Differential Regulation of the Cellular Response to DNA Double-strand Breaks in G1 (original) (raw)
. Author manuscript; available in PMC: 2009 Apr 11.
Summary
Double-strand breaks (DSBs) are potentially lethal DNA lesions that can be repaired by either homologous recombination (HR) or non-homologous end-joining (NHEJ). We show that DSBs induced by ionizing radiation (IR) are efficiently processed for HR and bound by Rfa1 during G1, while endonuclease-induced breaks are recognized by Rfa1 only after the cell enters S phase. This difference is dependent on the DNA end-binding Yku70/Yku80 complex. Cell cycle regulation is also observed in the DNA damage checkpoint response. Specifically, the 9-1-1 complex is required in G1 cells to recruit the Ddc2 checkpoint protein to damaged DNA, while upon entry into S phase, the cyclindependent kinase Cdc28 and the 9-1-1 complex both serve to recruit Ddc2 to foci. Together these results demonstrate that the DNA repair machinery distinguishes between different types of damage in G1, which translates into different modes of checkpoint activation in G1 and S/G2 cells.
Keywords: Homologous recombination, Rad52, Mre11, RPA, Ddc1, Ddc2, Mec3, Yku70, ionizing radiation, I-SceI endonuclease
Introduction
Cells encounter many kinds of DNA lesions, such as thymidine dimers created by ultraviolet radiation, single strand breaks and base changes from oxidative damage, DNA double-strand breaks resulting from ionizing radiation (IR) and collapsed forks from DNA replication stress. Many of these lesions utilize specific repair pathways, which require differential processing of the lesion. For example, damage affecting only one strand of the DNA duplex may be repaired most effectively via excision repair pathways, which remove the damaged strand and replace it with an intact copy using the undamaged complementary strand as a template. In contrast, double-strand breaks are repaired via either non-homologous endjoining (NHEJ) or homologous recombination (HR). Both NHEJ and HR require the Mre11- Rad50-Xrs2 (MRX) complex in S. cerevisiae. In addition, NHEJ requires the Yku70/Yku80 heterodimer, Lif1, Nej1, and ligase IV (Dnl4), while HR requires proteins of the RAD52 epistasis group (Krogh and Symington, 2004). NHEJ, which occurs preferentially in G1 (Moore and Haber, 1996; Takata et al., 1998), directly rejoins the DNA ends, and often results in loss of genetic information at the break site. HR, which occurs during S and G2 phase, requires a homologous template for repair and generally preserves genetic information at the break site (Aguilera and Rothstein, 2007; Moore and Haber, 1996; Paques and Haber, 1999). DNA repair pathways are coordinated with the cell cycle via checkpoint regulation as well as DNA processing events. The cyclin dependent kinase, Cdc28, is a key regulator of resection of DNA ends to create single-stranded DNA and the loading of RPA (ssDNA) at both telomeres and DSBs (Frank et al., 2006; Ira et al., 2004; Vodenicharov and Wellinger, 2006). Indeed, many members of the cell cycle and DNA replication machinery are implicated in DNA repair processes, and mutants show sensitivity to DNA damaging agents.
One of the first steps in DSB repair (DSBR) is binding of the exposed DNA ends by the MRX complex (Chen et al., 2001; Petrini and Stracker, 2003). Mre11 exhibits nuclease activity and aids in processing the ends into single-stranded DNA (ssDNA), which is next coated by replication protein A (RPA) (Alani et al., 1992; Krogh and Symington, 2004). RPA has multiple roles in DSBR; it acts to remove secondary structure in the ssDNA and it recruits the Ddc2 and Ddc1 checkpoint proteins as well as the Rad52 recombination protein to the lesion (Alani et al., 1992; Lisby et al., 2004; Zou and Elledge, 2003; Zou et al., 2003). Once a DSB has been recognized, the MRX-DNA and RPA-DNA complexes represent two independent checkpoint signals leading to the recruitment and activation of Tel1 and Mec1, respectively (Lisby et al., 2004; Usui et al., 2001). Subsequently, the heterotrimeric DNA repair clamp (9-1-1), composed of Rad17, Mec3 and Ddc1, is loaded onto DNA by the Rad24/Rfc2-5 clamp-loading complex (Majka and Burgers, 2003).
Here, we monitor the recruitment of checkpoint and recombination proteins into foci to analyze the recognition and processing of DSBs. We show that IR- and endonuclease-induced DSBs in G1 cells are differentially processed in a Ku-dependent fashion. Furthermore, the recruitment and activation of the checkpoint machinery is also cell cycle regulated. Rad53 activation absolutely requires the 9-1-1 complex in vivo and Cdc28 kinase activity is responsible for 9-1-1-independent recruitment of Ddc2 in S/G2 cells. These results demonstrate that the discrimination of different types of DSBs and the regulated recruitment of checkpoint proteins to the sites of DNA damage occur in a cell cycle-dependent manner.
Results
Multiple proteins recognize IR-induced DSBs in G1 cells
Previous experiments indicate that later steps of HR, such as DNA resection and recruitment of Rad52, may be inhibited in G1 cells (Ira et al., 2004; Lisby et al., 2001). However, we observed the induction of Rfa1, Mre11, Ddc1 and Ddc2 foci by IR at all phases of the cell cycle in asynchronously growing cells, suggesting that DSBs are recognized and resected in G1 phase (Lisby et al., 2004). Figure 1a shows that even when cells are held in G1 by α-factor, Mre11, Rfa1, Ddc1 and Ddc2 form foci in response to IR. On the other hand, significant levels of Rad52 foci do not form in G1 cells up to three hours after irradiation, indicating that the absence of Rad52 foci is due to cell cycle regulation rather than a temporal delay between the assembly of RPA and Rad52 into foci (Figure 1b). Consistently, when G1-irradiated cells are released into S phase, Rad52 foci readily form in more than 50% of the cells (Figure 1c). These results suggest that a substantial fraction of the lesions induced by IR in G1 are resected and not repaired by NHEJ. Rather, these DSBs persist until entry into S phase where they are recognized by the HR machinery (Aylon et al., 2004; Ira et al., 2004; Karathanasis and Wilson, 2002).
Figure 1. Cell cycle regulation of checkpoint and repair foci.
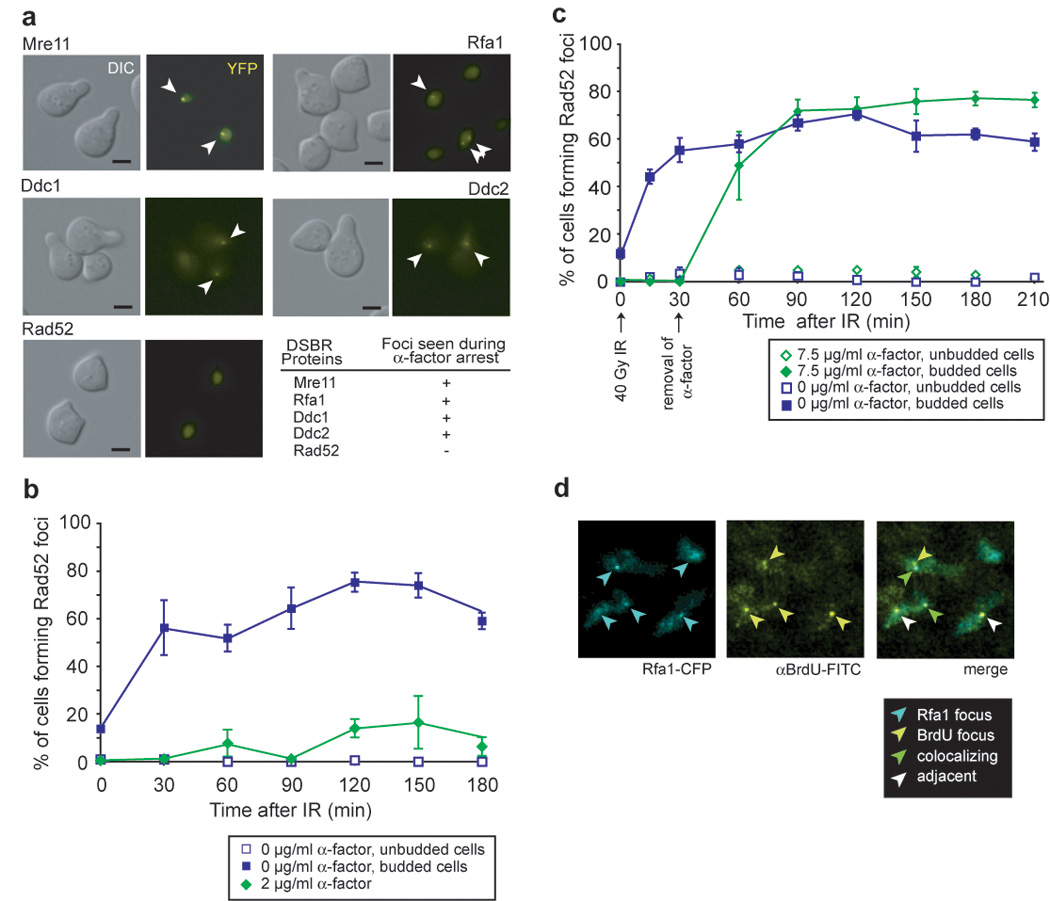
(a) IR-induction of foci in G1-arrested cells. Cells expressing Rfa1-YFP (W3775-12C), Mre11-YFP (W3483-10A), Ddc2-YFP (W3792-4B), Ddc1-YFP (W3923-12B) or Rad52-YFP (W3749-14C) were grown to OD600 ≈ 0.1 in the absence of exogenous DNA damage in SC medium supplemented with 2 µg/ml α-factor for 90 minutes and the cultures subsequently exposed to IR. Images were acquired 60 minutes after exposure to IR. The G1-arrest was confirmed by FACS analysis. Scale bar, 3 µm. Arrowheads mark foci in representative cells.
(b) Reduced efficiency of Rad52 focus formation in response to IR in G1-arrested cells. After 120 minutes, approximately 10% of cells form foci consistent with the percentage of the population escaping arrest as measured by FACS analysis. Points and error bars represent the mean and SEM, unless otherwise noted.
(c) Rad52 focus formation upon entry into S phase. Cells expressing Rad52-YFP (W4965-8B) were arrested with 7.5 µg/ml α-factor, exposed to IR, and then incubated for another 30 minutes before they were filtered and placed in fresh medium.
(d) IR exposure induces BrdU foci during G1. After exposure to IR, subnuclear BrdU foci are visible in G1-arrested cells (W8127-21B) and approximately 60% (18/29 at 30 minutes after exposure, time point shown) colocalize with Rfa1 foci.
RPA is recruited to ssDNA in G1 in response to IR
We were surprised to find that Rfa1 foci form in G1 cells in response to IR (Figure 1a), since previous observations showed that endonuclease-induced DSBs are inefficiently resected in G1-arrested cells (Frank-Vaillant and Marcand, 2002; Ira et al., 2004). To look directly at ssDNA formation in response to IR-mediated damage, we monitored the formation of singlestrand DNA under non-denaturing conditions using anti-BrdU antibodies that only detect BrdU in single-stranded DNA. Cells were labeled with BrdU for two generations and arrested in G1 by exposure to alpha factor and irradiated with 40 Gy IR. Before IR exposure, very few BrdU foci were observed (3/92 nuclei examined). At 30 and 120 minutes after exposure to IR, a significant number of cells contain BrdU foci (29/104 and 31/133, respectively) indicating that ssDNA is formed following gamma irradiation. Interestingly, more than 60% of the IR-induced BrdU foci co-localize with Rfa1 foci as illustrated in Figure 1d, further indicating that the ssDNA created following IR exposure is bound by Rfa1.
Differential response to endonuclease- and IR-induced DSBs in G1
We find that IR-induced damage can create ssDNA and recruit Rfa1 into foci during G1. However, only limited resection has been shown in response to HO endonuclease-mediated breaks in G1 (Frank-Vaillant and Marcand, 2002; Ira et al., 2004). To explore this difference in the cellular response to IR- and endonuclease-induced DSBs, we examined the recruitment of Mre11, Ddc1, Ddc2, Rfa1 and Rad52 to an I-SceI endonuclease-mediated DSB. Mre11 is rapidly recruited to the I-SceI-induced DSBs in either G1 or S/G2 phases of the cell cycle (Figure 2a). In contrast to what we find after IR, Rfa1, Ddc1 and Ddc2 are not efficiently recruited to the I-SceI-induced break in G1 cells (Figure 2a), even though I-SceI cutting is efficient (Supplemental Figure S1). On the other hand, Ddc1, Ddc2, Rfa1 and Rad52 all rapidly form foci in budded cells (S/G2) after DSB induction (Figure 2a; Lisby et al., 2004). These observations confirm that, unlike IR-induced DSBs (Figure 1a), endonuclease-induced DSBs are not resected in G1 (Ira et al., 2004). One explanation for this difference in processing is due to distinct molecular structures at DSB ends generated by I-SceI and IR. While endonuclease-mediated cleavage of the DNA phosphate backbone leaves a 3’ hydroxyl group to prime DNA synthesis or for ligation in NHEJ, IR can leave a number of structures not easily handled by the DNA repair machinery without further processing, including breaks along the phosphate backbone and even damaged or missing bases (Supplemental Figure S2). Such DNA ends likely require processing by the MRX complex before the recruitment of checkpoint machinery and subsequent repair.
Figure 2. Differential recruitment of checkpoint and repair proteins to IR- and endonuclease-induced DSBs.
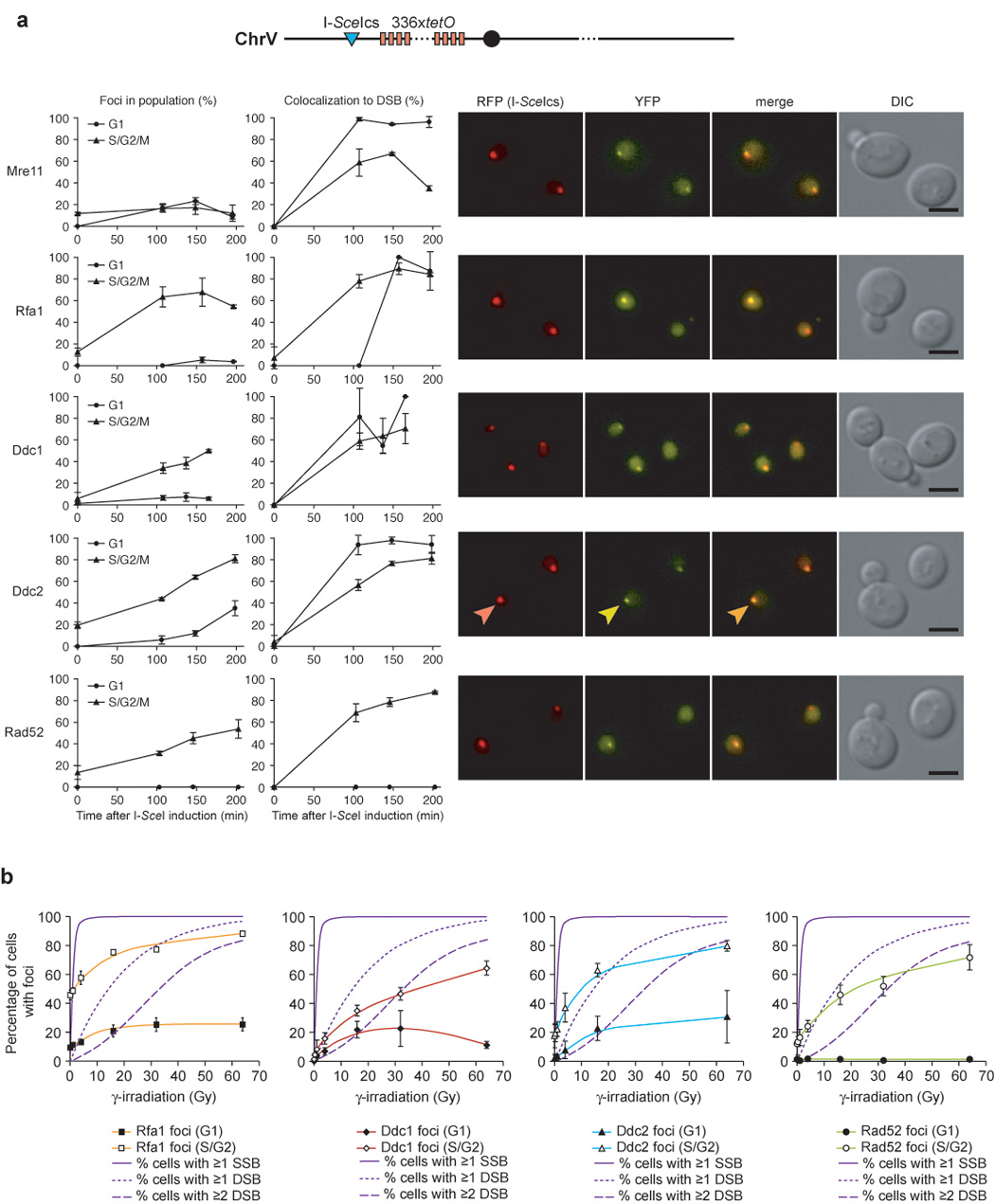
(a) Focus formation in response to an I-SceI-induced DSB. Expression of the I-SceI endonuclease was induced in cells harboring a fluorescently-marked I-SceI cut-site (I-SceIcs) and either Mre11-YFP (W4363-4B), Rfa1-YFP (W4362-1C), Ddc1-YFP (W4688-11D), Ddc2-YFP (W4364-9B) or Rad52-YFP (W4365-5B). The I-SceIcs is adjacent to an array of 336 copies of the Tet operator sequence in cells expressing the tet repressor fused to monomeric RFP, marking the locus as a discrete RFP dot. Focus formation and the percentage of colocalization with the I-SceI cut-site for each protein is plotted as a function of time after I-SceI was induced by addition of galactose. YFP, RFP, DIC and YFP/RFP merged images of representative cells are shown and selected foci indicated by arrowheads. Mre11 is recruited equally well to the I-SceI cut-site in G1 and S/G2/M. In contrast, Rfa1, Ddc1, and Ddc2 form very few foci in G1 cells in response to an I-SceI DSB.
(b) Proteins involved in DSBR do not respond to IR by forming foci at doses resulting only in SSBs. Dose dependency curves for Rfa1, Ddc1, Ddc2 and Rad52 foci, respectively, shown in the colored lines. The solid line indicates the predicted percentage of cells in the population receiving 1 or more SSBs, the dotted line indicates the predicted percentage of cells receiving 1 or more DSBs in response to IR, and the dashed line represents the predicted percentage of cells receiving 2 or more DSBs, assuming that 17 SSBs are generated for every DSB and that the number of breaks induced per cell follows a Poisson distribution (Friedland et al., 1999). None of the proteins form foci in response to low IR doses predominantly resulting in SSBs 60 minutes after exposure.
Low doses of IR that create single strand breaks do not induce DNA repair foci
Another possible explanation for the difference between IR and endonuclease-mediated DSBs is that IR creates damage other than DSBs. Exposure to IR leads to the formation of DNA strand breaks and hydroxyl radicals, which result in nicks in the DNA-phosphate backbone, as well as damage to the DNA bases themselves. Damage to the phosphate backbone has been reported to be the predominant form of DNA damage resulting in a 3’ terminal phosphate or a 3’ end that is not a hydroxyl, both of which require end processing before repair proceeds (Henner et al., 1982). The effect of photonic energy from a Cobalt 60 radiation source on human nuclei generates approximately 17 single-strand breaks (SSBs) for every DSB (Friedland et al., 1999). In addition, other studies in both yeast and mammalian cells show a linear relationship between the observed number of DNA breaks and the radiation dose received (Cundari and Averbeck, 1987; Ogorek and Bryant, 1985a, b). To distinguish between SSBs and DSBs induced during exposure to IR, we looked at focus formation at very low doses of IR (1–4 Gy) where most cells have SSBs but few cells have a DSB. None of the proteins examined—Rfa1, Ddc1, Ddc2 or Rad52—form foci above the background level at low doses of IR below 4 Gy (Figure 2b). Rather, proteins involved in DSBR form foci when exposed to IR at levels consistent with the population of cells receiving 1 or more DSBs (see Figure 2b). These results support the notion that the Rfa1, Ddc1 and Ddc2 foci induced by IR in G1 cells are due to DSBs and not to single-strand breaks.
Rfa1 Focus Intensity Increases upon Entry into S phase
Binding of RPA to ssDNA is an early step required to initiate HR. To monitor the kinetics of RPA recruitment to DSBs, we measured the frequency and brightness of individual Rfa1 foci after exposure to IR. In G2 cells, Rfa1 foci form rapidly (>90% 30 minutes after IR exposure), and the number decreases within 60 minutes, suggesting that ssDNA disappears concomitant with repair of the lesion (Figure 3b). In contrast, Rfa1 foci formed in G1- arrested cells persist, indicating that DSBs are not repaired (Figure 3a). Concomitantly, IR-induced Rfa1 foci formed during G1 contain 3- to 6-fold less Rfa1 than those formed in S/G2. Although the brightness of G1 Rfa1 foci increases over time, the rate is 4- to 6-fold slower than in S/G2 cells (Figure 3a and Figure 3b, dashed lines), suggesting that resection is slow and less extensive in G1. Upon entry into S phase, Rfa1 focus intensity dramatically increases (3- to 6-fold, Figure 3c), likely reflecting an increase in the rate of resection once the cells progress into S phase. This analysis of Rfa1 foci (Figure 2a and Figure 3a–c) suggests that initiation of resection is regulated in response to the type of DNA damage and the rate of resection increases at the G1 to S transition.
Figure 3. Regulation and kinetics of Rfa1 focus formation.
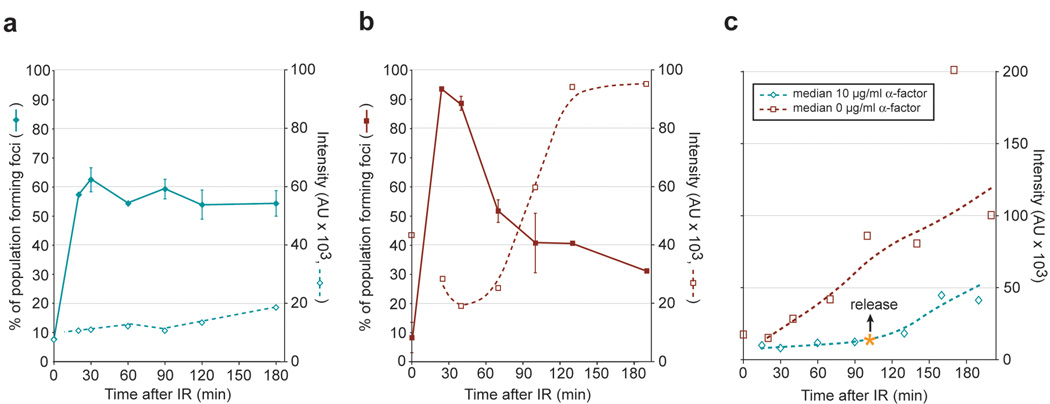
(a–c) Quantitation of Rfa1 foci in response to IR. Cells were arrested by either in G1 by α-factor (a and c) or in G2 by nocodazole (b) and then exposed to IR. Median values for the quantitation of individual Rfa1 focus intensity are plotted as dashed.
(a) Rfa1 focus formation in G1. Approximately 80% (0.6 / 0.75 = 0.80) of the cells receiving a DSB in G1 (75%) form an Rfa1 focus (60%) when cells are held in G1, however these foci are fainter than in budded cells.
(b) Rfa1 focus formation in G2. Rfa1 foci in G2-arrested cells are 4- to 6-fold brighter, and the focus intensity increases at a rate 3- to 6-fold faster than foci in G1.
(c) Cell cycle regulation of Rfa1 foci. The intensity of individual IR-induced Rfa1 foci was measured and the median plotted for cells arrested by α-factor in G1 and released at time = 100 min after exposure to IR. Upon entry into S phase, Rfa1 focus intensity increases at a rate that is 3- to 6-fold higher than in G1 cells, indicating that a faster rate of resection is taking place.
Yku70 inhibits Rfa1 focus formation at I-SceI-induced DNA ends
One possible explanation for the lack of recruitment of RPA to endonuclease-mediated DSBs in G1 is that these breaks are ideal substrates for NHEJ while those arising from IR are not. In NHEJ, the Yku70/Yku80 heterodimer first binds the DNA ends (Feldmann and Winnacker, 1993), followed by the ordered recruitment of the Nej1/Lif1 complex and the DNA ligase, Dnl4 (Critchlow and Jackson, 1998). Removal of the KU complex increases DNA resection in asynchronous cells (Lee et al., 1998) both at DSBs and at telomeres (Maringele and Lydall, 2002), suggesting that KU binding protects DNA ends from degradation. Therefore, we examined cells lacking a functional Yku70/Yku80 heterodimer. As shown in Figure 4a, yku70 Δ cells exhibit increased Rfa1 focus formation in G1 (17% of cells in WT compared to 43% in yku70Δ), indicating the presence of more free DNA ends available for resection. This result is consistent with a protection of nuclease-induced ends from resection by the Yku70/Yku80 complex. In contrast, Rfa1 focus formation is unaffected by deletion of DNA ligase IV (DNL4), a factor required for NHEJ that acts downstream of the Yku70/Yku80 complex (Supplemental Figure 2). Therefore, the increased Rfa1 focus formation in response to an endonuclease-mediated break in yku70Δ cells is not the result of lesions being processed for HR when NHEJ cannot be completed. Previous studies show that asynchronous populations of yku70Δ cells undergo ~2-fold more resection after a single endonuclease-mediated DSB (Lee et al., 1998). However, those studies did not distinguish between events in different phases of the cell cycle. Our results point to a direct effect of deleting Yku70/Yku80 on DNA end protection in G1 rather than an increase in the rate of resection.
Figure 4. G1 regulation of DSB resection.
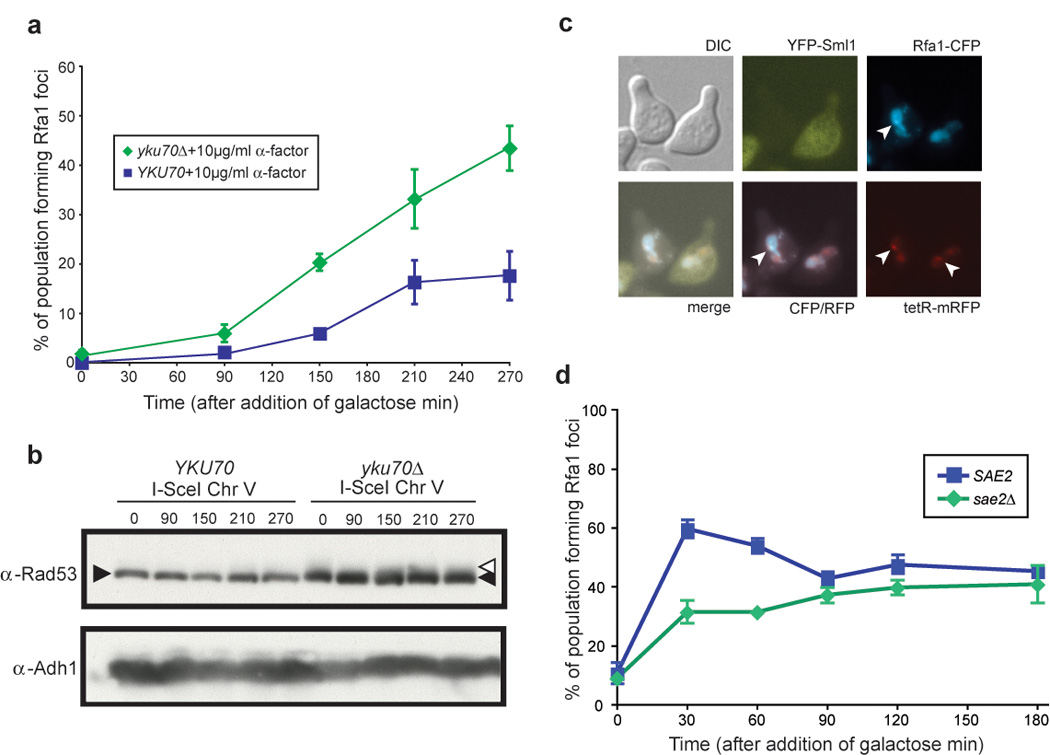
(a) Rfa1 focus formation in yku70Δ cells. Wild-type (W5713-18A) and yku70Δ (W5713-16D) cells were arrested with α-factor for 210 minutes after which an I-SceI-mediated DSB was induced by addition of galactose. Over the course of three hours, only 20% of wild-type cells accrue Rfa1 foci at the marked DSB. In contrast, 45% of yku70Δ cells have foci at the DSB by three hours after I-SceI induction.
(b) Rad53 phosphorylation is not activated in response to a single DSB, while there is constitutive checkpoint activation in yku70Δ cells, as indicated by the Rad53 smear at time 0.
(c) Rfa1 focus formation and Sml1 degradation in response to an endonuclease-mediated DSB in G1. As in (a), wild-type (W7542-11D) cells were arrested in G1, and followed by induction of I-SceI endonuclease. The cells forming Rfa1 foci in response to DSB induction also degrade Sml1, indicating downstream checkpoint induction though Rad53 phosphorylation is not visible by western blot.
(d) Rfa1 focus formation in sae2Δ cells. Cells were arrested in G1 then exposed to IR. The abrogation of SAE2 (W5071-5D), leads to a slower rate of Rfa1 focus formation in G1 cells, as sae2Δ cells have fewer Rfa1 foci at 30 and 60 minutes after exposure to IR compared to WT cells.
Sae2 and Mre11 are not responsible for the differential G1 processing of IR vs.endonuclease-mediated DSBs
Sae2, the S. cerevisiae homolog of human CtIP, has recently been shown to promote end resection, acting with the MRX complex to facilitate early steps in HR (Limbo et al., 2007; Sartori et al., 2007). To look specifically in G1 cells for an effect of Sae2 activity, we measured Rfa1 focus formation in cells arrested with α-factor. Cells lacking SAE2 show lower levels of Rfa1 foci at both 30 and 60 minutes after exposure to IR, indicating a delay in their appearance (Figure 4d). These results indicate a role for end processing by Sae2 prior to Rfa1 focus formation in response to IR-induced damage during G1. Sae2 acts in conjunction with the MRX complex, and Mre11 is recruited equally well to both IR-induced and endonuclease-mediated DSBs in G1 (Figure 1a and Figure 2a, respectively). To assess whether the MRX complex also has a role in the processing of DNA ends for resection in response to IR exposure in G1, we looked at the effect of mutating Mre11 nuclease activity on the appearance of Rfa1 foci in G1 cells. A histidine to asparagine change at position 125 abrogates both the endonuclease and exonuclease activities of Mre11 (Moreau et al., 1999). Cells harboring the mre11-H125N allele exhibit a moderate sensitivity to IR (10-fold decrease in survival at 500 Gy) yet show no obvious defect in processing DSBR intermediates into ssDNA (Moreau et al., 1999). Rfa1 focus formation is delayed in the mre11-H125N mutant, similar to sae2Δ cells, however the defect is less pronounced (Supplemental Figure S2). These results indicate that while Mre11 nuclease activity may facilitate the initial appearance of ssDNA in G1 cells, it is not the sole nuclease responsible for resection of DSBs. The similar phenotypes of mre11-H125N and SAE2 deletion on the kinetics of Rfa1 focus formation in response to IR corroborate the cooperative nature of Sae2 and the MRX complex to promote DSB end resection (Limbo et al., 2007; Sartori et al., 2007). However, deletion of SAE2 leads to a more striking delay in Rfa1 focus formation indicating that, while some lesions induced by IR may require the nuclease activity of Mre11 to create a structure recognizable by Sae2, others may not.
A single DSB is sufficient to generate a checkpoint response in G1
The 9-1-1 and Ddc2-Mec1 checkpoint complexes are recruited to foci in an RPA dependent fashion (Melo et al., 2001). To test whether appearance of these foci is accompanied by checkpoint activation, we investigated Rad53 phosphorylation and Sml1 degradation (Sanchez et al., 1999; Zhao et al., 2001; Figure 4b and 4c). We examined both WT and yku70Δ cells to determine if there is a difference in the checkpoint response following a single I-SceI cut in G1. Asynchronous yku70Δ cells show Rad53 activation even before endonuclease expression (Lee et al., 1998), and Sml1 is also degraded in virtually all yku70Δ cells, consistent with Rad53 activation (Corda et al., 2005). However, wild-type G1 cells do not show Rad53 phosphorylation, even 4.5 hours after I-SceI induction (Figure 4b, lane 5). Interestingly, Sml1 is degraded in ~20% of wild-type cells after I-SceI induction, the same population with Rfa1 foci (Figure 4b and 4c). To determine if the number of DNA DSBs in G1 affects the DNA damage response, we introduced two I-SceI target sites into a strain at two different loci. Even with two DSBs, Rad53 phosphorylation is not detected in G1 and Rfa1 focus formation does not occur at significantly higher levels (Supplemental Figure S3). Furthermore, Sml1 is degraded in those cells that contain Rfa1 foci. Together, these results indicate that Sml1 degradation is a more sensitive measure of checkpoint activation and that a single DSB is sufficient to induce a checkpoint response in G1.
The 9-1-1 complex is required for Ddc2 focus formation in G1
The 9-1-1 and Ddc2-Mec1 checkpoint complexes are recruited independently to sites of DNA damage by RPA in asynchronously growing cells (Lisby et al., 2004; Melo et al., 2001; Zou and Elledge, 2003). To determine if this holds true for G1 cells, α-factor arrested cells were examined for Ddc2 foci after gamma-irradiation. In G1 arrested cells where the 9-1-1 DNA damage clamp has been inactivated by deleting the MEC3 subunit, Ddc2 focus formation does not occur (Figure 5a). Similarly, Ddc2 foci do not form in the G1 population of asynchronously growing mec3Δ cells (Supplemental Figure S4). Indeed, in mec3Δ cells arrested in G1, Rad53, a downstream checkpoint kinase activated by Mec1, is not phosphorylated. These results demonstrate that the 9-1-1 complex is required for Mec1 activity in G1 cells (Figure 5b), which supports in vitro evidence showing that Mec1 kinase activity is greatly stimulated by RPA and the 9-1-1 complex (Majka et al., 2006). However, deletion of MEC3 has no effect on Rfa1 focus formation, indicating that the 9-1-1 clamp is not required to load RPA onto the DNA (Figure 5c). Together, these results demonstrate that Ddc2 focus formation is cell cycle regulated and the recruitment and activation of the Ddc2 Mec1 complex requires the 9-1-1 complex in G1.
Figure 5. Independent recruitment of checkpoint machinery 9-1-1 and Ddc2-Mec1 requiresCdc28 activity.
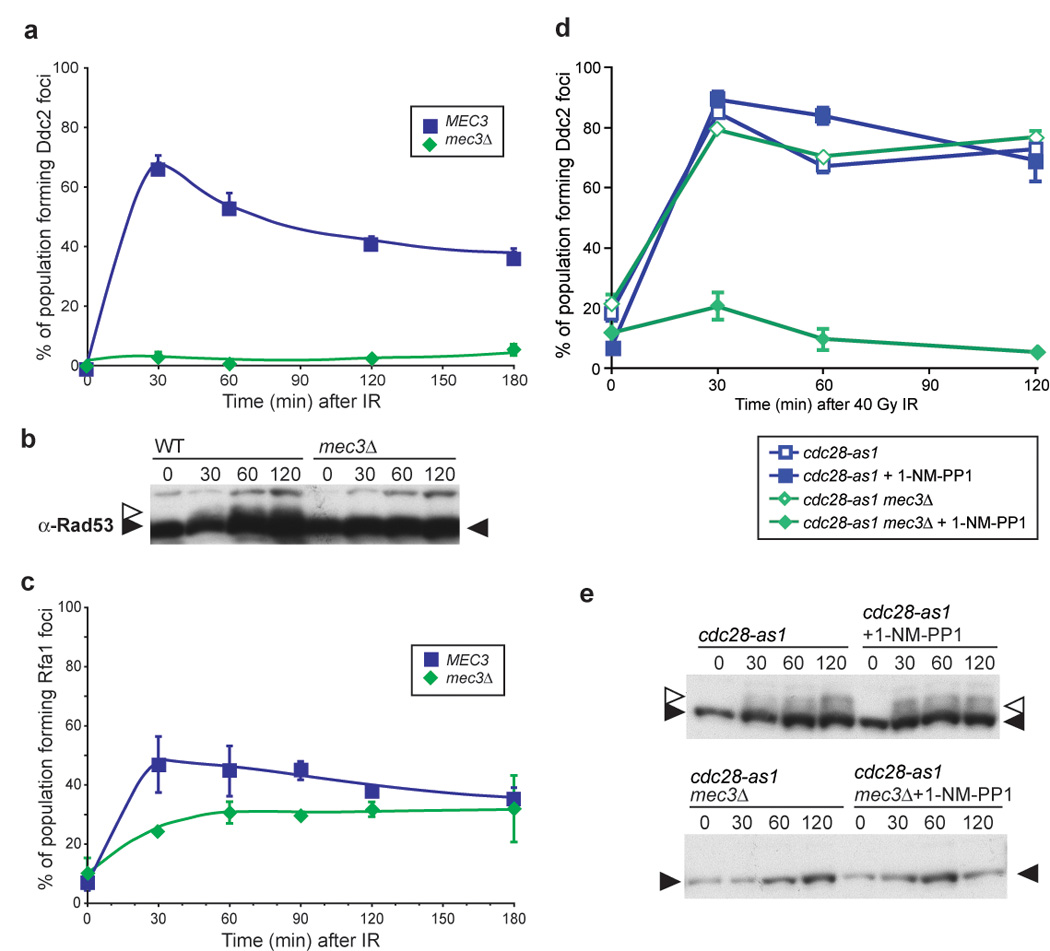
(a) Ddc2 focus formation in mec3Δ G1 cells. mec3Δ cells were arrested in 2 µg/ml α-factor for 90 minutes and the cultures subsequently exposed to IR, and images were acquired at the stated timepoints. Ddc2 focus formation does not occur in mec3Δ cells (W5358-9A) arrested in G1.
(b) Rad53 phosphorylation in WT and mec3Δ G1 cells. Rad53 is phosphorylated in WT G1 cells after exposure to 40 Gy IR, while mec3Δ cells do not.
(c) Rfa1 focus formation in mec3Δ G1 cells. Deletion of MEC3 does not prevent Rfa1 focus formation during G1 (W5793-10B).
(d) Ddc2 focus formation in S/G2 mec3Δ cells with and without Cdc28 kinase activity. Ddc2 forms foci in budded cells, regardless of the presence of an active DNA damage clamp. When Cdc28 kinase activity is abrogated by the addition of the inhibitor 1-NM-PP1 to a strain containing the cdc28-as1 allele (W7832-2A), no Ddc2 focus formation is observed after 40 Gy IR, indicating that Cdc28 kinase activity is required for recruitment of Ddc2-Mec1 to the sites of DNA damage when Mec3 is absent (W7832-1A).
(e) Rad53 phosphorylation in WT and mec3Δ cells. Inhibition of cdc28-as1 cells with 1-NM-PP1 does not alter Rad53 phosphorylation in WT or mec3Δ cells.
Cdc28 activity is required for Ddc2 focus formation in the absence of Mec3
The cyclin-dependent kinase Cdc28 is the major regulator of cell cycle processes. To determine if its activity governs Ddc2 focus formation, we utilized cdc28-as1, an allele of Cdc28 that contains a modification in the ATP binding pocket (Bishop et al., 2000). The kinase activity of Cdc28-as1 can be fully inhibited by addition of a non-hydrolyzable ATP analog 1-NM-PP1 that fits in the active site of the altered protein only and does not interfere with any other kinase activity present in the cell (Bishop et al., 2000). In response to IR, inhibition of Cdc28 alone does not affect Ddc2 focus formation. However, Cdc28 activity is essential for Ddc2 focus formation in S/G2 mec3Δ cells (Figure 5d). While the 9-1-1 complex alone recruits Ddc2 to DNA in G1, Cdc28 activity and 9-1-1 serve independently to recruit Ddc2 into foci in S/G2 cells, when Cdc28 kinase activity is highest.
To determine whether Ddc2 focus formation activates the damage checkpoint, we analyzed wild-type and mec3Δ cells for Rad53 phosphorylation. In response to 40 Gy IR, wild-type cells exhibit robust phosphorylation of Rad53. However, in _mec3Δ_-irradiated cells, Rad53 is not visibly phosphorylated even when Cdc28 is active (Figure 5e) and Ddc2 foci form (Figure 5d). These results indicate that even though Ddc2 forms foci in response to IR in mec3Δ cells, the downstream checkpoint is not activated without the presence of a functional DNA damage clamp.
RPA checkpoint functions maintain Ddc1 foci after DNA damage
RPA is required for the recruitment of Ddc1 and Ddc2 in vivo (Lisby et al., 2004; Melo et al., 2001), and has roles in both repair and checkpoint processes when cells suffer DNA damage (Umezu et al., 1998). All three subunits of the complex are modified as the cells enter S phase and in response to DNA damaging agents (Din et al., 1990; Kim and Brill, 2003; Ubersax et al., 2003). rfa1-t11 is a checkpoint-deficient allele of Rfa1 isolated for its sensitivity to UV and MMS, and it is also defective in the G2/M DNA damage checkpoint (Kim and Brill, 2001; Umezu et al., 1998). rfa1-t11 cells are fully functional in DNA replication, and the allele is characterized by a single amino acid change of lysine at position 45 to glutamic acid. We looked at Ddc1 and Ddc2 focus formation in the rfa1-t11 background to determine if the checkpoint functions of RPA are involved in recruiting these proteins after exposure to IR. Interestingly, we observe no effect of the rfa1-t11 allele on the initial assembly of Ddc1 foci, however the foci are transitory and disappear by 120 minutes after exposure to IR (Figure 6a). This result indicates that one of the checkpoint functions of wild-type Rfa1 is to maintain the localization of the DNA damage clamp at sites of damage. Furthermore, Ddc2 focus formation is also compromised in rfa1-t11 cells, as fewer cells form Ddc2 foci in response to 40 Gy IR (~60% compared to ~80% in wild-type cells) and the foci exhibit decreased intensity (Figure 6b, and data not shown). The effect of the rfa1-t11 allele on Ddc1 and Ddc2 focus formation suggests that RPA is important not only to recruit but also to maintain the presence of the 9-1-1 clamp and Ddc2-Mec1 checkpoint complexes at the damage site.
Figure 6. RPA checkpoint function is required to maintain Ddc1 foci.
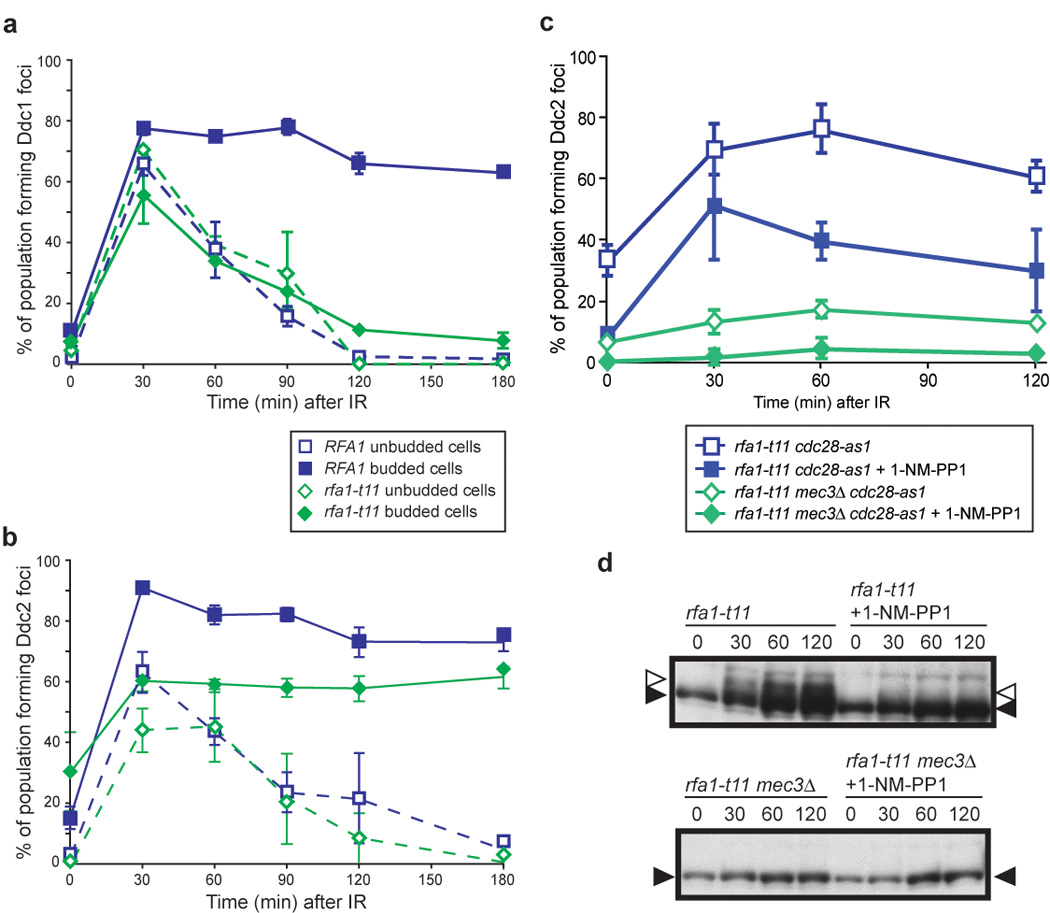
(a) Ddc1 focus formation in rfa1-t11 cells. Ddc1 forms foci in rfa1-t11 cells (W5872-3C), however these foci do not persist, and fully disappear by 120 min after exposure to IR, unlike in WT cells where the foci persist for over 3 hours.
(b) Ddc2 focus formation in rfa1-t11 cells. Ddc2 focus formation in rfa1-t11 cells (W5873-9B) is reduced from 80% to 60% compared to WT, but is not completely abrogated.
(c) Ddc2 focus formation in rfa1-t11 mec3Δ cells in the presence or absence of Cdc28 activity. We find that RPA and Cdc28 act independently to recruit Ddc2. Although rfa1-t11 cdc28-as1 cells (W7848-9B) are partially compromised for Ddc2 focus formation, inhibition of Cdc28 kinase activity further reduces Ddc2 focus formation in rfa1-t11 cells (~30% compared to ~70%), while Ddc2 foci still form in 1/3 of the population. On the other hand, rfa1-t11 mec3Δ cells (W7848-7A) are compromised for Ddc2 focus formation since rfa1-t11 mec3Δ cells, even in the presence of Cdc28 kinase activity, only form Ddc2 foci in 10% of budded cells in response to IR, compared to 70% in rfa1-t11 cells, and 40‐50% in rfa1-t11 cells where Cdc28 kinase activity is inhibited.
(d) Inhibition of Cdc28 activity reduces Rad53 phosphorylation in rfa1-t11 cells. After exposure to 40 Gy IR, Rad53 is phosphorylated in rfa1-t11 cells (left). Inhibition of Cdc28-as1 by addition of 1-NM-PP1 reduces Rad53 phosphorylation.
RPA and the 9-1-1 checkpoint complex independently recruit Ddc2 in S/G2 cells
To determine if Rfa1 and the 9-1-1 clamp act together to recruit Ddc2 in S/G2, we looked at Ddc2 focus formation in an rfa1-t11 mec3Δ double mutant. While Ddc2 foci form in response to IR in either single mutant (Figure 5d and Figure 6b), no foci are induced in the double mutant, indicating that the 9-1-1 complex and RPA govern two distinct pathways for Ddc2 focus formation in response to IR-induced DNA damage (~10% in Figure 6c compared to ~80% in wild-type cells in Figure 5d).
Cdc28 kinase activity regulates many aspects of DSBR including ssDNA formation and the recruitment of Rad52, a key HR protein, to the sites of DNA damage. We have shown that in the absence of a functional 9-1-1 complex, Cdc28 kinase activity is also responsible for Ddc2 focus formation in response to IR (Figure 5d). To understand further the role of Cdc28 in checkpoint regulation, we blocked its kinase activity in a checkpointdefective rfa1-t11 background. Inhibition of Cdc28 kinase activity has no effect on Ddc2 focus formation in rfa1-t11 cells during G1 (data not shown). In S/G2 rfa1-t11 cells however, loss of Cdc28 kinase activity decreases Ddc2 focus formation approximately two-fold (Figure 6c), yet 40% of cells are still able to form Ddc2 foci indicating that the rfa1-t11 checkpoint defects on Ddc1 focus formation require entry into S phase. Furthermore, inhibiting Cdc28 kinase activity reduces Rad53 phosphorylation in rfa1-t11 cells (Figure 6d), while no inhibition is observed in wild-type cells (Figure 5e). Together, these results indicate that in S/G2 cells, Cdc28 activity acts in parallel with RPA and the 9-1-1 DNA damage clamp to recruit and activate Ddc2 in response to DNA damage.
Discussion
Efficient recognition and the timely repair of DSBs are important for maintaining genomic integrity. Haploid cells are particularly vulnerable during G1, since they contain only a single complement of their genome. Here we show that budding yeast has developed mechanisms to respond to DSBs in a lesion-specific manner during G1. Specifically, endonuclease-mediated DNA breaks, which are suitable for NHEJ, are blocked from resection by the DNA-end binding Yku70/Yku80 complex, while the “ragged” ends of IR-induced breaks are readily resected. The rate of resection of IR-induced DSBs in G1 cells is 4- to 6-fold slower than in S/G2 (Figure 4a and 4b). Since most DSBs in G1 cells have no template for HR, reduced resection may facilitate repair by an alternative NHEJ pathway such as microhomology-mediated ligation (Lee et al., 2004). Other DNA-end binding proteins, the MRX complex and the associated Sae2 protein, also play roles in controlling resection of DSBs. In particular, we observe a delay in Rfa1 focus formation after irradiation in G1-arrested mre11-H125N and sae2Δ cells. These results are consistent with a role for MRX-Sae2 in the initial processing of non-ligatable IR-induced DSB ends for HR.
These observations lead us to the model in Figure 7a where we propose that, for ligatable DNA ends, Yku70/Yku80 competes with MRX for end-binding, thereby limiting the formation of ssDNA at the break site (Figure 7a). When the DNA ends are not ligated via NHEJ, nucleases produce 3’ single-stranded tails, which become substrates for HR in S phase (Figure 7a, part 5). In support of this notion, yku70Δ mutant cells display increased Mre11 focus formation both in the absence of exogenous damage and in response to IR (data not shown), and yku70Δ cells show no IR sensitivity unless HR is compromised (Siede et al., 1996). These results are also consistent with the observation that competition between Rad50, a member of the MRX complex, and Yku70 regulate Exo1 exonuclease activity at the ends of DSBs in S. pombe (Tomita et al., 2003). After gamma irradiation, the non-ligatable “ragged” ends are substrates for the MRX-Sae2 complex and nuclease activity generates singlestranded tails. Recent reports show that Sae2 acts in conjunction with the MRX complex to prepare the substrate for DNA end resection in vitro (Limbo et al., 2007; Sartori et al., 2007). Thus, in our model, the MRX-Sae2 and Ku complexes govern the processing of DNA ends that may arise in G1 cells.
Figure 7. Models for cell cycle regulation of DSB repair and checkpoint activation.
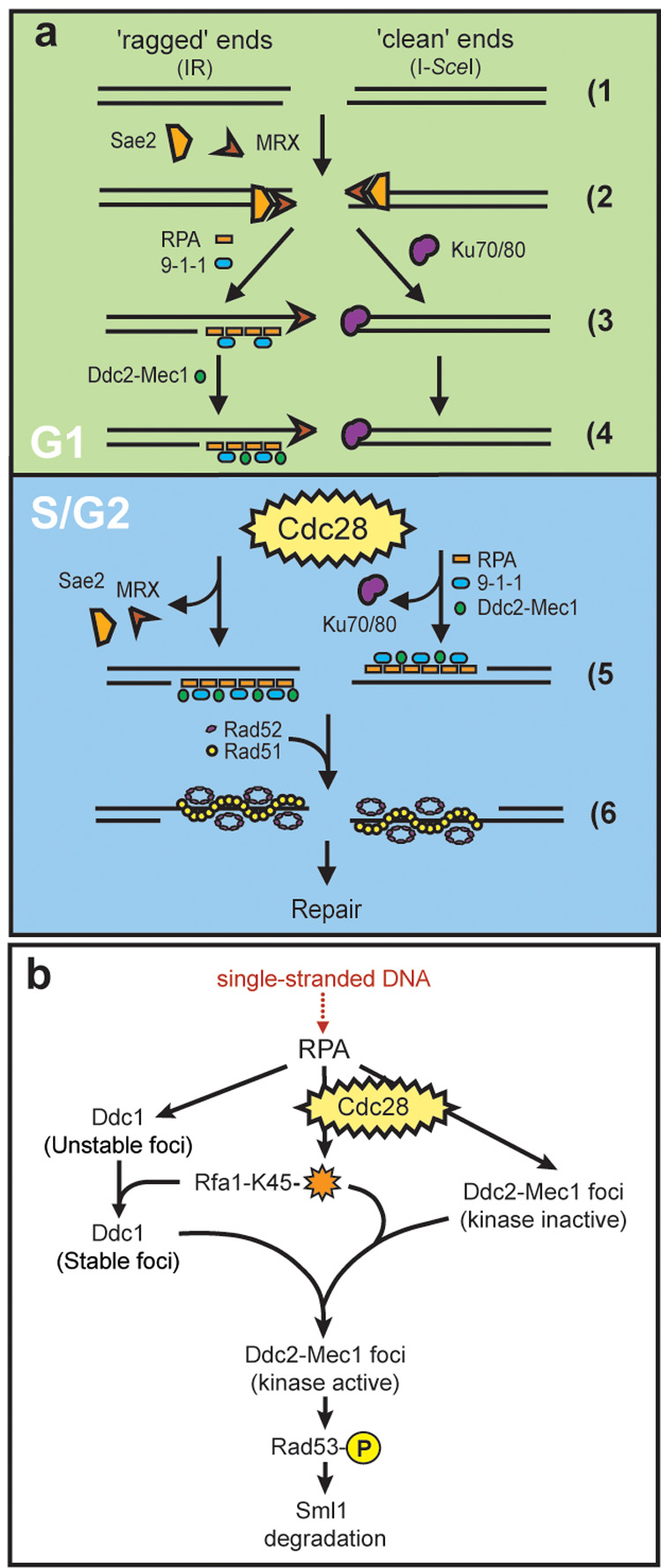
(a) Cell cycle regulation of DSB repair. When a DSB is generated (1), ends may be either ‘ragged’ as those produced following exposure to IR (left) or ‘clean’ such as those resulting from an endonuclease-mediated cleavage (right). The MRX complex recognizes both kinds of ends (2) while during G1, clean ends are preferentially bound by Yku70/Yku80 (3). Once Yku70/Yku80 is bound, the ends are not released until ligation is complete or until the cell has entered into S phase (4, 5). Upon entering S phase, Yku70/Yku80 dissociates from the ends, freeing them for digestion by nucleases resulting in ssDNA. RPA binds the ssDNA, independently recruiting the 9-1-1 (Ddc1-Mec3-Rad17) and Ddc2-Mec1 checkpoint complexes (5, right). ‘Ragged’ DNA ends, on the other hand, are not bound by Yku70/Yku80 during G1. Instead, these free ends are processed by one or more nucleases into 3’ ssDNA tails, recognized and bound by RPA. RPA then recruits Ddc1-Mec3-Rad17, which in turn is required for Ddc2-Mec1 focus formation in G1. (4, left). The homologous recombination machinery, here shown as Rad52 and Rad51, cannot be recruited however, until the cells have entered into S phase (6).
(b) Ddc2-Mec1 checkpoint activation. In G1 cells, 9-1-1(Ddc1-Mec3-Rad17) is absolutely required for recruitment of Ddc2 into foci and subsequent Rad53 phosphorylation. In S/G2, Cdc28 activity and RPA modification on Lysine 45 (orange star) act in conjunction with the 9-1-1 complex to promote Ddc2 focus formation and downstream checkpoint events. See discussion for details.
Finally, the recombination protein Rad52 is only recruited after cells have entered S phase (Lisby et al., 2001; Figure 7a, part f). Interestingly, deletion of CLB5 and CLB6, the S phase B-type cyclin activators of Cdc28 (Mendenhall and Hodge, 1998), slow the recruitment of this key recombination protein (data not shown). It is tempting to speculate that the S phase phosphorylation of Rad52 is important for this regulation (Antunez de Mayolo et al., 2006), although Cdc28 does not appear to be directly responsible (Ubersax et al., 2003).Thus, both robust DNA resection (Figure 4c) and Rad52 recruitment are initiated at the G1 to S transition, which is regulated by Cdc28 activation (Figure 7a).
Efficient checkpoint signaling requires the 9-1-1 DNA damage complex, comprised of Mec3, Ddc1 and Rad17, and the ATRIP-ATR homologs Ddc2 and Mec1. Previous work indicated that RPA independently recruits these two complexes to the sites of DNA damage (Lisby et al., 2004; Melo et al., 2001). Here we show that specifically during G1, Ddc2 focus formation requires the presence of the 9-1-1 DNA damage clamp (Figures 5a and Figure 5b). In S/G2 cells however, Ddc2 forms foci in response to DNA damage even in cells lacking 9-1-1, unless Cdc28 kinase activity is also abrogated (Figure 5d). We speculate that during S phase, PCNA substitutes for the 9-1-1 damage clamp and recruits Ddc2. Importantly, in the absence of 9-1-1, cells do not exhibit Rad53 phosphorylation, indicating that the DNA damage clamp is absolutely required for Mec1 kinase activity even when Ddc2 is recruited into foci during S/G2 (Figure 5e and Figure 7b).
As shown in Figure 7b, the Cdc28 kinase may act directly or indirectly to modify RPA activity during the DNA damage response. A checkpoint-defective allele of RPA, rfa1-t11 (Rfa1-K45E), leads to the destabilization of Ddc1 foci (Figure 6a) and also compromises Ddc2 focus formation, as shown by fewer foci (Figure 6b). Furthermore, rfa1-t11 mec3Δ cells form almost no Ddc2 foci in response to IR, demonstrating that both RPA and 9-1-1 contribute to the recruitment of Ddc2 after DNA damage in S/G2 cells. In the absence of Cdc28 kinase activity, Ddc2 focus formation is further reduced in rfa1-t11 mec3Δ cells (Figure 6c and Figure 7b), leading to a model where Cdc28, 9-1-1 and RPA cooperate to activate the DNA damage checkpoint in S/G2 (Figure 7b).
This study identifies multiple layers of cell cycle regulation for DNA repair, both in the recruitment of the ssDNA binding protein RPA to the sites of damage and the subsequent recruitment of the 9-1-1 and the Ddc2-Mec1 checkpoint complexes. In G1, the cell distinguishes between different types of DNA ends. Ku binds ligatable ends, while RPA binds to ssDNA at processed ends resulting in a checkpoint response. RPA collaborates with the 9-1-1 clamp to recruit Ddc2-Mec1 to DSBs. Upon entry into S phase, Cdc28 activity in conjunction with RPA checkpoint functions are sufficient to recruit Ddc2-Mec1, however 9-1-1 is absolutely required for Mec1 checkpoint activity. These results demonstrate that the cell relies on multiple cell cycle regulated pathways to sense DSBs and maintain DNA damage checkpoint activation. Integration of these pathways results in an effective DNA damage response.
Experimental Procedures
Yeast strains and media
Yeast strains used in this study are listed in the Supplemental Data, Table 1. Fluorescently-tagged proteins and chromosomal sites were described previously (Lisby et al., 2001; Lisby et al., 2004). Experiments were carried out at 23°aC in minimal media (SC) supplemented with 2% glucose, raffinose, or galactose as noted. Expression of the I-SceI enzyme from the Gal1-10 promoter was induced by the addition of galactose to a final concentration of 2% in cultures growing in 2% raffinose.
Cell synchronizations
Unless otherwise stated, cells were synchronized in G1 by the addition of 2 µg/ml α-factor and incubation for 90 minutes (in 2% glucose) or 3.5 hours (in 2% raffinose) at 23°C. For cell synchronization and release, cells were first synchronized in G1 with 10 µg/ml α-factor for 2 hours, filtered and released from G1 arrest by resuspension in fresh media without α-factor. Cells were arrested in G2 with 5 µg/ml nocodazole for 2 hours.
γ-irradiation
Cells analyzed by microscopy were pre-grown in SC at 23°C until OD600 reached 0.2. At this point, the liquid cultures were exposed to defined doses of irradiation using a Gammacell-220 60Co irradiator (Atomic Energy of Canada), and aliquots of the cultures were processed immediately for imaging. Cells for the low dose experiment in Figure 2b were exposed to different doses of γ-rays using a Gammacell-40 137Cs irradiator (Atomic Energy of Canada).
Genomic blot analysis
Genomic blot analysis was performed according to standard procedures (Maniatis et al., 1982). In brief, total yeast DNA was extracted and resuspended in TE buffer (Hoffman and Winston, 1987). DNA samples were digested with _Hin_dIII before agarose gel electrophoresis. Nitrocellulose membranes were probed with a PCR product generated using the primers iYEL023C_SQ1 (5’-TTAGTGAATGGCTGAGGTCC-3’) and iYEL023C_SQ2 (5’-GTATTTCCGCATCGCTGTCG-3’).
Live cell imaging and fluorescent microscopy
Cells were prepared for fluorescent microscopy as described previously (Lisby et al., 2004; Lisby et al., 2001).
BrdU Labeling
Yeast cells only able to use an exogenous source of thymidine (W8127-21B) were grown overnight in SC medium containing 2% galactose and 10 mM thymidine (Vernis et al., 2003). Overnight cultures were diluted into fresh medium supplemented with 6 mM thymidine and 4 mM BrdU and grown for additional two cell doublings, then arrested in α-factor. Cells were fixed with 4% paraformaldehyde (1 hour at RT), spheroplasted using 100T zymolase in sodium phosphate/sorbitol buffer (10–15 minutes at 30 °C), then washed in PBS buffer. BrdU detection was performed using the FITC-conjugated mouse α-BrdU antibody from BD Biosciences (cat. no. 347583) and the supplied protocol. Images were captured as described above, with a cooled Orca-ER CCD camera using a Leica HCX PL APO 100x objective 1.46 NA mounted on a Leica CTR5500 microscope.
Supplementary Material
01
Acknowledgments
We thank members of the Rothstein laboratory for discussions and in particular Peter Thorpe for helpful suggestions on the manuscript. We also thank Lorraine Symington and Jean Gautier for helpful discussions. We are grateful to Achille Pellicioli and Marco Foiani for sharing unpublished observations. This work was supported by GM50237 and GM67055 (RR), GM70088 and GM73568 (JHB) and The Danish Agency for Science, Technology, and Innovation (ML).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aguilera A, Rothstein R. Molecular Genetics of Recombination. Vol 17. Berlin Heidelberg New York: Springer; 2007. [Google Scholar]
- Alani E, Thresher R, Griffith JD, Kolodner RD. Characterization of DNA-binding and strand-exchange stimulation properties of y-RPA, a yeast single-strand-DNA-binding protein. J Mol Biol. 1992;227:54–71. doi: 10.1016/0022-2836(92)90681-9. [DOI] [PubMed] [Google Scholar]
- Antunez de Mayolo A, Lisby M, Erdeniz N, Thybo T, Mortensen UH, Rothstein R. Multiple start codons and phosphorylation result in discrete Rad52 protein species. Nucleic acids research. 2006;34:2587–2597. doi: 10.1093/nar/gkl280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aylon Y, Liefshitz B, Kupiec M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. Embo J. 2004;23:4868–4875. doi: 10.1038/sj.emboj.7600469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- Chen L, Trujillo K, Ramos W, Sung P, Tomkinson AE. Promotion of Dnl4-catalyzed DNA end-joining by the Rad50/Mre11/Xrs2 and Hdf1/Hdf2 complexes. Mol Cell. 2001;8:1105–1115. doi: 10.1016/s1097-2765(01)00388-4. [DOI] [PubMed] [Google Scholar]
- Corda Y, Lee SE, Guillot S, Walther A, Sollier J, Arbel-Eden A, Haber JE, Geli V. Inactivation of Ku-mediated end joining suppresses mec1Delta lethality by depleting the ribonucleotide reductase inhibitor Sml1 through a pathway controlled by Tel1 kinase and the Mrel1 complex. Mol Cell Biol. 2005;25:10652–10664. doi: 10.1128/MCB.25.23.10652-10664.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchlow SE, Jackson SP. DNA end-joining: from yeast to man. Trends Biochem Sci. 1998;23:394–398. doi: 10.1016/s0968-0004(98)01284-5. [DOI] [PubMed] [Google Scholar]
- Cundari E, Averbeck D. Alkaline step elution analysis of gamma-ray induced DNA strand breaks and repair in diploid yeast. International journal of radiation biology and related studies in physics, chemistry, and medicine. 1987;51:519–526. doi: 10.1080/09553008714550991. [DOI] [PubMed] [Google Scholar]
- Din S, Brill SJ, Fairman MP, Stillman B. Cell-cycle-regulated phosphorylation of DNA replication factor A from human and yeast cells. Genes Dev. 1990;4:968–977. doi: 10.1101/gad.4.6.968. [DOI] [PubMed] [Google Scholar]
- Feldmann H, Winnacker EL. A putative homologue of the human autoantigen Ku from Saccharomyces cerevisiae. J Biol Chem. 1993;268:12895–12900. [PubMed] [Google Scholar]
- Frank CJ, Hyde M, Greider CW. Regulation of telomere elongation by the cyclin-dependent kinase CDK1. Mol Cell. 2006;24:423–432. doi: 10.1016/j.molcel.2006.10.020. [DOI] [PubMed] [Google Scholar]
- Frank-Vaillant M, Marcand S. Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Mol Cell. 2002;10:1189–1199. doi: 10.1016/s1097-2765(02)00705-0. [DOI] [PubMed] [Google Scholar]
- Friedland W, Jacob P, Paretzke HG, Merzagora M, Ottolenghi A. Simulation of DNA fragment distributions after irradiation with photons. Radiat Environ Biophys. 1999;38:39–47. doi: 10.1007/s004110050136. [DOI] [PubMed] [Google Scholar]
- Henner WD, Grunberg SM, Haseltine WA. Sites and structure of gamma radiation-induced DNA strand breaks. J Biol Chem. 1982;257:11750–11754. [PubMed] [Google Scholar]
- Hoffman CS, Winston F. A ten-minute DNA preparation from yeast efficiently releases autonomous plasmids for transformation of Escherichia coli. Gene. 1987;57:267–272. doi: 10.1016/0378-1119(87)90131-4. [DOI] [PubMed] [Google Scholar]
- Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karathanasis E, Wilson TE. Enhancement of Saccharomyces cerevisiae end-joining efficiency by cell growth stage but not by impairment of recombination. Genetics. 2002;161:1015–1027. doi: 10.1093/genetics/161.3.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Brill SJ. Rfc4 interacts with Rpa1 and is required for both DNA replication and DNA damage checkpoints in Saccharomyces cerevisiae. Mol Cell Biol. 2001;21:3725–3737. doi: 10.1128/MCB.21.11.3725-3737.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HS, Brill SJ. MEC1-dependent phosphorylation of yeast RPA1 in vitro. DNA Repair (Amst) 2003;2:1321–1335. doi: 10.1016/j.dnarep.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Krogh BO, Symington LS. Recombination proteins in yeast. Annu Rev Genet. 2004;38:233–271. doi: 10.1146/annurev.genet.38.072902.091500. [DOI] [PubMed] [Google Scholar]
- Lee GS, Neiditch MB, Salus SS, Roth DB. RAG proteins shepherd double-strand breaks to a specific pathway, suppressing error-prone repair, but RAG nicking initiates homologous recombination. Cell. 2004;117:171–184. doi: 10.1016/s0092-8674(04)00301-0. [DOI] [PubMed] [Google Scholar]
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell. 1998;94:399–409. doi: 10.1016/s0092-8674(00)81482-8. [DOI] [PubMed] [Google Scholar]
- Limbo O, Chahwan C, Yamada Y, de Bruin RA, Wittenberg C, Russell P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell. 2007;28:134–146. doi: 10.1016/j.molcel.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell. 2004;118:699–713. doi: 10.1016/j.cell.2004.08.015. [DOI] [PubMed] [Google Scholar]
- Lisby M, Rothstein R, Mortensen UH. Rad52 forms DNA repair and recombination centers during S phase. Proc Natl Acad Sci U S A. 2001;98:8276–8282. doi: 10.1073/pnas.121006298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majka J, Binz SK, Wold MS, Burgers PM. Replication protein A directs loading of the DNA damage checkpoint clamp to 5'-DNA junctions. J Biol Chem. 2006;281:27855–27861. doi: 10.1074/jbc.M605176200. [DOI] [PubMed] [Google Scholar]
- Majka J, Burgers PM. Yeast Rad17/Mec3/Ddc1: a sliding clamp for the DNA damage checkpoint. Proc Natl Acad Sci U S A. 2003;100:2249–2254. doi: 10.1073/pnas.0437148100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, Fritsch EF, Sambrook J. Molecular cloning : a laboratory manual. Cold Spring Harbor, N.Y.: Cold Spring Harbor Laboratory; 1982. [Google Scholar]
- Maringele L, Lydall D. EXO1-dependent single-stranded DNA at telomeres activates subsets of DNA damage and spindle checkpoint pathways in budding yeast yku70Delta mutants. Genes Dev. 2002;16:1919–1933. doi: 10.1101/gad.225102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo JA, Cohen J, Toczyski DP. Two checkpoint complexes are independently recruited to sites of DNA damage in vivo. Genes Dev. 2001;15:2809–2821. doi: 10.1101/gad.903501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendenhall MD, Hodge AE. Regulation of Cdc28 cyclin-dependent protein kinase activity during the cell cycle of the yeast Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1998;62:1191–1243. doi: 10.1128/mmbr.62.4.1191-1243.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore JK, Haber JE. Cell cycle and genetic requirements of two pathways of nonhomologous end-joining repair of double-strand breaks in Saccharomyces cerevisiae. Mol Cell Biol. 1996;16:2164–2173. doi: 10.1128/mcb.16.5.2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau S, Ferguson JR, Symington LS. The nuclease activity of Mrel1 is required for meiosis but not for mating type switching, end joining, or telomere maintenance. Mol Cell Biol. 1999;19:556–566. doi: 10.1128/mcb.19.1.556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogorek B, Bryant PE. Repair of DNA single-strand breaks in X-irradiated yeast. I. Use of the DNA-unwinding method to measure DNA strand breaks. Mutat Res. 1985a;146:55–61. doi: 10.1016/0167-8817(85)90055-0. [DOI] [PubMed] [Google Scholar]
- Ogorek B, Bryant PE. Repair of DNA single-strand breaks in X-irradiated yeast. II. Kinetics of repair as measured by the DNA-unwinding method. Mutat Res. 1985b;146:63–70. doi: 10.1016/0167-8817(85)90056-2. [DOI] [PubMed] [Google Scholar]
- Paques F, Haber JE. Multiple pathways of recombination induced by double-strand breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol. 2003;13:458–462. doi: 10.1016/s0962-8924(03)00170-3. [DOI] [PubMed] [Google Scholar]
- Sanchez Y, Bachant J, Wang H, Hu F, Liu D, Tetzlaff M, Elledge SJ. Control of the DNA damage checkpoint by chk1 and rad53 protein kinases through distinct mechanisms. Science. 1999;286:1166–1171. doi: 10.1126/science.286.5442.1166. [DOI] [PubMed] [Google Scholar]
- Sartori AA, Lukas C, Coates J, Mistrik M, Fu S, Bartek J, Baer R, Lukas J, Jackson SP. Human CtIP promotes DNA end resection. Nature. 2007;450:509–514. doi: 10.1038/nature06337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siede W, Friedl AA, Dianova I, Eckardt-Schupp F, Friedberg EC. The Saccharomyces cerevisiae Ku autoantigen homologue affects radiosensitivity only in the absence of homologous recombination. Genetics. 1996;142:91–102. doi: 10.1093/genetics/142.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. Embo J. 1998;17:5497–5508. doi: 10.1093/emboj/17.18.5497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita K, Matsuura A, Caspari T, Carr AM, Akamatsu Y, Iwasaki H, Mizuno K, Ohta K, Uritani M, Ushimaru T, et al. Competition between the Rad50 complex and the Ku heterodimer reveals a role for Exo1 in processing double-strand breaks but not telomeres. Mol Cell Biol. 2003;23:5186–5197. doi: 10.1128/MCB.23.15.5186-5197.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubersax JA, Woodbury EL, Quang PN, Paraz M, Blethrow JD, Shah K, Shokat KM, Morgan DO. Targets of the cyclin-dependent kinase Cdk1. Nature. 2003;425:859–864. doi: 10.1038/nature02062. [DOI] [PubMed] [Google Scholar]
- Umezu K, Sugawara N, Chen C, Haber JE, Kolodner RD. Genetic analysis of yeast RPA1 reveals its multiple functions in DNA metabolism. Genetics. 1998;148:989–1005. doi: 10.1093/genetics/148.3.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T, Ogawa H, Petrini JH. A DNA damage response pathway controlled by Tel1 and the Mrel1 complex. Mol Cell. 2001;7:1255–1266. doi: 10.1016/s1097-2765(01)00270-2. [DOI] [PubMed] [Google Scholar]
- Vernis L, Piskur J, Diffley JF. Reconstitution of an efficient thymidine salvage pathway in Saccharomyces cerevisiae. Nucleic acids research. 2003;31:e120. doi: 10.1093/nar/gng121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vodenicharov MD, Wellinger RJ. DNA degradation at unprotected telomeres in yeast is regulated by the CDK1 (Cdc28/Clb) cell-cycle kinase. Mol Cell. 2006;24:127–137. doi: 10.1016/j.molcel.2006.07.035. [DOI] [PubMed] [Google Scholar]
- Zhao X, Chabes A, Domkin V, Thelander L, Rothstein R. The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. Embo J. 2001;20:3544–3553. doi: 10.1093/emboj/20.13.3544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- Zou L, Liu D, Elledge SJ. Replication protein A-mediated recruitment and activation of Rad17 complexes. Proc Natl Acad Sci U S A. 2003;100:13827–13832. doi: 10.1073/pnas.2336100100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01