Redirecting Specificity of T-Cell Populations For CD19 Using the Sleeping Beauty System (original) (raw)
. Author manuscript; available in PMC: 2009 Apr 15.
Abstract
Genetic modification of clinical-grade T cells is undertaken to augment function, including redirecting specificity for desired antigen. We and others have introduced a chimeric antigen receptor (CAR) to enable T cells to recognize lineage-specific tumor antigen, such as CD19, and early-phase human trials are currently assessing safety and feasibility. However, a significant barrier to next-generation clinical studies is developing a suitable CAR expression vector capable of genetically modifying a broad population of T cells. Transduction of T cells is relatively efficient but it requires specialized manufacture of expensive clinical grade recombinant virus. Electrotransfer of naked DNA plasmid offers a cost-effective alternative approach, but the inefficiency of transgene integration mandates ex vivo selection under cytocidal concentrations of drug to enforce expression of selection genes to achieve clinically meaningful numbers of CAR+ T cells. We report a new approach to efficiently generating T cells with redirected specificity, introducing DNA plasmids from the Sleeping Beauty transposon/transposase system to directly express a CD19-specific CAR in memory and effector T cells without drug selection. When coupled with numerical expansion on CD19+ artificial antigen-presenting cells, this gene transfer method results in rapid outgrowth of CD4+ and CD8+ T cells expressing CAR to redirect specificity for CD19+ tumor cells.
Introduction
The most robust example of successful T-cell therapy occurs following allogeneic hematopoietic stem-cell transplantation where the engrafted donor-derived T cells recognize recipient tumor-associated antigens in the context of MHC. However, the graft-versus-tumor effect after allogeneic-hematopoietic stem cell transplantation is incomplete, resulting in relapse as the major cause of mortality. To augment the graft-versus-tumor effect for B-lineage neoplasms, we have previously shown that genetically modified peripheral blood– and umbilical cord blood–derived T cells can be rendered specific for CD19, a molecule constitutively expressed on B-cell malignancies (1, 2). The redirected specificity was achieved by electrotransfer of a linearized DNA plasmid coding for a first-generation chimeric antigen receptor (CAR), designated CD19R, which recognizes CD19 via the scFv of a murine CD19-specific monoclonal antibody (mAb) fused to a chimeric CD3-ζ–derived activation endodomain. A phase I trial (BB-IND1141, clinicalTrials.gov identifier: NCT00182650; ref. 3) is currently evaluating the safety and feasibility of infusing autologous T cells electroporated to coexpress CD19R CAR and the hygromycin phosphotransferase (Hy) and herpes simplex virus-1 thymidine kinase selection/suicide fusion transgene (4).
We anticipated that the therapeutic efficacy of adoptive transfer of CD19-specific T cells would be improved by developing a CAR with a fully competent activation signal and introducing the CAR into central memory (CM) T cells. As a result, a second-generation CAR, designated CD19RCD28, has been developed that provides CD19-dependent signaling through chimeric CD3-ζ and CD28, resulting in improved in vivo persistence and antitumor effect, compared with CD19R+ T cells (5). To further optimize the clinical potential of CAR+ T cells, while taking advantage of the cost-efficiency of nonviral gene transfer, we desired a clinically feasible approach to the efficient propagation of CAR+ T-cell populations, including TCM, in the absence of expression immunogenic drug selection genes, such as Hy. We reasoned that genetically modified Tcells could be selectively propagated, upon activating T cells for sustained proliferation, through the introduced second-generation CAR. To maximize transgene expression, we codon-optimized (CoOp) the CAR as reports have shown that codon optimization of genes toward human consensus codon usage increases protein expression (6, 7).
The focus on developing nonviral gene transfer technologies is justified based on the cost and time savings compared with developing recombinant clinical-grade viral supernatant, which are subject to rigorous regulatory oversight and rely on specialized manufacturing experience of a limited number of production facilities. Although the transfection efficiency of nonviral gene transfer is inferior to viral-mediated transduction, naked DNA plasmids expressing desired transgenes such as CAR can be rapidly produced at a fraction of the cost compared with clinical grade γ-retrovirus and lentivirus. A potential drawback to nonviral gene transfer, compared with viral gene transfer, is the lengthy ex vivo manufacturing time to selectively propagate electroporated T cells with stable expression of transgene, during which time the cells may become susceptible to replicative senescence, lose expression of desired homing receptors, and furthermore be cleared in vivo due to recognition of immunogenic drug selection transgene (8, 9). What is needed is an approach that when coupled with nonviral gene transfer shortens the culture time to generate T cells with durably expressed transgene and maintains a desired T-cell immunophenotype.
To introduce the CAR, we evaluated whether the efficient transposition and long-lasting transgene expression of the Sleeping Beauty (SB) DNA transposon derived from Tc1/mariner superfamily of transposons (10, 11) can improve transgene transfer efficiency. The SB transposable element from a DNA donor plasmid can be adapted for nonviral gene transfer in T cells, using a SB transposase supplied in trans to mediate integration of a transposon CAR expression cassette flanked by terminal inverted repeats (IR), which each contain two copies of a short direct repeat (DR) that have binding sites for the transposase enzyme (Fig. 1_D_). The SB transposase mediates transposition by binding to IRs, excising a precise DNA sequence flanked by the IRs, and inserting the transposon into any of ∼200 million TA sites in a mammalian genome (12). Previously, the SB system has been used as a nonviral gene delivery into multiple murine and human cell lines, including liver, keratinocytes, endothelial cells, lung, hematopoietic progenitor cells, embryonic stem cells, and tumor cells (11). Of particular relevance is that _SB_-mediated integration has been shown in human T cells (13), signifying the potential application of this technology.
Figure 1.

Schematic of the expression plasmids and experimental design. A, CoOpCD19RCD28/pT-MNDU3 (Transposon). MNDU3 promoter, the constitutive promoter from the U3 region of the MND retrovirus; CoOp CD19RCD28, codon-optimized CD19RCD28 CAR; IR, _SB_-inverted/direct repeats; bGh pAn, polyadenylation signal from bovine growth hormone; AmpR, ampicillin resistance gene. B, pCMV-SB11 (Transposase). SB11, _SB_-transposase SB11; CMV promoter, CMV enhancer/promoter; SV40pAN, polyadenylation signals from SV40. C, CoOpCD19CD28/Hy-pVitro4. EF1α promoter, composite promoter comprising the elongation factor-1α (EF1α) core promoter and the R segment and part of the U5 sequence (R-U5′) of the human T-cell leukemia virus type 1 LTR; pMB1 ori, a minimal E. coli origin of replication; SpAn, synthetic pause; CAGp, a composite promoter that combines the human CMV immediate-early enhancer and a modified chicken β-actin promoter and first intron; Hy, hygromycin B resistance gene (hygromycin phosphotransferase); bGh pAn, polyadenylation signal from bovine growth hormone; EM7, synthetic prokaryotic promoter. D, an expression cassette in a plasmid (blue) provides only transient expression unless incorporated into an integrating transposon vector that can be cleaved from the plasmid and integrated into a host genome by a source of transposase (red).
We report that electrotransfer of a two-component DNA SB system into primary human T cells from umbilical cord blood and peripheral blood results in efficient and stable CAR gene transfer, which can be numerically expanded to clinically meaningful numbers within 4 weeks on CD19+ artificial antigen-presenting cell (aAPC), without the need for addition of cytocidal concentrations of drug for selection, and with the outgrowth of CD8+ and CD4+ CM and effector CAR+ T-cell subpopulations. This was achieved through the rationale design of (a) a next-generation codon-optimized CD19-specific CAR, (b) CD19+ aAPC expressing desired costimulatory and cytokine molecules, and (c) SB DNA plasmids expressing CAR transposon and an improved transposase. The relative ease of DNA plasmid production, electroporation, and outgrowth of stable integrants on a thawed γ-irradiated bank of aAPC can be readily transferred to the facilities operating in compliance with current good manufacturing practice (cGMP) for phase I/II trials. This is predicted to greatly facilitate trial design infusing CD4+ and CD8+ CAR+ T cells that have desired immunophenotype, including TCM.
Materials and Methods
Plasmids
The plasmid pT-MNDU3-eGFP containing salmonid fish–derived SB IR flanking the constitutive promoter, derived from the U3 region of the MND retrovirus (14), to drive an eGFP reporter gene (15), was derived from the plasmid pT-MCS (16) that was derived from pT/neo (10). The second-generation CD19RCD28 CAR (5) was human codon optimized (CoOp), substituting codons with those optimally used in mammals (GENEART) without altering anticipated amino acid sequence. The codon-optimized CD19RCD28 (CoOpCD19RCD28) CAR was subcloned into pT-MNDU3 DNA plasmid by replacing the eGFP sequence with the CAR to create CoOpCD19RCD28/pT-MNDU3 (Fig. 1_A_). The DNA plasmid pCMV-SB11 (Fig. 1_B_) expresses the SB11 transposase (17). Plasmid CoOpCD19RCD28/Hy-pVitro4 (Fig. 1_C_) was generated from pVitro4-mcs DNA vector (InvivoGen) by subcloning CoOpCD19RCD28 at _Nhe_I in multiple cloning site two and replacing the internal ribosome entry site (IRES) for the foot and mouth disease virus with that of the encephalomyocarditis virus ( from pMG vector described; ref. 6). To generate cell surface–bound human interleukin 15 (IL-15), the granulocyte macrophage colony-stimulating factor signal peptide sequence was fused to the coding sequence of mature human IL-15 at the 5′ end of a modified human IgG4 Fc region (5) fused in frame to human CD4 transmembrane domain and correct assembly was verified by DNA sequence analysis. The membrane-bound IL-15-Fc cytokine fusion gene was subcloned into pIRESpuro3 (Clontech) to obtain IL-15-Fc/pIRESpuro3.
Cell lines and primary human T cells
Daudi (Burkitt lymphoma) and HLAnull K562 (erythroleukemia) cells were obtained from American Type Culture Collection. Lymphoblastoid cells (LCL) were a kind gift of Dr. Helen Heslop (Cell and Gene Therapy, Baylor College of Medicine, Houston TX). These cell lines were cultured in HyQ RPMI 1640 (Hyclone) supplemented with 2 mmol/L Glutamax-1 (Life Technologies-Invitrogen) and 10% heat-inactivated defined FCS (Hyclone), referred to as culture medium (1). Human T cells were isolated by density gradient centrifugation over Ficoll-Paque-Plus (GE Healthcare Bio-Sciences AB), from umbilical cord blood or peripheral blood mononuclear cells (PBMC) after consent, and were cultured in culture medium.
Generation of aAPC
As previously reported, K562 cells were electroporated with DNA plasmids to enforce expression of all of the following: truncated CD19, 4−1BBL, and MICA fused to GFP (18). These aAPCs were further modified to express membrane-bound IL-15 to provide a cytokine stimulus at the site of CAR-binding and T-cell costimulation (19).
Electroporation and T-cell coculture with aAPC
On day 0, PBMCs and umbilical cord blood mononuclear cells (107) were suspended in 100 μL of Amaxa Nucleofector solution (CD34 kit) and mixed with 5 μg of supercoiled CoOpCD19RCD28/pT-MNDU3 and 5 μg pCMV-SB11 DNA plasmids, transferred to a cuvette, and electroporated (Program U-14). After a 10-min room temperature incubation, the cells were transferred to a six-well plate containing 3 to 4 mL incomplete phenol-free RPMI and rested for 2 to 3 h. The cells were cultured overnight in 6 to 7 mL 10% phenol-free RPMI and stimulated the next day (day 1) with γ-irradiated (100 Gy) aAPC at a 1:10 T cell/aAPC ratio. The γ-irradiated aAPC were re-added every 7 d. Recombinant human interleukin 2 (rhIL-2; Chiron) was added to the cultures at 50 units/mL on a Monday-Wednesday-Friday schedule, beginning day 1 of each 7-d expansion cycle. The supercoiled plasmid CoOpCD19RCD28/Hy-pVitro4 (expressing CAR under control of EF1α promoter and Hy under control of CAG promoter) was electroporated (10 μg) into PBMCs (107) using Nucleofector technology and T cells were propagated by cross-linking CD3 using an OKT3-mediated 14-d rapid expansion protocol (REP) as described previously using allogeneic γ-irradiated PBC and LCL feeder cells in the presence of exogenous (soluble) rhIL-2 (20). T cells were enumerated every 7 d, and viable cells were counted based on trypan blue exclusion.
Western blot
Expression of the chimeric 66-kD (CD19R) and 79-kD (CD19RCD28) CD3-ζ was accomplished using a primary mouse anti-human CD3-ζ mAb (1 μg/mL; BD Biosciences) and secondary horseradish peroxidase (HRP)–conjugated goat anti-mouse IgG (1:75,000; Pierce) under reducing conditions, based on methods previously described (20). Protein lysates were transferred onto nitrocellulose membrane using iBlot Dry Blotting System (Invitrogen) and developed with SuperSignal West Femto Maximum Sensitivity substrate (Pierce) per the manufacturer's instructions and chemiluminescence was captured after 1-min exposure using VersaDoc MP 4000 Imaging System (Bio-Rad).
Generation of monoclonal antibody recognizing a CD19-specific CAR
Female BALB/c mice were injected six times in the foot at 3-d intervals with syngeneic NS0 cells expressing CD19R CAR. Three days after the last immunization, mice were sacrificed, popliteal lymph nodes were removed, and cells were fused with P3−8AG-X653 myeloma cells at a ratio of 3:5, using 30% polyethylene glycol 1450 (in serum free RPMI containing 5% DMSO). After 10 d, hybridoma colonies were picked, cloned by limiting dilution in 96-well plates, and 100 μL of supernatants were screened by ELISA for differential binding to round-bottomed 96-plates containing adsorbed (105/well) CD19R+ and CD19Rneg Jurkat cells as detected by 1:500 dilution of HRP-conjugated goat anti-mouse IgG (Santa Cruz Biotechnology). Detection was achieved by TMB Microwell peroxidase substrate system (KPL). Protein G column (Roche) purified mAb was conjugated to Alexa Fluor 488 (Invitrogen-Molecular Probes) per manufacturer's instructions.
Flow cytometry
Fluorochrome-conjugated reagents were obtained from BD Biosciences: anti-CD4, anti-CD8, anti-CD25, anti-CD27, anti-CD28, anti-CD62L, anti-CD45RA, anti-CD45RO, and anti-CD95. Affinity-purified F(ab′)2 fragment of FITC-conjugated goat anti-human Fcg (Jackson Immunoresearch) was used at 1/20 dilution to detect cell surface expression of CD19-specific CAR. Purified CAR-specific mAb clone 2D3, conjugated to Alexa Fluor 488, was used at a dilution of 1/30, giving a concentration of ∼30 μg/mL. In some experiments, binding of this mAb to the Fc region of CAR was blocked (30 min at 4°C) using goat human Fc-specific antiserum (Sigma). Blocking of nonspecific antibody binding was achieved using FACS wash buffer (2% FCS in PBS). T-cell receptor (TCR)-Vβ expression was determined with a panel of 24 TCR-Vβ–specific mAbs (IO TEST Beta Mark TCR-Vβ repertoire kit, Beckman Coulter) used in association with anti-CD3 and appropriate isotype-matched control mAbs. Data acquisition was on a FACSCalibur (BD Biosciences) using CellQuest version 3.3 (BD Biosciences). Analyses and calculation of mean fluorescence intensity (MFI) was undertaken using FCS Express version 3.00.007 (Thornhill).
Intracellular IL-2 cytokine staining
Intracellular IL-2 was assayed using the Intracellular Cytokine Staining Starter Kit (BD PharMingen) per the manufacturer's instructions. Briefly, 105 T cells were incubated with 0.5 × 106 stimulator cells in 200 μL culture medium along with protein transport inhibitor (BD Golgi Plug containing Brefeldin A) in a 96-well plate. Following a 4- to 6-h incubation at 37°C, the cells were stained for CAR expression using hybridoma mAb clone 2D3 at 4°C for 30 min. After washing, the cells were fixed and permeabilized (100 μL, Cytofix/Cytoperm buffer) and phycoerythrin-conjugated mAb specific for IL-2 was added. Cells were further washed and analyzed by FACSCalibur. T cells were treated with a leukocyte activation cocktail (phorbol 12-myristate 13-acetate and ionomycin) as a positive control.
Confocal microscopy
Jurkat parental and CD19R+ Jurkat cells were stained with the hybridoma clone mAb 2D3, at a 1:50 dilution for 15 min at 4°C, washed in FACS wash buffer, and fixed with 0.1% paraformaldehyde. After fixing, the cells were washed twice with FACS wash buffer and transferred onto slides, and coverslips were mounted with Prolong Gold anti-fade agent (Invitrogen). Cells were examined under a confocal microscope (Leica TCS SP2-SE) using oil immersion lens (×63 objective). Single-scan images were obtained with a 4.76× zoom in a 1,024 × 1,024 format with a line averaging of 8.
Chromium release assay
The cytolytic activity of T cells was determined by 4-h chromium release assay (1). CD19-specific T cells were incubated with 5 × 103 51Cr-labeled target cells in a V-bottomed 96-well plate (Costar). The percentage of specific cytolysis was calculated from the release of 51Cr, as described earlier, using a TopCount NXT (Perkin-Elmer Life and Analytical Sciences, Inc.). Data are reported as mean ± SD.
DNA PCR for SB transposon and transposase
DNA was isolated from PBMC using the QIAmp DNA mini kit (Qiagen). PCR was carried out using CD19RCD28-specific forward primer 5′-AGATGACCCAGACCACCTCCAGC-3′ and reverse primer 5′-GGTATCCTTGGTGGCGGTGCT-3′ for the transposon. The PCR reaction used 1 μg of DNA/sample in a mix containing 10× PCR buffer, 2.5 mmol/L deoxynucleotide triphosphates, 3 μmol/L MgCl2, and 0.5 units of DNA polymerase (AmpliTaq Gold, Applied Biosystems) in a final volume of 50 μL amplified in a thermal Cycler (PTC-200 DNA Engine Cycler, Bio-Rad). After an initial denaturation at 95°C for 5 min, the samples underwent 34 cycles of 95°C for 30 s, 65°C for 30 s, 72°C for 1 min 15 s, followed by a prolonged extension step at 72°C for 7 min. For the transposase gene, PCR was carried out using SB11-specific forward primer 5′-ATGGGACCACGCAGCCG-3′ and reverse primer 5′-CGTTTCGGGTAGCCTTCCACA-3′. After an initial denaturation at 95°C for 5 min, the samples underwent 34 cycles of 95°C for 15 s, 58°C for 30 s, 74°C for 2 min followed by a prolonged extension step at 74°C for 7 min. The housekeeping gene GAPDH was also amplified in the same samples using the forward primer 5′-TCTCCAGAACATCATCCCTGCCAC-3′ and reverse primer 5′-TGGGCCATGAGGTCCACCACCCTG-3′.
Chromosome banding analysis
Exponentially growing _SB_-transfected T-cell cultures ( freshly fed 24 h earlier) were incubated for 2 h at 37°C with colcemid (20 μL of 0.04 μg/mL) per 10 mL medium followed by 0.075 mol/L KCl at room temperature for 15 min, fixed with acetic acid/ methanol (1:3), and washed thrice on a glass slide. For Giemsa (G) banding, 5- to 6-d-old slides treated with trypsin were stained with Giemsa stain following standard techniques described previously (21). A total of 15 G-banded metaphases were photographed and 5 complete karyotypes were prepared using a karyotyping system from Applied Imaging Corporation.
Results
We describe a new approach to using nonviral gene transfer of DNA plasmids to efficiently obtain populations of memory and effector T cells with desired specificity (Fig. 1_D_). The system we have devised provides for robust antigen-driven expansion of CD4+ and CD8+ CAR+ T cells to clinically meaningful numbers.
Monoclonal antibody with specificity for CD19-specific CAR
The cell surface expression of the introduced CAR was predicted to increase with outgrowth of T-cell populations that have undergone CAR-mediated numerical expansion on CD19+ aAPC. Currently, the only-commercially available flow cytometry reagents that recognize our CARs are polyclonal anti-Fc antibodies raised in goat, but we desired a homogeneous monoclonal product for use in the release of CAR+ T cells for clinical trials. To longitudinally follow the transgene expression, we developed a CAR-specific mAb by immunizing mice with syngeneic NS0 cells expressing CD19R. A hybridoma mAb clone 2D3 (IgG1) was selected by flow cytometry that selectively bound to CD19R+ Jurkat cells, but not parental Jurkat cells. The binding of 2D3 can be blocked using a Fc-specific antibody (Fig. 2A). The 2D3 clone bound a CD20-specific CAR that shares the IgG4 Fc region with CD19R and CD19RCD28 (data not shown). The pattern of staining by confocal microscopy showed 2D3 binding to CAR on the cell surface (Fig. 2_B_). These data are consistent with a mAb binding specifically to the CD19-specific CAR and recognizing the modified human IgG Fc region. Of note, the production of this mAb avoided the need to purity recombinant CAR protein as the immunogen was genetically modified NS0 cells and the ELISA screening used genetically modified Jurkat cells.
Figure 2.
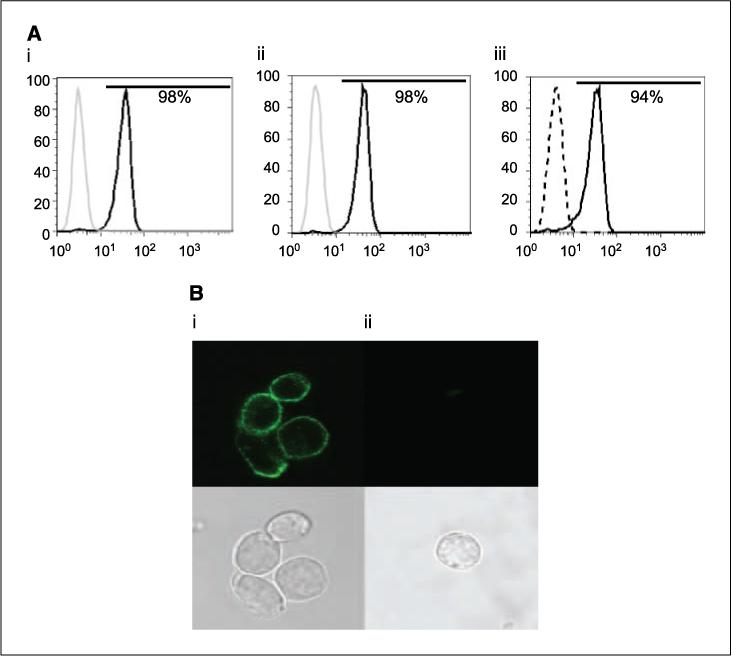
Specificity of mouse-derived CAR-specific mAb (clone 2D3). A, Jurkat cells were genetically modified and sorted to express CD19R. Jurkat parental (gray line) and CD19R+ (black line) cells were stained with (i) Alexa 488–conjugated clone 2D3 and (ii) F(ab′)2-fragment of goat-derived polyclonal antibody specific for human Fc; iii, binding of 2D3 (solid line) was blocked by polyclonal Fc-specific antisera (dashed line). B, cell surface staining of Alexa Fluor 488–conjugated clone 2D3 by confocal microscopy on (i) CD19R+ Jurkat cells and (ii) Jurkat parental cells. Cells were stained, fixed, and mounted as described in Materials and Methods.
Electrotransfer of SB two-plasmid DNA system
We have used a nonviral gene transfer approach to introduce codon optimized DNA expression plasmids because these expression vectors can be readily and cheaply manufactured to clinical grade. Although codon modification of TCR genes has been shown to enhance expression of transgenic TCR in primary human T cells (22), we now show the usage of a codon optimized second-generation CAR. Previously, our electroporation approach based on the Multiporator (Eppendorf; refs. 23, 24) used T cells that had been stimulated to proliferate by cross-linking CD3 with OKT3 to allow access of the introduced naked DNA to the nucleus after dissolution of the nuclear envelope during prometaphase. However, T cells nonspecially activated to proliferate, such as by cross-linking CD3 as occurs in the REP (25), would preclude subsequent immediate antigen-mediated propagation and thus directed outgrowth of CAR+ T cells. Nucleofector technology has been used to electroporate nonreplicating cells by direct transfer of DNA to the nucleus (26). Thus, we investigated whether this electrotransfer system could be used to genetically modify circulating T cells from peripheral blood and umbilical cord blood, which are in a quiescent state. To assist with subsequent translation to clinical practice, the Nucleofector solution is available for use in cGMP.
Both the SB transposase and IR have been independently manipulated to improve efficiency of transposition, but changes to both do not generally seem to be additive. In preliminary experiments, we too compared the relative transposition efficiency of the SB10 (10, 16) and SB11 transposases (the latter exhibiting improved enzymatic activity; ref. 17) in a two-by-two matrix using the Amaxa 96-well Shuttle system to introduce these transposases and pT (15, 16) and pT2 transposons (the latter exhibiting improved transposition; ref. 27) into Jurkat T cells. As observed with other cell lines, we found a similar increase in transposition using SB11 with pT and using SB10 with pT2 (27) although, as reported, overproduction of transposase inhibited transposition (13, 17). When the pT2-improved transposon was combined with SB11, no further increase in transgene expression was observed over that achieved when these components were used with SB10 or pT, respectively. In the present study, a combination of pT transposon (for integration) and SB11 transposase (for transient expression) was used for experiments with primary T cells.
Generation of CD19+ aAPC
We determined whether peripheral blood and umbilical cord blood–derived T cells could be selectively propagated by stimulating through an introduced immunoreceptor. This experiment would evaluate our underlying hypothesis of whether the presence of the SB transposase would improve efficiency of CAR transposon integration in T cells. Our initial attempts at CAR-dependent T-cell propagation after electrotransfer of the SB system used allogeneic LCL because these are widely available as master cell banks (including at M. D. Anderson Cancer Center) manufactured in compliance with cGMP for phase I/II trials. However, these LCL resulted in nonspecific outgrowth of CARnull T cells that had undergone electrotransfer of SB plasmids, independent of CAR expression (data not shown), presumably due to outgrowth of alloreactive T cells. Because our SB transposon by design does not include a drug resistance gene, we avoided nonspecific propagation of T cells using K562 as aAPC because these do not express classical HLA molecules. K562 cells are widely recognized as a platform suitable for the numerical expansion of lymphocytes because they (a) can be cultured in compliance with cGMP, (b) express desired endogenous T-cell costimulatory molecules, (c) secrete pro-inflammatory cytokines, and (d) can be readily modified to enforce the expression of antigen and desired endogenous T-cell costimulatory molecules (28, 29). To provide an IL-15–mediated growth stimulus coordinated with recognition of CD19 antigen, the aAPC expressing tCD19, 4−1BBL, and MICA were further modified to express the IL-15 cytokine on the cell surface (IL-15-Fc; Fig. 2A). Membrane-bound IL-15 has been used before to propagate natural killer (NK) cells on K562 (19). The ability of these K562 aAPCs to propagate CAR+ T cells after electrotransfer of SB transposon and transposase plasmids is described in the next section.
_SB_-mediated gene transfer of CAR transposon in primary T cells
After using the Nucleofector to import plasmid DNA into quiescent T cells, we observed that peripheral blood– and umbilical cord blood–derived electroporated CD4+ and CD8+ T cells readily expressed the CAR transposon (Table 1; Fig. 3_A_). Not surprisingly, the presence of the plasmid expressing the SB11 transposase did not increase transposon expression when measured 24 hours after electroporation (22% and 27% CAR expression with and without transposase, respectively), as this early time point for assessing transgene expression records transient nonintegrated CAR expression (Fig. 3_A_). The genotoxicity reported with excess expression of SB transposase (17) was apparently controlled in our two-plasmid system using a 1:1 ratio of transposon and transposase. To obtain peripheral blood– and umbilical cord blood–derived T cells with integrated transposon, the genetically modified cells were cocultured with γ-radiated aAPC (K562 genetically modified to express tCD19, 4−1BBL, MICA, IL-15-Fc) at a ratio of 1:10 (T cell to aAPC). After 5 weeks of continuous coculture (γ-radiated aAPC re-added every 7 days), the percentage of peripheral blood–derived T cells expressing CAR increased in the transposase-containing group (43%), whereas the CAR expression was lost (0.7%) when transposon was electro-transferred in the absence of transposase (Fig. 3_A_). Thus, after 28 to 35 days, the efficiency of two DNA plasmid _SB_-mediated gene transfer improved CAR expression by ∼49 to 60-fold, compared with a single plasmid transposon control (Table 1). The expression of the CAR was confirmed by Western blot of whole-cell lysates of propagated T cells probed using a mAb specific for CD3-ζ chain revealed the 79-kDa chimeric ζ chain in addition to the 21-kDa endogenous ζ chain (Fig. 3_C_). To monitor for the presence or absence of the integrated CD19RCD28 transgene, DNA from the numerically expanded T cells, electroporated with and without SB11, were PCR amplified using CAR-specific primers. A 1,900-bp band corresponding to the CD19RCD28 transgene was observed in T cells electroporated using the SB two-plasmid system, whereas no similar band was observed in cells electroporated with SB transposon in the absence of transposase, which is consistent with improved SB11-mediated transposition in T cells expressing CAR protein (Fig. 3_D_).
Table 1.
Percent CAR expression in T cells after electroporation of CAR transposon with or without SB11 transposase plasmid
| DNA plasmid(s) | Day 1 | Day 28 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD4+CAR+ | CD8+CAR+ | Total CAR+ | CD4+CAR+ | CD8+CAR+ | Total CAR+ | |||||||
| PBMC | UCB | PBMC | UCB | PBMC | UCB | PBMC | UCB | PBMC | UCB | PBMC | UCB | |
| No DNA | 0.6 | 1.0 | 0.2 | 0.1 | 1.6 | 1.3 | ||||||
| SB11 | 0.9 | 1.1 | 0.8 | 0.2 | 1.2 | 1.4 | ||||||
| Txpn* | 15.9 | 10.7 | 9.5 | 1.0 | 27.0 | 11.8 | 0.07 | 0.8 | 0.5 | 0.3 | 0.8 | 3.5 |
| Txpn* and SB11 | 13.3 | 4.9 | 7.9 | 0.8 | 22.0 | 5.4 | 27.5 | 24.8 | 13.5 | 1.9 | 38.9† | 29.7‡ |
Figure 3.
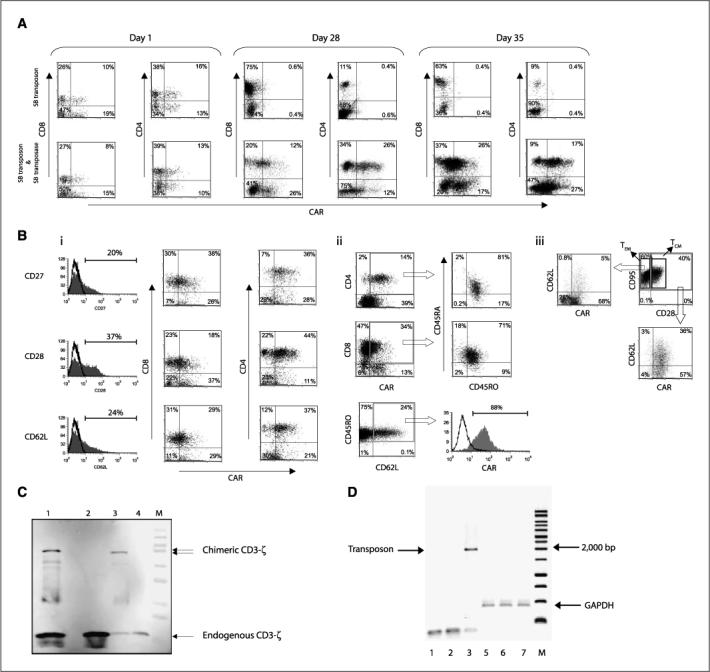
Characterization of CAR expression on peripheral blood–derived T cells after electrotransfer of SB plasmid system. A, expression of CAR on CD8+ and CD4+ T cells after electrotransfer of SB transposon with or without SB11 transposase at 24 h, and 4 and 5 wk of coculture on g-irradiated K562-derived aAPC expressing tCD19, IL-15-Fc, MICA, and 4−1BBL. B, i, immunophenotype of memory cell markers (CD27, CD28, CD62L) on genetically modified T cells generated after 5 wk of coculture on aAPC. The gray-filled histograms reveal the percentage of T cells expressing CD27, CD28, and CD62L in the lymphocyte-gated population. Those expressing the memory cell markers were analyzed for coexpression of CAR (detected by mAb clone 2D3) and CD8 or CD4. ii, expression of CD45RO, CD45RA, and CD62L on T cells generated after coculture. CAR+ CD4 or CD8 cells were analyzed for the expression of CD45RA and CD45RO. The MFI of the unmanipulated T cells was 867/50 (CD45RA/CD45RO) compared with 28/38 for the SB-transfected T cells. CD45RO and CD62L double-positive cells were also analyzed for coexpression of CAR. iii, TCM, defined as double-positive for CD28 and CD95 (TEM, CD28negCD95pos), were analyzed for coexpression of CD62L and CAR. C, Western blot analysis of CAR expression detected by mAb specific for CD3-ζ. Whole-cell protein (20 μg) lysates from primary T cells genetically modified with CoOpCD19RCD28 (lane 1, ∼79 kDa chimeric protein) or no plasmid control (lane 2); CD19R+ Jurkat cells (lane 3, ∼66 kDa chimeric protein) or parental Jurkat (lane 4) were resolved by SDS-PAGE under reducing conditions. D, integration of CoOpCD19RCD28 by PCR. DNA was isolated from T cells after mock electroporation (no DNA, lanes 1 and 4), from T cells 28 d after electroporation with SB transposon in the absence of transposase (lanes 2 and 5), and from T cells 28 d after electroporation with transposon in the presence of SB11 transposase (lanes 3 and 6). PCR was accomplished using transposon-specific primers (lanes 1–3) or GAPDH-specific primers (lanes 4−6). The data showing SB system in peripheral blood/cord blood are from a representative experiment.
Propagation of CAR+ T cells
The K562-derived aAPC was calculated to give a 20-fold growth of genetically modified T cells at the end of 4 weeks with continued and accelerated expansion thereafter (Fig. 4_A_). A subset analysis revealed that populations of both CD4+CAR+ and CD8+CAR+ T cells could be propagated (Table 1). Initially, the rates of CD4+ and CD8+ T-cell growth on aAPC were similar, but after ∼8 weeks there was an outgrowth of CD4+CAR+ T cells (Fig. 4_B_). Thus, continued time in tissue culture could be used to derive CAR+ T cells with an increased CD4 to CD8 ratio. We also followed the percentage expression and density of the CAR on the T-cell surface by flow cytometry. With coculture, there was outgrowth of percentage of T cells expressing the CD19-specific CAR (22% on day 1 and peaking at 99% on day 70). However, as the percentage of CAR+ T cells increased, there was a decrease in the density of CAR expression, as the MFI dropped from a peak of 109 arbitrary units at 21 days, early in the coculturing process, and then declined over culture time. The amount of CAR for the population peaked at ∼70 days after electroporation (percentage expression multiplied by MFI). Thus, adding a fixed ratio of aAPC (with a fixed density of CD19 antigen) to T cells seems to have supported the growth for populations of T cells that either expressed high density of CAR or high percentage of CAR.
Figure 4.

Sustained proliferation of genetically modified primary peripheral blood–derived T cells with analysis of CAR expression and TCR repertoire. A, kinetics of propagation of T cells in culture with aAPC. The average T-cell numerical expansion was 22-fold (range 20−31) every 7 d for up to 10 wk of continuous coculture with aAPC. B, percent expression (unbroken lines, left axis) and density as measured by MFI (dotted line, right axis) of CAR on total, CD4+, CD8+ T cells as measured by flow cytometry over culture time on aAPC. C, TCR Vβ analysis by flow cytometry 4 wk after electrotransfer of SB plasmids (filled columns) or CoOpCD19CD28/Hy-pVitro4 plasmid (open columns). Data are representative of two different experiments.
Immunophenotype of CAR+ memory T cells
Previously, T cells from healthy donors electroporated to express a CD19-specific CAR and nonspecifically activated for proliferation by cross-linking CD3 with OKT3 using REP have shown a predominant phenotype consistent with differentiated effector CD8+ T cells (20). In contrast, after electrotransfer of SB plasmids and numerical expansion on aAPC, T cells exhibited a heterogeneous immunophenotype and apparently included populations of CAR+ TCM. We showed that the CAR+ T cells expressed memory cell markers (CD27, CD28, CD62L; refs. 30–32) as well as determinants of an effector-cell phenotype (Fig. 3_Bi_). For example, over half of CD27+, CD28+, and CD62L+ T cells expressed CAR. Indeed, as a marker for TCM, 88% of the CD62L+CD45RO+ cells expressed the CAR (Fig. 3_Bii_; ref. 33). Upon gating CD45RO+ cells, we observed a preferential expansion of T cells with this memory-cell marker in the cultured _SB_-transfected T cells (90%; MFI, 38) compared with unmanipulated T cells obtained directly from PBMC (36%; MFI, 50). TEM and TCM have also been distinguished based on relative expression of CD28 and Fas (34). Using these markers, we were able to identify that genetically modified and propagated TCM constituted ∼40% of the total cell population and CD28negCD95+ TEM represented the remainder of the propagated T cells. Multiparameter flow cytometry further revealed that 39% of CAR+CD28+CD95+ TCM expressed CD62L. In comparison, only 7% of the CAR+ TEM expressed CD62L (Fig. 3_Biii_). These data reveal that CAR+ T cells are present in T cells that express markers consistent with TCM. Preferential expansion of T cells in tissue culture with an apparent memory phenotype can also be inferred by from the ratio of CD45RA/CD45RO, which decreased from 2.75 in unmanipulated freshly derived PBMC to 0.9 for SB-transfected and ex vivo propagated T cells (Fig. 3_Bii_). The relative percentage increase of observed CD45RO+ cells, or the decrease in CD45RA/CD45RO ratio, is presumably due to the repetitive antigenic stimulation of cultured T cells resulting in down-regulation of the high molecular weight CD45RA isoform and reciprocal up-regulation of the low molecular weight isoform CD45RO during time in culture. Coexpression of both CD45RA and CD45RO has been associated with the phenotype of effector T cells (35) but as in circulating peripheral blood–derived T cells express both CD45RA and CD45RO, the markers are presumably also present on memory cells. These data have implications for improved in vivo efficacy as TCM are associated with long-term persistence after adoptive transfer.
TCR Vβ repertoire
We tracked the expression of TCR Vβ usage by flow cytometry over time with the hypothesis that an improvement in DNA-plasmid integration would be reflected by maintenance of a broad pre-electroporation TCR Vβ repertoire. The pattern of TCR Vβ usage observed after electrotransfer of the two DNA SB plasmids and propagation on aAPC was much broader than when T cells were electroporated using the single CoOpCD19RCD28/Hy-pVitro4 plasmid and expanded by REP by cross-linking CD3 with OKT3 in cytocidal concentrations of hygromycin B. We observed that ∼80% of the T cells electroporated with CoOpCD19RCD28/HypVitro4 plasmid expressed a single TCR Vβ family (Vβ5.3). In contrast, ∼80% of the T cells electroporated with the complete SB system expressed 30% of the TCR Vβ families (Fig. 4_C_). This is consistent with less efficient integration of the CoOpCD19RCD28/Hy-pVitro4 plasmid compared with the SB system. These data have implications for design of adoptive immunotherapy trials as maintaining a broad TCR diversity is desired to restore immune reconstitution after myeloablative preparative regimens.
Redirected function of CAR+ T cells after electrotransfer of SB plasmids
The numerically expanded T cells were evaluated for redirected killing. The genetically modified T cells were able to lyse CD19+ targets, and specificity of killing was shown by the background lysis of CD19neg K562 cells (Fig. 5_A_). We showed a 25-fold increase in specific lysis of CD19+ K562 at effector-to-target ration of 50:1. The lack of killing of CD19neg K562 is consistent with absence of resident NK cell function in the culture, as these target cells are sensitive to NK cell–mediated lysis. Because the CAR contains a CD28 endodomain, we investigated whether T cell–derived IL-2 could be produced when CAR contacted CD19 antigen in the absence of binding CD80 or CD86. An intracellular cytokine assay showed that IL-2 could be detected in the CAR+ T cells only when cultured with CD19+ stimulator cells and not with CD19neg cells (Fig. 5_B_). There was an ∼4-fold increase in IL-2 expression when CAR+ T cells were stimulated by CD19+CD80negCD86neg K562 cells compared with CD19neg K562 parental controls. No significant IL-2 production was observed when T cells were cultured in absence of stimulator cells. These data are consistent with activation of T cells for killing and IL-2 cytokine production through the CAR.
Figure 5.
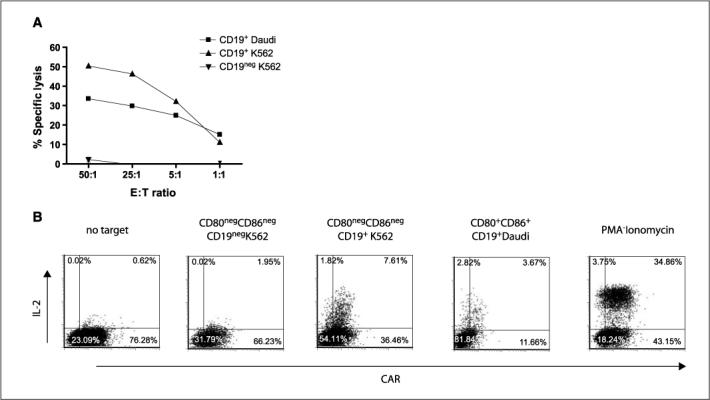
Redirected specificity of peripheral blood–derived T cells genetically modified with SB system. A, killing of CD19+ target cells (HLA Ineg Daudi; HLA class I/IIneg K562 cells transfected to express truncated CD19) in a standard 4-h chromium release assay. Background lysis of CD19neg (parental K562) cells is shown. Points, mean specific lysis of triplicate wells at effector to target (E:T) cell ratios; bars, SD. B, CAR and intracellular IL-2 expression after incubating with panel of stimulator cells by multiparameter flow cytometry gating on CD3+ lymphocytes. Phorbol 12-myristate 13-acetate (PMA) and ionomycin were added as a positive control.
Lack of integration of SB11 transposase in propagated T cells
Continued presence of the SB11 transposase in genetically modified T cells may cause genotoxicity. We evaluated for the presence of integrated transposase plasmid by genomic PCR. No band corresponding to the SB11 transposase gene (size ∼830 bp) was detected in T cells that were electroporated with the SB transposon and transposase and had undergone 4 weeks of coculture with aAPC (Fig. 6_A_), which is consistent with the rapid loss of transposase expression activity over the first few days postdelivery in mice (36). These results indicate that the SB11 transposase was not integrated into the genome of cells stably expressing the CD19RCD28 CAR.
Figure 6.
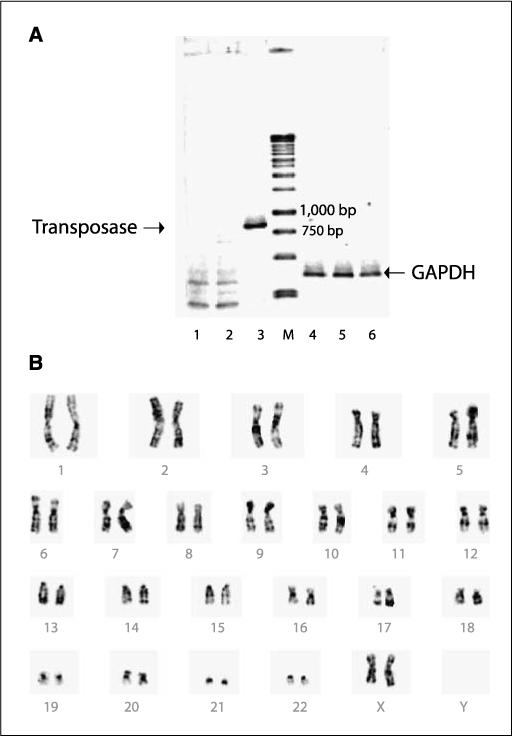
Safety issues regarding SB transposase and chromosomal aberrations. A, lack of integration of SB11 transposase by genomic PCR from genetically modified and propagated peripheral blood–derived T cells. DNA was isolated from T cells after mock electroporation (no DNA, lanes 1 and 4), from T cells 28 d after electroporation with the two-plasmid SB system (lanes 2 and 5), or from T cells 1 d after electroporation with the two-plasmid SB system (lanes 3 and 6). PCR was accomplished using transposase-specific primers (lanes 1−3) or GAPDH-specific primers (lanes 4−6). B, idiogram of a G-banded karyotype of the _SB_-transfected peripheral blood–derived T cells showing no apparent numerical or structural chromosome alterations.
Karyotype of genetically modified T cells
As a measure of global genotoxicity associated with undesired and continued transposition, we evaluated the integrity of the chromosome structure. G-banding analysis of the _SB_-transfected T cells showed a normal female karyotype, 46, XX with no apparent numerical or structural chromosome alterations (Fig. 6_B_). Although this does not exclude chromosomal damage below the limit of detection of this technique, it supports the premise that SB transposition in T cells is not associated with translocations and chromosomal aberrations.
Discussion
We have previously showed that peripheral blood– and umbilical cord blood–derived T cells can be rendered specific for CD19, based on using a CAR capable of providing a fully competent activation signal, development of aAPC-expressing antigen, and desired costimulatory signals. In this report, we describe the use of SB transposon/transposase plasmids to introduce CD19-specific CAR leading to efficient outgrowth of CAR+ T cells on aAPC with preservation of CD4+, CD8+, central memory, and effector-cell immunophenotypes. This is expected to be of widespread interest as many institutions are evaluating the clinical potential of genetically modified T cells with redirected specificity. The majority of these programs use recombinant viral vectors, which, although efficient at gene transfer, are generally cost-prohibitive to manufacture to clinical grade and still permit incremental changes to clinical trial design. Yet, at this early stage of gene therapy planning with clinical grade T cells, what is needed, and is provided here, is a cost-effective gene transfer system that encourages reiterative changes to expression vector and/or CAR design to be used in proof-of-concept clinical trials that support hypothesis testing from the bench to the bedside and back again. The approximate cost for manufacture and release of a clinical grade plasmid DNA is between 20,000and20,000 and 20,000and40,000 depending on supplier and degree of release testing needed. This release testing typically requires restriction enzyme analyses, sequencing, and measures of (a) homogeneity/purity/contamination (protein, RNA, and other DNA) and (b) sterility including endotoxin. For early-phase proof-of-concept trials, this pricing compares favorably with the relatively high cost of recombinant retrovirus, including lentivirus as manufacture and release of clinical grade viruses may exceed 10 times the cost of DNA-plasmid production. Furthermore, there is downward pressure on the unit cost for DNA because there are many vendors worldwide with the capability to produce clinical grade plasmids. The manufacture/release of recombinant retrovirus is highly specialized, requiring the expertise of a small number of GMP facilities that contributes to high cost and can introduce delays to production and thus availability for clinical use.
Previously, the relatively low levels of nonviral gene transfer efficiency to introduce naked DNA plasmid coding CAR transgene, compared with viral-mediated transduction, has been compensated by lengthy periods of ex vivo tissue culture to select-out T cells expressing drug-metabolizing enzymes. Thus, an attractive feature of the SB gene transfer system to introduce CAR into T cells is avoidance of the need to express immunogenic section genes, such as bacteria-derived Hy transgene. Some human-derived drug-resistant transgenes are available for use in hematopoietic cells (37, 38), but they typically incorporate amino acid changes from the native protein sequence that may compromise their inability to remain nonimmunogenic and the continued presence of chemo-selective drugs may slow kinetics of ex vivo numerical expansion and alter T-cell function.
Coupling electrotransfer of SB system with selective propagation of CAR+ T cells was made possible using K562 cells that had been genetically modified to express costimulatory molecules to function as aAPC. We have previously shown that the presence of 4−1BBL and MICA on CD19+ K562 could propagate CD19R+ T cells (18). However, sustained antigen-driven numerical expansion of genetically modified T cells on aAPC has required the presence of rhIL-15 (18). In our current experiments, we found that the exogenous addition of this soluble cytokine led to nonspecific stimulation of T cells after electrotransfer, especially because there was no concomitant drug selection, resulting in the outgrowth of T cells that did not maintain CAR expression (data not shown). This could be corrected by expression of IL-15 at the interface between aAPC and T cells, using membrane-bound IL-15, as has been shown for the survival/propagation of NK cells (19, 39). This approach of expressing IL-15 on the cell surface has the further advantage of avoiding rhIL-15 protein that is not yet readily/widely available for use in clinical trials. Allogeneic LCL are another source of CD19+ aAPC to propagate CD19-specific T cells and are available to many centers operating facilities in compliance with cGMP as a master cell bank. However, the presence of HLA led to stimulation of T cells through activation of allospecific TCR and subsequent outgrowth of T cells that lacked CAR expression. This alloimmune response could be avoided using the K562 as aAPC, as these cells lack endogenous class I and II MHC (29).
Next-generation clinical trials using genetically modified T cells are expected to infuse predefined populations of T cells with defined characteristics such as the inclusion of both CD4+ and CD8+ T cells and TCM. There is convincing clinical data that the presence of CD4+ T-helper cells improves the persistence of CD8+ antigen-specific T cells (40). Furthermore, clinical trials using melanoma-specific T cells have shown in vivo long-term persistence of subpopulations of infused CD28+ memory T cells (41) and human experience has shown a preference for the selective survival of autologous HIV-specific CD27+ versus CD27neg adoptively transferred T cells (31). These data are supported by nonhuman experiments in which adoptive transfer of ex vivo propagated macaque CD28+CD95+CD62L+ TCM resulted in longer in vivo persistence compared with infusion of numerically expanded effector T cells (34). We note that the electrotransfer of SB plasmids and subsequent CAR-mediated propagation on aAPC supports the outgrowth of T cells with these desired phenotypes as our CAR+ T cells maintain expression of CD27, CD28, CD45RO, CD95, and CD62L. Clinical trials will be needed to determine whether adoptive transfer of these CAR+ T cells with an apparent central memory immunophenotype (CD28+CD95+CD62L+) results in long-term in vivo persistence of genetically modified T cells or whether these cells, despite being maintained for weeks in culture, will differentiate after infusion into effector T cells with limited in vivo survival. Clinical experience will also be needed to assess whether the presence of CD62L (L-selectin) on genetically modified T cells enables CAR+ T cells to traffic to sites of minimal residual disease for B-lineage malignancies, such as secondary lymphoid organs (42, 43).
Although there are a variety of transposase/transposon expression vectors available, we elected to combine the improved enzymatic activity of the SB11 transposase with pT IR sequences, rather than less efficient SB10 transposase with pT2 plasmid containing IR with improved IR activity. This was based on (a) the observation that integrated SB11 transposase could not be detected after T-cell culture on aAPC and (b) the assumption that because the CAR transposon with flanking IR is to be integrated, we wished to reduce the potential for introducing an element with increased potential for retransposition and potential deleterious chromosomal rearrangement. We note that the majority of viral vectors currently used in human gene therapy trials also contain elements flanking transgene to be integrated, such as the long terminal repeat (LTR) termini of recombinant retrovirus, with binding sites for enzymes with integrase activity. The transfer of retroviral-derived LTR has not been associated with deleterious host genome chromosome rearrangements, especially in T cells (44), and the low risk of genotoxicity due to the integrated presence of SB IR should be on par with retrovirally mediated transduction.
A gene transfer event with stable integration could result in deleterious insertional mutagenesis, but for SB transposition this seems to be less than retrovirally mediated transduction given the observed preference for random chromosomal integration at TA-dinucleotide base pairs areas. Although the safety of SB transposition can only be adequately addressed in clinical trials, we have not seen major chromosomal aberrations after electro-transfer of SB plasmids. Furthermore, to safeguard against the emergence of genetically modified T cells with autonomous growth, we routinely culture T cells after electroporation without aAPC and we are yet to observe evidence of antigen-independent proliferation. The risks for first-in-human trials using SB system would seem to be ameliorated when using T cells, rather than hematopoietic progenitor cells and in the setting of high-risk malignancies in which patients are expected to succumb to underlying relapsed malignancies. The risks of genotoxicity may be reduced in the future using a transposase with directed integration (45, 46), coupling persistent transposase activity with a transgene mediating conditional suicide, or introducing mRNA (47) rather than DNA coding for the transposase.
The future for clinical therapy infusing genetically modified T cells with redirected specificity looks promising. There are published reports on the therapeutic effect of T cells genetically modified to express full-length αβTCR (48) and clinical studies using T cells expressing chimeric receptors to redirect specificity have been reported or are under way (35, 49, 50). With the results of the first in-human trial infusing CD19-specific T cells being reported (3), the next step (the so-called second translational hurdle) will be expanding these single-institution experiences to multi-institution trials powered for efficacy. The platform we describe for producing CAR+ T cells should be appealing to investigators undertaking single-site as well as multisite trials using gene transfer of immunoreceptor(s) to redirect the specificity of T cells, including TCM. The system we have developed uses technology that is readily accessible and practiced in compliance with cGMP for phase I/II trials because we use (a) DNA plasmids, (b) electroporation using a commercial device, (c) weekly addition of irradiated immortalized aAPC feeder cells derived from K562 (which are available for use in cGMP), and (d) addition of exogenous rhIL-2 purchased through pharmacy stores.
In conclusion, we report a new gene transfer approach for the clinical application of T cells with redirected specificity for desired antigens. It is anticipated that this approach will be of interest not just for generating clinical grade T cells with specificity for CD19, but for genetically modifying T cells to express CAR with alternative specificities as well as for introducing TCR transgenes. Most adoptive immunotherapy trials that have shown therapeutic efficacy, e.g., to melanoma, CMV, EBV, and adenoviral antigens, have all used an in vitro antigen-driven proliferation step to propagate antigen-specific T cells before infusion. We have now incorporated ex vivo CAR-dependent proliferation to derive genetically modified T cells and will evaluate the CD19-specific T cells, using SB transposition and aAPC, in a next-generation clinical trial.
Acknowledgments
Grant support: Cancer Center Core Grant CA16672; RO1 CA124782 and CA120956; R21 CA129390 and CA116127; Department of Defense grant PR064229; The Alliance for Cancer Gene Therapy; The Alex's Lemonade Stand Foundation; The Carl C. Anderson, Sr. and Marie Jo Anderson Charitable Foundation; The Gillson Longenbaugh Foundation; The J.P. McCarthy Fund Developmental Grant Program; The Leukemia and Lymphoma Society; The Lymphoma Research Foundation; The Miller Foundation, The National Foundation for Cancer Research; The National Marrow Donor Program; and The Pediatric Cancer Research Foundation.
We thank Dr. Mark Kay at Stanford School of Medicine, Stanford, CA, for providing the pT plasmid; Dr. John Rossi at City of Hope Cancer Center, Duarte, CA, for his valuable suggestions; Karen Ramirez and David He from Flow Cytometry Core Laboratory (NIH grant 5P30CA016672-32) for their help with flow cytometry; Jim Wygant and Dr. Brad McIntrye in the Immunology Core for their help with the monoclonal antibody production; and Dr. Asha Multani from T.C. Hsu Molecular Cytogenetics Core for her assistance with G-banding.
References
- 1.Cooper LJ, Topp MS, Serrano LM, et al. T-cell clones can be rendered specific for CD19: toward the selective augmentation of the graft-versus-B-lineage leukemia effect. Blood. 2003;101:1637–44. doi: 10.1182/blood-2002-07-1989. [DOI] [PubMed] [Google Scholar]
- 2.Serrano LM, Pfeiffer T, Olivares S, et al. Differentiation of naive cord-blood T cells into CD19-specific cytolytic effectors for posttransplantation adoptive immunotherapy. Blood. 2006;107:2643–52. doi: 10.1182/blood-2005-09-3904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jensen MC, Popplewell L, DiGiusto DL, et al. A first-n-human clinical trial of adoptive therapy using CD19-specific chimeric antigen receptor re-directed T cells for recurrent/refractory follicular lymphoma. Mol Ther. 2007;15:S142. [Google Scholar]
- 4.Lupton SD, Brunton LL, Kalberg VA, Overell RW. Dominant positive and negative selection using a hygromycin phosphotransferase-thymidine kinase fusion gene. Mol Cell Biol. 1991;11:3374–8. doi: 10.1128/mcb.11.6.3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kowolik CM, Topp MS, Gonzalez S, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–1004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- 6.Cid-Arregui A, Juarez V, zur Hausen H. A synthetic E7 gene of human papillomavirus type 16 that yields enhanced expression of the protein in mammalian cells and is useful for DNA immunization studies. J Virol. 2003;77:4928–37. doi: 10.1128/JVI.77.8.4928-4937.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patterson SS, Dionisi HM, Gupta RK, Sayler GS. Codon optimization of bacterial luciferase (lux) for expression in mammalian cells. J Ind Microbiol Biotechnol. 2005;32:115–23. doi: 10.1007/s10295-005-0211-8. [DOI] [PubMed] [Google Scholar]
- 8.Berger C, Huang ML, Gough M, Greenberg PD, Riddell SR, Kiem HP. Nonmyeloablative immunosuppressive regimen prolongs in vivo persistence of gene-modified autologous T cells in a nonhuman primate model. J Virol. 2001;75:799–808. doi: 10.1128/JVI.75.2.799-808.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jung D, Jaeger E, Cayeux S, et al. Strong immunogenic potential of a B7 retroviral expression vector: generation of HLA-B7-restricted CTL response against selectable marker genes. Hum Gene Ther. 1998;9:53–62. doi: 10.1089/hum.1998.9.1-53. [DOI] [PubMed] [Google Scholar]
- 10.Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–10. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- 11.Izsvak Z, Ivics Z. Sleeping beauty transposition: biology and applications for molecular therapy. Mol Ther. 2004;9:147–56. doi: 10.1016/j.ymthe.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 12.Geurts AM, Hackett CS, Bell JB, et al. Structure-based prediction of insertion-site preferences of transposons into chromosomes. Nucleic Acids Res. 2006;34:2803–11. doi: 10.1093/nar/gkl301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang X, Wilber AC, Bao L, et al. Stable gene transfer and expression in human primary T cells by the Sleeping Beauty transposon system. Blood. 2006;107:483–91. doi: 10.1182/blood-2005-05-2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robbin PB, Yu XJ, Skelton DM, et al. Increased probability of expression from modified retroviral vectors in embryonal stem cells and embryonal carcinoma cells. J Virol. 1997;71:9466–74. doi: 10.1128/jvi.71.12.9466-9474.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holli RP, Nightingal SJ, Wan X, et al. Stable gene transfer to human CD34(+) hematopoietic cells using the Sleeping Beauty transposon. Exp Hematol. 2006;34:1333–43. doi: 10.1016/j.exphem.2006.05.023. [DOI] [PubMed] [Google Scholar]
- 16.Yan SR, Meus L, Chi W, Ivic Z, Izsva Z, Ka MA. Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat Genet. 2000;25:35–41. doi: 10.1038/75568. [DOI] [PubMed] [Google Scholar]
- 17.Geurts AM, Yang Y, Clar KJ, et al. Gene transfer into genomes of human cells by the sleeping beauty transposon system. Mol Ther. 2003;8:108–17. doi: 10.1016/s1525-0016(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 18.Numbenjapon T, Serrano LM, Singh H, et al. Characterization of an artificial antigen-presenting cell to propagate cytolytic CD19-specific T cells. Leukemia. 2006;20:1889–92. doi: 10.1038/sj.leu.2404329. [DOI] [PubMed] [Google Scholar]
- 19.Imai C, Iwamoto S, Campana D. Genetic modification of primary natural killer cells overcomes inhibitory signals and induces specific killing of leukemic cells. Blood. 2005;106:376–83. doi: 10.1182/blood-2004-12-4797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cooper LJ, Al-Kadhimi Z, Serrano LM, et al. Enhanced antilymphoma efficacy of CD19-redirected influenza MP1-specific CTLs by cotransfer of T cells modified to present influenza MP1. Blood. 2005;105:1622–31. doi: 10.1182/blood-2004-03-1208. [DOI] [PubMed] [Google Scholar]
- 21.Pathak S. Chromosome banding techniques. J Reprod Med. 1976;17:25–8. [PubMed] [Google Scholar]
- 22.Scholten KB, Kramer D, Kueter EW, et al. Codon modification of T cell receptors allows enhanced functional expression in transgenic human T cells. Clin Immunol. 2006;119:135–45. doi: 10.1016/j.clim.2005.12.009. [DOI] [PubMed] [Google Scholar]
- 23.Cooper LJ, Ausubel L, Gutierrez M, et al. Manufacturing of gene-modified cytotoxic T lymphocytes for autologous cellular therapy for lymphoma. Cytotherapy. 2006;8:105–17. doi: 10.1080/14653240600620176. [DOI] [PubMed] [Google Scholar]
- 24.Jensen MC, Clarke P, Tan G, et al. Human T lymphocyte genetic modification with naked DNA. Mol Ther. 2000;1:49–55. doi: 10.1006/mthe.1999.0012. [DOI] [PubMed] [Google Scholar]
- 25.Riddell SR, Greenberg PD. The use of anti-CD3 and anti-CD28 monoclonal antibodies to clone and expand human antigen-specific T cells. J Immunol Methods. 1990;128:189–201. doi: 10.1016/0022-1759(90)90210-m. [DOI] [PubMed] [Google Scholar]
- 26.Gresch O, Engel FB, Nesic D, et al. New non-viral method for gene transfer into primary cells. Methods. 2004;33:151–63. doi: 10.1016/j.ymeth.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 27.Cui Z, Geurts AM, Liu G, Kaufman CD, Hackett PB. Structure-function analysis of the inverted terminal repeats of the sleeping beauty transposon. J Mol Biol. 2002;318:1221–35. doi: 10.1016/s0022-2836(02)00237-1. [DOI] [PubMed] [Google Scholar]
- 28.Butler MO, Lee JS, Ansen S, et al. Long-lived antitumor CD8+ lymphocytes for adoptive therapy generated using an artificial antigen-presenting cell. Clin Cancer Res. 2007;13:1857–67. doi: 10.1158/1078-0432.CCR-06-1905. [DOI] [PubMed] [Google Scholar]
- 29.Suhoski MM, Golovina TN, Aqui NA, et al. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther. 2007;15:981–8. doi: 10.1038/mt.sj.6300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bachmann MF, Wolint P, Schwarz K, Jager P, Oxenius A. Functional properties and lineage relationship of CD8+ T cell subsets identified by expression of IL-7 receptor α and CD62L. J Immunol. 2005;175:4686–96. doi: 10.4049/jimmunol.175.7.4686. [DOI] [PubMed] [Google Scholar]
- 31.Ochsenbein AF, Riddell SR, Brown M, et al. CD27 expression promotes long-term survival of functional effector-memory CD8+ cytotoxic T lymphocytes in HIV-infected patients. J Exp Med. 2004;200:1407–17. doi: 10.1084/jem.20040717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–12. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 33.Baron V, Bouneaud C, Cumano A, et al. The repertoires of circulating human CD8(+) central and effector memory T cell subsets are largely distinct. Immunity. 2003;18:193–204. doi: 10.1016/s1074-7613(03)00020-7. [DOI] [PubMed] [Google Scholar]
- 34.Berger C, Jensen MC, Lansdorp PM, Gough M, Elliott C, Riddell SR. Adoptive transfer of effector CD8 T cells derived from central memory cells establishes persistent T cell memory in primates. J Clin Invest. 2008;118:294–305. doi: 10.1172/JCI32103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Park JR, Digiusto DL, Slovak M, et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Mol Ther. 2007;15:825–33. doi: 10.1038/sj.mt.6300104. [DOI] [PubMed] [Google Scholar]
- 36.Bell JB, Aronvovich EL, Schriefels JM, Clifford AM, Hoekstra ND, Whitley CB, Hackett PB. Duration of expression of Sleeping Beauty transposase in mouse liver following hydrodynamic delivery. Mol Ther. 2006;13(Suppl 1):S150. doi: 10.1038/mt.2010.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sato T, Neschadim A, Konrad M, Fowler DH, Lavie A, Medin JA. Engineered human tmpk/AZT as a novel enzyme/prodrug axis for suicide gene therapy. Mol Ther. 2007;15:962–70. doi: 10.1038/mt.sj.6300122. [DOI] [PubMed] [Google Scholar]
- 38.Yam P, Jensen M, Akkina R, et al. Ex vivo selection and expansion of cells based on expression of a mutated inosine monophosphate dehydrogenase 2 after HIV vector transduction: effects on lymphocytes, monocytes, and CD34+ stem cells. Mol Ther. 2006;14:236–44. doi: 10.1016/j.ymthe.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 39.Wittnebel S, Da Rocha S, Giron-Michel J, et al. Membrane-bound interleukin (IL)-15 on renal tumor cells rescues natural killer cells from IL-2 starvation-induced apoptosis. Cancer Res. 2007;67:5594–9. doi: 10.1158/0008-5472.CAN-06-4406. [DOI] [PubMed] [Google Scholar]
- 40.Rooney CM, Smith CA, Ng CY, et al. Infusion of cytotoxic T cells for the prevention and treatment of Epstein-Barr virus-induced lymphoma in allogeneic transplant recipients. Blood. 1998;92:1549–55. [PubMed] [Google Scholar]
- 41.Powell DJ, Jr., Dudley ME, Robbins PF, Rosenberg SA. Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood. 2005;105:241–50. doi: 10.1182/blood-2004-06-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galkina E, Florey O, Zarbock A, et al. T lymphocyte rolling and recruitment into peripheral lymph nodes is regulated by a saturable density of L-selectin (CD62L). Eur J Immunol. 2007;37:1243–53. doi: 10.1002/eji.200636481. [DOI] [PubMed] [Google Scholar]
- 43.Mitoma J, Bao X, Petryanik B, et al. Critical functions of N-glycans in L-selectin-mediated lymphocyte homing and recruitment. Nat Immunol. 2007;8:409–18. doi: 10.1038/ni1442. [DOI] [PubMed] [Google Scholar]
- 44.Bonini C, Bondanza A, Perna SK, et al. The suicide gene therapy challenge: how to improve a successful gene therapy approach. Mol Ther. 2007;15:1248–52. doi: 10.1038/sj.mt.6300190. [DOI] [PubMed] [Google Scholar]
- 45.Ivics Z, Katzer A, Stuwe EE, Fiedler D, Knespel S, Izsvak Z. Targeted sleeping beauty transposition in human cells. Mol Ther. 2007;15:1137–44. doi: 10.1038/sj.mt.6300169. [DOI] [PubMed] [Google Scholar]
- 46.Yant SR, Huang Y, Akache B, Kay MA. Site-directed transposon integration in human cells. Nucleic Acids Res. 2007;35:e50. doi: 10.1093/nar/gkm089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wilber A, Wangensteen KJ, Chen Y, et al. Messenger RNA as a source of transposase for Sleeping Beauty transposon-mediated correction of hereditary tyrosinemia type I. Mol Ther. 2007;15:1280–7. doi: 10.1038/sj.mt.6300160. [DOI] [PubMed] [Google Scholar]
- 48.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–9. doi: 10.1126/science.1129003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lamers CH, Langeveld SC, Groot-van Ruijven CM, Debets R, Sleijfer S, Gratama JW. Gene-modified T cells for adoptive immunotherapy of renal cell cancer maintain transgene-specific immune functions in vivo. Cancer Immunol Immunother. 2007;56:1875–83. doi: 10.1007/s00262-007-0330-3. [DOI] [PMC free article] [PubMed] [Google Scholar]