Multifunctional Roles of a Bacteriophage ϕ29 Morphogenetic Factor in Assembly and Infection (original) (raw)
. Author manuscript; available in PMC: 2009 May 9.
Published in final edited form as: J Mol Biol. 2008 Mar 7;378(4):802–815. doi: 10.1016/j.jmb.2008.02.068
SUMMARY
Low copy number proteins within macromolecular complexes, such as viruses, can be critical to biological function while comprising a minimal mass fraction of the complex. The Bacillus subtilis dsDNA bacteriophage ϕ29 gene 13 product (gp13), previously undetected in the virion, was identified and localized to the distal tip of the tail knob. Western blots and immuno-electron microscopy detected a few copies of gp13 in ϕ29, DNA-free particles, purified tails, and defective particles produced in suppressor-sensitive (sus) mutant _sus_13(330) infections. Particles assembled in the absence of intact gp13 (_sus_13(342) and _sus_13(330)) had the gross morphology of ϕ29 but were noninfectious. gp13 has predicted structural homology and sequence similarity to the M23 metalloprotease LytM. Poised at the tip of the ϕ29 tail knob, gp13 may serve as a plug to help restrain the highly pressurized packaged genome. Also, in this position, gp13 may be the first virion protein to contact the cell wall in infection, acting as a pilot protein to depolymerize the cell wall. gp13 may facilitate juxtaposing of the tail knob onto the cytoplasmic membrane and the triggering of genome injection.
Keywords: bacteriophage ϕ29, morphogenetic factor, immuno-electron microscopy, tail protein, endopeptidase
INTRODUCTION
To understand in detail the events of morphogenesis and infection of viruses, such as the bacteriophage ϕ29, all components of the virion involved in these processes must be identified. This is not trivial, as minor proteins present in only a few copies may be confused with contaminants or be obscured by major proteins. Here the ϕ29 gene product 13 (gp13), previously identified as a morphogenetic factor in ϕ29 assembly,1 is identified as a minor ϕ29 tail component that has escaped detection for more than three decades since identification of the major structural components of the virion.2, 3
Phage ϕ29 morphogenesis begins with formation of the dodecameric head-tail connector (gene product 10, gp10), onto which the scaffolding (gp7), capsid (gp8) and head fiber (gp8.5) proteins assemble to form the 54 × 42 nm prolate prohead shell4-6 (Figure 1). Attachment of an oligomer of prohead RNA (pRNA) to the connector yields the mature prohead.7, 8 Multiple copies of the ATPase gp16 attach to the pRNA to constitute the packaging motor, and hydrolysis of ATP powers the translocation of the dsDNA genome with its covalently-bound gp3 (DNA-gp3)9 into the prohead with scaffold (gp7) exit.10-12 Assembly of the lower collar (gp11) to the DNA-filled head13-15 triggers pRNA/gp16 release.16 Finally, assembly of the tail knob (gp9) and twelve appendages (gp12*, cleaved from the gp12 precursor)17, 18 yields the infectious virion.2, 4, 19, 20
Figure 1.
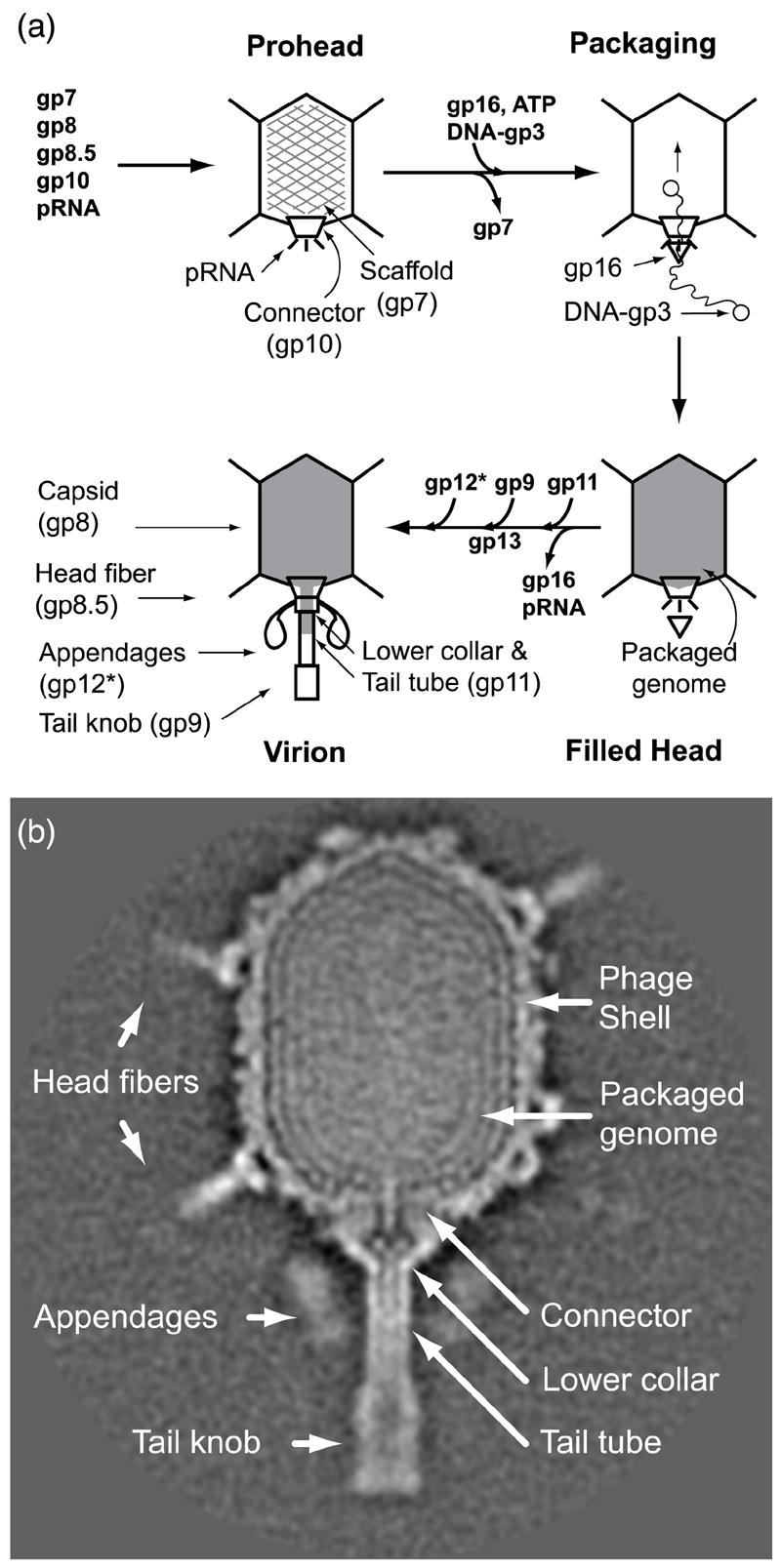
ϕ29 assembly pathway and cryo-EM 3D reconstruction. (a) Assembly begins with the formation of the head-tail connector (gp10) onto which the scaffolding (gp7) (hatched particle), major capsid (gp8) and head fiber (gp8.5) proteins and prohead RNA (pRNA) assemble to form the prohead. The DNA packaging motor is complete on assembly of the ATPase gp16 to pRNA, and on hydrolysis of ATP, drives translocation of DNA-gp3 into the prohead concurrent with scaffold extrusion (un-hatched particle). Lower collar and tail tube (gp11) assembly triggers release of gp16/pRNA after complete packaging (filled grey particle). The tail knob (gp9), pre-assembled with gp13 (this study), is fixed onto the tail tube. Lastly, the appendages (gp12*), which serve as adsorption organelles, are attached at the connector-lower collar junction to form the infectious virion. (b) ϕ29 structures revealed in a cryo-EM 3D reconstruction.22
gp13 is required for ϕ29 assembly, and its activity as a morphogenetic factor, which occurs in vivo but not in extract complementation, co-purifies with the tail knob (gp9).1 Using restrictive infections with suppressor-sensitive (sus) ϕ29 mutants, purified particles from a tail knob-defective lysate (_sus_9(756)) with functional gp13 (9-13+) do not form infectious virions when mixed with an extract (_sus_13(342)) that lacks functional gp13 (13- 9+) and vice versa.1 In contrast, complementation does occur with most virion proteins: filled heads isolated from a lower collar-defective lysate (11- 8+) form infectious phages when incubated with a capsid-defective extract (8- 11+) that provides gene products 11, 9, 13 and 12*, showing that gp8 activity in morphogenesis is not gp11 dependent nor vice versa.
gp13 was hypothesized to be a previously undetected tail component because the lower collar and tail tube of ϕ29 (Figure 1), reported to consist of gp11,21 appears to have more mass in cryo-electron microscopy reconstruction22 than can be accounted for by gp11 alone (compared to the dodecameric connector comprised of subunits of similar mass23). Here, identification of gp13 as a ϕ29 structural component and its localization to the distal end of the tail knob was accomplished by the use of gp13-specific antibodies in Western blots and by immuno-electron microscopy of ϕ29, isolated tails, and defective particles produced in restrictive infection with _sus_13(342) and _sus_13(330) mutants. Previous investigations1 found that assembly of an active tail knob requires both gp9 and gp13, and the present study demonstrates that the active knob contains both gp9 and gp13. Therefore the knob is a gp9/gp13 complex that is likely fixed en bloc onto the lower collar and tail tube before gp12* addition, defining a sub-assembly pathway for ϕ29. The use of specific antibody and electron microscopy to localize gp13 at the distal tip of the tail knob of ϕ29, a method pioneered to identify gene products on phage T4,24 is applicable to the identification and localization of minor protein components with critical roles in assembly and function of other viruses and macromolecular complexes. Moreover, the method is applicable to detecting minor proteins in assembly intermediates and as a tool in unraveling assembly pathways.
Secondary structure predictions of gp13 suggest N-terminal α-helices and C-terminal β-strands. The C-terminal region shows sequence alignment to the zinc-metalloprotease LytM of Staphylococcus aureus (PDB: 1qwy) and a similarity in arrangement of β-strands. Biochemical identification of gp13 as an endopeptidase (Cohen et al., in preparation) provides direct evidence that gp13 has enzymatic activity analogous to LytM activity on the Staphylococcal peptidoglycan peptide cross-link that may be needed for the tail to penetrate the Bacillus subtilis cell wall during ϕ29 infection.
RESULTS
gp13 is a previously unidentified structural component of ϕ29
An abbreviated ϕ29 assembly pathway is shown in Figure 1(a), and a cryo-electron microscopy three-dimensional reconstruction of ϕ29 is shown in Figure 1(b), which illustrates the six visible protein components and the packaged genome: capsid (gp8), head fibers (gp8.5), head-tail connector (gp10), lower collar and tail tube (gp11), knob (gp9), appendages (gp12*) and DNA-gp3. gp13 was demonstrated to be a seventh structural component of ϕ29 by the use of gp13-specific polyclonal antiserum (anti-gp13 serum) in Western blot analysis (Figure 2). Following LDS-PAGE, blots of various amounts of ϕ29, proheads, and purified gp13 were incubated with anti-gp13 serum (Figure 2(a)) and developed with enzyme-linked secondary antibody (Methods). Mature ϕ29 particles showed gp13-specific signal (lanes 1-3), while an equivalent number of proheads did not (lanes 4-6). Dilutions of purified gp13 served as a positive control (lanes 7-10). Re-probing with anti-connector (anti-gp10) serum served as a loading control. These results demonstrated that gp13 is present in ϕ29 and is assembled after prohead formation and during tail assembly.
Figure 2.
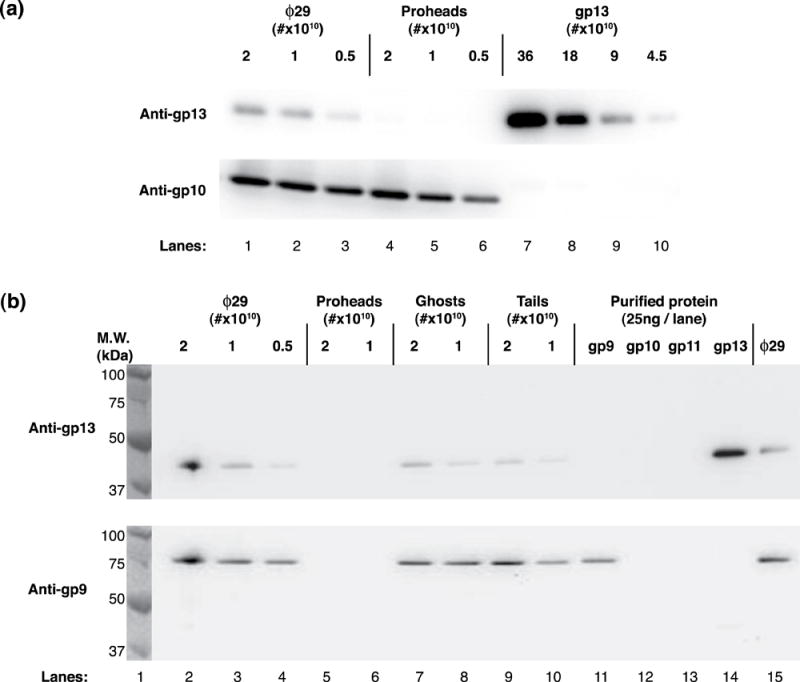
Western blot analysis of ϕ29 particles and tails using gp13-specific antiserum. gp13 is demonstrated in ϕ29 but not the prohead (a, top). Particle number is controlled and shown with connector-specific (anti-gp10) serum (a, bottom). gp13 is shown to be present in ϕ29 (b, top, lanes 2-4), ghosts (DNA-free particles) (lanes 7-8) and isolated tails (gp10, gp11, gp9, gp12*) (lanes 9-10) but not in proheads (lanes 5-6). gp9 does not react with anti-gp13 serum (b, top, lane 11), and gp13 does not react with anti-gp9 serum (b, bottom, lane 14). Anti-gp9 serum was used as a loading control, and ϕ29, ghosts and tails show equivalent signal (b, bottom, lanes 2-4 and 7-10).
To demonstrate that gp13 is a tail component, a blot that included ϕ29, proheads, particles emptied of DNA (ghosts), and isolated tails (gp10, 11, 12*, and 9) was probed with anti-gp13 serum (Figure 2(b) top). gp13 signal was observed in ϕ29 (lanes 2-4 and 15), ghosts (lanes 7 and 8) and tails (lanes 9 and 10) but not in proheads (lanes 5 and 6). Signal with purified gp13 (lane 14) at approximately 41kDa marked the appropriate migration distance of gp13, in accordance with its sequence-predicted mass (Table 2). The purified proteins gp9, gp11 and gp13 (lanes 11, 13, 14) and purified connectors (gp10) (lane 12) served as controls both for loading and for cross-reactivity. Approximately equivalent numbers of ϕ29, ghosts and tails were loaded as shown by gp9 signal with anti-gp9 serum (Figure 2(b) bottom). Additionally, anti-gp9 serum did not cross-react with gp13 (Figure 2(b) bottom), nor did anti-gp13 serum cross-react with gp9 (Figure 2(b) top). Tail preparations were confirmed by TEM and SDS-PAGE to lack proheads and phages (data not shown). The results demonstrated that gp13 is a structural component of the ϕ29 tail, not previously recognized,1, 13 and led to experiments to localize gp13 in the ϕ29 tail.
Table 2.
Sequence analysis of gene 13 suppressor-sensitive mutants
| Mutant | Positiona | Fragment (aa length) | Fragment (Da mass)b |
|---|---|---|---|
| _sus_13(342) | Q33* | 32 | 3777.3 |
| _sus_13(44) | Q43* | 42 | 4796.6 |
| _sus_13(336) | Q83* | 82 | 9189.3 |
| _sus_13(53) | Q167* | 166 | 18883.2 |
| _sus_13(55) | Q332* | 331 | 37314.9 |
| _sus_13(330) | Q332* | 331 | 37314.9 |
| wild-type | n/a | 365 | 40924.7 |
gp13 was localized in the ϕ29 tail by immuno-electron microscopy
To localize gp13 in the tail, immuno-electron microscopy was utilized as described (Methods). Anti-gp13 IgGs were used to decorate mutant particles lacking the gp12* appendages, which serve as adsorption organelles (12- particles; Figure 1), and this permitted access of the antibodies to the lower collar and tail tube. Particle/antibody complexes were observed by negative staining and transmission electron microscopy (TEM) (Figure 3 (a, c)). Appendageless 12- particles formed antibody-dependent tail-bound aggregates when incubated with anti-gp13 IgG (Figure 3(a)), while 12- particles incubated with pre-immune IgG (Figure 3(b)) and 12- particles alone (data not shown) adsorbed randomly to the support film. Wild-type ϕ29 particles behave similarly (Figure 7(b)). Additionally, the distal ends of the tail knobs were obscured in the presence of anti-gp13 IgG (Figure 3(a, i)), while particles treated with pre-immune IgG retained the sharp knob ends (Figure 3(b, i)). To confirm that antibodies caused the aggregation 12- particles decorated with biotinylated antibodies were separated from excess antibody by HPLC (Methods), labeled with streptavidin-gold (6 nm), and observed by TEM. Electron dense gold spheres were observed only closely associated with ϕ29 on the distal end of tails (Figure 3(c)). As a control, 12- particles were decorated with biotinylated anti-capsid (anti-gp8) IgG and labeled with streptavidin-gold; no tail bound aggregates were observed, and particles aggregated by their heads were tagged with gold (data not shown). The results demonstrated that gp13 epitopes are present at the distal end of the ϕ29 tail knob.
Figure 3.
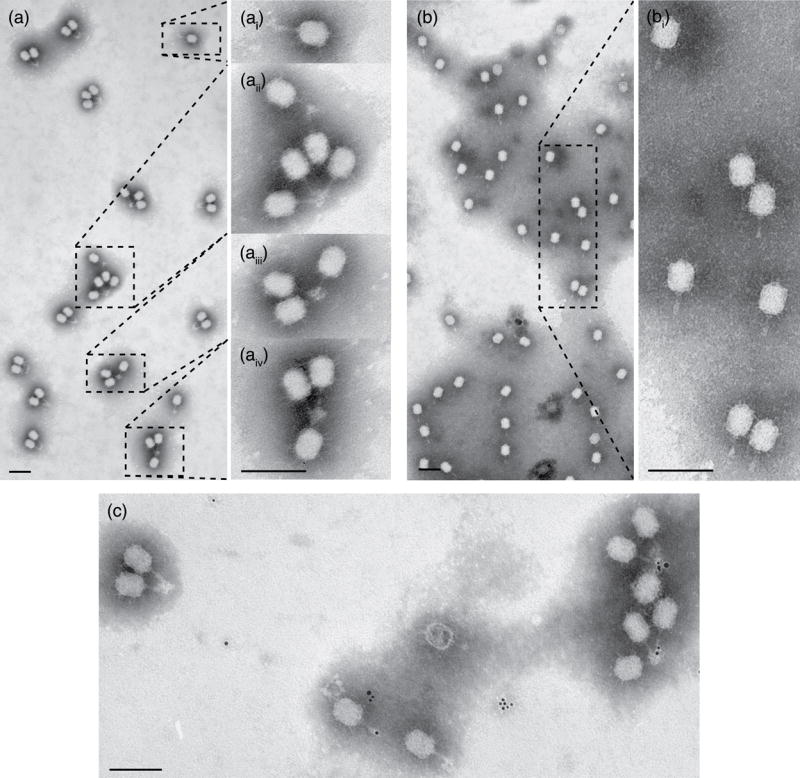
Immuno-electron microscopy localization of gp13 in ϕ29 using anti-gp13 serum. Particles without appendages (12-) form antibody-dependent tail-bound aggregates in the presence of anti-gp13 serum (a) while 12- particles incubated with pre-immune serum (b) do not. Insets (a, i-iii) show magnified aggregates and isolated particles, demonstrating that tail knobs are obscured in the presence of anti-gp13 serum but not in the presence of non-specific IgG (b, i). Immuno-gold localization targets the tail in the presence of anti-gp13 IgG (c), as the only phage-associated gold is tail bound. Negative staining with phosphotungstate. Magnification bars = 100 nm.
Figure 7.
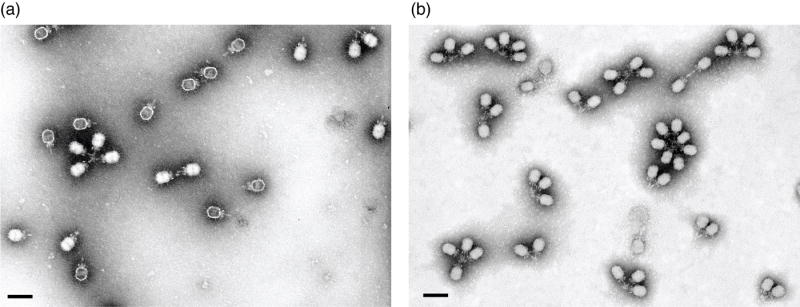
Immuno-electron microscopy detects the short _sus_13(342) ochre fragment of gp13 in DNA-filled but not empty particles. (A) Representative micrographs of DNA-containing and empty _sus_13(342) mutant particles incubated with anti-gp13 IgG. (B) ϕ29 aggregated with anti-gp13 IgG. Negative staining with phosphotungstate. Magnification bar = 100 nm.
Prediction of gp13 secondary structure
The gp13 secondary structure was predicted by use of the Psipred and Profsec algorithms through the BioInfobank Metaserver (Methods) (Figure 4, α-helices [‘H’, red], β-strands [‘E’, blue]). A crystal structure database search using mGenTHREADER and 3D Jury yielded a significant homology hit by both methods. LytM (Figure 4, line 4) has 14% identity with gp13 and scored 0.764 (p-value = 3×10-8) by mGenTHREADER analysis.
Figure 4.
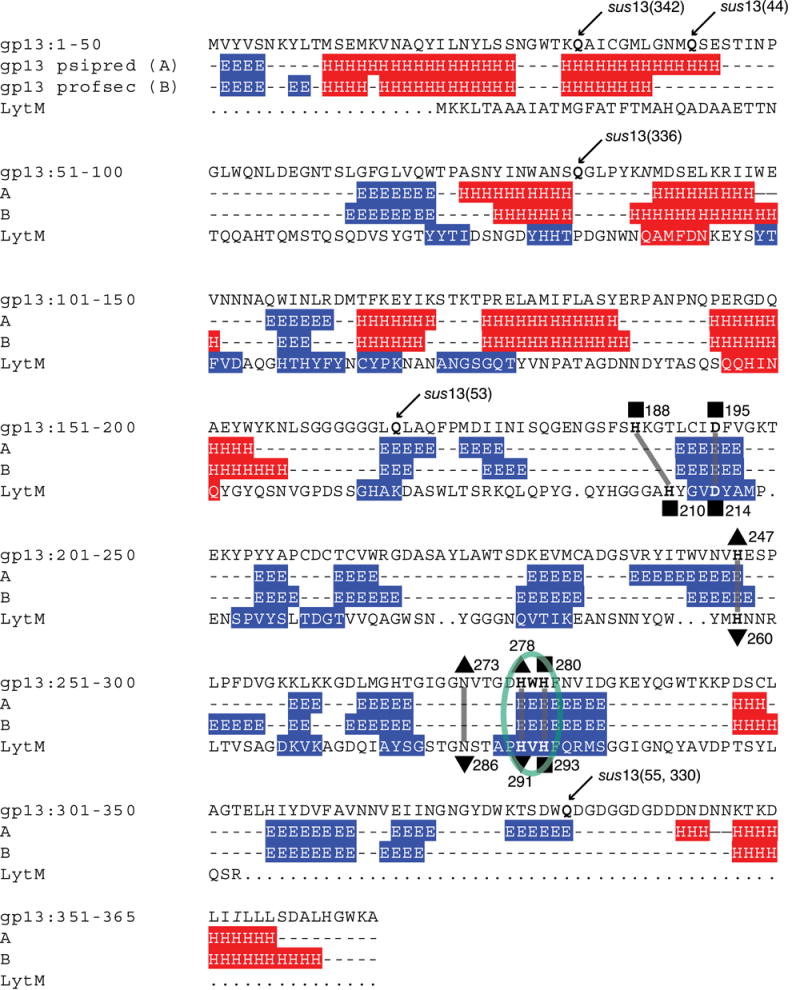
Secondary structure prediction of the gp13 primary amino acid sequence (line 1), showing mutant glutamine (Q) residues, ochre mutations (arrows) and a His-x-His motif (circled green). Two secondary structure prediction models (psipred, line 2, and profsec, line 3) show predicted N-terminal α-helices (‘H’, red) and C-terminal β-sheets (‘E’, blue). Three putative Zn2+ ligands align with LytM-Zn2+ ligands (squares) following the canonical motif structure H-x(3,6)-D and H-x-H, and three residues provide putative anchoring for the fourth Zn2+ ligand as either a phosphate or water molecule in LytM (triangles).27 Zn2+-ligand residues in LytM are numbered below each residue. Amino acids N89 and I353 are italicized to denote differences compared to the published gp13 sequence.29
LytM is a Zn2+ metalloendopeptidase that has a His-x-His motif common to the M23 proteases.25, 26 gp13 has a His-Trp-His motif (Figure 4, circled), and alignment of this motif with the His-Val-His of LytM places all eight beta-strands of LytM in register with predicted beta-strands in gp13 (Figure 4, lines 2-4). This may suggest a role for gp13 as a Zn2+-binding metalloendopeptidase. The Zn2+ ligands of active LytM have been demonstrated to involve three amino acids (His210, Asp214 and His293 in the His-x-His) and a fourth ligand that is a phosphate ion coordinated by three additional residues (His260, Asn286 and His291 in the His-x-His).27 Three putative gp13 Zn-binding ligands were identified in gene 13 by sequence homology to LytM (His188, Asp195, His280 in the His-x-His) (Figure 4, squares). Homologous putative phosphate binding residues that enable formation of the fourth Zn2+ ligand were also observed in the gene 13 sequence (His247, Asn273 and His278 in the His-x-His) (Figure 4, triangles).
Sequencing of gene 13 suppressor-sensitive mutants
To assess further the role of gp13 in ϕ29 morphogenesis and infection, six suppressor-sensitive (sus) mutants that map to gene 13 were sequenced. Notably, 51 of 133 sus mutants reported28 were in gene 13. Purified mutant genomes were sequenced by the use of four primers (Methods), permitting bidirectional double coverage through gene 13. The results (Table 2; Figure 4, top line) demonstrated that each mutant contained a C/T transition within the glutamine codon CAA, generating the nonsense codon TAA (ochre) and resulting in a truncated polypeptide. The _sus_13(55) and _sus_13(330) mutants were isolated separately but contained the same mutation. The mutants were predicted to produce protein fragments of variable length (ochre fragments) (Table 2), and mutants predicted to produce the longest (_sus_13(330)) and shortest (_sus_13(342)) ochre fragments were chosen for further study.
In comparison with the published gene 13 sequence,29 two amino acid substitutions were detected in all eight gp13 genes sequenced for this study (two wt, from _sus_8.5(900)_sus_14(1241) and _sus_12(305)_sus_14(1241) and the six mutants of Figure 4). The aspartic acid at position 89 is asparagine, and the threonine at position 353 is isoleucine. The difference between the published sequence and our data may have arisen from lab strain differences or advancements in DNA sequencing fidelities during the last 20 years. These differences are present as italic letters in Figure 4 and do not change the secondary structure prediction (both the published and newly determined gene 13 sequence produced the same result [data not shown]).
_sus_13(330) mutant particles contain the long gp13 ochre fragment
To detect assembly of ochre fragments of gp13 into particles produced in restrictive sus mutant infections, DNA-filled particles were isolated by isopycnic CsCl density gradient centrifugation and their gp13 content assessed by Western blot analysis using anti-gp13 serum. gp13-specific signal was observed in the defective _sus_13(330) particles (Figure 5, lanes 4 and 5) above 37 kDa (see lane 1 marker), consistent with the predicted size of the ochre fragment (Table 2). In contrast, the defective _sus_13(342) particles did not show a gp13 signal, although the predicted 3.8kDa fragment (Table 2) might have run off the gel, passed through the membrane in blotting, or have fewer epitopes. Signal from the connector (gp10) on interaction with anti-gp10 serum served as a loading control (Figure 5, bottom).
Figure 5.
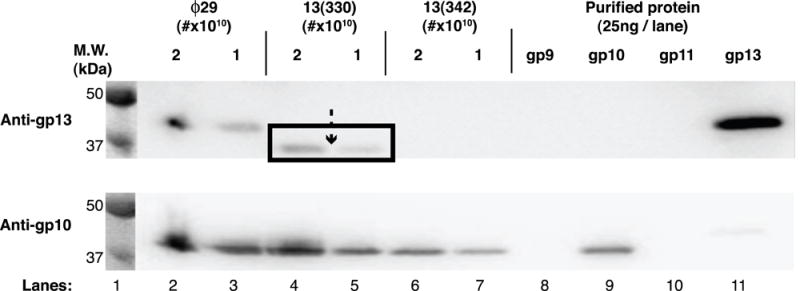
Western blot analysis of gp13-defective particles produced in restrictive infection with the mutants _sus_13(330) and _sus_13(342). _sus_13(330) particles contain a long ochre fragment of gp13 (top panel, arrow with box down-shifted). _sus_13(342) particles contain no detectible gp13 (top panel). Reprobing of the blot with anti-connector (anti-gp10) serum serves as a loading control (bottom panel). Molecular weight standards are with visible light illumination.
_sus_13(330) mutant particles have a distinctive tail knob structure
The phenotype of gp13-defective particles produced in restrictive infection with the mutants _sus_13(342) and _sus_13(330) is a DNA-filled particle with all of the structural proteins of earlier ϕ29 models,4 but is unstable, losing DNA and sometimes the tail tube-knob complex in sucrose density gradient centrifugation and in negative staining for TEM.1, 3 DNA-containing particles were preserved and purified for TEM, albeit with variable efficiency, by the use of isopycnic density gradient centrifugation in CsCl.19, 30 The defective particles of both mutants resembled ϕ29 (Figure 6). However, the _sus_13(330) particles differed in having “notched” tail knobs (Figure 6, dashed arrows). The mass distal to the notch has a cone-shaped structure in a cryo-EM 3D-reconstruction of the _sus_13(330) defective particle (Xiang et al., in preparation) that resembles the structure observed in chemically emptied (ghosted) wt ϕ29.22 Since the _sus_13(330) particle is aggregated by anti-gp9 IgG but not anti-gp13 IgG (Table 3, expt. 2 and 3), a possible explanation is that the cone-shaped structure represents a misassembly or rearrangement of a gp9 knob subunit that blocks access of antibody to the truncated gp13.
Figure 6.
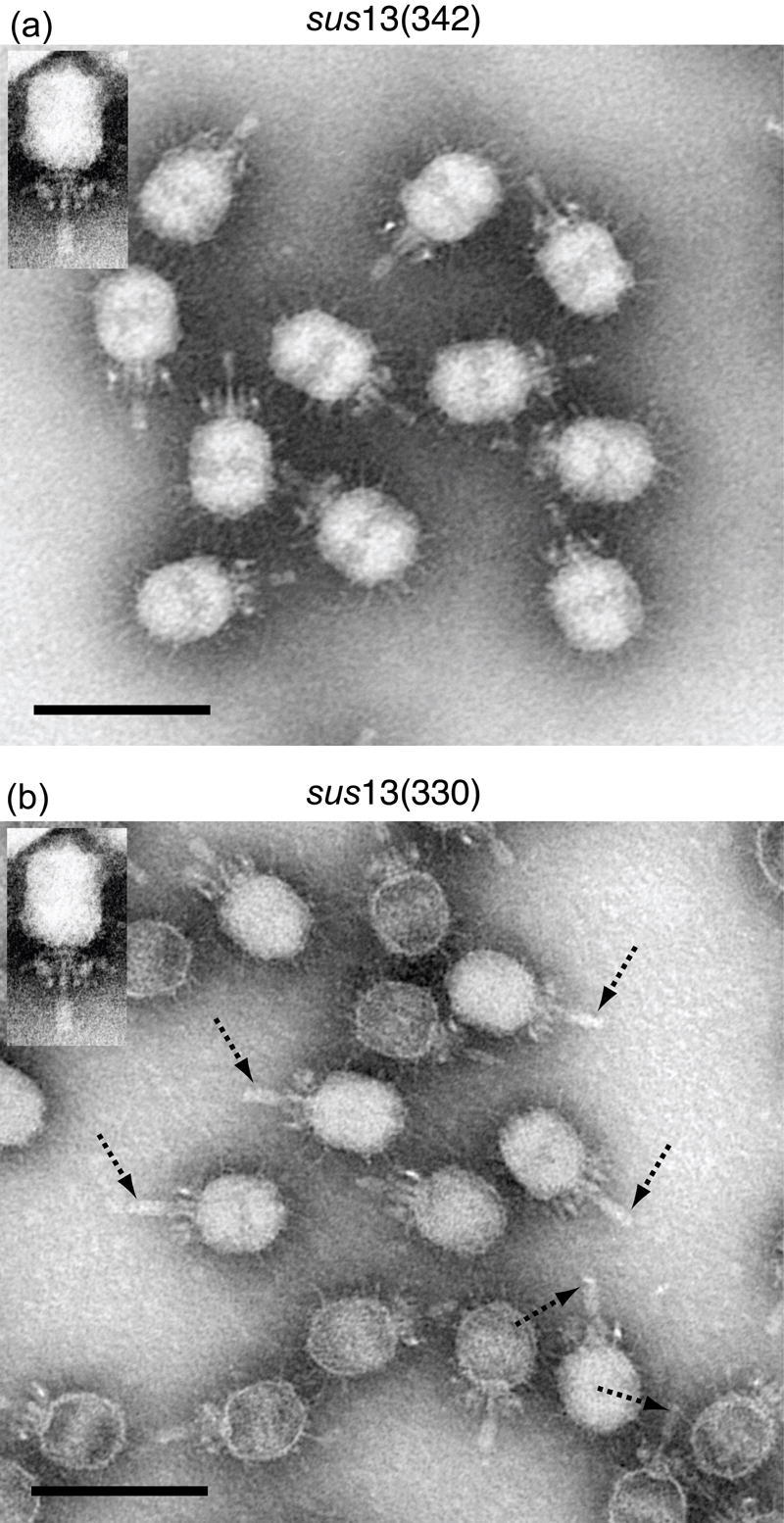
Transmission electron microscopy of gp13-defective mutant particles. Particles produced in restrictive infection with the mutants _sus_13(342) (left panel) and _sus_13(330) (right panel) look grossly normal. However, on close inspection, _sus_13(330) particles have notched tail knobs (right panel, dashed arrows). For reference, ϕ29 is shown (insets). Negative staining with phosphotungstate. ϕ29 inset was stained with uranyl acetate. Magnification bar = 100 nm.
Table 3.
Aggregation of ϕ29 and gp13-defective mutants with antibodies to tail components
| Expt. | Particlea | Antibody | Total Particles | DNA Filled (%) | Particles Aggregated (%) |
|---|---|---|---|---|---|
| 1 | WT (1241) | gp13 | 574 | 549 (96) | 552 (96) |
| 13- (342) | gp13 | 272 | 64 (24) | 53 (20) | |
| 2 | 13- (330) | gp13 | 299 | 201 (67) | 6 (2) |
| 13- (342) | gp13 | 300 | 243 (81) | 189 (63) | |
| 3 | 13- (330) | gp13 | 355 | 257 (72) | 5 (1) |
| 13- (330) | gp9 | 501 | 444 (89) | 322 (64) | |
| 13- (330) | p.i.b | 305 | 215 (71) | 9 (3) |
Tails of _sus_13(342) mutant particles contain the short ochre fragment of gp13
Because _sus_13(342) particles did not show gp13 signal by Western blot (Figure 5), attempts were made to detect gp13 by immuno-EM. _sus_13(342) particles were incubated with anti-gp13 IgG-biotin and observed by negative staining in TEM (Figure 7(a)). As a control, wt ϕ29 particles were incubated with anti-gp13 IgG-biotin (Figure 7(b)). An average of 96% of wild-type particles aggregated, while an average of only 20% of _sus_13(342) particles aggregated, a difference of greater than 4-fold (Table 3). In addition to the aggregation frequency, genome retention capacity was measured for each type of particle (Table 3). While 96% of wt ϕ29 retained the genome under EM staining conditions, gp13 defective particles variably lost the packaged genome (Table 3). In two EM preparations of _sus_13(342) particles, mounts of freshly prepared particles on support films (within two days of isolation) showed 81% DNA-retention (Table 3, expt. 2, 13(342)) while 24% of thirty day-old particles retained the packaged genome (Table 3, expt. 1, 13(342)). Freshly-mounted _sus_13(342) particles aggregated at 63% (Table 3, expt. 2), and thus the aggregation frequency was similar to the genome retention. The aggregation of the sus13(342) particles suggested that the short 3.8kDa fragment (Table 2) of gp13 from the N-terminus, although expected to display fewer epitopes, is sufficiently exposed in the assembled particle. However, of the _sus_13(342) particles observed, generally only those that contained DNA participated in aggregation; empty _sus_13(342) particles aggregated infrequently (Figure 7(a); Table 3). Thus, the 3.8 kDa _sus_13(342) ochre fragment may exit with the DNA. Taken together, these observations suggest that gp13 also serves as a plug that aids genome retention in ϕ29.
DISCUSSION
A sensitive method with general applicability for identifying and localizing a minor protein structural constituent of a virus or other macromolecular complex is described. Western blot analyses and immuno-electron microscopy were used to identify gp13 as a previously undetected structural component of the phage ϕ29 tail, in addition to its role as a morphogenetic factor in assembly of the tail knob.1 gp13 was identified in ϕ29, ghosts, isolated tails and defective sus mutant particles by the use of gp13-specific antibodies, but not in proheads, becoming the eighth virion protein in the linear ϕ29 assembly pathway first proposed by Mendez et al.2 Mutant studies confirmed that gp13 is in the tail, as the predicted long ochre fragment of gp13 produced in restrictive infection with the mutant _sus_13(330) was identified in defective particles by Western blot analysis. Previous studies of gp131, 3, 15, 31 likely did not identify gp13 in virion or knob preparations because in some PAGE conditions tested for this study, purified gp13 co-migrated with gp10 and gp11 despite a 5-7kDa predicted difference in size (data not shown). A similar co-migration artifact has been reported for T4 gp11 and gp2632 and T7 proteins 6.7 and 1333 whose predicted mass differences are also ~6kDa. The use of gp13-specific antibodies provided the sensitivity required for gp13 detection. Historical literature review demonstrated an unidentified band between the connector (gp10) and lower collar (gp11) proteins in an autoradiogram of purified ϕ29 that may, in hindsight, have been gp13.13
The in vitro assembly activity of gp13 copurifies with gp9, yet the ϕ29 tail knob (gp9) assembles in cells infected with the mutant _sus_13(342), which produces a small 3.8kDa ochre fragment of gp13, to yield noninfectious particles that resemble ϕ291 (Figs. 4, 6 and 7; Table 1). [Similarly, the T7 gene product 6.7 has been suggested to be essential for infectivity but not for formation of morphologically normal particles.33] To produce infectious ϕ29, it was found that gp9 must interact with gp13 in vivo, suggesting that gp13 is a morphogenetic assembly factor.1 The λ tail protein gpM appears as a morphogenetic factor by the same rationale: gpM- lysates (gpG+ gpH+) cannot complement gpG- (gpM+ gpH+) nor gpH- (gpM+ gpG+) extracts from λ̃.34 While gpG is not a virion component, the presence or absence of gpM in the λ virion remains unclear35 and may be studied with antibody localization.
Table 1.
ϕ29 phages used in this study
| Phages | Source |
|---|---|
| _sus_8.5(900)_sus_14(1241) | 38 |
| _sus_12(305)_sus_14(1241) | 70 |
| _sus_12(305)_sus_13(55)_sus_14(1241) | Reilly et al., unpublished |
| _sus_13(336) | 28 |
| _sus_13(44) | 28 |
| _sus_13(53) | 28 |
| _sus_13(330)_sus_14(1241) | 3, 28 |
| _sus_13(342) | 1, 59 |
| _sus_13(342)_sus_14(1242) | 1 |
| _sus_14(1241)a | 59 |
| _sus_14(1241)_sus_16(300) | 10 |
The present work shows that gp13 is a virion protein that likely co-assembles with gp9 to produce the tail knob. The gp13/gp9 knob complex then attaches en bloc to the tail tube during ϕ29 morphogenesis. Attempted sequential addition of tail components gp9 and gp13 to assembly intermediates has utilized expression of ϕ29 genes in E. coli from restriction fragments that contain flanking genes.36, 37 The authors have argued for the ordering of gp9 activity prior to gp13; however, because partially assembled complexes were not purified away from residual proteins in the lysates, unreacted gp9 or gp13 may have remained available.36, 37 In previous reports involving gp13 defective lysates1, 3 the defective gp13 protein may have acted as a dominant-negative inactivating functional gp9 from subsequent reaction with wt gp13 and vice versa. The purported purified component experiments of Lee et al.36, 37 did not test if defective gp13 blocks gp9 action or vice versa.
Incorporation of gp13 into ϕ29 occurs before appendage (gp12*) attachment, because 12- particles aggregate with anti-gp13 IgG (Figure 3), consistent with prior reports.15 With the gp13/gp9 knob attached, the twelve appendages that function in adsorption assemble to yield the infectious virion. Since purified ϕ29 proteins such as head fibers38 and appendages18 are readily assembled to intermediates having DNA-filled heads in vitro, the requirement for in vivo assembly of the gp13/gp9 knob appears to define a distinct multi-protein tail sub-assembly pathway in a podophage. A model for this gp13/gp9 accessory or sub-assembly pathway is shown in Figure 8. The myo- and sipho-viridae (T4-like and λ-like, respectively) have tail assembly pathways independent of prohead/capsid assembly. In contrast, tails of the podophage T3 are assembled only on the head.39 However, assembly of the lambdoid podophage phage P22 gp9 into tail spikes in P22 virions40 can be considered a tail sub-assembly pathway,41 and similarly, oligomers of the ϕ29 appendages (gp12*) are preassembled.1 The ϕ29 gp13/gp9 sub-assembly proposed here involves multiple copies of two polypeptides, suggesting that ϕ29 uses a mixture of traditional podophage and myo/siphoviridae assembly pathway processes.
Figure 8.
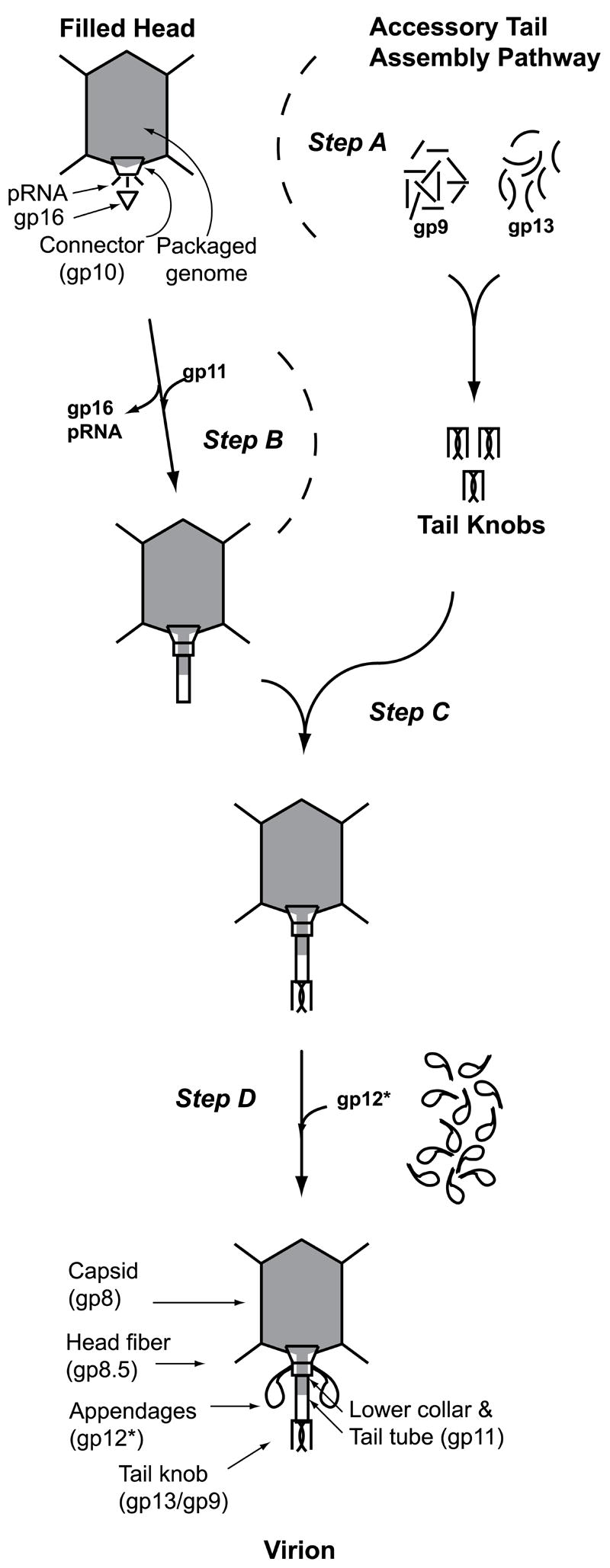
The ϕ29 Accessory Tail Assembly Pathway. gp13 and gp9 pre-assemble to form the tail knob (step A), and this complex is then attached en bloc onto the lower collar and tail tube (gp11) of parallel assembling filled heads (step B) as the penultimate morphogenesis step (C) that precedes attachment of the adsorption organelle appendages (gp12*) (step D) to form the infectious virion.
Secondary structure prediction and alignment to known crystal structures shows similarity between the gp13 C-terminal domain and LytM. LytM is a Zn2+-containing metalloprotease autolysin from Staphylococcus aureus that cleaves the pentaglycine cross-links of the staphylococcal cell wall.25, 27 The HxH Zn2+-binding motif of the M23 family (e.g. LytM [_Staphylococcus aureus_], Lysostaphin [_S. simulans_]) is present in ϕ29 gp13 and in the ϕ29 relatives GA-1 and B103.20 Other phage proteins, such as T4 gp12, contain the HxH motif and have been shown to bind Zn2+ in a fashion that aids oligomerization.42-44 Additionally, the tape measure proteins in λ and mycobacteriophages45 as well as the λ Ur short-tail fiber (stf gene product) contain HxH motifs (data not shown). The metal-chelating role for these HxH motifs that aids protein oligomerization or enzymatic activity has yet to be demonstrated.
Bacteriophage tail-associated enzymes exemplify highly evolved virus entry machines. Biochemical studies of the T7-like phage K1-E,46 the P22 endorhamnosidase tail spike (gp9)47, 48 and the ϕ29 relative GA-1 appendages (gp12*)49 have demonstrated the critical role of oligomeric adsorption/cell wall degrading complexes for infectivity. Low copy number enzymes can also facilitate cell wall puncture. The tail-associated lysozyme gp5 of T450 is present in only three copies in each virion and is critically located on the tail axis near the bottom of the tail tube.51 T4 does not appear to have the radially oriented enzymatic complexes that are present in phages K1-E, P22 and the appendages of GA-1/ϕ29.49 Conversely, the phage tails of P22 and K1-E do not appear to have axially positioned enzymes like T450, 51 or ϕ29 (this study). Thus ϕ29 emerges as hybrid of structures and functions with both axially and radially positioned enzymes required for virus puncture of the cell wall.
The ϕ29 tail protein gp13 closely resembles a metalloendopeptidase, and low copy number virion proteins are likely present in other viruses. In seeking a gp13 counterpart in T4, gp26 of T452 may be a candidate. gp26, which has been recovered from isolated T4 baseplates and studied with gp26-specific antibodies,32 is likely present in only a few copies and has been suggested to be attached to the distal gp5 trimer at the tip of the T4 tail tube.53
Immuno-EM of ϕ29 and _sus_13(342) particles localized gp13 to the distal tip of the tail knob (Figs. 3 and 7). Further, aggregation of _sus_13(342) defective particles with anti-gp13 IgG (Table 3) demonstrated that gp13 epitopes are accessible, suggesting that the short 3.8 kDa fragment, the gp13 N-terminus, is exposed at the tip of the knob. In this position, full length gp13 likely serves as a plug to help restrain the highly pressurized packaged genome. The defective particles produced in the restrictive gene 13 sus mutant infections were unstable and readily lost their DNA (Figure 7, Table 3). Also, prior work attributed ϕ29 genome stability to gp13,1, 3 in a similar fashion to gp4 and gp10 of phage P22.54 In the absence of these proteins, ϕ29 and P22 particles lose their DNA and are rendered non-infectious. Non-functional gpZ in λ results in particles defective for DNA injection;55 this defect is because the DNA does not descend into the tail in preparation for injection.56 Immuno-EM may aid the localization of gpZ in a λ pretail assembly intermediate.
In addition to its function of in stabilizing the particle, gp13 is likely the first viral protein to contact the B. subtilis cell wall. Both purified gp13 and ϕ29 show enzymatic activity on the Bacillus subtilis peptidoglycan (Cohen et al., in preparation). The mass observed distal to the “notch” in the tail knob of the defective _sus_13(330) particle by negative stain TEM (Figure 6(b)) likely corresponds to a cone-shaped protrusion in a cryo-EM reconstruction of the particle (Xiang et al., in preparation). The cone is similar to the knob protrusion in a cryo-EM reconstruction of ϕ29 ghosts (particles emptied of DNA by chemical treatment).22 This structure might form a channel for passage of DNA through the cell membrane. The dimensions of the cone suggest that it would have a mass approximately equivalent to one copy of gp9 (67 kDa) or two copies of gp13 (2×41 kDa). Whereas the immuno-EM data suggest that the cone of the sus13(330) particle is gp9 (Table 3), a crystal structure of gp13 may fit into the cryo-EM density (Xiang et al., in preparation). Atomic structures of distal knobs with and without gp13 will be needed to determine the molecular boundaries of gp9 and gp13 in this important infection organelle. The degradation of peptidoglycan by gp13 (Cohen et al., in preparation) likely enables tail knob penetration of the wall, access of the tail appendages (gp12*, Figure 1) to the outer cell wall to stabilize adsorption, and juxtaposing of the tail and cell membrane that triggers genome ejection.
METHODS
Phage Particle Preparation
Stocks of sus mutants were prepared by infection of the permissive host Bacillus subtilis su+44 (spoA- met- thr- sup-).57 ϕ29 with and without head fibers were produced in infections of the nonpermissive host Bacillus subtilis strain SpoOA12 (trpC2, spoOA12, _sup_-)58 with the suppressor-sensitive mutants _sus_14(1241),59 which confers the delayed lysis phenotype and assembles wild-type (wt) ϕ29 progeny, and _sus_8.5(900)_sus_14(1241) (fiberless ϕ29).28 Wild-type and fiberless particles were purified as described.3 DNA-free particles (ghosts) and tails were produced from chemically treated particles and separated by sucrose density gradient centrifugation (S. Grimes, personal communication).
gp13-defective particles were produced in infection of SpoOA12 with the mutants _sus_13(342)_sus_14(1242)1, 59 and _sus_13(330)_sus_14(1241).3 Briefly, 2×109 SpoOA12 cells per ml of 416 medium [2% Bactotryptone, 1% yeast extract, 170 mM NaCl] were infected at a multiplicity of infection (MOI) of 15, diluted ten-fold in fresh 416 and concentrated 20-fold by centrifugation 5-10 min prior to lysis. The lysates were treated with DNase I (50 μg/ml) and clarified by centrifugation. DNA containing particles were isolated by two successive isopycnic centrifugations in 60% (w/v) CsCl gradients, first in 416 medium and then in TMS buffer [50 mM Tris-HCl (pH 7.8), 10 mM MgCl2 and 100 mM NaCl] at 60,000 g for 30-40 hr at 4°C in the SW55 rotor. DNA-filled particles were stored at 4°C in CsCl and dialyzed on 0.025 μm filters (Millipore) against TMS buffer prior to use.
Proheads were isolated from lysates of SpoOA12 cells infected with the mutant _sus_14(1241)_sus_16(300) as described.60 Particles without appendages (gp12*-minus, 12- particles), were prepared after infection of SpoOA12 cells with the mutant _sus_12(305)_sus_14(1241),18 and the DNA-containing particles were isolated by centrifugation in 10-30% (w/v) sucrose density gradients at 45,000 rpm for 60 min at 4°C in the SW55 rotor.
Expression and purification of gp9, gp10, gp11 and gp13
ϕ29 gene product 10 (gp10) was expressed in Escherichia coli (pPLc28D1) and purified as described.61 gp9, gp11 and gp13 were expressed from cloned genes. Briefly, purified PCR products of gp13 amplified from purified ϕ29 DNA were cloned into pTYB1 (New England Biolabs, NEB, Ipswich, MA, NEB) using the NdeI-SapI sites, which generated a C-terminal intein fusion protein (NEB). The recombinant proteins were expressed in E. coli BL21(DE3) CodonPlus-RP cells (Stratagene, La Jolla, CA) and purified to homogeneity using chitin-affinity chromatography (NEB) per manufacture guidelines. Self-cleavage activity of the intein tag was induced by 50mM DTT. Free gp13 released from the chitin beads were further purified with a Superdex 200 column (GE Healthcare, Piscataway, NJ) and then concentrated to ~20 mg/ml. Similarly, purified PCR products of gp9 and gp11 amplified from purified ϕ29 DNA were cloned into pET30b (Novagen, EMD, Madison, WI) using the NcoI-XbaI and the NdeI-XbaI sites, respectively, which generated C-terminal hexa-histidine fusion proteins. The recombinant proteins of gp9 and gp11 were expressed in E. coli BL21(DE3) CodonPlus-RP cells and purified to homogeneity using NTA-Cobalt resin (BD) and a Superdex 200 column.
Antibody preparation and IgG Purification
Polyclonal antisera against purified ϕ29 proteins (gp9, gp11, gp13) were produced in rabbits by Rockland Immunochemicals Inc. (Gilbertsville, PA). Terminal bleed serum was delipidated by centrifugation at 10,000 g for 15 min; immunoglobulins were precipitated with 40% ammonium sulfate62 and resolubilized in PBS buffer, pH 8.0. The IgG fraction from each antiserum (gp9, gp11, gp13 and pre-immune) was isolated on separate protein A columns (Pierce, Rockford, IL). Polyclonal antiserum specific for gp8 was raised in rabbits against fiberless (gp8.5-), connector-free (gp10-) isometric particles produced in restrictive infection with the mutant _sus_8.5(900)_sus_10(302). Polyclonal antiserum prepared against purified connectors (gp10) has been described.61, 63 Biotinylation of purified IgG was performed using EZ Link NHS-LC-LC-Biotin (Pierce) following the product recommendations. Biotin-IgG was purified from free biotin-NHS using a PD10 column (Pharmacia, Piscataway, NJ), eluted with PBS and stored at 4°C.
Electrophoresis and Western blotting
Polyacylamide gel electrophoresis (PAGE) utilized 12% acrylamide precast Invitrogen Novex gels in MOPS buffer with samples prepared in lithium dodecylsulfate (LDS) buffer (Invitrogen, Carlsbad, CA) according to the manufacturer’s guidelines, with 10 mM dithiothreitol. Transfer of proteins to nitrocellulose membranes (Biotrace NT, Pall Corp, NY) was performed by standard procedures,64 and membranes were blocked in TS buffer [20 mM Tris-HCl (pH 7.4), 150 mM NaCl] with 1% w/v BSA (TS-B) and 0.1% v/v Tween-20 (TS-BT) at 4°C for at least two hours. Primary antisera raised to recombinant proteins (described above) was diluted 1:10,000 or 1:20,000 (final serum protein concentration 4-8 μg/ml) in TS-B and 0.05% Tween-20 and used to probe the blot for 2-3 hr at 4°C or 1 hr at 23°C. Antisera against isometric particles (anti-gp8) or connectors (anti-gp10) were diluted 1:100,000 in TS-B and 0.05% Tween-20 as above. Blots were washed free of serum with TS-BT and probed with goat anti-rabbit-horseradish peroxidase secondary antibody diluted 1:20,000 in TS-BT for 1 hr at 4°C or 23°C, washed with TS-T and imaged in Super-Signal West Pico-enhanced chemiluminescent (ECL) reagent. CCD imaging of 10 sec delayed frames was performed using the Chemidoc system (Biorad, Hercules, CA), and unsaturated images were used for all analyses. Primary antibodies to purified proteins were from Rockland Immuno Chemicals (Gilbertsville, PA), and secondary antibody and ECL reagents from Pierce (Rockland, IL).
Electron microscopy
Electron microscopy of specimens mounted on thin carbon films over fenestrated supports or 0.15% polyvinyl formal (Formvar) films on 400-mesh copper grids (Fullam Inc., Clifton Park, NY) was performed at 80 keV using a Philips EM301 instrument.
Immuno-electron microscopy was performed with purified immunoglobulin type G (IgG) specific to gp13, gp9 and gp8 diluted in TMS or pre-immune non-specific IgG from the same animal used for gp13 specific preparation, as adapted from prior work.24 Immuno-gold labeling of 2×1010 phage particles was conducted with 4 μg biotin-conjugated IgG and strepavidin-6 nm gold (Aurion Inc., Costerwegs, The Netherlands), adapted from prior studies.65 Phage particle/antibody complexes were incubated on support films and stained with phosphotungstate (PTA), pH 7. Streptavidin-gold labeled complexes were prepared from particle-IgG complexes separated from free immunoglobulins by size exclusion chromatography using HPLC, System Gold (Beckman, Fullerton, CA).
Aggregation frequency
The frequency of aggregation was determined either from electron micrographs or by direct grid visualization. Tail aggregation was defined as two or more virus particles that had coincident tails on the surface where the knobs were within a knob-width from each other. Particles 1) with deformed or otherwise abnormal tail morphologies, 2) lacking sufficient staining to visualize the entire virion or 3) with tails incompletely visible in the micrograph or grid space were excluded from measurements.
Secondary structure prediction modeling
Secondary structure predictions of gp13 (GenBank accession no. P15132) and crystal structure searches were performed via the Bioinfobank metaserver with Profsec and the 3D Jury approach (<http://bioinfo.pl/meta>)66 and with Psipred (<http://bioinf.cs.ucl.ac.uk/psipred/>)67 and mGenTHREADER.68
Phage genome isolation and sequencing
Phages containing sus mutations were purified by isopycnic CsCl gradient centrifugation19, 30 and used for genome extraction. 1010 particles were heated at 65°C in TE buffer [50 mM Tris, pH 7.8, 10 mM EDTA] and 10 mg/ml proteinase K to release the genome as described.69 Nucleic acid was extracted twice with phenol:isoamyl alcohol:Tris-HCl (pH 7.8), EtOH-precipitated and EtOH washed, lyophilized and resuspended in water. Gene 13 sequencing utilized 2 direct (5’AGACGGCGATAACTTTATT3’, 5’AATTAGCACAATTCCCAA3’) and 2 complementary (5’TGGGAATTGTGCTAATTG3’, 5’ACTGTATCAACAACCATACAA3’) primers that bind relative to the gene 13 start site at -99--81, +499-+516, +498-+515 and +1196-+1216 bp, respectively.
Mutants _sus_13(44), _sus_13(53) and _sus_13(336) were sequenced from single mutants, while the _sus_13(330) and _sus_13(55) mutants were sequenced from mutants that contained the additional _sus_14(1241) mutation that provides delayed lysis.59 The _sus_13(342) mutant was sequenced both from the single mutant background and the double mutant background with _sus_14(1242) which also provides the delayed lysis phenotype.59 The mutant _sus_13(55) was a triple mutant (_sus_12-_sus_13-_sus_14) and was not used further. Sequencing was performed at the University of Minnesota Biomedical Genomics core facility.
Acknowledgments
The authors are grateful to Dr. Margarita Salas for providing the gp13-defective mutants _sus_13(342) and _sus_13(342)14(1242). We thank Dr. Gary Dunny, Dr. Louis Mansky and members of our laboratories for critical reading of previous versions of the manuscript. We thank Dr. Shelley Grimes, Dr. Jaya Koti and Jesse Nease for providing some materials and Rockney Atz for bioinformatics assistance. This work was supported by NIH grant DE003606.
Abbreviations used
PAGE
polyacrylamide gel electrophoresis
TEM
transmission electron microscopy
cryo-EM
cryo-electron microscopy
gp
gene product
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Garcia JA, Carrascosa JL, Salas M. Assembly of the tail protein of the Bacillus subtilis phage ϕ29. Virology. 1983;125:18–30. doi: 10.1016/0042-6822(83)90060-0. [DOI] [PubMed] [Google Scholar]
- 2.Mendez E, Ramirez G, Salas M, Vinuela E. Structural proteins of bacteriophage ϕ29. Virology. 1971;45:567–576. doi: 10.1016/0042-6822(71)90172-3. [DOI] [PubMed] [Google Scholar]
- 3.Hagen EW, Reilly BE, Tosi ME, Anderson DL. Analysis of gene function of bacteriophage ϕ29 of Bacillus subtilis: identification of cistrons essential for viral assembly. J Virol. 1976;19:501–517. doi: 10.1128/jvi.19.2.501-517.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Anderson DL, Reilly BE. Morphogenesis of bacteriophage ϕ29. In: Sonenshein AL, Hoch JA, Losick R, editors. Bacillus subtilis and other gram-positive bacteria: Biochemistry, physiology, and molecular genetics. 2. American Society for Microbiology; Washington, DC: 1993. pp. 859–867. [Google Scholar]
- 5.Tao Y, Olson NH, Xu W, Anderson DL, Rossmann MG, Baker TS. Assembly of a tailed bacterial virus and its genome release studied in three dimensions. Cell. 1998;95:431–437. doi: 10.1016/s0092-8674(00)81773-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morais MC, Choi KH, Koti JS, Chipman PR, Anderson DL, Rossmann MG. Conservation of the capsid structure in tailed dsDNA bacteriophages: the pseudoatomic structure of ϕ29. Mol Cell. 2005;18:149–159. doi: 10.1016/j.molcel.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 7.Guo PX, Bailey S, Bodley JW, Anderson D. Characterization of the small RNA of the bacteriophage ϕ29 DNA packaging machine. Nucleic Acids Res. 1987;15:7081–7090. doi: 10.1093/nar/15.17.7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guo PX, Erickson S, Anderson D. A small viral RNA is required for in vitro packaging of bacteriophage ϕ29 DNA. Science. 1987;236:690–694. doi: 10.1126/science.3107124. [DOI] [PubMed] [Google Scholar]
- 9.Salas M, Mellado RP, Vinuela E, Sogo JM. Characterization of a protein covalently linked to the 5’ termini of the DNA of Bacillus subtilis phage ϕ29. J Mol Biol. 1978;119:269–291. doi: 10.1016/0022-2836(78)90438-2. [DOI] [PubMed] [Google Scholar]
- 10.Bjornsti MA, Reilly BE, Anderson DL. In vitro assembly of the Bacillus subtilis bacteriophage ϕ29. Proc Natl Acad Sci U S A. 1981;78:5861–5865. doi: 10.1073/pnas.78.9.5861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bjornsti MA, Reilly BE, Anderson DL. Bacteriophage ϕ29 proteins required for in vitro DNA-gp3 packaging. J Virol. 1984;50:766–772. doi: 10.1128/jvi.50.3.766-772.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo P, Grimes S, Anderson D. A defined system for in vitro packaging of DNA-gp3 of the Bacillus subtilis bacteriophage ϕ29. Proc Natl Acad Sci U S A. 1986;83:3505–3509. doi: 10.1073/pnas.83.10.3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nelson RA, Reilly BE, Anderson DL. Morphogenesis of bacteriophage ϕ29 of Bacillus subtilis: preliminary isolation and characterization of intermediate particles of the assembly pathway. J Virol. 1976;19:518–532. doi: 10.1128/jvi.19.2.518-532.1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Camacho A, Jimenez F, Vinuela E, Salas M. Order of assembly of the lower collar and the tail proteins of Bacillus subtilis bacteriophage ϕ29. J Virol. 1979;29:540–545. doi: 10.1128/jvi.29.2.540-545.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Krawiec S, Jimenez F, Garcia JA, Villanueva N, Sogo JM, Salas M. The orderly, in vitro emergence of DNA from bacteriophage ϕ29 particles. Virology. 1981;111:440–454. doi: 10.1016/0042-6822(81)90347-0. [DOI] [PubMed] [Google Scholar]
- 16.Bjornsti MA, Reilly BE, Anderson DL. Morphogenesis of bacteriophage ϕ29 of Bacillus subtilis: oriented and quantized in vitro packaging of DNA protein gp3. J Virol. 1983;45:383–396. doi: 10.1128/jvi.45.1.383-396.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carrascosa JL, Camacho A, Vinuela E, Salas M. A precursor of the neck appendage protein of B. subtilis phage ϕ29. FEBS Lett. 1974;44:317–321. [PubMed] [Google Scholar]
- 18.Tosi ME, Reilly BE, Anderson DL. Morphogenesis of bacteriophage ϕ29 of Bacillus subtilis: cleavage and assembly of the neck appendage protein. J Virol. 1975;16:1282–1295. doi: 10.1128/jvi.16.5.1282-1295.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anderson DL, Hickman DD, Reilly BE. Structure of Bacillus subtilis bacteriophage ϕ29 and the length of ϕ29 deoxyribonucleic acid. J Bacteriol. 1966;91:2081–2089. doi: 10.1128/jb.91.5.2081-2089.1966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Salas M. ϕ29 and its relatives. In: Calendar R, editor. The Bacteriophages. 2. Oxford University Press; New York, New York: 2006. pp. 315–330. [Google Scholar]
- 21.Carrascosa JL, Vinuela E, Garcia N, Santisteban A. Structure of the head-tail connector of bacteriophage ϕ29. J Mol Biol. 1982;154:311–324. doi: 10.1016/0022-2836(82)90066-3. [DOI] [PubMed] [Google Scholar]
- 22.Xiang Y, Morais MC, Battisti AJ, Grimes S, Jardine PJ, Anderson DL, Rossmann MG. Structural changes of bacteriophage ϕ29 upon DNA packaging and release. EMBO J. 2006;25:5229–5239. doi: 10.1038/sj.emboj.7601386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simpson AA, Tao Y, Leiman PG, Badasso MO, He Y, Jardine PJ, Olson NH, Morais MC, Grimes S, Anderson DL, Baker TS, Rossmann MG. Structure of the bacteriophage ϕ29 DNA packaging motor. Nature. 2000;408:745–750. doi: 10.1038/35047129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yanagida M, Ahmad-Zadeh C. Determination of gene product positions in bacteriophage T4 by specific antibody association. J Mol Biol. 1970;51:411–421. doi: 10.1016/0022-2836(70)90151-8. [DOI] [PubMed] [Google Scholar]
- 25.Odintsov SG, Sabala I, Marcyjaniak M, Bochtler M. Latent LytM at 1.3Å resolution. J Mol Biol. 2004;335:775–785. doi: 10.1016/j.jmb.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 26.Rawlings ND, Morton FR, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res. 2006;34:D270–2. doi: 10.1093/nar/gkj089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Firczuk M, Mucha A, Bochtler M. Crystal structures of active LytM. J Mol Biol. 2005;354:578–590. doi: 10.1016/j.jmb.2005.09.082. [DOI] [PubMed] [Google Scholar]
- 28.Reilly BE, Zeece VM, Anderson DL. Genetic study of suppressor-sensitive mutants of the Bacillus subtilis bacteriophage ϕ29. J Virol. 1973;11:756–760. doi: 10.1128/jvi.11.5.756-760.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vlcek C, Paces V. Nucleotide sequence of the late region of Bacillus phage ϕ29 completes the 19,285-bp sequence of ϕ29 genome. Comparison with the homologous sequence of phage PZA. Gene. 1986;46:215–225. doi: 10.1016/0378-1119(86)90406-3. [DOI] [PubMed] [Google Scholar]
- 30.Peterson C, Simon M, Hodges J, Mertens P, Higgins L, Egelman E, Anderson D. Composition and mass of the bacteriophage ϕ29 prohead and virion. J Struct Biol. 2001;135:18–25. doi: 10.1006/jsbi.2001.4375. [DOI] [PubMed] [Google Scholar]
- 31.Jimenez F, Camacho A, De La Torre J, Vinuela E, Salas M. Assembly of Bacillus subtilis phage ϕ29. 2. Mutants in the cistrons coding for the non-structural proteins. Eur J Biochem. 1977;73:57–72. doi: 10.1111/j.1432-1033.1977.tb11291.x. [DOI] [PubMed] [Google Scholar]
- 32.Ye N, Nemoto N. Processing of the tail lysozyme (gp5) of bacteriophage T4. J Bacteriol. 2004;186:6335–6339. doi: 10.1128/JB.186.18.6335-6339.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Molineux IJ. The T7 group. In: Calendar R, editor. The Bacteriophages. 2. Oxford University Press; New York, New York: 2006. pp. 277–301. [Google Scholar]
- 34.Katsura I. Tail assembly and injection. In: Hendrix RW, Roberts JW, Stahl FW, Weisberg RA, editors. Lambda II. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 1983. pp. 331–346. [Google Scholar]
- 35.Hendrix RW, Casjens S. Bacteriophage lambda and its genetic neighborhood. In: Calendar R, editor. The Bacteriophages. 2. Oxford University Press; New York: 2006. pp. 409–447. [Google Scholar]
- 36.Lee CS, Guo P. A highly sensitive system for the in vitro assembly of bacteriophage ϕ29 of Bacillus subtilis. Virology. 1994;202:1039–1042. doi: 10.1006/viro.1994.1434. [DOI] [PubMed] [Google Scholar]
- 37.Lee CS, Guo P. In vitro assembly of infectious virions of double-stranded DNA phage ϕ29 from cloned gene products and synthetic nucleic acids. J Virol. 1995;69:5018–5023. doi: 10.1128/jvi.69.8.5018-5023.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reilly BE, Nelson RA, Anderson DL. Morphogenesis of bacteriophage ϕ29 of Bacillus subtilis: mapping and functional analysis of the head fiber gene. J Virol. 1977;24:363–377. doi: 10.1128/jvi.24.1.363-377.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsuo-Kato H, Fujisawa H, Minagawa T. Structure and assembly of bacteriophage T3 tails. Virology. 1981;109:157–164. doi: 10.1016/0042-6822(81)90480-3. [DOI] [PubMed] [Google Scholar]
- 40.Goldenberg DP, Berget PB, King J. Maturation of the tail spike endorhamnosidase of Salmonella phage P22. J Biol Chem. 1982;257:7864–7871. [PubMed] [Google Scholar]
- 41.Prevelige, Peter E., Jr . Bacteriophage P22. In: Calendar R, editor. The Bacteriophages. 2. Oxford University Press; New York, New York: 2006. pp. 457–468. [Google Scholar]
- 42.Zorzopulos J, Kozloff LM. Identification of T4D bacteriophage gene product 12 as the baseplate zinc metalloprotein. J Biol Chem. 1978;253:5543–5547. [PubMed] [Google Scholar]
- 43.Kozloff LM, Zorzopulos J. Zinc uptake and incorporation into proteins in T4D bacteriophage-infected Escherichia coli. J Biol Chem. 1978;253:5548–5550. [PubMed] [Google Scholar]
- 44.Thomassen E, Gielen G, Schutz M, Schoehn G, Abrahams JP, Miller S, van Raaij MJ. The structure of the receptor-binding domain of the bacteriophage T4 short tail fiber reveals a knitted trimeric metal-binding fold. J Mol Biol. 2003;331:361–373. doi: 10.1016/s0022-2836(03)00755-1. [DOI] [PubMed] [Google Scholar]
- 45.Piuri M, Hatfull GF. A peptidoglycan hydrolase motif within the mycobacteriophage TM4 tape measure protein promotes efficient infection of stationary phase cells. Mol Microbiol. 2006;62:1569–1585. doi: 10.1111/j.1365-2958.2006.05473.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stummeyer K, Schwarzer D, Claus H, Vogel U, Gerardy-Schahn R, Muhlenhoff M. Evolution of bacteriophages infecting encapsulated bacteria: lessons from Escherichia coli K1-specific phages. Mol Microbiol. 2006;60:1123–1135. doi: 10.1111/j.1365-2958.2006.05173.x. [DOI] [PubMed] [Google Scholar]
- 47.Iwashita S, Kanegasaki S. Smooth specific phage adsorption: endorhamnosidase activity of tail parts of P22. Biochem Biophys Res Commun. 1973;55:403–409. doi: 10.1016/0006-291x(73)91101-7. [DOI] [PubMed] [Google Scholar]
- 48.Baxa U, Steinbacher S, Miller S, Weintraub A, Huber R, Seckler R. Interactions of phage P22 tails with their cellular receptor, Salmonella O-antigen polysaccharide. Biophys J. 1996;71:2040–2048. doi: 10.1016/S0006-3495(96)79402-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwarzer D, Stummeyer K, Gerardy-Schahn R, Muhlenhoff M. Characterization of a novel intramolecular chaperone domain conserved in endosialidases and other bacteriophage tail spike and fiber proteins. J Biol Chem. 2007;282:2821–2831. doi: 10.1074/jbc.M609543200. [DOI] [PubMed] [Google Scholar]
- 50.Mosig G, Lin GW, Franklin J, Fan WH. Functional relationships and structural determinants of two bacteriophage T4 lysozymes: a soluble (gene e) and a baseplate-associated (gene 5) protein. New Biol. 1989;1:171–179. [PubMed] [Google Scholar]
- 51.Kanamaru S, Leiman PG, Kostyuchenko VA, Chipman PR, Mesyanzhinov VV, Arisaka F, Rossmann MG. Structure of the cell-puncturing device of bacteriophage T4. Nature. 2002;415:553–557. doi: 10.1038/415553a. [DOI] [PubMed] [Google Scholar]
- 52.Coombs DH, Arisaka F. T4 tail structure and function. In: Karam JD, editor. Molecular Biology of Bacteriophage T4. American Society for Microbiology; Washington, DC: 1994. pp. 259–281. [Google Scholar]
- 53.Rossmann MG, Mesyanzhinov VV, Arisaka F, Leiman PG. The bacteriophage T4 DNA injection machine. Curr Opin Struct Biol. 2004;14:171–180. doi: 10.1016/j.sbi.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 54.Strauss H, King J. Steps in the stabilization of newly packaged DNA during phage P22 morphogenesis. J Mol Biol. 1984;172:523–543. doi: 10.1016/s0022-2836(84)80021-2. [DOI] [PubMed] [Google Scholar]
- 55.Casjens S, Hendrix R. Comments on the arrangement of the morphogenetic genes of bacteriophage lambda. J Mol Biol. 1974;90:20–25. doi: 10.1016/0022-2836(74)90253-8. [DOI] [PubMed] [Google Scholar]
- 56.Thomas JO, Sternberg N, Weisberg R. Altered arrangement of the DNA in injection-defective lambda bacteriophage. J Mol Biol. 1978;123:149–161. doi: 10.1016/0022-2836(78)90318-2. [DOI] [PubMed] [Google Scholar]
- 57.Mellado RP, Vinuela E, Salas M. Isolation of a strong suppressor of nonsense mutations in Bacillus subtilis. Eur J Biochem. 1976;65:213–223. doi: 10.1111/j.1432-1033.1976.tb10408.x. [DOI] [PubMed] [Google Scholar]
- 58.Spizizen J. Analysis of asporogenic mutants in Bacillus subtilis by genetic transformation. In: Campbell LL, Halvorson HO, editors. Spores III. American Society for Microbiology; Ann Arbor, MI: 1965. pp. 125–137. [Google Scholar]
- 59.Moreno F, Camacho A, Vinuela E, Salas M. Suppressor-sensitive mutants and genetic map of Bacillus subtilis bacteriophage ϕ29. Virology. 1974;62:1–16. doi: 10.1016/0042-6822(74)90298-0. [DOI] [PubMed] [Google Scholar]
- 60.Grimes S, Anderson D. The bacteriophage ϕ29 packaging proteins supercoil the DNA ends. J Mol Biol. 1997;266:901–914. doi: 10.1006/jmbi.1996.0843. [DOI] [PubMed] [Google Scholar]
- 61.Turnquist S, Simon M, Egelman E, Anderson D. Supercoiled DNA wraps around the bacteriophage ϕ29 head-tail connector. Proc Natl Acad Sci U S A. 1992;89:10479–10483. doi: 10.1073/pnas.89.21.10479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Harlow E, Lane D. Antibodies: A laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York, USA: 1988. [Google Scholar]
- 63.Rajagopal BS, Reilly BE, Anderson DL. Bacillus subtilis mutants defective in bacteriophage ϕ29 head assembly. J Bacteriol. 1993;175:2357–2362. doi: 10.1128/jb.175.8.2357-2362.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sambrook J, Fritsch E, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York, USA: 1989. [Google Scholar]
- 65.McGrath S, Neve H, Seegers JF, Eijlander R, Vegge CS, Brondsted L, Heller KJ, Fitzgerald GF, Vogensen FK, van Sinderen D. Anatomy of a lactococcal phage tail. J Bacteriol. 2006;188:3972–3982. doi: 10.1128/JB.00024-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ginalski K, Elofsson A, Fischer D, Rychlewski L. 3D-Jury: a simple approach to improve protein structure predictions. Bioinformatics. 2003;19:1015–1018. doi: 10.1093/bioinformatics/btg124. [DOI] [PubMed] [Google Scholar]
- 67.Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292:195–202. doi: 10.1006/jmbi.1999.3091. [DOI] [PubMed] [Google Scholar]
- 68.McGuffin LJ, Jones DT. Improvement of the GenTHREADER method for genomic fold recognition. Bioinformatics. 2003;19:874–881. doi: 10.1093/bioinformatics/btg097. [DOI] [PubMed] [Google Scholar]
- 69.Grimes S, Anderson D. In vitro packaging of bacteriophage ϕ29 DNA restriction fragments and the role of the terminal protein gp3. J Mol Biol. 1989;209:91–100. doi: 10.1016/0022-2836(89)90172-1. [DOI] [PubMed] [Google Scholar]
- 70.Tosi M, Anderson DL. Antigenic properties of bacteriophage ϕ29 structural proteins. J Virol. 1973;12:1548–1559. doi: 10.1128/jvi.12.6.1548-1559.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In: Walker JM, editor. The proteomics protocols handbook. Humana Press; Totowa, NJ: 2005. pp. 571–607. [Google Scholar]