The trans-envelope Tol–Pal complex is part of the cell division machinery and required for proper outer-membrane invagination during cell constriction in E. coli (original) (raw)
. Author manuscript; available in PMC: 2015 May 12.
Summary
Fission of bacterial cells involves the co-ordinated invagination of the envelope layers. Invagination of the cytoplasmic membrane (IM) and peptidoglycan (PG) layer is likely driven by the septal ring organelle. Invagination of the outer membrane (OM) in Gram-negative species is thought to occur passively via its tethering to the underlying PG layer with generally distributed PG-binding OM (lipo)proteins. The Tol–Pal system is energized by proton motive force and is well conserved in Gram-negative bacteria. It consists of five proteins that can connect the OM to both the PG and IM layers via protein–PG and protein–protein interactions. Although the system is needed to maintain full OM integrity, and for class A colicins and filamentous phages to enter cells, its precise role has remained unclear. We show that all five components accumulate at constriction sites in Escherichia coli and that mutants lacking an intact system suffer delayed OM invagination and contain large OM blebs at constriction sites and cell poles. We propose that Tol–Pal constitutes a dynamic subcomplex of the division apparatus in Gram-negative bacteria that consumes energy to establish transient _trans_-envelope connections at/near the septal ring to draw the OM onto the invaginating PG and IM layers during constriction.
Introduction
Cytokinesis of bacteria is mediated by the septal ring (SR) (divisome, septasome), a ring-shaped organelle associated with the invaginating envelope layers during cell constriction. Components of the SR include essential division proteins, without which cells cannot divide at all, as well as nonessential factors that may serve critical but redundant functions, or nonessential accessory functions that improve the efficiency of the constriction process. To date, the SR of Escherichia coli is known to contain 10 essential division proteins that make up the core of the organelle. Core proteins are either cytoplasmic (FtsZ, A), or integral inner-membrane (IM) species of bitopic (ZipA, FtsQ, B, L, I, N) or polytopic (FtsK, W) topology. Assembly of the core SR starts well before division and is thought to initiate with the polymerization of FtsZ just underneath the IM. These polymers are joined by the FtsZ binding proteins FtsA, ZipA and ZapA, resulting in a mostly cytoplasmic intermediate assembly referred to as the Z-ring. The Z-ring is then joined by the remaining core components in a certain order to form a mature constriction-competent SR. FtsN is the last known essential core protein to join the ring, and its recruitment occurs just prior to or coincident with the onset of cell constriction. (For recent reviews, see Addinall et al., 1997; Chen and Beckwith, 2001; Errington et al., 2003; Goehring and Beckwith, 2005; Margolin, 2005; Vicente et al., 2006.)
In addition to core proteins, a number of nonessential SR components have been identified. These include ZapA and the chaperone GroEL in the cytoplasm (Gueiros-Filho and Losick, 2002; Ogino et al., 2004), the IM-associated (predicted) ABC transporter FtsE/FtsX (Schmidt et al., 2004), and the periplasmic murein hydrolases AmiC and EnvC (Bernhardt and de Boer, 2003; 2004).
How the mature SR drives cell envelope invagination is an important unsolved question. Inward movement of the IM is closely co-ordinated with inward growth of septal peptidoglycan (PG, murein) during invagination (Woldringh, 1976; Rothfield et al., 1986; MacAlister et al., 1987). In principle, the IM could be pushed inwards by septal PG synthesis, pulled inwards by contraction of the Z-ring in the cytoplasm, or move by a combination of the two. One of the latter two scenarios is most likely as membrane invagination can occur without septal PG ingrowth in certain mutants of E. coli and Bacillus subtilis (Daniel et al., 2000; Heidrich et al., 2002; Siddiqui et al., 2006). In turn, Z-ring contraction is likely coupled to some ordered rearrangement or depolymerization of FtsZ polymers, but the molecular mechanisms behind this important step remain obscure (Addinall and Holland, 2002; Ryan and Shapiro, 2003; Goehring and Beckwith, 2005).
Gram-negative bacteria such as E. coli face the additional task of ensuring proper invagination of the outer membrane (OM). OM invagination requires separation of the underlying septal murein into two daughter layers by murein hydrolases (Heidrich et al., 2001; 2002), at least two of which (AmiC and EnvC) specifically localize to the SR during constriction (Bernhardt and de Boer, 2003; 2004). Thin-section electron microscopic studies on E. coli and Salmonella typhimurium indicate that invagination of all three envelope layers is normally tightly co-ordinated in time and space (Weigand et al., 1976; Fung et al., 1978; Rothfield et al., 1986; MacAlister et al., 1987; Bi and Lutkenhaus, 1991; Lutkenhaus, 1993). This implies that there are only small intervals between septal PG ingrowth, its splitting from the OM proximal end, and coverage of the separating daughter layers by inward moving OM in these species
It is notable that none of the known SR components are OM proteins, and it has long been suspected that OM invagination occurs passively via generally distributed (lipo)protein linkages between the OM and PG layers (Rothfield and Justice, 1997; Weiss, 2004). The major murein lipoprotein (Lpp, Braun's lipoprotein), in particular, plays an important role in maintaining contacts between the OM and PG, including at constriction sites. Besides a general weakening of OM barrier functions, cells defective for Lpp also show a distinct defect in OM invagination under limiting Mg++ conditions. This is accompanied by the formation of large OM blebs at constriction sites and cell poles, and by a mild cell chaining phenotype due to delayed daughter cell separation (Weigand et al., 1976; Fung et al., 1978; Suzuki et al., 1978; Yem and Wu, 1978). Even though Lpp contributes to proper OM invagination, it appears homogenously distributed along the cell envelope (Hiemstra et al., 1986; 1987), and is generally not considered to be a specific component of the division machinery.
The Tol–Pal system is well conserved in Gram-negative bacteria (Sturgis, 2001) and is also required for maintaining OM integrity. The system consists of (at least) five proteins. TolA, TolQ and TolR are IM proteins, TolB is periplasmic, and Pal (peptidoglycan-associated lipoprotein) is an abundant OM lipoprotein (Lloubes et al., 2001; Cascales et al., 2002; Lazzaroni et al., 2002). TolA, Q and R interact via their _trans_-membrane helices to form a complex in the IM (Derouiche et al., 1995; Lazzaroni et al., 1995; Germon et al., 1998; Journet et al., 1999) while Pal and TolB interact near the OM (Bouveret et al., 1995; 1999; Clavel et al., 1998; Ray et al., 2000; Cascales and Lloubes, 2004). The system can bridge the IM and OM via a specific interaction between the extended periplasmic C-terminus of TolA with Pal (Cascales et al., 2000; Cascales and Lloubes, 2004) and, possibly, via an interaction between TolA and TolB as well (Dubuisson et al., 2002; Walburger et al., 2002). Interestingly, the TolA–Pal interaction requires work as it depends on both the proton motive force (pmf) and TolQ and TolR activities (Cascales et al., 2000; 2001). The pmf and TolQ/R induce a conformational change in the periplasmic portion of TolA that likely allows it to engage Pal (Germon et al., 2001). TolQ and TolR show similarities to ExbB/MotA and ExbD/MotB respectively, suggesting they directly transduce membrane ion potential to ‘energize’ TolA into capturing a Pal partner in the OM (Braun and Herrmann, 1993; Cascales et al., 2001).
In addition to connecting the IM and OM as part of the TolA–Pal complex, Pal lipoprotein can also directly connect the OM and PG layers via a strong, non-covalent, interaction between a C-terminal domain and the peptide moieties of PG (Mizuno, 1981; Lazzaroni and Portalier, 1992; Clavel et al., 1998; Bouveret et al., 1999; Parsons et al., 2006). This interaction does not require the other Tol proteins, but it is likely modulated by them, as TolB competes with PG for binding Pal (Clavel et al., 1998; Bouveret et al., 1999; Ray et al., 2000; Cascales and Lloubes, 2004).
Although the Tol–Pal system has been intensely studied, its physiological role(s) has remained unclear. The system was first identified for its abuse by both group A colicins (Bernstein et al., 1972; Benedetti et al., 1991; Lazdunski, 1995; 1998; Cao and Klebba, 2002; Lazzaroni et al., 2002; Pommier et al., 2005) and filamentous ssDNA phages (such as f1, fd, M13 and CTXphi) (Sun and Webster, 1986; Click and Webster, 1997; 1998; Riechmann and Holliger, 1997; Lubkowski et al., 1999; Heilpern and Waldor, 2000) to intoxicate/infect cells. Both agents mimic features of Pal suggesting they fool TolA and/or TolB in engaging them instead of the lipoprotein (Deprez et al., 2002; Cascales and Lloubes, 2004; Pommier et al., 2005). The Tol proteins likely aid in positioning the colicins or phage particles close to the IM, but the precise mechanisms whereby these agents cross the envelope and gain access to the cytoplasm are still incompletely understood (Bouveret et al., 2002; James et al., 2002; Lazzaroni et al., 2002).
Clues as to the physiologically relevant role of the Tol–Pal system are that mutations in the tol-pal genes lead to increased sensitivity of cells to a variety of drugs and detergents, leakage of periplasmic components, prolific shedding of OM vesicles in the medium, and upregulation of the RpoE-mediated extracytoplasmic stress response (Bernadac et al., 1998; Lazzaroni et al., 1999; Llamas et al., 2000; Cascales et al., 2002; Prouty et al., 2002; Henry et al., 2004; Dubuisson et al., 2005; Vines et al., 2005). Moreover, Tol–Pal mutants show a pronounced cell division phenotype indicative of defects in OM invagination and cell separation. Thus, E. coli tolA mutants form long multi-septate cell chains in rich medium of low osmolarity or high ionic strength even when Mg++ is not limiting (Meury and Devilliers, 1999). Similar chaining has been observed in mutants of any of the tol-pal genes of Vibrio Cholerae (Heilpern and Waldor, 2000), Pseudomonas putida (Llamas et al., 2000) and/or Erwinia chrysanthemi (Dubuisson et al., 2005).
These phenotypes are reminiscent of those of lpp mutants (Fung et al., 1978; Suzuki et al., 1978; Yem and Wu, 1978; Bernadac et al., 1998; Cascales et al., 2002) and the Tol–Pal system could play a general structural role similar to that of Lpp in maintaining OM integrity. However, the complexity of the system plus the facts that the Tol–Pal proteins: (i) can connect the OM with both the PG and IM layers, (ii) constitute a pmf-energized system, and (iii) prevent chaining phenotypes that are more severe than those of lpp cells, suggest that the Tol–Pal system may serve a more active role in the division process.
Here we used fluorescent fusions to show that all five Tol–Pal proteins accumulate at cell constriction sites in E. coli. Recruitment to these sites is dependent on FtsN activity and may be coincident with the onset of envelope invagination. TolQ and TolA localize independently of any of the other four Tol–Pal proteins, while TolR and Pal require other components of the system. Studies with a fluorescent periplasmic probe (TTGFP) showed a localized expansion of the periplasm at constriction sites in tolA, pal and tolQ-pal mutant cells. In addition, they revealed the prolific generation of large OM blebs/vesicles specifically at division sites and cell poles in these cells. We propose that the Tol–Pal system forms a subcomplex of the division apparatus in Gram-negative bacteria, and that one of its primary functions is to draw the OM into the space created by the separation of septal murein during the cell constriction process. The dynamic localization of the Tol–Pal proteins in live cells implies that the interactions of Pal with TolA and with PG must be transient in nature. We suggest that the system is energized to allow for rapid Pal–TolA and Pal–PG engagement/disengagement cycles as Tol–Pal complexes are recruited to sites of constriction and as they move along with the contracting SR to establish new OM–IM bridges across, and OM–PG bridges with, freshly separated septal murein. The wave of transient Tol–Pal-mediated OM–IM and OM–PG connections at/near the SR is proposed to keep the OM closely associated with the underlying PG layer during the constriction process. In the wake of this wave, other PG-binding OM (lipo)proteins could efficiently secure the OM to the PG of the nascent poles in a more permanent manner.
Results
Discordant invagination of IMs and OMs during cell constriction in tol-pal mutants
In a survey of the localization patterns of E. coli proteins that are fused at their C-terminus to GFP [ASKA library (Kitagawa et al., 2005)], we noticed accumulation of a TolQ–GFP fusion at sites of cell constriction. Because tol-pal mutations can cause cell chaining in several Gram-negative species (Meury and Devilliers, 1999; Heilpern and Waldor, 2000; Llamas et al., 2000; Dubuisson et al., 2005), this observation prompted us to further explore the possibility of a direct role of the Tol–Pal system in the division process. We examined the morphology of three mutant strains. FB20229 [_tolA_] carries a transposon insertion in tolA, MG5 [_pal_] is deleted for pal, and MG4 [_tolQ-pal_] lacks all five genes of the Tol–Pal system (Fig. 1, Table 1). E. coli tolA mutants were previously shown to form cell chains in medium of low osmolarity (Meury and Devilliers, 1999). Accordingly, FB20229 cells formed long multi-septate chains in Luria–Bertani (LB) medium that lacked added NaCl (Fig. 2A), and chaining was suppressed by addition of NaCl to 0.5% (not shown). In addition, MG4 and MG5 showed the same phenotypes as FB20229 (Fig. 2C, and data not shown), suggesting the division defect in the mutants is due to the inability to form a complete _trans_-envelope complex.
Fig. 1.
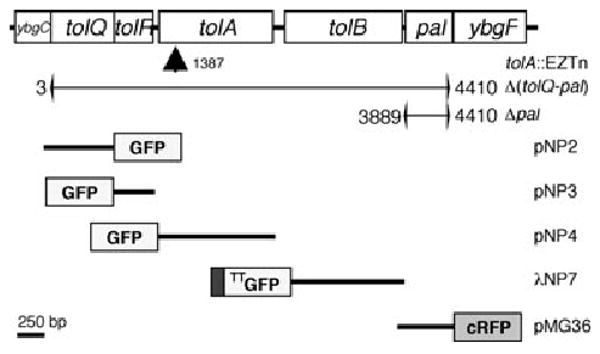
Map of chromosomal tol-pal cluster, mutations and constructs. The E. coli tol-pal gene cluster is arranged in two operons (ybgC-tolA and tolB-ybgF) the transcription of which proceeds from left to right (Vianney et al., 1996). Indicated are the site of EZTn insertion in strain FB20229 [_tolA_], the deletions in strains MG4 [_tolQ-pal_] and MG5 [_pal_], and the inserts present in the plasmids or phage encoding GFP or RFP fusions. Numbers refer to base pairs counting from the start codon of tolQ. GFP refers to GFPmut2 (Cormack et al., 1996), and RFP to the cherry variant of monomeric RFP (Shaner et al., 2004). The immature form of the TTGFP–TolB fusion encoded by λNP7 lacks the native signal peptide of TolB but contains the N-terminal signal sequence of TorA such that it is routed to the periplasm via the Tat system. The other four fusions contain complete native polypeptide appended to GFP (TolQ, R, A) or RFP (Pal), as indicated.
Table 1.
E. coli strains, plasmids and phages used in this study.
| Strain, plasmid or phage | Relevant genotypea | Source or reference |
|---|---|---|
| MG1655 | _rph_1 _ilvG rfb_-50 | Guyer et al. (1981) |
| TB28 | MG1655 lacIZYA <>frt | Bernhardt and de Boer (2003) |
| FB20229 | MG1655 _tolA_∷EZTN | ASAP (Glasner et al. 2003) |
| MG4 | TB28 tolQ-pal<>aph | This study |
| MG5 | TB28 pal<>aph | This study |
| CH34b | TB28 ftsN<>aph | Laboratory collection |
| pKD46 | bla repA_101(ts) araC PBAD∷_γ β exo | Datsenko and Wanner (2000) |
| pNP2 | bla lacI_q Plac∷_tolQ-gfp | This study |
| pNP3 | bla lacI_q Plac∷_gfp-t-tolR | This study |
| pNP4 | bla lacI_q Plac∷_gfp-t-tolA | This study |
| pNP7 | bla lacI_q Plac∷ss_torA-gfp-t-tolB | This study |
| pMG20 | cat araC_PBAD∷ss_torA-bfp-ftsN(71–105) | Laboratory collection |
| pMG36 | bla lacI_q Plac∷_pal-crfp | This study |
| pTB6 | bla lacI_q Plac∷ss_torA-gfp-t | Bernhardt and de Boer (2004) |
| pJE80 | cat araC PBAD∷sfiA | Johnson et al. (2002) |
| λNP7 | bla lacI_q Plac∷ss_torA-gfp-t-tolB | This study |
| λCH151 | imm_21_bla lacI_q Plac∷_zipA-gfp | Bernhardt and de Boer (2004) |
| λTB6 | imm_21_bla lacI_q Plac∷ss_torA-gfp-t | Bernhardt and de Boer (2004) |
Fig. 2.
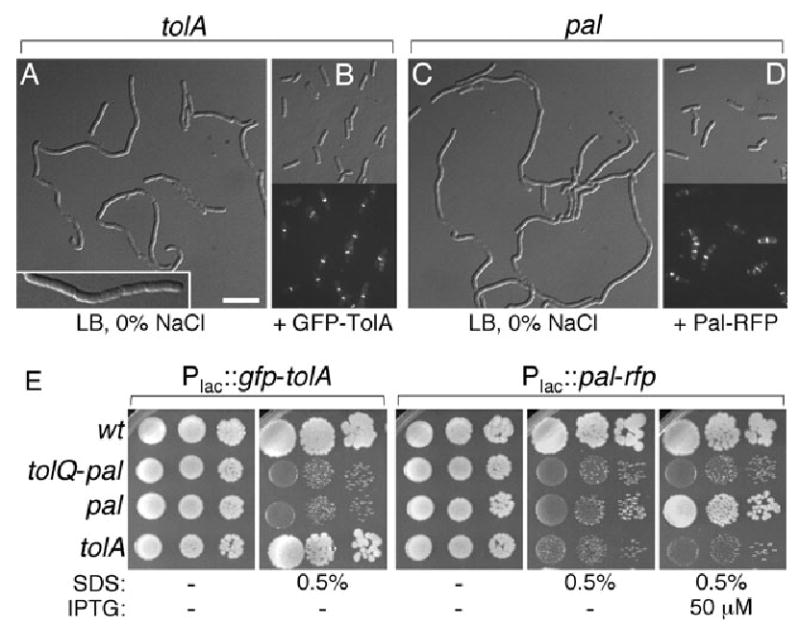
Functionality of GFP–TolA and Pal–RFP. Panels A–D show suppression of cell chaining in tolA cells by GFP–TolA, and in pal cells by Pal–RFP. Strains FB20229/pMLB1113ΔH (A), FB20229/pNP4 (B), MG5/pMLB1113ΔH (C) and MG5/pMG36 (D) were grown in LB (0% NaCl) medium supplemented with 5 μM (A–C) or 100 μM (D) IPTG. Plasmid pMLB1113ΔH is an appropriate vector control for both pNP4 and pMG36. Panels A and C show DIC images, and panels B and D show both DIC and corresponding fluorescence images. The insert in A shows part of a tolA chain at higher magnification. Bar equals 2.5 (insert in A) or 5 μm. Panel E shows suppression of hypersensitivity to SDS by GFP–TolA in tolA cells, and by Pal–RFP in pal cells. Overnight cultures of wt and mutant strains carrying pNP4 [Plac∷_gfp-tolA_] or pMG36 [Plac∷_pal-rfp_] were diluted to optical densities (600 nm) of 2 × 10−3, 2 × 10−4 and 2 × 10−5 (left to right), and 5 μl aliquots were spotted on LB agar containing SDS and/or IPTG as indicated. Plates were incubated at 30°C for 24 h and then at 20°C for 72 h. Strains used were TB28 [wt], MG4 [_tolQ-pal_], MG5 [_pal_] and FB20229 [_tolA_].
Chaining of the three mutants was also largely suppressed by growth in M9 minimal medium (Figs 3–7, panels B–D). Even under suppressing growth conditions, however, cell morphology was abnormal in several respects. First, cells of all three mutants were both moderately shorter and wider than wild-type (wt) cells, leading to a reduction in the average length/width ratio from 2.6 (wt) to 2.0 (Figs 3–7, Table 2). Second, while the accuracy of septal placement at midcell was not affected, the fraction of cells with a visible constriction was significantly higher in the three mutants compared with the wt control, indicating the constriction process takes longer to complete (Table 2). Third, mutant cells showed pronounced local expansions of their periplasmic space at sites of constriction, as inferred from fluorescence patterns created by Twin-arginine transport system (Tat)-targeted GFP (TTGFP). This version of GFP is routed to the periplasm via Tat, and is a useful soluble probe of periplasmic space (Bernhardt and de Boer, 2004; Mullineaux et al., 2006). As expected (Santini et al., 2001; Thomas et al., 2001; Bernhardt and de Boer, 2003; 2004), TTGFP created an even halo of fluorescence along the periphery of wt cells (Fig. 3A). In any of the three mutants, however, it additionally produced strong ring-like signals at sites of constriction (Fig. 3B–D). We previously observed such TTGFP ‘rings’ in another chain-forming mutant (envC) and take it to mean that invagination of the OM lags behind that of the IM, creating a relatively large periplasmic volume around the leading edge of cell constriction (Bernhardt and de Boer, 2004).
Fig. 3.
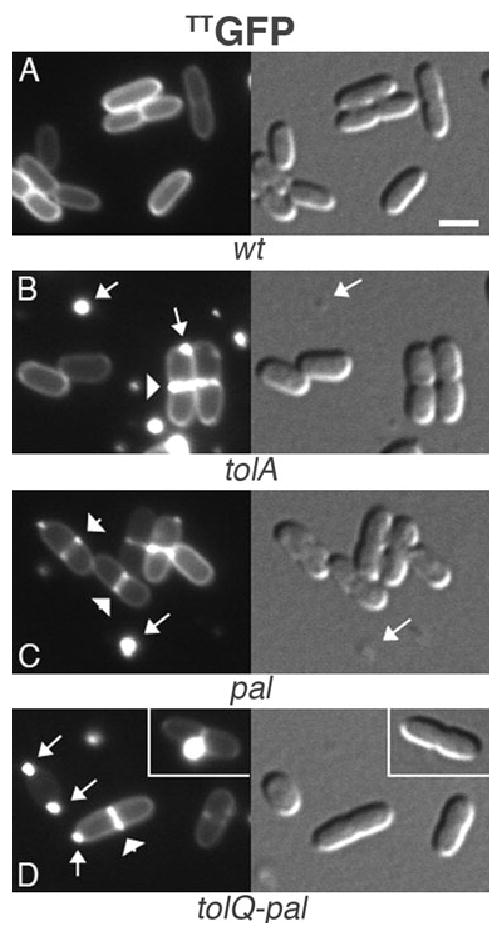
Increased periplasmic volume at the constriction site of tol-pal mutants, and release of OM vesicles. Periplasmic, Tat-targeted, GFP (TTGFP) in wt (A), tolA (B), pal (C) and tolQ-pal (D) cells. Note: (i) the squatness of mutant versus wt cells, (ii) the pronounced accumulation of TTGFP around constriction sites (some are indicated by arrowheads) in mutant cells versus the even peripheral distribution of TTGFP in wt cells, and (iii) the presence of fluorescence-filled blebs at the poles of mutant cells, and numerous free vesicles in the culture media. Arrows point to some of the cell-associated and free vesicles. The inset in D shows a prominent bleb associated with the constriction site. Strains used were TB28 (A), FB20229 (B), MG5 (C) and MG4 (D), each harbouring pTB6 [Plac∷TT_gfp_]. Cells were grown at 30°C in M9-glucose medium supplemented with 5 μM IPTG. Bar equals 2 μm. DIC images in this and following figures are shown immediately to the right, or below, the corresponding fluorescence images.
Fig. 7.
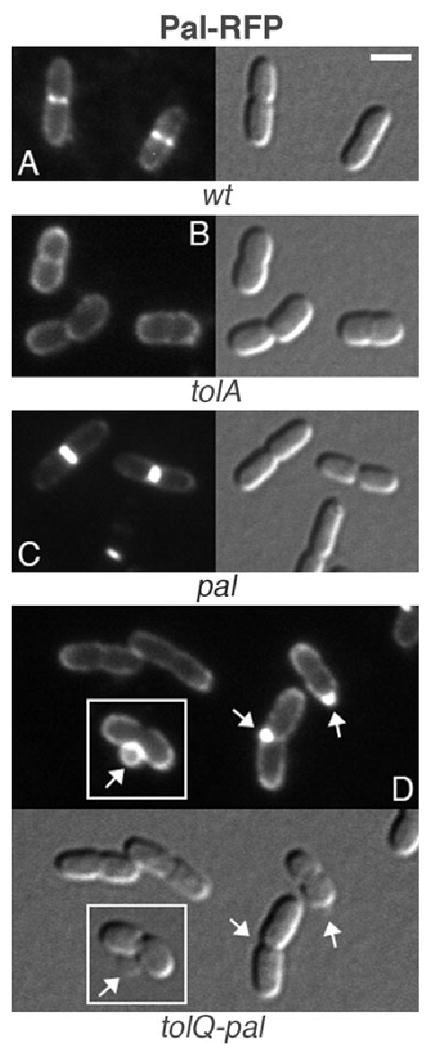
Localization of Pal–RFP in wt and mutant cells. Pal–RFP in wt (A), tolA (B), pal (C) and tolQ-pal (D) cells. Note the normal morphology of cells in C, and vesicle formation in D. The inset in D shows the formation of a large vesicle at the constriction site of a cell, and the absence of Pal–RFP in the lumen of the vesicle. Strains used were TB28 (A), FB20229 (B), MG5 (C) and MG4 (D), each harbouring pMG36 [Plac∷_pal-rfp_]. Cells were grown at 30°C in M9-glucose medium supplemented with 50 μM IPTG. Bar equals 2 μm.
Table 2.
Cellular parameters of wt and tol-pal mutant cells.
| Straina | TB28/pTB6 | FB20229/pTB6 | MG5/pTB6 | MG4/pTB6 | FB20229/pNP4 | MG5/pMG36 |
|---|---|---|---|---|---|---|
| Genotype | wt/Plac∷TT_gfp_ | tolA/Plac∷TT_gfp_ | pal/Plac∷TT_gfp_ | tolQ-pal/Plac∷TT_gfp_ | tolA/Plac∷gfp-tolA | pal/Plac∷pal-rfp |
| Number of cells | 343 | 381 | 230 | 290 | 367 | 266 |
| Width (SD) in μm | 1.33 (0.08) | 1.64 (0.11) | 1.58 (0.10) | 1.57 (0.12) | 1.42 (0.09) | 1.40 (0.08) |
| Length (SD) in μm | 3.51 (0.73) | 3.36 (0.85) | 3.12 (0.68) | 3.25 (0.73) | 3.60 (0.73) | 4.24 (0.94) |
| Length/width | 2.64 | 2.05 | 1.98 | 2.07 | 2.53 | 3.09 |
| % cells with single constr. | 43 | 60 | 86 | 68 | 44 | 78 |
| Constriction position (SD) | 0.49 (0.03) | 0.49 (0.03) | 0.48 (0.03) | 0.48 (0.03) | 0.48 (0.02) | 0.49 (0.03) |
| % cells w. multiple constr. | 0 | 3 | 1 | 1 | 0 | 0 |
| Number of OM blebs | 0 | 207 | 246 | 218 | ND | 2 |
| % cells with OM blebs | 0 | 44 | 81 | 54 | ND | <1 |
| % of blebs at constr. site | NA | 42 | 31 | 37 | ND | NA |
| % of blebs at polar caps | NA | 53 | 64 | 56 | ND | NA |
| % of blebs elsewhere | NA | 5 | 5 | 7 | ND | NA |
Finally, although the prolific shedding of OM vesicles is a known property of tol-pal mutants (Bernadac et al., 1998; Henry et al., 2004), the TTGFP probe allowed us to monitor the production of such vesicles for the first time in live cells. In contrast to wt cells (Fig. 3A), cells of all three mutants frequently appeared in the process of budding-off one or two prominent fluorescent vesicles, and their culture media contained numerous fluorescent vesicles already released (Fig. 3B–D). Notably, over 90% of all OM blebs readily scoreable by fluorescence microscopy (diameter > 300 nm) were associated with either the site of constriction or the cell poles (Table 2). These results suggested that OM blebbing in the tol-pal mutants is closely related to a defect in the division process.
Fluorescent versions of Tol–Pal components
Fusions from the ASKA collection (Kitagawa et al., 2005) of TolA, TolB, TolR and Pal carrying GFP at their C-termini were not useful for sublocalization studies (not shown). This was not unexpected because the C-termini of these four proteins reside in the periplasm and periplasmic GFP fails to become fluorescent after export by the Sec general secretory system (Feilmeier et al., 2000). Therefore, we constructed additional fusions allowing sublocalization of all five known components of the Tol–Pal complex in live cells (Fig. 1). GFPmut2 (Cormack et al., 1996) was fused to the cytoplasmic N- (TolR, TolA), or C-termini (TolQ) of the integral IM proteins of the complex. The wholly periplasmic TolB protein was visualized by exploiting the ability of Tat to export pre-folded fluorescent GFP (-fusions) (Santini et al., 2001; Thomas et al., 2001; Bernhardt and de Boer, 2003; 2004). To this end, the native Sec-specific export signal of TolB was substituted with TTGFP.
In contrast to Aequorea GFP, a monomeric derivative of Discosoma DsRed (Campbell et al., 2002) was recently shown capable of becoming fluorescent upon Sec-mediated export to the periplasm in both E. coli and Caulobacter crecentus (Chen et al., 2005). Hence, to sub-localize the OM lipoprotein Pal, we fused mCherry RFP, an improved variant of mRFP1 (Shaner et al., 2004), to its C-terminus. Fusions were expressed from a plasmid or lysogenic phage under control of the lac regulatory region (Table 1).
Tol–Pal mutants are hypersensitive to detergents (Davies and Reeves, 1975; Lazzaroni et al., 2002). To assess functionality of the GFP–TolA and Pal–RFP fusions, we tested their abilities to restore detergent resistance to FB20229 [_tolA_], MG5 [_pal_] or MG4 [_tolQ-pal_]. Plasmids pNP4 [Plac∷_gfp-tolA_] or pMG36 [Plac∷_pal-rfp_] were introduced into TB28 [wt] and the three mutant strains, and transformant cultures were spot-titered on LB agar containing either no or 0.5% SDS, and various concentrations (0, 5, 50 and 100 μM) of IPTG. Plasmid pNP4 [Plac∷_gfp-tolA_] conferred SDS resistance to FB20229 [_tolA_], and did so even when no inducer was added. As expected, however, it failed to rescue growth of MG5 [_pal_] and MG4 [_tolQ-pal_] at any concentration of IPTG (Fig. 2E, and not shown). Likewise, plasmid pMG36 [Plac∷_pal-rfp_] specifically allowed growth of MG5 [_pal_] in the presence of detergent, but only when IPTG was present at 50 μM or more (Fig. 2E, and not shown). The requirement for added inducer in this case is consistent with the relatively high abundance of Pal in wt cells (Cascales et al., 2002).
The GFP–TolA and Pal–RFP fusions also specifically corrected the chaining and shape phenotypes of tolA and pal cells respectively. Thus, plasmid pNP4 [Plac∷_gfp-tolA_] fully corrected the chaining phenotype of FB20229, and pMG36 [Plac∷_pal-rfp_] that of MG5, in low osmotic LB medium containing minimally 5 μM (pNP4) or 100 μM (pMG36) IPTG (Fig. 2B and D, and not shown). Moreover, GFP–TolA and Pal–RFP specifically suppressed the short and fat morphology of M9-grown FB20229 and MG5 cells respectively (Table 2).
We conclude that the GFP–TolA and Pal–RFP fusions retained function. Although we did not test GFP–TolQ, GFP–TolR and TTGFP–TolB in a similar manner, their localization patterns described below suggests that these fusions retained at least part of their respective Tol functions as well.
Tol–Pal proteins localize to the division site
Production of the five fluorescent fusions in wt strain TB28 revealed that each specifically accumulated at sites of cell constriction (Figs 4–8, panels A). Given that the Tol–Pal system forms _trans_-envelope complexes, and that tol-pal mutants suffer discordant invagination of the IM and OM during cell constriction (see above), this finding strongly supports a specific and direct role for the Tol–Pal proteins in the normal cell division process. As such, this finding increases the number of nonessential division proteins by five, and identifies Pal as the first OM-associated component of the division machinery in Gram-negative bacteria.
Fig. 4.
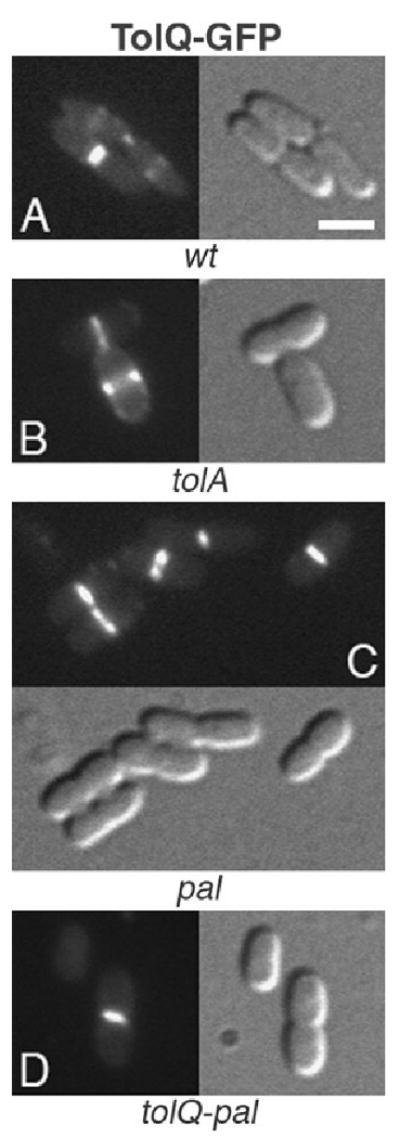
Localization of TolQ–GFP to the division site in wt and mutant cells. TolQ–GFP in wt (A), tolA (B), pal (C) and tolQ-pal (D) cells. Strains used were TB28 (A), FB20229 (B), MG5 (C) and MG4 (D), each harbouring pNP2 [Plac∷_tolQ-gfp_]. Cells were grown at 30°C in M9-glucose medium supplemented with 25 μM IPTG. Bar equals 2 μm.
Fig. 8.
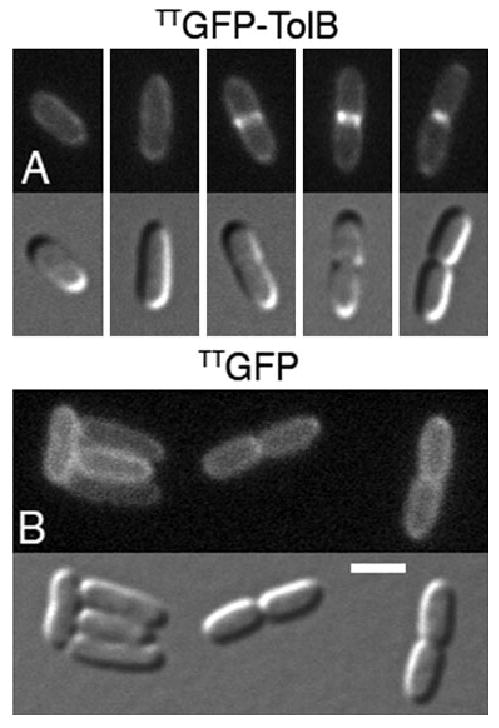
Localization of TTGFP–TolB to division sites. Panel A shows the accumulation of TTGFP–TolB at sites of constriction in wt cells. Cells from a single field were rearranged to highlight the localization pattern of the fusion during the division cycle. Panel B shows the localization of unfused TTGFP as a control. Note the absence of accumulation at constriction sites of TTGFP. Strain TB28 lysogenic for λNP7 [Plac∷TT_gfp-tolB_] (A) or λTB6 [Plac∷TT_gfp_] (B) was grown at 30°C in M9-glucose medium supplemented with 50 μM IPTG. Bar equals 2 μm.
As Tol–Pal complexes form via multiple interactions among its components (Lazzaroni et al., 2002; Cascales and Lloubes, 2004), the specific accumulation at the division site of one or more of the Tol–Pal proteins might be interdependent. To start exploring this possibility, we also monitored the localization patterns of TolQ–GFP, GFP–TolR, GFP–TolA and Pal–RFP in the tolA, pal and tolQ-pal mutant strains. The TTGFP–TolB fusion was not included in these experiments because the localized expansion of the periplasm at division sites in these mutants (Fig. 3) renders results with this fully periplasmic fusion difficult to interpret.
Interestingly, TolQ–GFP still associated with division sites in all three mutants (Fig. 4B–D), showing that TolQ recognizes these sites in the absence of the four other proteins. GFP–TolR did similarly not require TolA or Pal to associate with division sites (Fig. 5B and C). However, while still intact as judged by Western analyses (Fig. S1), the fusion appeared dispersed around the periphery of MG4 [_tolQ-pal_] cells (Fig. 5D), indicating that recruitment of TolR to division sites requires TolQ, and/or possibly TolB.
Fig. 5.
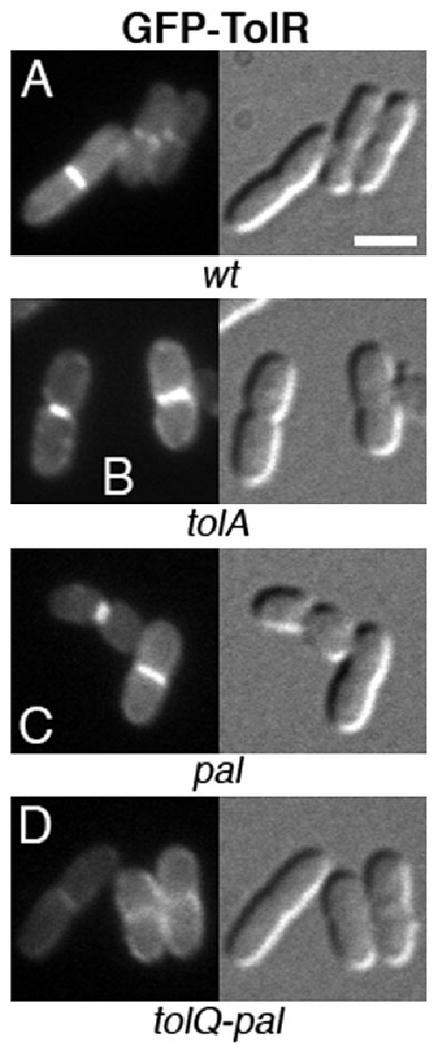
Localization of GFP–TolR in wt and mutant cells. GFP–TolR in wt (A), tolA (B), pal (C) and tolQ-pal (D) cells. Note the failure of the fusion to accumulate at constriction sites in cells that lack the other four Tol–Pal proteins (D). Strains used were TB28 (A), FB20229 (B), MG5 (C) and MG4 (D), each harbouring pNP3 [Plac∷_gfp-tolR_]. Cells were grown at 30°C in M9-glucose medium supplemented with 5 μM IPTG. Bar equals 2 μm.
As expected, GFP–TolA localized to division sites in tolA cells while correcting their detergent sensitivity, chaining and shape defects (Figs 2B and 6B). Like TolQ–GFP, moreover, GFP–TolA still localized to division sites in the absence of the other four proteins (Fig. 6C and D). Pal–RFP accumulated sharply at division sites in pal cells (Figs 2D and 7C), but failed to localize in the other two mutants. Though intact (Fig. S1), the Pal–RFP fusion appeared dispersed along the periphery of tolA and tolQ-pal cells, indicating its interaction with TolA (Cascales et al., 2000; Cascales and Lloubes, 2004) is important for its accumulation at sites of constriction (Fig. 7B and D). In addition, and consistent with its localization in the OM, Pal–RFP also decorated the OM blebs and vesicles produced by these cells (Fig. 7D, and not shown). No such blebbing was observed in the wt or complemented pal cells (Fig. 7A and C, Table 2, and not shown). Thus, as might be expected, the fusion not only suppressed cell chaining (Fig. 2D), SDS sensitivity (Fig. 2E) and shape alteration (Fig. 7C, Table 2), but also OM blebbing in the pal mutant.
Fig. 6.
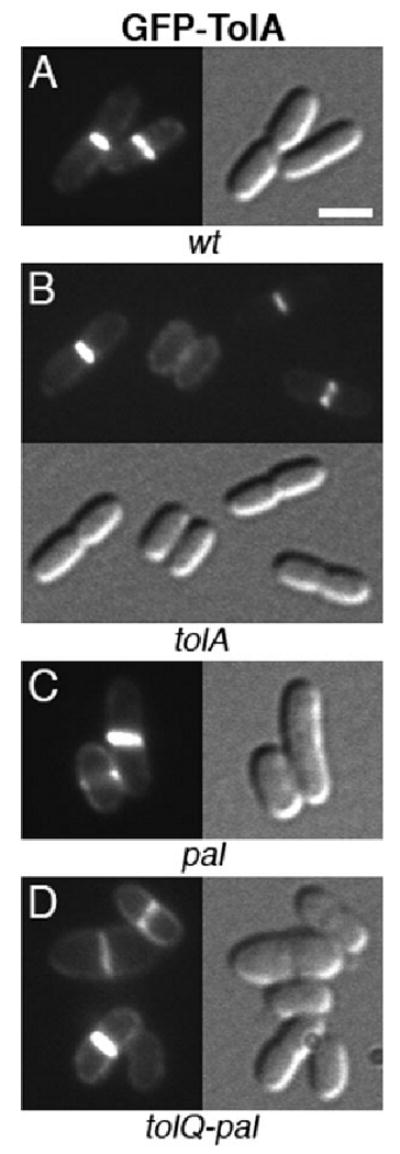
Localization of GFP–TolA to the division site in wt and mutant cells. GFP–TolA in wt (A), tolA (B), pal (C) and tolQ-pal (D) cells. Note the normal morphology of cells in B. Strains used were TB28 (A), FB20229 (B), MG5 (C) and MG4 (D), each harbouring pNP4 [Plac∷_gfp-tolA_]. Cells were grown at 30°C in M9-glucose medium supplemented with 5 μM IPTG. Bar equals 2 μm.
Localization of Tol–Pal complexes to the division site requires FtsN
The finding that TolQ and TolA localized to division sites in the absence of each other, and of the other Tol–Pal components, showed that each is independently attracted by some non-Tol/Pal feature at these sites. Candidates for recruitment of the Tol–Pal complex to the division site include any of the known division proteins that make up the SR. Polymerization of the FtsZ protein underneath the IM is an initiating step in assembly of the SR apparatus at the prospective site of division. We determined that localization of functional GFP–TolA requires this step by observing the fusion in filaments expressing the FtsZ polymerization antagonist SfiA(SulA) (Mukherjee et al., 1998; Trusca et al., 1998; Cordell et al., 2003). GFP–TolA accumulated normally at division sites in cells of strain TB28/pNP4/pJE80 [wt/Plac∷gfp-tolA/PBAD∷_sfiA_] growing in the presence of glucose (SfiA−) (Fig. 9A). Division ceased in the presence of arabinose (SfiA+), however, and GFP–TolA dispersed along the periphery of the resulting filaments (Fig. 9B).
Fig. 9.
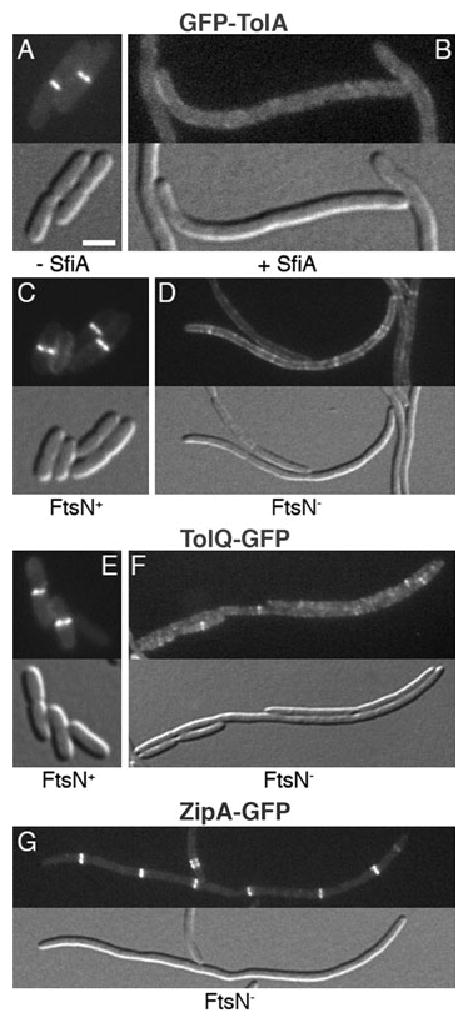
Recruitment of Tol–Pal to division sites requires FtsN. Panels A and B show the dispersal of GFP–TolA in SfiA(SulA)-induced filaments of strain TB28/pJE80/pNP4 [wt/PBAD∷sfiA/Plac∷gfp-tolA_]. Cells were grown in LB supplemented with 25 μM IPTG and either 0.1% glucose (A), or 0.005% arabinose (B). Panels C–G illustrate the localization of GFP–TolA (C and D), TolQ–GFP (E and F) and ZipA–GFP (G) in FtsN+ cells (C and E) and FtsN-depleted filaments (D, F and G). Note the sharp rings in C, E and G versus the peripherally dispersed signals in D and F. Fusions to TolR, TolB and Pal similarly failed to localize to rings in FtsN− filaments (not shown). Strain CH34/pMG20 [Δ_ftsN/PBAD∷TT_bfp_-ED_ftsN_] carrying pNP4 [Plac∷_gfp-tolA_] (C and D), pNP2 [Plac∷_tolQ-gfp_] (E and F), or lysogenic for λCH151 [Plac∷_zipA-gfp_] (G), was grown in M9-maltose (0.2%) supplemented with 5 (C and D), 25 (G), or 50 (E and F) μM IPTG, and with either 0.1% arabinose (C and E) or 0.1% glucose (D, F and G). Bar equals 2 μm.
A large fraction (85% or more) of wt cells that showed accumulation of any of the Tol–Pal fusions at midcell also showed a corresponding constriction, suggesting that Tol–Pal complexes join the division apparatus at a relatively late stage. In the SR assembly pathway, FtsN is the last known essential division protein recruited. It is required for the onset of visible cell constriction and joins the SR at, or just prior to, this stage (Addinall et al., 1997; Chen and Beckwith, 2001; Wissel and Weiss, 2004). To determine if localization of the Tol–Pal proteins depends on FtsN, we used an FtsN-depletion strain, CH34/pMG20 [Δ_ftsN_/PBAD∷TT_bfp_-E_ftsN_], which lacks chromosomal ftsN and is rescued by expression of a Tat-targeted periplasmic fusion containing the essential periplasmic domain of FtsN. When grown in the presence of arabinose (FtsN+), CH34/pNP4/pMG20 cells divided normally and GFP–TolA accumulated normally at division sites (Fig. 9C). Cells growing in the presence of glucose (FtsN−) formed filaments as expected, and these often still contained some constrictions. However, GFP–TolA notably failed to localize sharply in these filaments. Rather, the fusion dispersed along the periphery, in addition to forming some weak bands and/or spots along the length of the filaments (Fig. 9D). We similarly tested the localization of TolQ–GFP, GFP–TolR, TTGFP–TolB and Pal–RFP in filaments of CH34/pMG20. None of the fusions localized to rings (Fig. 9F, and not shown). The ZipA protein is an early SR component whose recruitment to the ring does not require FtsN (Hale and de Boer, 1999; 2002; Liu et al., 1999). Accordingly, and in contrast to the Tol–Pal fusions, a ZipA–GFP fusion localized sharply to regularly spaced rings in FtsN-depleted CH34(λCH151)/pMG20 filaments (Fig. 9G).
We conclude that accumulation of Tol–Pal complexes at the division site requires FtsN activity.
Discussion
It has long been unclear what the physiological role(s) of the Tol–Pal system is (Lazdunski et al., 1998; 2002; Bouveret et al., 2002). We discovered that the Tol–Pal proteins specifically localize to the site of cell constriction in E. coli, and propose that the _trans_-envelope Tol–Pal system constitutes a subcomplex of the division machinery in Gram-negative bacteria that is specifically employed to ensure proper invagination of the OM.
Such a role of the Tol–Pal proteins in division is now supported by their localization, by the phenotypes of tol-pal mutant cells, and by the genetic and biochemical evidence that the proteins form complexes that span the envelope (see above) (Lazdunski et al., 1998; 2002; Bouveret et al., 2002). It was recognized early on that the Tol–Pal proteins play some role in OM integrity (Nagel de Zwaig and Luria, 1967; Bernstein et al., 1972; Lazzaroni and Portalier, 1992), and the release of OM vesicles by mutants indicated a failure to properly connect the OM with the two other layers (Bernadac et al., 1998). Our results suggest that tol-pal mutants more specifically fail to properly connect the OM to the other two envelope layers during cell constriction. Localization of TTGFP revealed a pronounced expansion of the periplasmic space at constriction sites of live mutant cells. In addition, a large fraction (> 44%, Table 2) of these cells displayed readily visible (> 300 nm) TTGFP-filled OM blebs. Over 90% of these blebs localized to sites of constriction or polar caps, indicating that a substantial fraction of OM and periplasm shed by tol-pal mutants is released from these sites. It is possible that most or all blebs initially form at the division site during constriction and then are inherited by new poles upon completion of the process. Alternatively, some blebs may form at poles after sister cells have already separated. Negative stain electron microscopy images of tol-pal mutant cells in a previous study showed the presence of relatively small (20–200 nm) OM blebs/vesicles on the surface of cells, but failed to reveal any preferential location of these structures (Bernadac et al., 1998). The reason for this discrepancy is unclear. As noted before with lpp mutants (Fung et al., 1978), however, relatively large septal/polar OM blebs, such as those scored in this study (> 300 nm), are fragile and may not have survived sample preparation.
A failure to properly connect the OM with the IM and/or PG layers during constriction is also consistent with the chaining phenotype of tol-pal mutants (Meury and Devilliers, 1999; Heilpern and Waldor, 2000; Llamas et al., 2000; Dubuisson et al., 2005). This phenotype was first described for tolA mutants of E. coli growing in medium of low osmolarity or high ionic strength (Meury and Devilliers, 1999). We found that E. coli pal and tolQ-pal mutants behave identically to tolA mutants in this regard, which is consistent with results obtained in other Gram-negative species (Heilpern and Waldor, 2000; Llamas et al., 2000; Dubuisson et al., 2005). A defect in septal murein separation can be a primary cause of delayed OM invagination and cell chaining (Rodolakis et al., 1973; Heidrich et al., 2001; 2002; Bernhardt and de Boer, 2004). However, such a defect is not apparent in thin-section transmission electron microscopy images of tol-pal mutants (Bernadac et al., 1998; Meury and Devilliers, 1999; Llamas et al., 2000), suggesting that their primary division defect lies in the inability of the OM to keep up with the invagination of the other two layers during constriction.
A straightforward interpretation of the results is that Tol–Pal stimulates OM invagination during cell constriction by tethering the OM to the inward moving IM and PG layers. This is likely accomplished via multiple linkages involving the Pal lipoprotein. First, Pal binds the peptide moieties of PG, establishing non-covalent OM–PG links (Mizuno, 1981; Cascales et al., 2002; Parsons et al., 2006). Second, Pal binds the C-terminus of TolA in a pmf-, and TolQ/R-dependent fashion, establishing OM–IM linkages that cross the PG meshwork (Cascales et al., 2000; 2001; Cascales and Lloubes, 2004). Third, TolB can bind TolA via its N-terminal domain (Dubuisson et al., 2002; Walburger et al., 2002) and Pal via its C-terminal one (Ray et al., 2000). If TolB were able to engage both partners simultaneously, this might create a second type IM–OM bridge. However, a tripartite Pal–TolB–TolA complex has not yet been observed in vivo or in vitro (Walburger et al., 2002). This could be due to technical issues, or it might mean that such a complex is not made or short-lived.
Our images reveal that the Tol–Pal proteins localize dynamically during the cell cycle in a fashion very similar to the previously studied non-cytoplasmic SR proteins (Errington et al., 2003; Aarsman et al., 2005; Goehring and Beckwith, 2005; Margolin, 2005; Vicente et al., 2006). The proteins appear dispersed along the entire cell envelope in young non-constricting cells. They then accumulate in a ring at the division site near the time of constriction initiation. The ring remains at/near the leading edge of the invaginating envelope as constriction progresses, and the proteins disperse again after cell separation is complete.
As it seems impossible for PG-bound/trapped complexes to move in this fashion, these Tol–Pal dynamics indicate that the interactions of Pal with PG and with TolA must be readily reversible in vivo. In this regard, it is interesting that the Pal–PG and Pal–TolB interactions are mutually exclusive (Bouveret et al., 1995; 1999; Clavel et al., 1998; Cascales and Lloubes, 2004). Therefore, it is tempting to speculate that TolB plays a role in reversing Pal–PG interactions to permit lateral movement of the lipoprotein in the OM. TolB might similarly play a role in breaking up the Pal–TolA interaction, which would permit lateral displacement of the lipoprotein in the OM as well as that of the TolQRA subcomplex in the IM.
We envision a mechanism for Tol/Pal-mediated OM invagination as depicted in Fig. 10. As the core SR begins to constrict, it directs synthesis of septal murein just behind the inward moving IM, and Tol–Pal proteins move from other sites in the cell to the SR (Fig. 10A). As soon as murein hydrolases split the septal PG from the OM-proximal side into two separate mesh works, energized TolA molecules reach through the freshly created meshes to grab hold of free Pal molecules in the OM (Fig. 10B). After a Pal partner is caught, it is pulled inwards, drawing the OM over the PG and IM layers (Fig. 10C). Provided that the Tol–Pal complexes are physically associated with cytoplasmic and/or IM components of the core SR, the inward pull could be generated by the contracting SR itself. However, it is tempting to speculate that the Tol proteins play a more active role in drawing in Pal. For example, TolA could act like a chameleon's tongue that in its energized state is in an elongated conformation allowing it to reach for Pal over some distance (Fig. 10B and E). After catching a Pal partner, it may then snap back to a less extended conformation and pull Pal inwards (Fig. 10C and F). The idea of TolA as a retractable grabbing device is attractive because: (i) it is consistent with its energy requirement for engaging Pal (Cascales et al., 2000; 2001; Germon et al., 2001), (ii) it allows for the generation of a pulling motion independently of the core SR (see below), and (iii) it renders it easy to picture why colicins and phages would hijack the device to be drawn through the periplasm towards the IM (Bouveret et al., 2002; Cao and Klebba, 2002; Lazzaroni et al., 2002).
Fig. 10.
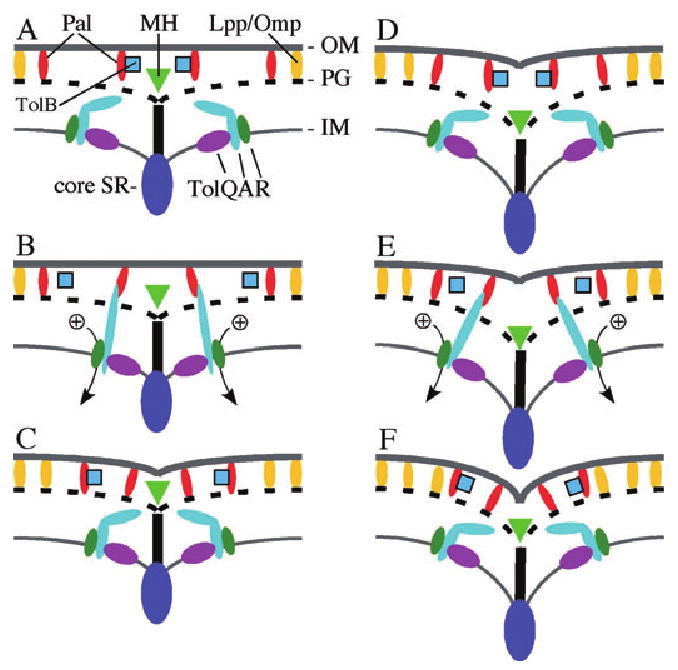
Model of Tol–Pal action in OM invagination during cell constriction. Panels represent a longitudinal section through the SR apparatus at the site of constriction. Indicated are the core machinery (core SR), representing all essential division proteins in the SR (dark-blue oval), the periplasmic murein hydrolases responsible for splitting septal murein in separate PG layers (green triangle), and general OM (lipo)proteins, such as Lpp and OmpA, with affinity for PG (yellow ovals). Superimposed are the IM-associated TolQ (purple ovals), TolR (green ovals) and TolA (straight or kinked light-blue ovals) proteins, the periplasmic TolB protein (blue squares), and the Pal OM lipoprotein (red ovals). The results of this study indicate that Pal–PG and Pal–TolA interactions must be readily reversible in vivo. Panels A–C represent a TolA–Pal engagement–disengagement cycle near the start of OM invagination, and D–F represent a cycle when constriction has progressed further. Ion potential over the IM is converted by TolQ and R to allow TolA to engage Pal (Cascales et al., 2000; 2001; Germon et al., 2001). It is proposed that TolA acts as a grabbing device that in its energized form extends through meshes of newly split murein to reach for free Pal in the OM (B and E). Upon catching a Pal partner, TolA snaps back, drawing Pal inwards, and then disengages, allowing Pal to engage the PG layer (C and F). TolA repeats this action as the TolQRA subcomplexes in the IM move along with the constricting core machinery (D–F). The Pal–PG interaction is also transient, and TolB may play a role in dislodging Pal to be reused in a subsequent TolA–Pal interaction event. The resulting waves of Tol–Pal mediated OM–PG and OM–IM connections at/near the core SR allow other OM (lipo)proteins to secure the OM to the PG in a more permanent manner. See the text for additional discussion.
As the SR and murein hydrolases generate freshly split murein adjacent to the just-established TolA–Pal bridges, the proteins disengage again (Fig. 10C and D), and TolA reconnects with free Pal through the newer meshes more proximal to the core SR (Fig. 10E). Cycles of Tol–Pal binding and unbinding continue as the proteins follow the core SR until the OM fuses at the tip of the new cell poles. Even though each TolA–Pal bridge is transient, the wave of interactions at/near the constricting SR ensures that the OM invaginates co-ordinately with the other two layers. In addition, by maintaining the OM and PG in close proximity at/near the SR, the transient OM–IM bridges would likely increase the rate by which other PG-binding OM (lipo)proteins, such as Lpp and OmpA, can secure the OM to the PG layer at the nascent polar caps more permanently. It is intriguing in this regard that both Lpp and OmpA can be specifically cross-linked in vivo with Pal and TolB (Palva, 1979; Clavel et al., 1998; Cascales et al., 2002; Cascales and Lloubes, 2004). Perhaps the Pal and/or TolB protein helps to recruit free Lpp and/or OmpA to form more permanent connections, following the wave of TolA–Pal interactions.
Even though the results of this study support a specific role for Tol–Pal in OM invagination during cell fission, the system is not essential. OM invagination may be delayed in cells lacking an intact Tol–Pal system, but they do manage to complete division in a time frame that is dependent on the osmolarity and/or ionic strength of the medium. Then how does the OM eventually invaginate in these cells? The simplest possibility is that other PG-binding OM (lipo)proteins are responsible. Perhaps additional such proteins accumulate specifically at the SR to help connect the OM to the PG during constriction. Alternatively, generally distributed OM (lipo)proteins such as Lpp and OmpA (Hiemstra et al., 1986; 1987; Lai et al., 2004) may be able to partially compensate for the absence of SR-specific proteins. By establishing new stable OM–PG contacts next to existing ones in the cylindrical portion of the cell, the affinity of such proteins for PG might eventually connect the OM to the PG over the entire polar cap. This process might be too slow to support a well-coordinated invagination of the OM, leading to slow OM invagination and OM blebbing at the division site in the absence of Tol–Pal. Conversely, cells lacking Lpp might be prone to OM blebbing at the division site (Weigand et al., 1976; Fung et al., 1978) because the wave of transient Tol–Pal connections at the SR is insufficiently followed up by more permanent OM–PG linkages.
Defective OM invagination, cell chaining, and the formation of large OM blebs at constriction sites and cell poles also occurred in lkyD mutants of Salmonella enterica typhimurium (Weigand et al., 1976; Fung et al., 1978; MacAlister et al., 1987). Unpublished mapping data render it highly unlikely that lkyD is allelic with any of the tol-pal genes (J. Fung and L. Rothfield, pers. comm.). As lkyD might have activities that are partially redundant with that of the Tol–Pal system, it will be interesting to determine its precise identity.
To prevent cell lysis, there must be a certain lag between the synthesis of septal PG at the ingrowing edge and its processing into separate daughter layers from the OM-proximal end. The extent of this lag will determine a minimal distance between the two membranes at the constricting SR. Although it is not clear whether the intermembrane distance at the SR in E. coli is much different from that elsewhere in the cell (Weigand et al., 1976; Fung et al., 1978; Rothfield et al., 1986; MacAlister et al., 1987; Bi and Lutkenhaus, 1991; Lutkenhaus, 1993), this distance is enlarged in deeply constricted cells of Caulobacter crecentus (Judd et al., 2005). We suggest two non-exclusive ways in which the Tol–Pal system might compensate for such an increased distance. One is that energized TolA may be able to reach out for Pal sufficiently far to overcome a modest increase in intermembrane distance. The other is that the Tol–Pal proteins may form the IM–OM bridges a compensatory distance away from the core SR. In this case, ‘rings’ of dynamic Tol–Pal complexes that are not necessarily physically connected to the core SR might flank the core.
Additional studies will be needed to test these possibilities, as well as to shed light on the related issues of what attracts the Tol–Pal proteins to the division site, and keeps them there during constriction? It is notable that both TolQ and TolA accumulate at the site independently of each other and of the other Tol–Pal partners. Localization of all five proteins depends on active FtsN; however, and it is conceivable that TolQ and TolA are each directly recruited by FtsN. Alternatively, the Tol–Pal proteins may not contact any of the core SR proteins directly, but recognize septal murein or some other product of the constricting SR.
The results of this study may also have interesting implications for the mechanisms by which the colicins and phages that use the Tol proteins infect cells. For example, the high local density of Tol proteins at division sites may render these also the preferred sites of toxin/phage entry. Whether this is so, and whether such localized entry has any mechanistic implications for the infection processes remains to be seen.
Experimental procedures
E. coli plasmids, phages and strains
The most relevant plasmids, phages and strains used in this study are listed in Table 1, and depicted in Fig. 1.
Plasmids pMLB1113 (de Boer et al., 1989), pDR120 (Hale and de Boer, 1999), pKD13 and pKD46 (Datsenko and Wanner, 2000), pmRFP1 (Campbell et al., 2002), pJE80 (Johnson et al., 2002), pCH151 (Bernhardt and de Boer, 2003), pTB6 (Bernhardt and de Boer, 2004) and pmCherry (Shaner et al., 2004), and phages λCH151 and λTB6 (Bernhardt and de Boer, 2004) were described previously.
To create pNP2 [Plac∷_tolQ-gfpmut2_], we performed a PCR with primers 5′-CCTGTCTAGAAATGAAGCCTCGTGCGCTTCC-3′ and 5′-CCTGCTCGAGCCCCTTGTTGCTCTCGCTA-3′. The product was digested with XbaI and XhoI (sites underlined), and the 744 bp fragment was used to replace the 1025 bp XbaI-XhoI fragment of pCH151 [Plac∷_zipA-gfpmut2_].
For pNP3 [Plac∷_gfpmut2-t-tolR_], tolR was amplified with primers 5′-GTACGGATCCATGGCCAGAGCGCGTGG-3′ and 5′-CGATGTCGACTTAGATAGGCTGCGTCATTAAACCAAC-3′. The product was treated with BamHI and SalI (sites underlined), and the 434 bp fragment was used to replace the 1163 bp BamHI-SalI fragment of pDR120 [Plac∷_gfpmut2-t-ftsZ_].
For pNP4 [Plac∷_gfpmut2-t-tolA_], tolA was amplified with primers 5′-CCTGGGATCCGTGTCAAAGGCAACCGAACAAAAC-3′ and 5′-CGACGTCGACTTACGGTTTGAAGTCCAATGGCGCG-3′. The product was treated with BamHI and SalI (sites underlined), and the 1272 bp fragment was used to replace the 1163 bp BamHI-SalI fragment of pDR120 [Plac∷_gfpmut2-t-ftsZ_].
To obtain pNP7 [Plac∷SS_torA-gfpmut2-t-tolB_], we performed a PCR with primers 5′-TCGAGAATTCGAAGTCCGCATT GTGATCGACAGC-3′ and 5′-GTCGAAGCTTTTATCACAGATACGGCGACCAGG-3′. The product was cut with EcoRI and HindIII (sites underlined), and the 1239 bp fragment was used to replace the 30 bp EcoRI-HindIII fragment of pTB6 [Plac∷SS_torA-gfpmut2-t_].
Phage λNP7 was obtained by crossing λNT5 with pNP7 as described (de Boer et al., 1989). This lysogenic phage encodes a fusion in which the signal sequence of pre-TolB (residues 1–21) is replaced by a signal peptide (SSTorA) that is specific for the Tat system, fused to GFP and the T7.tag peptide.
Plasmid pMG36 [Plac∷_pal-rfp_] was obtained after several steps. Using pmRFP1 as template, _mrfp_1 was amplified with primers 5′-CCTGCTCGAGATGGCCTCCTCCGAGGACG-3′ and 5′-CGCGCGAAGCTTTTAGGCGCCGGTGGAGTGG-3′. The product was digested with XhoI and HindIII (sites underlined), and the 684 bp fragment was used to replace the 753 bp XhoI-HindIII fragment of pCH151, yielding pTB64 [Plac∷_zipA-mrfp_1]. pal was amplified using primers 5′-TCCCTCTAGACCCTGCCTGGTCGCCGTATCTGTG-3′ and 5′-AATGCTCGAGGTAAACCAGTACCGCACGACGGTTTTTGG-3′. The product was treated with XbaI and XhoI (sites underlined), and the 584 bp fragment was used to replace the 1025 bp XbaI-XhoI fragment of pTB64, resulting in pMG10 [Plac∷_pal-mrfp_1]. Next, pCherry was used as template to amplify mCherry rfp with primers 5′-CCGGGATCCCCCCGCTGAATTCATGGTGAGCAAGGGCGAGG-3′ and 5′-CCCAAGCTTGTCGACTTACTTGTACAGCTCGTCC-3′. The product was cut with BamHI and HindIII (sites underlined), and the 736 bp fragment was used to replace the 748 bp BamHI-HindIII fragment of pCH151, yielding pMG30 [Plac∷_zipA-rfp_]. Finally, the 684 bp XhoI-HindIII fragment of pMG10 was replaced with the 741 bp XhoI-HindIII fragment of pMG30, resulting in pMG36.
Plasmid pMLB1113ΔH was obtained by digestion of the vector plasmid pMLB1113 with HindIII, incubation with Klenow enzyme and dNTP's, and recircularization.
Construction of the FtsN-depletion strain CH34/pMG20 will be detailed elsewhere. Strains MG4 and MG5 were constructed by λ Red recombineering (Datsenko and Wanner, 2000; Yu et al., 2000). Plasmid pKD13 was used as a template for amplification of a (frt aph frt) cassette flanked by suitable tolQ and/or pal sequences. Primer sets (chromosomal sequences are underlined) used were 5′-CGTGCGCTTCCCAAGTCTATTGTCGCGGAGTTTAAGCAGTAATTCCGGGGATCCGTCGACC-3′ and 5′-CTGCTCATGCAATTCTCTTAGTAAACCAGTACCGCACGACGGTGTAGGCTGGAGCTGCTTCG-3′ for tolQ-pal <> aph (MG4), and 5′-AGGTCAAATTCCCTGCCTGGTCGCCGTATCTGTGATAATAATTCCGGGGATCCGTCGACC-3′ and 5′-CTGCTCATGCAATTCTCTTAGTAAACCAGTACCGCACGACGGTGTAGGCTGGAGCTGCTTCG-3′ for pal <> aph (MG5).
The linear tolQ-pal and pal knockout fragments were recombined with the chromosome of strain TB28 carrying plasmid pKD46. Recombinants were cured of the plasmid by growth at 37°C (Datsenko and Wanner, 2000).
Growth conditions, microscopy and image analyses
Unless mentioned otherwise, cells were grown at 30°C in standard (Sambrook et al., 1989) LB medium (containing 1% NaCl), or M9 minimal medium (containing MgSO4 at a non-limiting concentration of 2 mM) supplemented with 0.2% casamino acids and 0.2% of either glucose or maltose. When appropriate, medium was supplemented with 50 μg ml−1 ampicillin (Amp), 20 μg ml−1 kanamycin (Kan) and/or 25 μg ml−1 chloramphenicol (Cam). In addition, medium was sometimes supplemented with glucose, arabinose, IPTG and/or SDS, as indicated.
For cell imaging experiments, cultures were grown to density overnight in M9 medium supplemented with 0.2% maltose and 0.01% arabinose (CH34/pMG20 derivatives), or in LB supplemented with 0.1% or 0.2% glucose (all other strains). Cultures were diluted 1:50 or 1:100 in the indicated medium, and grown to an OD600 between 0.63 and 0.71. Cells were applied to microscope slides coated with a thin layer of solidified agarose (1% in water), covered with a coverslip, and imaged immediately. Fluorescence and differential interference contrast (DIC) microscopy were performed essentially as described (Johnson et al., 2002), except that Pal–RFP was visualized using a rhodamine filter set (565 nm dichroic mirror, 513–558 nm excitation filter and a 590 nm long pass barrier filter).
Cell axes, the presence and positions of constrictions, fluorescent rings, and/or membrane blebs were measured using Object-Image (Vischer et al., 1994). Only fluorescent blebs with a diameter of 335 nm (five pixels) or larger were scored.
Supplementary Material
s1
Acknowledgments
We thank Fred Blattner, Roger Tsien and Patrick Viollier for materials, Yuko Kurita and Cynthia Hale for technical assistance, Larry Rothfield for communication of unpublished results on lkyD, and Tom Bernhardt, Arne Rietsch and Patrick Viollier for comments on the manuscript. This work was supported by grants from the NIH (GM57059) and the Human Frontiers Science Program (RGP0001-C103) (to P.dB.), by NIH training Grant T32 G07250 (N.P.), and by MEXT and JSPS KAKENHI grants and a Grant-in-Aid for Scientific Research on Priority Areas or for Scientific Research (to H.N.).
Footnotes
The following supplementary material is available for this article online:
Fig. S1. No excessive degradation of GFP–TolR or Pal–RFP in tolA or tolQ-pal cells.
This material is available as part of the online article from http://www.blackwell-synergy.com
References
- Aarsman ME, Piette A, Fraipont C, Vinkenvleugel TM, Nguyen-Disteche M, den Blaauwen T. Maturation of the Escherichia coli divisome occurs in two steps. Mol Microbiol. 2005;55:1631–1645. doi: 10.1111/j.1365-2958.2005.04502.x. [DOI] [PubMed] [Google Scholar]
- Addinall SG, Holland B. The tubulin ancestor, FtsZ, draughtsman, designer and driving force for bacterial cytokinesis. J Mol Biol. 2002;318:219–236. doi: 10.1016/S0022-2836(02)00024-4. [DOI] [PubMed] [Google Scholar]
- Addinall SG, Cao C, Lutkenhaus J. FtsN, a late recruit to the septum in Escherichia coli. Mol Microbiol. 1997;25:303–309. doi: 10.1046/j.1365-2958.1997.4641833.x. [DOI] [PubMed] [Google Scholar]
- Benedetti H, Lazdunski C, Lloubes R. Protein import into Escherichia coli: colicins A and E1 interact with a component of their translocation system. EMBO J. 1991;10:1989–1995. doi: 10.1002/j.1460-2075.1991.tb07728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernadac A, Gavioli M, Lazzaroni JC, Raina S, Lloubes R. Escherichia coli tol-pal mutants form outer membrane vesicles. J Bacteriol. 1998;180:4872–4878. doi: 10.1128/jb.180.18.4872-4878.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PAJ. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol Microbiol. 2003;48:1171–1182. doi: 10.1046/j.1365-2958.2003.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PA. Screening for synthetic lethal mutants in Escherichia coli and identification of EnvC (YibP) as a periplasmic septal ring factor with murein hydrolase activity. Mol Microbiol. 2004;52:1255–1269. doi: 10.1111/j.1365-2958.2004.04063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein A, Rolfe B, Onodera K. Pleiotropic properties and genetic organization of the tolA,B locus of Escherichia coli K-12. J Bacteriol. 1972;112:74–83. doi: 10.1128/jb.112.1.74-83.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi E, Lutkenhaus J. FtsZ ring structure associated with division in Escherichia coli. Nature. 1991;354:161–164. doi: 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- de Boer PAJ, Crossley RE, Rothfield LI. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell. 1989;56:641–649. doi: 10.1016/0092-8674(89)90586-2. [DOI] [PubMed] [Google Scholar]
- Bouveret E, Derouiche R, Rigal A, Lloubes R, Lazdunski C, Benedetti H. Peptidoglycan-associated lipoprotein–TolB interaction. A possible key to explaining the formation of contact sites between the inner and outer membranes of Escherichia coli. J Biol Chem. 1995;270:11071–11077. doi: 10.1074/jbc.270.19.11071. [DOI] [PubMed] [Google Scholar]
- Bouveret E, Benedetti H, Rigal A, Loret E, Lazdunski C. In vitro characterization of peptidoglycan-associated lipoprotein (PAL)–peptidoglycan and PAL–TolB interactions. J Bacteriol. 1999;181:6306–6311. doi: 10.1128/jb.181.20.6306-6311.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouveret E, Journet L, Walburger A, Cascales E, Benedetti H, Lloubes R. Analysis of the Escherichia coli Tol-Pal and TonB systems by periplasmic production of Tol, TonB, colicin, or phage capsid soluble domains. Biochimie. 2002;84:413–421. doi: 10.1016/s0300-9084(02)01423-2. [DOI] [PubMed] [Google Scholar]
- Braun V, Herrmann C. Evolutionary relationship of uptake systems for biopolymers in Escherichia coli: cross-complementation between the TonB-ExbB-ExbD and the TolA-TolQ-TolR proteins. Mol Microbiol. 1993;8:261–268. doi: 10.1111/j.1365-2958.1993.tb01570.x. [DOI] [PubMed] [Google Scholar]
- Campbell RE, Tour O, Palmer AE, Steinbach PA, Baird GS, Zacharias DA, Tsien RY. A monomeric red fluorescent protein. Proc Natl Acad Sci USA. 2002;99:7877–7882. doi: 10.1073/pnas.082243699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Klebba PE. Mechanisms of colicin binding and transport through outer membrane porins. Biochimie. 2002;84:399–412. doi: 10.1016/s0300-9084(02)01455-4. [DOI] [PubMed] [Google Scholar]
- Cascales E, Lloubes R. Deletion analyses of the peptidoglycan-associated lipoprotein Pal reveals three independent binding sequences including a TolA box. Mol Microbiol. 2004;51:873–885. doi: 10.1046/j.1365-2958.2003.03881.x. [DOI] [PubMed] [Google Scholar]
- Cascales E, Gavioli M, Sturgis JN, Lloubes R. Proton motive force drives the interaction of the inner membrane TolA and outer membrane pal proteins in Escherichia coli. Mol Microbiol. 2000;38:904–915. doi: 10.1046/j.1365-2958.2000.02190.x. [DOI] [PubMed] [Google Scholar]
- Cascales E, Lloubes R, Sturgis JN. The TolQ-TolR proteins energize TolA and share homologies with the flagellar motor proteins MotA-MotB. Mol Microbiol. 2001;42:795–807. doi: 10.1046/j.1365-2958.2001.02673.x. [DOI] [PubMed] [Google Scholar]
- Cascales E, Bernadac A, Gavioli M, Lazzaroni JC, Lloubes R. Pal lipoprotein of Escherichia coli plays a major role in outer membrane integrity. J Bacteriol. 2002;184:754–759. doi: 10.1128/JB.184.3.754-759.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JC, Beckwith J. FtsQ, FtsL and FtsI require FtsK, but not FtsN, for co-localization with FtsZ during Escherichia coli cell division. Mol Microbiol. 2001;42:395–413. doi: 10.1046/j.1365-2958.2001.02640.x. [DOI] [PubMed] [Google Scholar]
- Chen JC, Viollier PH, Shapiro L. A membrane metalloprotease participates in the sequential degradation of a Caulobacter polarity determinant. Mol Microbiol. 2005;55:1085–1103. doi: 10.1111/j.1365-2958.2004.04443.x. [DOI] [PubMed] [Google Scholar]
- Clavel T, Germon P, Vianney A, Portalier R, Lazzaroni JC. TolB protein of Escherichia coli K-12 interacts with the outer membrane peptidoglycan-associated proteins Pal, Lpp and OmpA. Mol Microbiol. 1998;29:359–367. doi: 10.1046/j.1365-2958.1998.00945.x. [DOI] [PubMed] [Google Scholar]
- Click EM, Webster RE. Filamentous phage infection: required interactions with the TolA protein. J Bacteriol. 1997;179:6464–6471. doi: 10.1128/jb.179.20.6464-6471.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Click EM, Webster RE. The TolQRA proteins are required for membrane insertion of the major capsid protein of the filamentous phage f1 during infection. J Bacteriol. 1998;180:1723–1728. doi: 10.1128/jb.180.7.1723-1728.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordell SC, Robinson EJ, Lowe J. Crystal structure of the SOS cell division inhibitor SulA and in complex with FtsZ. Proc Natl Acad Sci USA. 2003;100:7889–7894. doi: 10.1073/pnas.1330742100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- Daniel RA, Harry EJ, Errington J. Role of penicillin-binding protein PBP 2B in assembly and functioning of the division machinery of Bacillus subtilis. Mol Microbiol. 2000;35:299–311. doi: 10.1046/j.1365-2958.2000.01724.x. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JK, Reeves P. Genetics of resistance to colicins in Escherichia coli K-12: cross-resistance among colicins of group A. J Bacteriol. 1975;123:102–117. doi: 10.1128/jb.123.1.102-117.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deprez C, Blanchard L, Guerlesquin F, Gavioli M, Simorre JP, Lazdunski C, et al. Macromolecular import into Escherichia coli: the TolA C-terminal domain changes conformation when interacting with the colicin A toxin. Biochemistry. 2002;41:2589–2598. doi: 10.1021/bi0157262. [DOI] [PubMed] [Google Scholar]
- Derouiche R, Benedetti H, Lazzaroni JC, Lazdunski C, Lloubes R. Protein complex within Escherichia coli inner membrane. TolA N-terminal domain interacts with TolQ and TolR proteins. J Biol Chem. 1995;270:11078–11084. doi: 10.1074/jbc.270.19.11078. [DOI] [PubMed] [Google Scholar]
- Dubuisson JF, Vianney A, Lazzaroni JC. Mutational analysis of the TolA C-terminal domain of Escherichia coli and genetic evidence for an interaction between TolA and TolB. J Bacteriol. 2002;184:4620–4625. doi: 10.1128/JB.184.16.4620-4625.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubuisson JF, Vianney A, Hugouvieux-Cotte-Pattat N, Lazzaroni JC. Tol-Pal proteins are critical cell envelope components of Erwinia chrysanthemi affecting cell morphology and virulence. Microbiology. 2005;151:3337–3347. doi: 10.1099/mic.0.28237-0. [DOI] [PubMed] [Google Scholar]
- Errington J, Daniel RA, Scheffers DJ. Cytokinesis in bacteria. Microbiol Mol Biol Rev. 2003;67:52–65. doi: 10.1128/MMBR.67.1.52-65.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feilmeier BJ, Iseminger G, Schroeder D, Webber H, Phillips GJ. Green fluorescent protein functions as a reporter for protein localization in Escherichia coli. J Bacteriol. 2000;182:4068–4076. doi: 10.1128/jb.182.14.4068-4076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung J, MacAlister TJ, Rothfield LI. Role of murein-lipoprotein in morphogenesis of the bacterial division septum; phenotypic similarity of lkyD and lpo mutants. J Bacteriol. 1978;133:1467–1471. doi: 10.1128/jb.133.3.1467-1471.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germon P, Clavel T, Vianney A, Portalier R, Lazzaroni JC. Mutational analysis of the Escherichia coli K-12 TolA N-terminal region and characterization of its TolQ-interacting domain by genetic suppression. J Bacteriol. 1998;180:6433–6439. doi: 10.1128/jb.180.24.6433-6439.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germon P, Ray MC, Vianney A, Lazzaroni JC. Energy-dependent conformational change in the TolA protein of Escherichia coli involves its N-terminal domain, TolQ, and TolR. J Bacteriol. 2001;183:4110–4114. doi: 10.1128/JB.183.14.4110-4114.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasner JD, Liss P, Plunkett G, 3rd, Darling A, Prasad T, Rusch M, et al. ASAP, a systematic annotation package for community analysis of genomes. Nucleic Acids Res. 2003;31:147–151. doi: 10.1093/nar/gkg125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring NW, Beckwith J. Diverse paths to midcell: assembly of the bacterial cell division machinery. Curr Biol. 2005;15:R514–R526. doi: 10.1016/j.cub.2005.06.038. [DOI] [PubMed] [Google Scholar]
- Gueiros-Filho FJ, Losick R. A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes Dev. 2002;16:2544–2556. doi: 10.1101/gad.1014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyer MS, Reed RR, Steitz JA, Low KB. Identification of a sex-factor-affinity site in E. coli as gamma delta. Cold Spring Harb Symp Quant Biol. 1981;45:135–140. doi: 10.1101/sqb.1981.045.01.022. [DOI] [PubMed] [Google Scholar]
- Hale CA, de Boer PAJ. Recruitment of ZipA to the septal ring of Escherichia coli is dependent on FtsZ, and independent of FtsA. J Bacteriol. 1999;181:167–176. doi: 10.1128/jb.181.1.167-176.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, de Boer PAJ. ZipA is required for recruitment of FtsK, FtsQ, FtsL, and FtsN to the septal ring in Escherichia coli. J Bacteriol. 2002;184:2552–2556. doi: 10.1128/JB.184.9.2552-2556.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, et al. Involvement of N-acetylmuramyl-L-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol Microbiol. 2001;41:167–178. doi: 10.1046/j.1365-2958.2001.02499.x. [DOI] [PubMed] [Google Scholar]
- Heidrich C, Ursinus A, Berger J, Schwarz H, Höltje JV. Effects of multiple deletions of murein hydrolases on viability, septum cleavage, and sensitivity to large toxic molecules in Escherichia coli. J Bacteriol. 2002;184:6093–6099. doi: 10.1128/JB.184.22.6093-6099.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilpern AJ, Waldor MK. CTXphi infection of Vibrio cholerae requires the tolQRA gene products. J Bacteriol. 2000;182:1739–1747. doi: 10.1128/jb.182.6.1739-1747.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry T, Pommier S, Journet L, Bernadac A, Gorvel JP, Lloubes R. Improved methods for producing outer membrane vesicles in Gram-negative bacteria. Res Microbiol. 2004;155:437–446. doi: 10.1016/j.resmic.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Hiemstra H, de Hoop MJ, Inouye M, Witholt B. Induction kinetics and cell surface distribution of Escherichia coli lipoprotein under lac promoter control. J Bacteriol. 1986;168:140–151. doi: 10.1128/jb.168.1.140-151.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiemstra H, Nanninga N, Woldringh CL, Inouye M, Witholt B. Distribution of newly synthesized lipoprotein over the outer membrane and the peptidoglycan sacculus of an Escherichia coli lac-lpp strain. J Bacteriol. 1987;169:5434–5444. doi: 10.1128/jb.169.12.5434-5444.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James R, Penfold CN, Moore GR, Kleanthous C. Killing of E coli cells by E group nuclease colicins. Biochimie. 2002;84:381–389. doi: 10.1016/s0300-9084(02)01450-5. [DOI] [PubMed] [Google Scholar]
- Johnson JE, Lackner LL, de Boer PAJ. Targeting of DMinC/MinD and DMinC/DicB complexes to septal rings in Escherichia coli suggests a multistep mechanism for MinC-mediated destruction of nascent FtsZ-rings. J Bacteriol. 2002;184:2951–2962. doi: 10.1128/JB.184.11.2951-2962.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Journet L, Rigal A, Lazdunski C, Benedetti H. Role of TolR N-terminal, central, and C-terminal domains in dimerization and interaction with TolA and TolQ. J Bacteriol. 1999;181:4476–4484. doi: 10.1128/jb.181.15.4476-4484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judd EM, Comolli LR, Chen JC, Downing KH, Moerner WE, McAdams HH. Distinct constrictive processes, separated in time and space, divide Caulobacter inner and outer membranes. J Bacteriol. 2005;187:6874–6882. doi: 10.1128/JB.187.20.6874-6882.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M, Ara T, Arifuzzaman M, Ioka-Nakamichi T, Inamoto E, Toyonaga H, Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K-12 ORF archive): unique resources for biological research. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- Lai EM, Nair U, Phadke ND, Maddock JR. Proteomic screening and identification of differentially distributed membrane proteins in Escherichia coli. Mol Microbiol. 2004;52:1029–1044. doi: 10.1111/j.1365-2958.2004.04040.x. [DOI] [PubMed] [Google Scholar]
- Lazdunski CJ. Colicin import and pore formation: a system for studying protein transport across membranes? Mol Microbiol. 1995;16:1059–1066. doi: 10.1111/j.1365-2958.1995.tb02331.x. [DOI] [PubMed] [Google Scholar]
- Lazdunski CJ, Bouveret E, Rigal A, Journet L, Lloubes R, Benedetti H. Colicin import into Escherichia coli cells. J Bacteriol. 1998;180:4993–5002. doi: 10.1128/jb.180.19.4993-5002.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazzaroni JC, Portalier R. The excC gene of Escherichia coli K-12 required for cell envelope integrity encodes the peptidoglycan-associated lipoprotein (PAL) Mol Microbiol. 1992;6:735–742. doi: 10.1111/j.1365-2958.1992.tb01523.x. [DOI] [PubMed] [Google Scholar]
- Lazzaroni JC, Vianney A, Popot JL, Benedetti H, Samatey F, Lazdunski C, et al. Transmembrane alpha-helix interactions are required for the functional assembly of the Escherichia coli Tol complex. J Mol Biol. 1995;246:1–7. doi: 10.1006/jmbi.1994.0058. [DOI] [PubMed] [Google Scholar]
- Lazzaroni JC, Germon P, Ray MC, Vianney A. The Tol proteins of Escherichia coli and their involvement in the uptake of biomolecules and outer membrane stability. FEMS Microbiol Lett. 1999;177:191–197. doi: 10.1111/j.1574-6968.1999.tb13731.x. [DOI] [PubMed] [Google Scholar]
- Lazzaroni JC, Dubuisson JF, Vianney A. The Tol proteins of Escherichia coli and their involvement in the translocation of group A colicins. Biochimie. 2002;84:391–397. doi: 10.1016/s0300-9084(02)01419-0. [DOI] [PubMed] [Google Scholar]
- Liu Z, Mukherjee A, Lutkenhaus J. Recruitment of ZipA to the division site by interaction with FtsZ. Mol Microbiol. 1999;31:1853–1861. doi: 10.1046/j.1365-2958.1999.01322.x. [DOI] [PubMed] [Google Scholar]
- Llamas MA, Ramos JL, Rodriguez-Herva JJ. Mutations in each of the tol genes of Pseudomonas putida reveal that they are critical for maintenance of outer membrane stability. J Bacteriol. 2000;182:4764–4772. doi: 10.1128/jb.182.17.4764-4772.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloubes R, Cascales E, Walburger A, Bouveret E, Lazdunski C, Bernadac A, Journet L. The Tol-Pal proteins of the Escherichia coli cell envelope: an energized system required for outer membrane integrity? Res Microbiol. 2001;152:523–529. doi: 10.1016/s0923-2508(01)01226-8. [DOI] [PubMed] [Google Scholar]
- Lubkowski J, Hennecke F, Pluckthun A, Wlodawer A. Filamentous phage infection: crystal structure of g3p in complex with its coreceptor, the C-terminal domain of TolA. Structure. 1999;7:711–722. doi: 10.1016/s0969-2126(99)80092-6. [DOI] [PubMed] [Google Scholar]
- Lutkenhaus J. FtsZ ring in bacterial cytokinesis. Mol Microbiol. 1993;9:403–409. doi: 10.1111/j.1365-2958.1993.tb01701.x. [DOI] [PubMed] [Google Scholar]
- MacAlister TJ, Cook WR, Weigand R, Rothfield LI. Membrane-murein attachment at the leading edge of the division septum: a second membrane-murein structure associated with morphogenesis of the gram-negative bacterial division septum. J Bacteriol. 1987;169:3945–3951. doi: 10.1128/jb.169.9.3945-3951.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin W. FtsZ and the division of prokaryotic cells and organelles. Nat Rev Mol Cell Biol. 2005;6:862–871. doi: 10.1038/nrm1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meury J, Devilliers G. Impairment of cell division in tolA mutants of Escherichia coli at low and high medium osmolarities. Biol Cell. 1999;91:67–75. [PubMed] [Google Scholar]
- Mizuno T. A novel peptidoglycan-associated lipoprotein (PAL) found in the outer membrane of Proteus mirabilis and other Gram-negative bacteria. J Biochem (Tokyo) 1981;89:1039–1049. [PubMed] [Google Scholar]
- Mukherjee A, Cao C, Lutkenhaus J. Inhibition of FtsZ polymerization by SulA, an inhibitor of septation in Escherichia coli. Proc Natl Acad Sci USA. 1998;95:2885–2890. doi: 10.1073/pnas.95.6.2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullineaux CW, Nenninger A, Ray N, Robinson C. Diffusion of green fluorescent protein in three cell environments in Escherichia coli. J Bacteriol. 2006;188:3442–3448. doi: 10.1128/JB.188.10.3442-3448.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel de Zwaig R, Luria SE. Genetics and physiology of colicin-tolerant mutants of Escherichia coli. J Bacteriol. 1967;94:1112–1123. doi: 10.1128/jb.94.4.1112-1123.1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino H, Wachi M, Ishii A, Iwai N, Nishida T, Yamada S, et al. FtsZ-dependent localization of GroEL protein at possible division sites. Genes Cells. 2004;9:765–771. doi: 10.1111/j.1365-2443.2004.00770.x. [DOI] [PubMed] [Google Scholar]
- Palva ET. Protein interactions in the outer membrane of Escherichia coli. Eur J Biochem. 1979;93:495–503. doi: 10.1111/j.1432-1033.1979.tb12848.x. [DOI] [PubMed] [Google Scholar]
- Parsons LM, Lin F, Orban J. Peptidoglycan recognition by Pal, an outer membrane lipoprotein. Biochemistry. 2006;45:2122–2128. doi: 10.1021/bi052227i. [DOI] [PubMed] [Google Scholar]
- Pommier S, Gavioli M, Cascales E, Lloubes R. Tol-dependent macromolecule import through the Escherichia coli cell envelope requires the presence of an exposed TolA binding motif. J Bacteriol. 2005;187:7526–7534. doi: 10.1128/JB.187.21.7526-7534.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prouty AM, Van Velkinburgh JC, Gunn JS. Salmonella enterica serovar typhimurium resistance to bile: identification and characterization of the tolQRA cluster. J Bacteriol. 2002;184:1270–1276. doi: 10.1128/JB.184.5.1270-1276.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray MC, Germon P, Vianney A, Portalier R, Lazzaroni JC. Identification by genetic suppression of Escherichia coli TolB residues important for TolB–Pal interaction. J Bacteriol. 2000;182:821–824. doi: 10.1128/jb.182.3.821-824.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riechmann L, Holliger P. The C-terminal domain of TolA is the coreceptor for filamentous phage infection of E. coli. Cell. 1997;90:351–360. doi: 10.1016/s0092-8674(00)80342-6. [DOI] [PubMed] [Google Scholar]
- Rodolakis A, Thomas P, Starka J. Morphological mutants of Escherichia coli. Isolation and ultrastructure of a chain-forming envC mutant. J Gen Microbiol. 1973;75:409–416. doi: 10.1099/00221287-75-2-409. [DOI] [PubMed] [Google Scholar]
- Rothfield LI, Justice SS. Bacterial cell division: the cycle of the ring. Cell. 1997;88:581–584. doi: 10.1016/s0092-8674(00)81899-1. [DOI] [PubMed] [Google Scholar]
- Rothfield LI, MacAlister TJ, Cook WR. Murein–membrane interactions in cell division. In: Inouye M, editor. Bacterial Outer Membranes as Model Systems. New York, NY: John Wiley & Sons; 1986. pp. 247–275. [Google Scholar]
- Ryan KR, Shapiro L. Temporal and spatial regulation in prokaryotic cell cycle progression and development. Annu Rev Biochem. 2003;72:367–394. doi: 10.1146/annurev.biochem.72.121801.161824. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Santini CL, Bernadac A, Zhang M, Chanal A, Ize B, Blanco C, Wu LF. Translocation of jellyfish green fluorescent protein via the Tat system of Escherichia coli and change of its periplasmic localization in response to osmotic up-shock. J Biol Chem. 2001;276:8159–8164. doi: 10.1074/jbc.C000833200. [DOI] [PubMed] [Google Scholar]
- Schmidt KL, Peterson ND, Kustusch RJ, Wissel MC, Graham B, Phillips GJ, Weiss DS. A predicted ABC transporter, FtsEX, is needed for cell division in Escherichia coli. J Bacteriol. 2004;186:785–793. doi: 10.1128/JB.186.3.785-793.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY. Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol. 2004;22:1567–1572. doi: 10.1038/nbt1037. [DOI] [PubMed] [Google Scholar]
- Siddiqui RA, Hoischen C, Holst O, Heinze I, Schlott B, Gumpert J, et al. The analysis of cell division and cell wall synthesis genes reveals mutationally inactivated ftsQ and mraY in a protoplast-type 1-form of Escherichia coli. FEMS Microbiol Lett. 2006;258:305–311. doi: 10.1111/j.1574-6968.2006.00237.x. [DOI] [PubMed] [Google Scholar]
- Sturgis JN. Organisation and evolution of the tol-pal gene cluster. J Mol Microbiol Biotechnol. 2001;3:113–122. [PubMed] [Google Scholar]
- Sun TP, Webster RE. fii, a bacterial locus required for filamentous phage infection and its relation to colicin-tolerant tolA and tolB. J Bacteriol. 1986;165:107–115. doi: 10.1128/jb.165.1.107-115.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Nishimura Y, Yasuda S, Nishimura A, Yamada M, Hirota Y. Murein-lipoprotein of Escherichia coli: a protein involved in the stabilization of bacterial cell envelope. Mol Gen Genet. 1978;167:1–9. doi: 10.1007/BF00270315. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Daniel RA, Errington J, Robinson C. Export of active green fluorescent protein to the periplasm by the twin- arginine translocase (Tat) pathway in Escherichia coli. Mol Microbiol. 2001;39:47–53. doi: 10.1046/j.1365-2958.2001.02253.x. [DOI] [PubMed] [Google Scholar]
- Trusca D, Scott S, Thompson C, Bramhill D. Bacterial SOS checkpoint protein SulA inhibits polymerization of purified FtsZ cell division protein. J Bacteriol. 1998;180:3946–3953. doi: 10.1128/jb.180.15.3946-3953.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vianney A, Muller MM, Clavel T, Lazzaroni JC, Portalier R, Webster RE. Characterization of the tol-pal region of Escherichia coli K-12: translational control of tolR expression by TolQ and identification of a new open reading frame downstream of pal encoding a periplasmic protein. J Bacteriol. 1996;178:4031–4038. doi: 10.1128/jb.178.14.4031-4038.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente M, Rico AI, Martinez-Arteaga R, Mingorance J. Septum enlightenment: assembly of bacterial division proteins. J Bacteriol. 2006;188:19–27. doi: 10.1128/JB.188.1.19-27.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vines ED, Marolda CL, Balachandran A, Valvano MA. Defective O-antigen polymerization in tolA and pal mutants of Escherichia coli in response to extracytoplasmic stress. J Bacteriol. 2005;187:3359–3368. doi: 10.1128/JB.187.10.3359-3368.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vischer NOE, Huls PG, Woldringh CL. Object-Image: an interactive image analysis program using structured point collection. Binary. 1994;6:160–166. [Google Scholar]
- Walburger A, Lazdunski C, Corda Y. The Tol/Pal system function requires an interaction between the C-terminal domain of TolA and the N-terminal domain of TolB. Mol Microbiol. 2002;44:695–708. doi: 10.1046/j.1365-2958.2002.02895.x. [DOI] [PubMed] [Google Scholar]
- Weigand RA, Vinci KD, Rothfield LI. Morphogenesis of the bacterial division septum: a new class of septation-defective mutants. Proc Natl Acad Sci USA. 1976;73:1882–1886. doi: 10.1073/pnas.73.6.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss DS. Bacterial cell division and the septal ring. Mol Microbiol. 2004;54:588–597. doi: 10.1111/j.1365-2958.2004.04283.x. [DOI] [PubMed] [Google Scholar]
- Wissel MC, Weiss DS. Genetic analysis of the cell division protein FtsI (PBP3): amino acid substitutions that impair septal localization of FtsI and recruitment of FtsN. J Bacteriol. 2004;186:490–502. doi: 10.1128/JB.186.2.490-502.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woldringh CL. Morphological analysis of nuclear separation and cell division during the life cycle of Escherichia coli. J Bacteriol. 1976;125:248–257. doi: 10.1128/jb.125.1.248-257.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yem DW, Wu HC. Physiological characterization of an Escherichia coli mutant altered in the structure of murein lipoprotein. J Bacteriol. 1978;133:1419–1426. doi: 10.1128/jb.133.3.1419-1426.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci USA. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
s1