PCDH8, the human homolog of PAPC, is a candidate tumor suppressor of breast cancer (original) (raw)
. Author manuscript; available in PMC: 2010 Dec 31.
Published in final edited form as: Oncogene. 2008 Apr 14;27(34):4657–4665. doi: 10.1038/onc.2008.101
Abstract
Carcinoma is an altered state of tissue differentiation in which epithelial cells no longer respond to cues that keep them in their proper position. A break down in these cues has disastrous consequences not only in cancer but also in embryonic development when cells of various lineages must organize into discrete entities to form a body plan. Paraxial protocadherin (PAPC) is an adhesion protein with six cadherin repeats that organizes the formation and polarity of developing cellular structures in frog, fish and mouse embryos. Here we show that protocadherin-8 (PCDH8), the human ortholog of PAPC, is inactivated through either mutation or epigenetic silencing in a high fraction of breast carcinomas. Los s of PCDH8 expression is associated with loss of heterozygosity, partial promoter methylation, and increased proliferation. Complementation of mutant tumor cell line HCC2218 with wild-type PCDH8 inhibited its growth. Two tumor mutants, E146K and R343H, were defective for inhibition of cell growth and migration. Surprisingly, the E146K mutant transformed the human mammary epithelial cell line MCF10A and sustained the expression of cyclin D1 and MYC without epidermal growth factor. We propose that loss of PCDH8 promotes oncogenesis in epithelial human cancers by disrupting cell–cell communication dedicated to tissue organization and repression of mitogenic signaling.
Keywords: PCDH8, protocadherin, breast cancer, tumor suppressor
Introduction
Cadherin molecules are known to be critical for creating and maintaining proper tissue architecture in cancer and development (Zhong et al., 1999; Gumbiner, 2005). E-cadherin is a classical tumor suppressor that is mutated in lobular breast carcinoma and gastric carcinoma (Berx et al., 1998; Guilford et al., 1998; Batlle et al., 2000). E-cadherin can also be silenced by SNAIL in a variety of different tumors and is a critical barrier for migration and metastasis (Cano et al., 2000; Yang et al., 2004). During the epithelial to mesenchymal transition, E-cadherin is switched off while N-cadherin is induced. One of the features of this switch is the stimulation of fibroblast growth factor (FGF) signaling due to the direct binding of N-cadherin to FGF receptor (Suyama et al., 2002).
Accumulating evidence suggests that protocadherins can function as tumor suppressors. Two members of the protocadherin family (protocadherin-10 and -20) are frequently silenced in carcinomas of the nasopharynx and lung due to promoter methylation and inhibit cell migration and proliferation (Imoto et al., 2006; Ying et al., 2006). Paraxial protocadherin (PAPC) is capable of homotypic binding and is a critical mediator of blastocyst somite organization, cell movement and cell polarity during embryogenesis, but its involvement in cancer development is not known (Kim et al., 1998, 2000; Rhee et al., 2003; Unterseher et al., 2004). Given the importance of PAPC in vertebrate development, we decided to evaluate its human ortholog PCDH8 for a role in tumor progression after finding a homozygous deletion of the gene in a breast cancer cell line. In this report, we show that PCDH8 is mutated and epigenetically silenced in a large proportion of breast tumors and that PCDH8 functions to suppress breast epithelial migration and proliferation. Interestingly, we show that a point mutation of PCDH8 is able to transform the normal mammary epithelial cell line MCF10A.
Results
PCDH8 is deleted in a breast cancer line and expressed in normal breast cells
To find genomic alterations that might contribute to tumor development, we performed genomic subtraction on the breast tumor cell line HCC1395 to define a homozygous deletion encompassing several genes including a cadherin family member PCDH8 at chromosome 13q14.3–21.2, a candidate tumor suppressor locus distinct from BRCA2 and RB (Figures 1a and b; Melamed et al., 1997; Eiriksdottir et al., 1998; Yin et al., 1999).
Figure 1.

Homozygous deletion in a breast tumor cell line and expression of PCDH8 in normal breast and breast tumors. (a) A homozygous deletion of 13q14–21 in HCC1395 was found by RDA analysis and corroborated by SNP chip and PCR analysis. Genes that are deleted include CHM1, PCDH8, GW112, PCDH17, DIAPH3 and TDRD3. A short stretch of DNA between PCDH8 and GW112 was retained. Other homozygous deletions in this vicinity do not appear to affect PCDH8 or any other gene expressed in breast tissue (Cox et al., 2005). (b) Southern analysis confirming deletion of 13q21 in HCC1395 tumor (T) DNA but not corresponding normal (N) DNA. The blot was stripped and hybridized with a chromosome X probe to demonstrate equal loading. Arrows denote deleted DNA. (c) Reverse transcription (RT)–PCR and western blot analysis reveals PCDH8 expression in a control cell line (MCF7) and the immortalized lines M2E6E7, M3E6E7, and MCF10A (10A), and loss of expression of PCDH8 in HCC1395 (1395). (d) PCDH8 mRNA is expressed in murine hippocampus and breast duct by in situ hybridization. Hematoxylin and eosin stain of breast duct (× 1000). (e) PCDH8 expression is lost in multiple breast cancer cell lines, HCC1395, ZR75-30 (75–30), MDA-MB-435 s (435 s) and MDA-MB-436 (436), by RT–PCR and western blot.
We detected mRNA and protein expression of PCDH8 in two breast luminal epithelial cells lines (M2 and M3), a spontaneously immortalized breast epithelial line MCF10A (10A) and the breast tumor line MCF7, but not in HCC1395 the line with the homozygous deletion (Figure 1c; Wazer et al., 1995). As an initial screen to determine whether PCDH8 mRNA was produced in mammary epithelium in vivo, we designed an in situ hybridization probe and determined that mouse PCDH8 message was expressed in mouse mammary ducts and brain (Figure 1d).
Reduced expression of PCDH8 in breast cancer
To look for changes in PCDH8 expression in breast cancer, we screened a panel of 85 cancer cell lines and tumor biopsies for PCDH8 message. As shown in Figure 1e, we did not detect PCDH8 in ZR75-30 (75-30), MDA-MB-435s (435s) or MDA-MB-436 (436). The overall frequency of PCDH8 mRNA downregulation was 32% in tumors and 18% in cell lines (Figure 1e; Table 1). In addition, tumor cell lines such as ZR75-30, MDA-MB-435s and MDA-MB-436 that exhibited no message for PCDH8 also expressed little to no protein (Figure 1e).
Table 1.
Summary of inactivation of PCDH8 in breast cancers
| Breast tumors | Tumors | Cell lines | Total |
|---|---|---|---|
| Reduced message | 13/41 (31.7%) | 8/44 (18.2%) | 21/85 (24.7%) |
| Reduced protein | |||
| Carcinoma biopsies | 8/35 (22.9%) | NA | 8/35 (22.9%) |
| Carcinoma (TMA) | 26/64 (40.6%) | NA | 26/64 (40.6%) |
| DCIS | 3/10 (30.0%) | NA | 3/10 (30.0%) |
| Somatic mutations | 2/116 (1.7%) | 2/21 (9.5%) | 4/137 (2.9%) |
| Methylation | 6/21 (28.6%) | 4/12 (33.3%) | 10/33 (30.3%) |
Somatic mutations of PCDH8 in breast carcinoma
Loss of expression of PCDH8 in breast tumors suggests that PCDH8 may be a tumor suppressor gene. To test this hypothesis, we screened 116 breast tumors as well as 21 additional breast tumor cell lines for mutations. In a subset of cases, we screened for loss of heterozygosity (LOH), which was present in 39% of cases. We found four cancer-specific somatic mutations that were all associated with loss of the wild-type allele.
The PCDH8 gene is predicted to encode an open reading frame with a signal peptide sequence, six extracellular cadherin repeats (EC), a transmembrane domain and a cytoplasmic tail (Table 2; Supplementary Figure S1). In one tumor biopsy, we found a G436A:E146K mutation in EC2; in another tumor biopsy, a C2089T:R697C mutation in EC6; and in HCC1599, a G1028A:R343H mutation in EC3 (Figures 2a and b; Table 2). In addition, we confirmed a previously reported somatic G2868C:K956C mutation in the intracellular portion of PCDH8 in HCC2218 (Sjoblom et al., 2006). Interestingly, this mutation is in an intracellular region of PCDH8 that is evolutionarily conserved among several different protocadherin family members (Supplementary Figure S1).
Table 2.
Summary of somatic mutations of PCDH8 in breast tumors
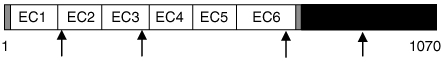 |
|||
|---|---|---|---|
| Sample | Mutation | LOH | Predicted Effect |
| HCC1395 | HD | Yes | No protein |
| 68T | G436A | Yes | E146K (EC2) |
| HCC1599 | G1028A | Yes | R343H (EC3) |
| 355T | C2089T | Yes | R697C (EC6) |
| HCC2218 | G2868C | Yes | K956C (intracellular) |
Figure 2.

Downregulation of PCDH8 in breast tumors. (a) Loss of heterozygosity (LOH) at markers D13S1305, D13S155 and D13S1228 is found in tumor 68T. Heterozygosity is seen in the corresponding normal tissue, 68N. A 50% or greater reduction in peak intensity was scored as a loss. The position of markers relative to the PCDH8 locus is mapped. (b) Inactivation of PCDH8 by somatic mutation in two tumors: one missense mutation G436A:E146K is found in the extracellular domain of PCDH8 in tumor 68T, and another missense mutation, G1028A:R343H, in breast cancer cell line HCC1599. (c) Southern analysis of methylation of the PCDH8 promoter. DNA was digested with one or more restriction enzymes and electrophoresis performed in lanes 1–4, where lane 1 corresponds to digestion with RsaI, lane 2 to RsaI and CfoI, lane 3 to RsaI and HpaII and lane 4 to RsaI and MspI. CfoI and HpaII are methylation sensitive enzymes; MspI is the methylation insensitive isoschizomer of HpaII. Methylation is detected in the breast cancer cell lines ZR-75-30 (75-30) and MDA-MB-435s (MDA-435s), and breast tumors 21T, 95T and 584T. Normal breast samples and tumor 33T lack methylation of PCDH8. An ANKRD3 control blot shows completion of digestion and serves as a loading control. (d) Restriction map of PCDH8 promoter. RsaI sites are denoted by tall vertical lines labeled ‘R’. CfoI, HpaII and MspI sites containing CpGs are denoted by short vertical lines. Site of probe for Southern blotting is indicated by horizontal line. (e) PCDH8 is reactivated in MDA-MB-435s treated with 5-aza-deoxycytidine (5AdC) but not PBS control. (f) Loss of expression of PCDH8 in tumors 21T and 95T correlates with promoter methylation, as shown in (c). Adjacent normal breast lobules and ducts exhibit membranous and cytoplasmic staining of PCDH8 in breast epithelial cells (× 400). (g) PCDH8 is downregulated in breast cancer cells relative to adjacent normal breast duct cells in a breast tumor biopsy (× 100). Arrowhead = normal cells. Arrow = tumor cells.
The genetic changes in PCDH8 found in the breast cancer samples were consistent with the tumor suppressor hypothesis. All of the missense changes clustered in conserved domains, suggesting that they may disrupt adhesive and/or signaling function. Of particular note, alignment of the E146K mutation to the analogous glutamic acid residue in C-cadherin predicts that it coordinates calcium ions, a function that is required for proper adhesive function (Shapiro et al., 1995; Nagar et al., 1996; Boggon et al., 2002).
DNA methylation analysis of PCDH8 promoter and regulation of expression
To determine the basis for PCDH8 silencing seen in some breast cancer cases, we assessed PCDH8 cytosine phosphate guanine (CpG) island methylation by Southern blot. The PCDH8 CpG island was not methylated in normal breast (Figures 2c and d). However, complete methylation was present in the cancer cell line ZR75-30 and partial methylation was detected in the cell line MDA-MB-435s and breast tumors 21T, 95T and 584T, but not 33T. The same blots were stripped and probed with ankyrin repeat domain-containing protein 3 (ANKRD3), which demonstrated that the DNA was completely digested. Evidence of PCDH8 methylation was seen in 6 of 21 (29%) breast tumor biopsies and 4 of 12 (33%) cell lines (Table 1; Supplementary Table S1). Partial or full methylation in each case correlated with reduction of PCDH8 expression (Figures 1e, 2c and f). In patient biopsies, PCDH8 protein was expressed in the cytoplasm and on the membrane of the luminal and basal epithelial layers of normal breast ducts and lobules but was markedly reduced in tumors with a methylated promoter (n = 6, P = 0.0456; Figures 2c and f; Supplementary Table S2). To study the relationship between partial promoter methylation and PCDH8 silencing, we treated MDA-MB-435s cells with a DNA methyl-transferase inhibitor, 5-aza-deoxycytidine. Treatment restored expression of PCDH8, suggesting that chromatin modification of the CpG island is involved in gene silencing in tumors (Figure 2e).
Correlative analysis of PCDH8 protein expression in breast tumors
We next screened 35 of the breast tumors that had been evaluated for LOH and PCDH8 mutation for loss of protein expression. Levels of PCDH8 protein were reduced, relative to adjacent normal ducts and lobules in 8/35 (23%) tumors (Figure 2g; Table 1). Downregulation of PCDH8 correlated with LOH for 13q14 (P = 0.0178), suggesting that two hits are required for PCDH8 inactivation (Supplementary Table S2). In addition, reduced PCDH8 correlated with reduced estrogen receptor (P = 0.001) and progesterone receptor (P = 0.001), and increased S-phase tumors (P = 0.0454), raising the possibility that PCDH8 may regulate cell proliferation in estrogen receptor negative tumors (Supplementary Table S2). Cases with missense mutations showed no evidence of reduced PCDH8 expression by staining. This observation suggests that the mutants are stable. Thus, our data support a model of tumor formation in which PCDH8 is commonly inactivated by a combination of LOH of one allele and promoter silencing or missense mutation of the remaining allele. In a separate series of breast tumors on a tissue microarray, we found reduced PCDH8 expression in tumor cells in 26/64 invasive ductal breast cancers (41%) and 3/10 ductal carcinomas in situ (30%; Table 1). These data show that PCDH8 reduction occurs prior to invasion of the basement membrane but is associated with altered epithelial organization found in ductal carcinoma in situ (DCIS).
Complementation of a mutant tumor cell line
Having established that PCDH8 is a candidate tumor suppressor, we wanted to determine the effect of expressing wild-type PCDH8 in a mutant tumor cell line. For HCC2218, which grows in suspension, we successfully generated stable pools of cells infected with retroviruses expressing either wild type or two of the somatic mutant forms of PCDH8, E146K (PCDH8K) and R343H (PCDH8H; Figure 3a). Wild-type PCDH8 suppressed the growth of HCC2218 relative to empty vector and tumor-derived mutants (Figure 3b). No changes in cell morphology or cell clumping were observed (data not shown).
Figure 3.

Retroviral expression of wild-type PCDH8 suppresses growth of a mutant breast cell line HCC2218. (a) Stable pools of HCC2218 express two species of PCDH8 expressed via the pBABE-puro retroviral vectors containing wild type and mutant forms of PCDH8 as detected by immunoblot. Tubulin is used as a loading control. (b) After plating 50 000 cells, cells were resuspended and counted on the indicated days. Wild-type PCDH8 reduced the number of cells relative to empty vector control. The mutant expressing cells (PCDH8K and PCDH8H) had an intermediate effect.
Evaluation of the effect of PCDH8 on cell migration
We next wondered whether introducing wild-type and mutant PCDH8 into untransformed mammary cells could alter the growth and differentiation of normal mammary cells. We used MCF10A, which expresses endogenous PCDH8 at low levels (Figure 1c). MCF10A cells grow as a monolayer on plastic and arrest after contact inhibition. When grown in Matrigel, a single MCF10A cell can develop into a multi-cellular acinus that exits the cell cycle, recapitulating in many ways the development of a normal breast duct (Debnath et al., 2003a). Perturbation of mitogenic pathways by overexpression of ErbB2/HER2/Neu, for example, produces disorganized structures resembling breast cancers (Muthuswamy et al., 2001; Debnath et al., 2003b). In addition, these cells are extremely useful for studying epithelial cell migration.
MCF10A cells were infected with retroviruses expressing myc epitope-tagged wild-type PCDH8, the somatic mutants E146K (PCDH8K) and R343H (PCDH8H) or the empty vector pBABEpuro. The expression of exogenous PCDH8 was readily detectable and was higher than the endogenous level expressed in MCF10A (Figure 4a). Interestingly, exogenous PCDH8 and the PCDH8H mutant migrated at two different molecular weights, but the PCDH8K mutant only expressed the smaller species. Indirect immunofluorescence using an anti-myc antibody revealed that wild-type PCDH8 but not the E146K mutant was concentrated in delicate connections between cells (Figure 4b). These data suggest that the E146K mutation affects the posttranslational processing of PCDH8 and transport to the membrane in MCF10A.
Figure 4.

Wild-type, but not mutant, PCDH8 inhibits migration of normal breast epithelial cells. (a) Protein expression of PCDH8 (10A-PCDH8), PCDH8K (10A-PCDH8K) and PCDH8H (10A-PCDH8H) in MCF10A detected by immunoblot. (b) Subcellular localization of PCDH8 and PCDH8K were determined by immunofluorescence using anti-MYC 9E10 antibodies. While PCDH8 localizes at cell processes and cell–cell junctions, PCDH8K localizes to the cytoplasm and is concentrated in perinuclear regions. Corresponding 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI) stain of PCDH8 and PCDH8K transfected cells (× 400). (c) PCDH8 expression inhibits migration. Wound healing assay reveals reduced migration of 10A-PCDH8 relative to empty vector control and 10A-PCDH8 mutant cells (× 100). At 24 h 10A-PCDH8 cells continue to have an open wound, while control and mutant cells have already repaired the wound.
Considering that PAPC regulates embryonic cell movements and PCDH8 is expressed in cell membranes, we asked whether PCDH8 could alter migration. In wound healing assays, cells overexpressing PCDH8 showed reduced ability to migrate into the wound relative to empty vector control cells (Figure 4c). In this context, both PCDH8K and PCDH8H mutants behaved like empty vector and closed the wound completely within 24 h. These observations suggest that PCDH8 diminishes migration and that the mutations are deficient in this capacity.
E146K mutation of PCDH8 triggers transformation of breast epithelial cells
To our surprise, under both enriched and limited growth factor conditions, the E146K mutation of PCDH8 (PCDHK) transformed MCF10A cells. A subset of MCF10A-PCDH8K cells was able to form foci when grown on plastic, whereas empty vector, wild-type PCDH8 and PCDH8H cells were not (Figure 5a, left column and b). When grown in reduced growth factor Matrigel, MCF10A-PCDH8K cells formed large spiculated colonies, at an average incidence of 1:1000 while such growth was not observed for other infectants (Figure 5a, middle column and b). While the transformation of MCF10A-PCDH8K cells was highly induced above base-line control cells, transformation only occurred in a portion of the infected cell population. Similar results were observed from multiple independent infections of MCF10A cells with the PCDH8K retroviral vector.
Figure 5.

PCDH8K transforms normal breast epithelial cells and increases expression of MYC and cyclin D1 in the absence of serum. (a) PCDH8K (E146K) transforms MCF10A in 2-dimensional culture on plastic (left column, × 40) and 3-dimensional culture in Matrigel (center column, × 40). PCDH8K accelerates acinus size relative to control cells, 10A-PCDH8, and 10A-PCDH8H (right column, × 400). (b, c) Quantification of aberrant 10A-PCDH8K foci. (b) When cultured on plastic, all foci visible to the naked eye were counted in 75 cm2 flasks. (c) In reduced growth factor Matrigel, spiculated acini were counted and quantified as a ratio of spiculated acini to the number of cells originally suspended in matrigel (spiculated acini per 5000 suspended cells). (d) MCF10A derivatives were grown in the absence of epidermal growth factor (EGF) and in low serum for up to 48 h and cell lysates harvested at the indicated time points. Cyclin D1 and MYC proteins expression measured by immunoblot persists in 10A-PCDH8K (10A-P8K) and 10A-RasV12 after withdrawal of EGF.
At the same time we noticed a more subtle growth alteration in the PCDH8K infectants that occurred in all of the acini grown in Matrigel. While wild-type PCDH8, mutant PCDH8H and empty vector acini were small as expected (Figure 5a, right column), PCDH8K acini were larger and contained more cells (Figure 5a, right column). This suggested that the entire population of PCDH8K cells was receiving a pro-oncogenic signal that was not present in the other infected populations.
Since cyclin D1 and MYC can transform human and mouse mammary cells, we asked whether PCDH8K could alter the expression of these proteins after the withdrawal of growth factors (Chou et al., 1999). As expected, vector and wild-type PCDH8 had low levels of cyclin D1 and MYC, 16 h after epidermal growth factor withdrawal (Figure 5d). PCDH8K cells, on the other hand, showed elevated levels of cyclin D1 and MYC over a 48 h period, similar to those induced by expression of H-RasV12. Thus, the E146K mutation of PCDH8 is likely to promote cellular transformation through its ability to reduce the growth factor requirements for the expression of cyclin D1 and MYC. To examine the possibility that wild-type PCDH8 could suppress H-RasV12, we introduced PCDH8 into H-RasV12 MCF10A cells and found that it was unable to affect cyclin D1 (data not shown).
Discussion
We have shown that PCDH8 is inactivated in a large proportion of epithelial tumors through either genetic alteration or epigenetic silencing of expression (Figures 1 and 2; Tables 1 and 2). Somatic mutations clustered in highly conserved domains of the gene and were associated with LOH, while partial methylation of the promoter was associated with LOH and reduced gene expression. With the exception of one tumor cell line, methylation was partial, which suggests that it may be a consequence of gene silencing rather than a cause. Overall, approximately one third of all breast carcinomas had either evidence of genetic or epigenetic inactivation of PCDH8. Loss of PCDH8 occurred early in tumor development in DCIS and correlated with increased S-phase and loss of estrogen receptor expression. Based upon these data, we conclude that PCDH8 is a candidate tumor suppressor.
Consistent with this hypothesis, we have found that PCDH8 suppresses tumor cell proliferation and inhibits cell migration (Figures 3 and 4). PCDH8 inhibited the proliferation of the mutant tumor cell line HCC2218 and migration of the mammary cell line MCF10A, while the tumor-derived mutants E146K and R343H were defective in these assays. The E146K tumor-derived mutant promoted acini expansion in Matrigel and sustained elevated levels of cyclin D1 and MYC expression in the absence of growth factors (Figure 5). Moreover, the E146K mutation had transforming properties of its own suggesting that this mutation functions in a dominant-negative manner, either through interfering with endogenous PCDH8 or potentially other protocadherin proteins. E146K transformation is likely to require at least one additional independent event in MCF10A cells since only a minority of expressing cells exhibited the transformed phenotype.
Understanding how PCDH8 suppresses tumor growth is an interesting question. Our results suggest that PCDH8 has a role in morphogenesis and cell growth. We suspect that PCDH8 mutations, such as E146K, that occur on the cell surface disrupt the interaction of one PCDH8 molecule with other PCDH8 molecules in the same or neighboring cells that serve to restrain and organize clusters of breast epithelial cells. On the other hand, the mutation (K956C) observed in the intracellular domain of PCDH8 probably affects an intracellular signaling pathway. Unlike other protocadherins, the intracellular domain of PCDH8 has no homology to cadherins and therefore is not likely to interact with catenins. However, there is a highly conserved region (>40%) that is shared among several different human protocadherin paralogs (PCDH1, PCDH7, PCDH9, PCDH10, PCDH11, PCDH17, PCDH18, PCDH19) that could be responsible for transmitting signals within cells (Supplementary Figure S1). Reintroduction of wild-type PCDH8 into a cell line expressing a mutation of this region was able to suppress cell growth in vitro (Figure 3). This finding suggests that the cytoplasmic domain makes a critical contribution to the tumor suppressor function of PCDH8. It will be interesting to dissect the intracellular signals that PCDH8 regulates in breast epithelial cells.
Inactivating PCDH8 appears to be an early step in breast tumor progression that may be related to its role in regulating cellular polarity and tissue organization during vertebrate embryogenesis (Hukriede et al., 2003; Medina et al., 2004; Unterseher et al., 2004). Given its high frequency of inactivation, it is likely to represent a key step in the evolution of breast epithelial malignancy. Our findings suggest that PCDH8 cell–cell communication restrains the expansion of epithelial cells present in breast tissue and provides a mechanism for maintaining normal breast epithelial architecture and homeostatis. PCDH8 is located on chromosome 13q14.3 and is within a cluster of protocadherins (PCDH8, PCDH9, PCDH17 and PCDH20) spanning 13q14–21 that is conserved between humans and mice. It is interesting to note that PCDH20 is methylated and homozygously deleted in lung cancer, and when reintroduced into an altered tumor cell line reduces proliferation (Imoto et al., 2006). Based upon our findings and these, we suggest that the chromosome 13q14–21 protocadherin cluster may be broadly involved in tumor suppression in a range of tumor types.
Materials and methods
Representational difference analysis
Genomic subtraction was performed on the normal/tumor cell line pair (HCC1395) using representational difference analysis (Lisitsyn and Wigler, 1993). Unique sequence was identified in 18 of 150 clones. Six fragments were derived from the Epstein–Barr virus genome. Two of the fragments mapped to chromosome 13q21 and were absent in the tumor line.
Cell lines
HCC1395 and HCC1395BL were obtained from Dr Adi Gazdar (University of Texas, Southwestern). UACC-812, UACC-893, MDA-MB-453, MDA-MB-175vii, MDA-MB-468, MDA-MB-361, MDA-MB-231, MDA-MB-436, MDA-MB-415, MDA-MB-330, MDA-MB-157, MDA-MB-134vi, MDA-MB-435s, ZR75-30, ZR75-1, BT-549, BT-483, T-47D, BT-474, DU-4475, MCF7, SK-BR-3, Hs578t, HCC38, HCC1143, HCC1187, HCC1428, HCC1806, HCC1937, HCC2157, HCC1500, HCC1599, HCC2218, HCC1419, HCC70, HCC202, HCC1954, HCC1569, HCC1008 and MCF10A were purchased from the ATCC. SUM44, SUM52, SUM102, SUM149, SUM159, SUM185, SUM225, SUM190 and SUM1315 were acquired from Dr Stephen Ethier (Karamanos Cancer Center). M2 and M3 are luminal breast cell lines derived from human milk immortalized with E6 and E7 and were gifts from Dr Vimla Band (Northwestern University). Breast tumor samples were from the Herbert Irving Comprehensive Cancer Center Tumor Bank and were obtained with permission of the IRB.
RNA isolation, cDNA synthesis and RT–PCR
cDNA was synthesized from RNA primed with random hexamers (Amersham Biosciences, Piscataway, NJ, USA). Primers used for reverse transcription (RT)–PCR: DIAPH3, ATCTCCCTGATCAAGACTCAAT, ACTGTGAGAAAGT GGAAAGTA; PCDH8, TGGCGGTGTGGAAAGGACA, CGGAGTGACCTGTATATGTG. For reactivation studies, cells were treated with 1 µm 5-aza-deoxycytidine for 72 h.
LOH and mutation analysis
Markers D13S1305, D13S155 and D13S1228 were amplified from genomic DNA. A 50% or greater reduction in peak intensity was scored as a loss. Primers used for mutation analysis and sequencing are listed in Supplementary Table S3. Sequences were analysed using Mutation Surveyor (SoftGenetics LLC., State College, PA, USA). Tumors were obtained from Columbia University Medical Center with permission from the Institutional Review Board. Cell lines screened for mutation include UACC812, UACC893, MDA-MB-453, MDA-MB-231, MDA-MB-436, MDA-MB-415, BT-483, T47D, BT474, DU4475, CAMA1, HCC2157, SUM102, SUM185, SUM1315, HCC1599, HCC1008, HCC1806, HCC1187, MCF7, HCC2218.
Southern blotting
Probes were PCR amplified and labeled randomly. PCR primers are 13q21 probe: 13q21F, AGGCTTTTGAGTTCAAGGTG; 13q21R, GTAAGTCTCAGTCTCAACA; PCDH8 probe: PCDH8-CpG-F3, AGAGGCTATTCCAGGCACCG; PCDH8-CpG-R3, CTCTCGGAATCACGCTCTTTG; ANKRD3 probe: ANKRD3-F, GGACGACCTACGGAAGTGAC; ANKRD3-R, CTAACTCCACTCACAAAGCC.
In situ hybridization
Tissue was fixed in 4% paraformaldehyde overnight at 4 °C and dehydrated in 30% sucrose. Sections were hybridized with DIG-labeled cRNA probe from mouse clone ID 3813893 and incubated with anti-DIG-AP antibody (Roche Diagnostics Corp., Indianapolis, IN, USA). Alkaline phosphatase activity was visualized with NBT/BCIP (Vector Laboratories, Burlingame, CA, USA).
Cloning and mutagenesis
A human PCDH8 clone was purchased from OriGene (OriGene Technologies Inc., Rockville, MD, USA), clone ID FB1851_H03, pCMV6-XL4-PCDH8. This clone contained a missense change. The wild-type sequence was created using QuikChange XL Site-Directed Mutagenesis Kit (Stratagene Corp., La Jolla, CA, USA), primers CAGGACACCTACGAGCTGGACGTGCG and CGCACGTCCAGCTCGTAGGTGTCCTG. pBABE-PCDH8-myc was generated by PCR amplification and cloned into pBABEpuro between _Eco_RI and _Sal_I sites. PCDH8-E146K mutant was generated using primers GGTAGAAGGTGTCCAAGGGTGCGGCAGTG and CACTGCCGCACCCTTGGACACCTCTACC; PCDH8-R343H mutant with primers GCAAGGTCATCGTGCACATCCGAGACGTC and ATTGACGTCTCGGATGTGCACGATGACCT. pBABE-RasV12 was a gift from Dr Scott Lowe.
Retrovirus production and infection
Phoenix-ampho cells for retrovirus production were provided by Dr Gary Nolan. A T75 flask of cells was transfected with 21 µg plasmid using Lipofectamine 2000 (Invitrogen Corporation, Carlsbad, CA, USA). Virus was harvested 48–72 h post transfection, stabilized with FBS, and passed through a 0.45 µm filter. Cells were infected with viruses in the presence of 8 µg/ml polybrene and selected with 1 µg/ml puromycin.
Immunofluorescence
MCF10A cells expressing MYC epitope-tagged proteins were plated onto sterile cover slips in a six-well dish. Sixteen hours after plating, cells were fixed in 2% paraformaldehyde in phosphate-buffered saline (PBS) pH 7.4 for 30 min at room temperature. Cells were washed for 20 min in PBS, permeabilized for 1 h in buffer A (5% goat serum, 0.1% Triton X-100 in PBS), and incubated with 1:1000 dilution of mouse monoclonal anti-MYC (9E10) antibody (Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA) in buffer A. Cells were washed in PBS, and incubated with 1:600 dilution of Alexafluor 568 antimouse antibody (Molecular Probes, Invitrogen Corporation, Carlsbad, CA, USA) and counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; 0.15 µg/ml in water).
Western blotting and immunohistochemistry
Whole cell lysates were used in all western blots. Paraffin sections were stained with 1:5000 dilution of the anti-PCDH8 antibody. Slides were developed with ABC-DAB (Vector, Biogenics, NAPA, CA, USA). Antibodies: anti-PCDH8 was raised against amino acids 1052–1070 (YQSPPGRYLSPKK-GANENV) in rabbits and affinity purified (NCBI accession number AAC70009). Other antibodies were commercially available: anti-tubulin (Tu27; Covance Research Products, Berkeley, CA, USA), anti-vinculin (hVIN-1; Sigma-Aldrich, St Louis, MO, USA), anti-Myc (9E10; Santa Cruz), anti-E-cadherin (BD Biosciences, San Jose, CA, USA), anti-v-H-ras (Ab-1; EMD Chemicals Inc., San Diego, CA, USA).
Morphogenesis assay
Growth-factor (40 µl) reduced Matrigel (BD Biosciences) was plated on eight-chamber slides (Corning Incorporated, Lowell, MA, USA). MCF10A cell lines were grown in each chamber as described (Debnath et al., 2003a).
Migration assays
Equal numbers of cells were plated on a six-well plate. A single wound was introduced using a P20 pipette tip and media was replaced. Migration was assessed at indicated times.
Supplementary Material
Fig S1
Figure Legend Sup Fig S1
Table S1
Table S2
Table S3
Acknowledgements
We thank Tom Jessell and Stephen Price for suggestions and assistance with in situ hybridization. We thank Michael R Stratton and Graham R Bignell for their help in mapping the break points of the homozygous deletion in HCC1395. We also thank Vimla Band, Gary Nolan, Scott Lowe, Joan Brugge, Nancy Hynes for reagents and Larry Shapiro for assistance in the analysis of coding changes. JSY received support from NIH and NCI and RP received support from Avon Foundation, Octoberwoman Foundation, and NCI. HH received support from Avon Foundation.
Footnotes
References
- Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat Cell Biol. 2000;2:84–89. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- Berx G, Becker KF, Hofler H, van Roy F. Mutations of the human E-cadherin (CDH1) gene. Hum Mutat. 1998;12:226–237. doi: 10.1002/(SICI)1098-1004(1998)12:4<226::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–1313. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat Cell Biol. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- Chou JL, Fan Z, DeBlasio T, Koff A, Rosen N, Mendelsohn J. Constitutive overexpression of cyclin D1 in human breast epithelial cells does not prevent G1 arrest induced by deprivation of epidermal growth factor. Breast Cancer Res Treat. 1999;55:267–283. doi: 10.1023/a:1006217413089. [DOI] [PubMed] [Google Scholar]
- Cox C, Bignell G, Greenman C, Stabenau A, Warren W, Stephens P, et al. A survey of homozygous deletions in human cancer genomes. Proc Natl Acad Sci USA. 2005;102:4542–4547. doi: 10.1073/pnas.0408593102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003a;30:256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]
- Debnath J, Walker SJ, Brugge JS. Akt activation disrupts mammary acinar architecture and enhances proliferation in an mTOR-dependent manner. J Cell Biol. 2003b;163:315–326. doi: 10.1083/jcb.200304159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eiriksdottir G, Johannesdottir G, Ingvarsson S, Bjornsdottir IB, Jonasson JG, Agnarsson BA, et al. Mapping loss of heterozygosity at chromosome 13q: loss at 13q12–q13 is associated with breast tumour progression and poor prognosis. Eur J Cancer. 1998;34:2076–2081. doi: 10.1016/s0959-8049(98)00241-x. [DOI] [PubMed] [Google Scholar]
- Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, et al. E-cadherin germline mutations in familial gastric cancer. Nature. 1998;392:402–405. doi: 10.1038/32918. [DOI] [PubMed] [Google Scholar]
- Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–634. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- Hukriede NA, Tsang TE, Habas R, Khoo PL, Steiner K, Weeks DL, et al. Conserved requirement of Lim1 function for cell movements during gastrulation. Dev Cell. 2003;4:83–94. doi: 10.1016/s1534-5807(02)00398-2. [DOI] [PubMed] [Google Scholar]
- Imoto I, Izumi H, Yokoi S, Hosoda H, Shibata T, Hosoda F, et al. Frequent silencing of the candidate tumor suppressor PCDH20 by epigenetic mechanism in non-small-cell lung cancers. Cancer Res. 2006;66:4617–4626. doi: 10.1158/0008-5472.CAN-05-4437. [DOI] [PubMed] [Google Scholar]
- Kim SH, Jen WC, De Robertis EM, Kintner C. The protocadherin PAPC establishes segmental boundaries during somitogenesis in xenopus embryos. Curr Biol. 2000;10:821–830. doi: 10.1016/s0960-9822(00)00580-7. [DOI] [PubMed] [Google Scholar]
- Kim SH, Yamamoto A, Bouwmeester T, Agius E, Robertis EM. The role of paraxial protocadherin in selective adhesion and cell movements of the mesoderm during xenopus gastrulation. Development. 1998;125:4681–4690. doi: 10.1242/dev.125.23.4681. [DOI] [PubMed] [Google Scholar]
- Lisitsyn N, Wigler M. Cloning the differences between two complex genomes. Science. 1993;259:946–951. doi: 10.1126/science.8438152. [DOI] [PubMed] [Google Scholar]
- Medina A, Swain RK, Kuerner KM, Steinbeisser H. Xenopus paraxial protocadherin has signaling functions and is involved in tissue separation. EMBO J. 2004;23:3249–3258. doi: 10.1038/sj.emboj.7600329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamed J, Einhorn JM, Ittmann MM. Allelic loss on chromosome 13q in human prostate carcinoma. Clin Cancer Res. 1997;3:1867–1872. [PubMed] [Google Scholar]
- Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2, but not ErbB1, reinitiates proliferation and induces luminal repopulation in epithelial acini. Nat Cell Biol. 2001;3:785–792. doi: 10.1038/ncb0901-785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagar B, Overduin M, Ikura M, Rini JM. Structural basis of calcium-induced E-cadherin rigidification and dimerization. Nature. 1996;380:360–364. doi: 10.1038/380360a0. [DOI] [PubMed] [Google Scholar]
- Rhee J, Takahashi Y, Saga Y, Wilson-Rawls J, Rawls A. The protocadherin papc is involved in the organization of the epithelium along the segmental border during mouse somitogenesis. Dev Biol. 2003;254:248–261. doi: 10.1016/s0012-1606(02)00085-4. [DOI] [PubMed] [Google Scholar]
- Shapiro L, Fannon AM, Kwong PD, Thompson A, Lehmann MS, Grubel G, et al. Structural basis of cell-cell adhesion by cadherins. Nature. 1995;374:327–337. doi: 10.1038/374327a0. [DOI] [PubMed] [Google Scholar]
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- Suyama K, Shapiro I, Guttman M, Hazan RB. A signaling pathway leading to metastasis is controlled by N-cadherin and the FGF receptor. Cancer Cell. 2002;2:301–314. doi: 10.1016/s1535-6108(02)00150-2. [DOI] [PubMed] [Google Scholar]
- Unterseher F, Hefele JA, Giehl K, De Robertis EM, Wedlich D, Schambony A. Paraxial protocadherin coordinates cell polarity during convergent extension via Rho A and JNK. EMBO J. 2004;23:3259–3269. doi: 10.1038/sj.emboj.7600332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wazer DE, Liu XL, Chu Q, Gao Q, Band V. Immortalization of distinct human mammary epithelial cell types by human papilloma virus 16 E6 or E7. Proc Natl Acad Sci USA. 1995;92:3687–3691. doi: 10.1073/pnas.92.9.3687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, et al. Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell. 2004;117:927–939. doi: 10.1016/j.cell.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Yin Z, Spitz MR, Babaian RJ, Strom SS, Troncoso P, Kagan J. Limiting the location of a putative human prostate cancer tumor suppressor gene at chromosome 13q14.3. Oncogene. 1999;18:7576–7583. doi: 10.1038/sj.onc.1203203. [DOI] [PubMed] [Google Scholar]
- Ying J, Li H, Seng TJ, Langford C, Srivastava G, Tsao SW, et al. Functional epigenetics identifies a protocadherin PCDH10 as a candidate tumor suppressor for nasopharyngeal, esophageal and multiple other carcinomas with frequent methylation. Oncogene. 2006;25:1070–1080. doi: 10.1038/sj.onc.1209154. [DOI] [PubMed] [Google Scholar]
- Zhong Y, Brieher WM, Gumbiner BM. Analysis of C-cadherin regulation during tissue morphogenesis with an activating antibody. J Cell Biol. 1999;144:351–359. doi: 10.1083/jcb.144.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Figure Legend Sup Fig S1
Table S1
Table S2
Table S3