected by autoradiography.
In vitro chemotaxis assay and chemokine measurement
Conditioned media before and after 12-h TNF stimulation were used. Peripheral mononuclear cells were freshly isolated after dextran sedimentation and Ficoll gradient centrifugation, as previously described (Diamond et al, 1991). The cells were then fluorescently labeled with Calcein-AM (Sigma) and chemotactic migration across a membrane and toward conditioned media was determined utilizing a 96-well FluoroBlock plate pre-coated with fibronectin, according to the manufacturer's instructions (BD Biosciences). The plate was placed at 37°C on a fluorometer (ABI Cytofluor 4000) and cell migration was monitored every 15 min for a total of 2 h. CCL2 levels in conditioned media were determined by ELISA according to the manufacturer's instructions (R&D systems).
In vitro ubiquitination reaction
Each reaction mixture consisted of recombinant ubiquitin (2.5 μg/reaction), E1 (Uba1, 50 ng/reaction), E2 (UbcH5a, 100 ng/reaction), and ATP regenerating buffer (Boston Biochem), resuspended in reaction buffer (40 mM HEPES pH 7.9, 60 mM potassium acetate, 2 mM DTT, 5 mM MgCl2, 10% glycerol). The material obtained from immunoprecipitation of COMMD1 (or control) was added as a source for E3 ubiquitin ligase activity. Recombinant GST-RelA was used as the substrate in these reactions and was prepared by expression in 293 cells and affinity purification using standard protocols.
Supplementary Material
Supplementary Figure Legends
Supplementary Figures
Acknowledgments
We are grateful to David Baltimore, Colin Duckett, Warner Greene, Michele Pagano, Liangyou Rui, and Ranjeny Thomas for sharing plasmids used in our studies. We also thank Robert Rottapel and Tadamitsu Kishimoto for sharing SOCS1 antibodies, and Nicholas Lukacs and Lilli Petruzzelli for their technical assistance. This work was supported by an American Gastroenterological Association Research Scholar Award, a Merit Review Entry Program Award, a Crohn's and Colitis Foundation of America Senior Research Award, and a Veterans Education and Research Association of Michigan Award to EB.
References
- Burstein E, Ganesh L, Dick RD, van De Sluis B, Wilkinson JC, Klomp LW, Wijmenga C, Brewer GJ, Nabel GJ, Duckett CS (2004) A novel role for XIAP in copper homeostasis through regulation of MURR1. EMBO J 23: 244–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein E, Hoberg JE, Wilkinson AS, Rumble JM, Csomos RA, Komarck CM, Maine GN, Wilkinson JC, Mayo MW, Duckett CS (2005) COMMD proteins: a novel family of structural and functional homologs of MURR1. J Biol Chem 280: 22222–22232 [DOI] [PubMed] [Google Scholar]
- Chen L, Fischle W, Verdin E, Greene WC (2001) Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293: 1653–1657 [DOI] [PubMed] [Google Scholar]
- Chen XP, Losman JA, Cowan S, Donahue E, Fay S, Vuong BQ, Nawijn MC, Capece D, Cohan VL, Rothman PB (2002) Pim serine/threonine kinases regulate the stability of Socs-1 protein. Proc Natl Acad Sci USA 99: 2175–2180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Hagler J, Palombella VJ, Melandri F, Scherer D, Ballard D, Maniatis T (1995) Signal-induced site-specific phosphorylation targets IκBα to the ubiquitin–proteasome pathway. Genes Dev 9: 1586–1597 [DOI] [PubMed] [Google Scholar]
- Diamond MS, Staunton DE, Marlin SD, Springer TA (1991) Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell 65: 961–971 [DOI] [PubMed] [Google Scholar]
- Endo TA, Masuhara M, Yokouchi M, Suzuki R, Sakamoto H, Mitsui K, Matsumoto A, Tanimura S, Ohtsubo M, Misawa H, Miyazaki T, Leonor N, Taniguchi T, Fujita T, Kanakura Y, Komiya S, Yoshimura A (1997) A new protein containing an SH2 domain that inhibits JAK kinases. Nature 387: 921–924 [DOI] [PubMed] [Google Scholar]
- Ganesh L, Burstein E, Guha-Niyogi A, Louder MK, Mascola JR, Klomp LW, Wijmenga C, Duckett CS, Nabel GJ (2003) The gene product Murr1 restricts HIV-1 replication in resting CD4+ lymphocytes. Nature 426: 853–857 [DOI] [PubMed] [Google Scholar]
- Henkel T, Machleidt T, Alkalay I, Krönke M, Ben-Neriah Y, Baeuerle PA (1993) Rapid proteolysis of IκB-α is necessary for activation of transcription factor NF-κB. Nature 365: 182–185 [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Levchenko A, Scott ML, Baltimore D (2002) The IκB–NF-κB signaling module: temporal control and selective gene activation. Science 298: 11241–11245 [DOI] [PubMed] [Google Scholar]
- Karin M, Lin A (2002) NF-κB at the crossroads of life and death. Nat Immunol 3: 221–227 [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D (2002) Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295: 868–872 [DOI] [PubMed] [Google Scholar]
- Naka T, Narazaki M, Hirata M, Matsumoto T, Minamoto S, Aono A, Nishimoto N, Kajita T, Taga T, Yoshizaki K, Akira S, Kishimoto T (1997) Structure and function of a new STAT-induced STAT inhibitor. Nature 387: 924–929 [DOI] [PubMed] [Google Scholar]
- Pause A, Lee S, Worrell RA, Chen DY, Burgess WH, Linehan WM, Klausner RD (1997) The von Hippel–Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci USA 94: 2156–2161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui L, Yuan M, Frantz D, Shoelson S, White MF (2002) SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem 277: 42394–42398 [DOI] [PubMed] [Google Scholar]
- Ryo A, Suizu F, Yoshida Y, Perrem K, Liou YC, Wulf G, Rottapel R, Yamaoka S, Lu KP (2003) Regulation of NF-κB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol Cell 12: 1413–1426 [DOI] [PubMed] [Google Scholar]
- Saccani S, Marazzi I, Beg AA, Natoli G (2004) Degradation of promoter-bound p65/RelA is essential for the prompt termination of the nuclear factor κB response. J Exp Med 200: 107–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakurai H, Chiba H, Miyoshi H, Sugita T, Toriumi W (1999) IκB kinases phosphorylate NF-κB p65 subunit on serine 536 in the transactivation domain. J Biol Chem 274: 30353–30356 [DOI] [PubMed] [Google Scholar]
- Tergaonkar V, Correa RG, Ikawa M, Verma IM (2005) Distinct roles of IκB proteins in regulating constitutive NF-κB activity. Nat Cell Biol 7: 921–923 [DOI] [PubMed] [Google Scholar]
- Ungureanu D, Saharinen P, Junttila I, Hilton DJ, Silvennoinen O (2002) Regulation of Jak2 through the ubiquitin–proteasome pathway involves phosphorylation of Jak2 on Y1007 and interaction with SOCS-1. Mol Cell Biol 22: 3316–3326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Sluis B, Rothuizen J, Pearson PL, van Oost BA, Wijmenga C (2002) Identification of a new copper metabolism gene by positional cloning in a purebred dog population. Hum Mol Genet 11: 165–173 [DOI] [PubMed] [Google Scholar]
- von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, Hydbring P, Weidung I, Nakayama K, Nakayama KI, Soderberg O, Kerppola TK, Larsson LG (2003) The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell 11: 1189–1200 [DOI] [PubMed] [Google Scholar]
- Vuong BQ, Arenzana TL, Showalter BM, Losman J, Chen XP, Mostecki J, Banks AS, Limnander A, Fernandez N, Rothman PB (2004) SOCS-1 localizes to the microtubule organizing complex-associated 20S proteasome. Mol Cell Biol 24: 9092–9101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willems AR, Schwab M, Tyers M (2004) A hitchhiker's guide to the cullin ubiquitin ligases: SCF and its kin. Biochim Biophys Acta 1695: 133–170 [DOI] [PubMed] [Google Scholar]
- Yaron A, Hatzubai A, Davis M, Lavon I, Amit S, Manning AM, Andersen JS, Mann M, Mercurio F, Ben-Neriah Y (1998) Identification of the receptor component of the IκBα-ubiquitin ligase. Nature 396: 590–594 [DOI] [PubMed] [Google Scholar]
- Zhang Y, Broser M, Rom WN (1994) Activation of the interleukin 6 gene by Mycobacterium tuberculosis or lipopolysaccharide is mediated by nuclear factors NF-IL6 and NF-κB. Proc Natl Acad Sci USA 91: 2225–2229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong H, May MJ, Jimi E, Ghosh S (2002) The phosphorylation status of nuclear NF-κB determines its association with CBP/p300 or HDAC-1. Mol Cell 9: 625–636 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure Legends
Supplementary Figures