Structures of DNA Polymerase β with Active Site Mismatches Suggest a Transient Abasic Site Intermediate During Misincorporation (original) (raw)
. Author manuscript; available in PMC: 2008 May 29.
SUMMARY
We report the crystallographic structures of DNA polymerase β with dG—dAMPCPP and dC— dAMPCPP mismatches in the active site. These pre-mutagenic structures were obtained with a non-hydrolysable incoming nucleotide analog, dAMPCPP, and Mn+2. Substituting Mn2+ for Mg2+ significantly decreases the fidelity of DNA synthesis. The structures reveal that the enzyme is in a closed conformation like that observed with a matched Watson-Crick base pair. The incorrect dAMPCPP binds in a conformation identical to that observed with the correct nucleotide. To accommodate the incorrect nucleotide and closed protein conformation, the template strand in the vicinity of the active site has shifted upstream over 3 Å, permitting the coding base to vacate the active site generating an abasic templating pocket. The primer terminus rotates as its complementary template base is repositioned. This rotation moves O3’ of the primer terminus away from the α-phosphate of the incoming nucleotide thereby deterring misincorporation.
INTRODUCTION
DNA polymerases must select the proper nucleoside triphosphate from a pool of similar molecules to preserve the integrity of the genome (Kunkel and Bebenek, 2000). Failure to faithfully copy the genome during replication and DNA repair results in mutations that hasten aging and human disease (Loeb et al., 1974). Structural and biochemical data support the hypothesis that DNA polymerases discriminate between alternate substrates through an induced-fit mechanism where binding of the correct nucleotide leads to substrate/protein conformational adjustments that align catalytic groups to optimize chemistry (Joyce and Benkovic, 2004). In contrast, re-alignment of catalytic groups upon binding the wrong nucleotide is sub-optimal. The fidelity and catalytic efficiency for correct nucleotide insertion is dependent on the identity of the DNA polymerase. However, the efficiencies of incorrect nucleotide insertion by high- and low-fidelity DNA polymerases are similar suggesting that structural characterization of mismatched structures for one DNA polymerase could lead to general strategies utilized by all DNA polymerases to deter misinsertion (Beard et al., 2002a).
DNA polymerase β (Pol β) is a member of the X-family of DNA polymerases and, because of its size (39-kDa), is considered the simplest mammalian polymerase making it an ideal model to study the nucleotidyl transferase reaction at atomic detail. Pol β contributes two critical activities during the repair of simple DNA lesions through the base excision repair pathway: DNA synthesis and 5′-deoxyribose phosphate removal (Beard and Wilson, 2006). The extensive biological, kinetic and structural characterization of this enzyme provides a foundation to interpret new and novel structures.
Crystallographic structures of DNA polymerases derived from different polymerase families and biological sources indicate that they are composed of functionally distinct domains (Beard and Wilson, 2003). The polymerase domain is further generally comprised of three subdomains. The catalytic subdomain includes three carboxylates that coordinate two divalent metals, usually Mg2+, that are necessary for the nucleotidyl transferase reaction. Even though the structure of the catalytic subdomain is not conserved among the polymerase families, the divalent metals, primer terminus, and incoming correct deoxynucleoside triphosphate (dNTP) have a similar three-dimensional arrangement consistent with a two-metal ion mechanism for nucleotidyl transfer (Steitz et al., 1994).
Comparing structures of DNA polymerases with and without substrates indicates that polymerases display numerous conformational differences between liganded and non-liganded states. The largest protein conformational adjustment is often observed to be subdomain motions that allow the enzyme to close around the correct nascent base pair (Doublié et al., 1999). Previous structures of mismatches in the confines of the Pol β active site employed binary nicked DNA complexes with a terminal mispair (Krahn et al., 2004). These product complexes indicate that the polymerase approaches a fully open conformation and that the Watson-Crick edges of the mispair do not form hydrogen bonds. Additionally, a ternary complex structure of a dT— dGTP mispair in the confines of a Y-family polymerase, Dpo4 DNA polymerase, was trapped employing a dideoxy-terminated primer and Ca2+ (Vaisman et al., 2005). In this case, the triphosphate of the incoming dGTP and active site metals are distorted and stacking interactions of the ribose/base with the primer terminus decreased. It is unclear if the active site distortions are due to the lack of the primer O3’ and/or an alternate metal (i.e., Ca2+) with a much larger coordination sphere than Mg2+. Due to lack of ternary complex structures with an incorrect incoming nucleoside triphosphate and activating catalytic metal, the mechanism of mismatch incorporation has remained speculative.
RESULTS
Mn2+ Lowers DNA Synthesis Fidelity
We have found it necessary to use non-hydrolysable dNTP analogs to capture ternary Pol β structures with bound Mg2+ (Batra et al., 2006). Apparently, the catalytic Mg2+ can activate a coordinated water molecule that can hydrolyze the incoming correct nucleotide resulting in dNMP and PPi (Krahn et al., 2003). The weak binding of an incorrect incoming nucleotide is an added obstacle to forming a ternary substrate complex with a mismatched nascent base pair. Attempts to soak an incorrect nucleotide into crystals of binary polymerase/DNA complexes have failed to form ternary complex structures in the presence of Mg2+. Substituting Mn2+ for Mg2+ increases the binding affinity of the incoming nucleotide, both right (Table S1 in Supplemental Data) and wrong (Table 1), significantly and increases the rate of misinsertion, but not correct insertion, resulting in a significant decrease (>35-fold) in fidelity. Finally, Mn2+ serves as an excellent metal surrogate for Mg2+ since both have similar ionic radii and coordination geometries.
Table 1.
Steady State Misinsertion Kinetics of Human DNA Polymerase βa
| dX—dNTPb | _k_cat | _K_m,dNTP | _k_cat/_K_m | Fidelityc |
|---|---|---|---|---|
| 10_−_2 s−1 | μM | 10_−_4μM_−_1s−1 | ||
| Mg2+,d | ||||
| dC—dGTP | 34 (3) | 0.34 (0.06) | 10000 (2000) | 1 |
| dC—dATP | 0.27 (0.07) | 235 (34) | 0.115 (0.034) | 86960 (31040) |
| dG—dCTP | 30 (0.3) | 0.4 (0.1) | 7500 (2020) | 1 |
| dG—dATP | 0.14 (0.01) | 450 (50) | 0.031 (0.004) | 241900 (72200) |
| Mn2+ | ||||
| dC—dGTP | nde | nd | nd | 1 |
| dC—dATP | 1.8 (<0.1) | 14.1 (1.1) | 12.8 (1.2) | 2200 (210) |
| dG—dCTP | 5.2 (0.4) | 0.009 (<0.001) | 58000 (4400) | 1 |
| dG—dATP | 1.4 (0.2) | 16.5 (0.5) | 8.5 (1.2) | 6800 (1100) |
Closed Polymerase Conformation with a Nascent Base Pair Mismatch
A ternary complex structure with a dG—dAMPCPP (G—A) mismatch in the Pol β active site was obtained by soaking crystals of the polymerase binary single-nucleotide gapped DNA complex with dAMPCPP and Mn2+. A methylene group replaces the bridging oxygen between the α- and β-phosphates rendering the nucleotide inert toward hydrolysis and nucleotidyl transfer. Accordingly, this analog is an excellent inhibitor of Pol β-dependent DNA synthesis (Figure S1). Additionally, this approach permits the primer terminus to include O3’ that provides an important coordinating ligand with the catalytic metal upon binding the correct incoming nucleotide (Batra et al., 2006). The resultant crystal diffracted to 2.15 Å (Table 2). Two active site Mn2+ can easily be discerned from their anomalous signals (Figure S2). Compared to the open binary DNA complex (Figure 1A), the polymerase structure reveals that the enzyme is in a similar overall closed conformation as that seen with a correct Watson-Crick base pair (Figure 1B). The root mean square deviation (rmsd) of 314 Cα atoms between the structure with a correct incoming nucleotide (PDB ID 2FMS) and the G—A mismatch structure is 0.62 Å. As observed previously with the product mismatch complex (Krahn et al., 2004), the mispaired bases are not co-planar, but staggered (Figure 2A). However, a single hydrogen bond can be formed between O6(G) and N6(A). Using the same procedure, a crystal of a ternary complex structure with a pyrimidine–purine mismatch (dC—dAMPCPP) was also examined. The overall characteristics are very similar to those observed with the purine–purine (G—A) mismatch. The rmsd for the C—A mismatch structure and the closed complex with a Watson-Crick base pair is 0.70 Å (Figure S3). In contrast to the G—A mismatch, there are no hydrogen bonds between the mismatched bases. Instead, the templating cytosine (N4) may form a weak hydrogen bond with N1 of the primer terminal cytosine (Figure 2B).
Table 2.
Crystallographic Statistics
| Incoming dNTP | dUMPNPP | dAMPCPP | |
|---|---|---|---|
| Templating nucleotide | dA | dC | dG |
| Data collection | |||
| a (Å) | 50.79 | 54.44 | 54.75 |
| b (Å) | 80.68 | 78.24 | 78.37 |
| c (Å) | 55.06 | 54.85 | 55.17 |
| β (˚) | 108.38 | 113.19 | 113.36 |
| dmin (Å) | 2.40 | 2.60 | 2.15 |
| Rmerge (%)a | 8.1 (27.2)b | 9.6 (51.9) | 6.9 (34.4) |
| Completeness (%) | 99.3 (96.9) | 99.5 (99.4) | 98.3 (95.6) |
| I/σI | 17.4 (3.4) | 15.1 (2.2) | 9.3 (3.0) |
| Number of observed reflections | 58115 | 47096 | 82664 |
| Number of unique reflections | 16452 (1578) | 13062 (1280) | 22971 (2218) |
| Refinement | |||
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.006 | 0.007 | 0.006 |
| Bond angles (˚) | 1.094 | 1.256 | 1.084 |
| Rwork (%)c | 21.03 | 22.46 | 21.42 |
| Rfree (%)d | 27.83 | 30.73 | 27.04 |
| Average B factor (Å2) | |||
| Protein | 32.12 | 38.77 | 30.19 |
| DNA | 32.82 | 44.47 | 35.50 |
| Analogue | 15.71 | 44.11 | 31.41 |
| Ramachandran analysis (%)e | |||
| Favored | 94.4 | 89.0 | 96.0 |
| Allowed | 100 | 98.2 | 98.5 |
Figure 1. Closed Conformation of the Ternary Substrate Complex with an Active Site Mismatched Nascent Base Pair.
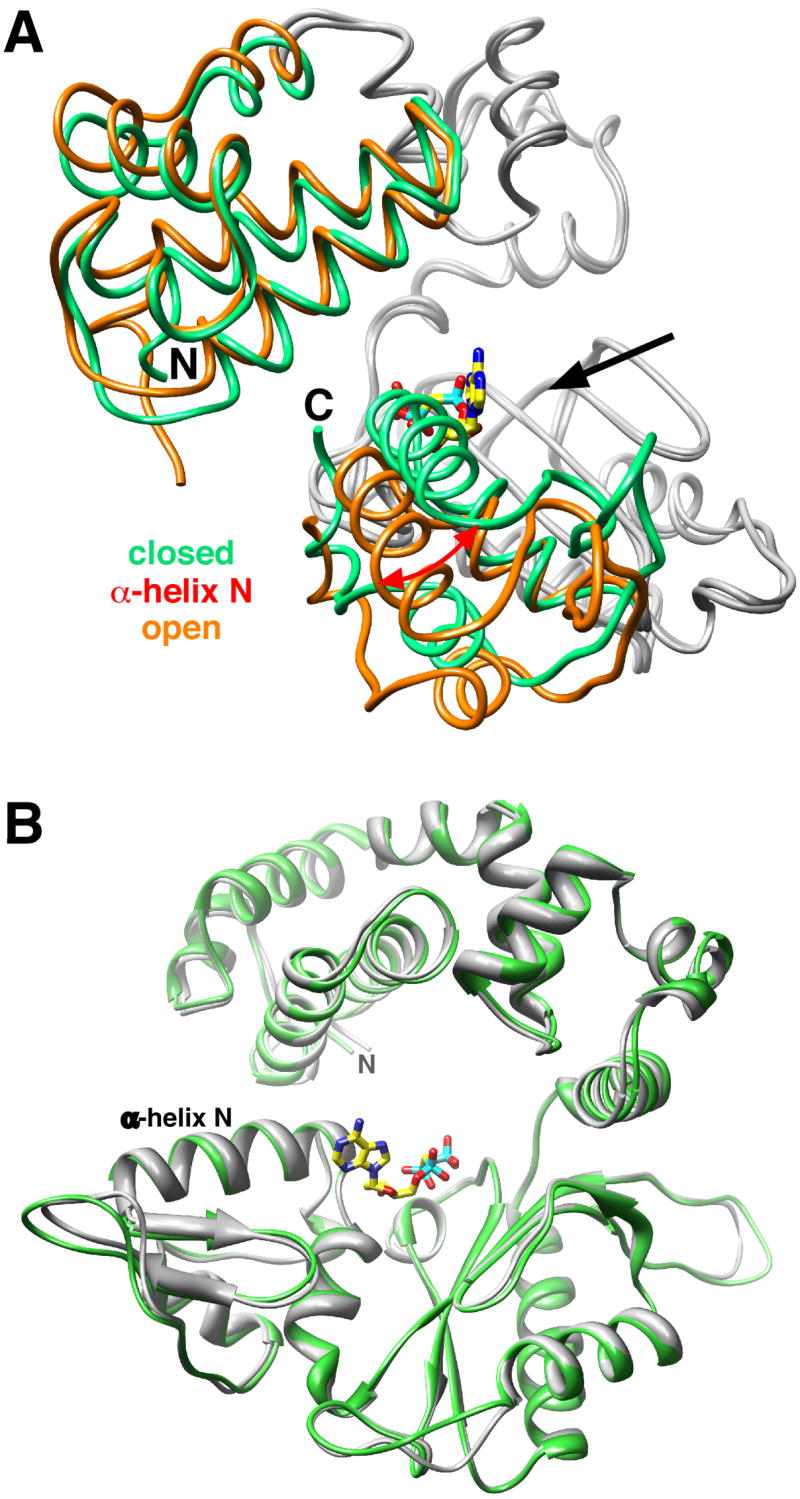
(A) Licorice representation of the Pol β backbone of the binary DNA complex (PDB ID 1BPX; orange) and ternary substrate complex (green) with an incorrect incoming nucleotide (dAMPCPP; yellow carbons) with a templating guanine. The catalytic and DNA-binding subdomains superimposed (gray backbone) with an rmsd of 0.56 Å (177 Cα). The DNA is omitted for clarity, but the 5′—3′ direction of the primer entry into the active site is indicated with a solid arrow. The open and closed position of α-helix N is shown. The amino-terminal lyase domain and carboxyl-terminal N—subdomain (colored) move in response to binding an incorrect dNTP. The amino– (N) and carboxyl-terminal (C) ends are labeled.
(B) Ribbon representation of the Pol β backbone of the ternary substrate complex with correct (gray) or incorrect (green) incoming nucleotides. The polymerase domain with a correct (dA—dUMPNPP; PDB ID 2FMS) and incorrect (dG—dAMPCPP) nascent base pair were superimposed with an rmsd of 0.62 Å (314 Cα). The superimposed structures indicate that α-helix N is in a ‘closed’ position like that observed with a Watson-Crick nascent base pair. The incoming dAMPCPP of the mismatch structure is shown (yellow carbons), but the DNA is omitted for clarity. The amino-terminus (N) of the lyase domain is also indicated.
Figure 2. Nucleotides of the Mismatch are in a Staggered Conformation.
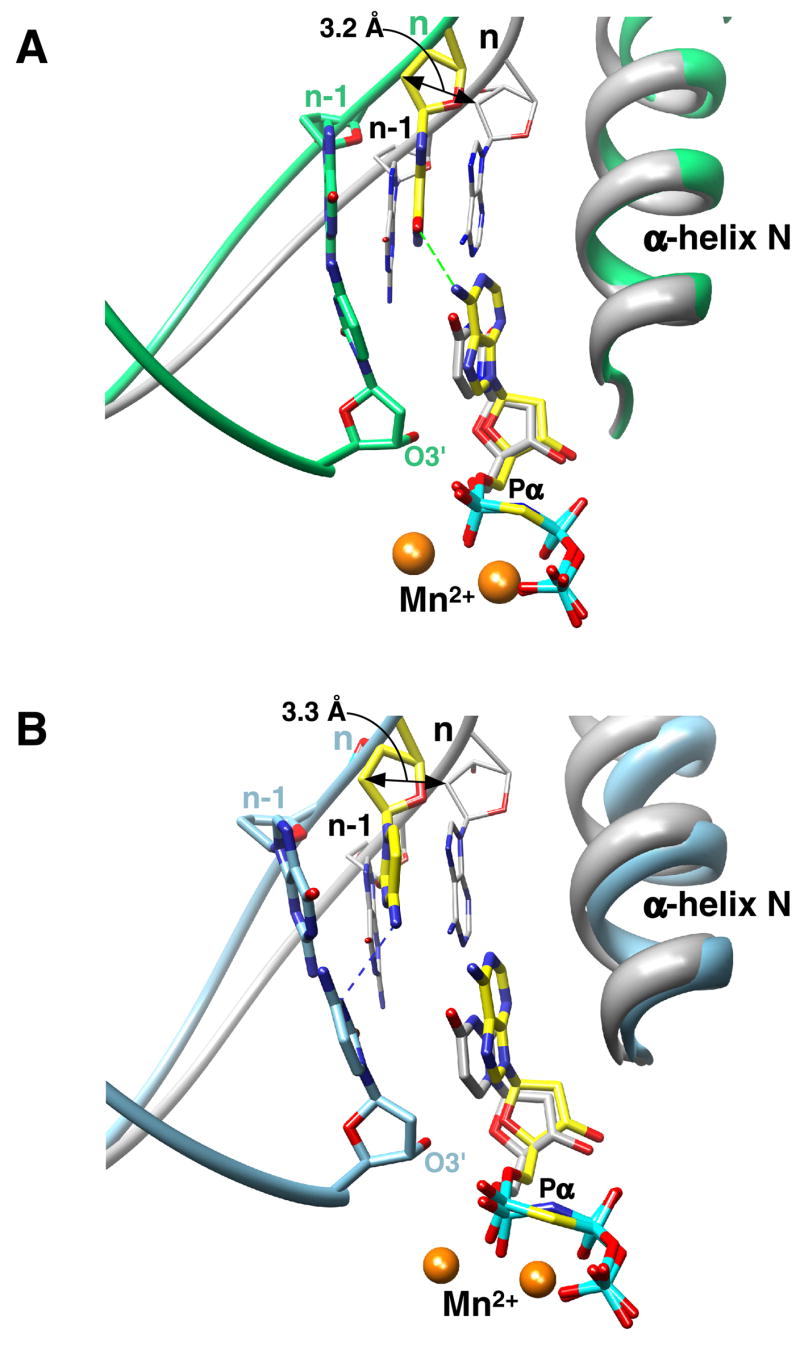
(A) The carbons of the mismatch (dG—dAMPCPP) are colored yellow and those of the primer terminus and α-helix N are green. The coding template base (n) is shifted upstream 3.2 Å while the incoming nucleotide is positioned appropriately in the dNTP binding pocket as illustrated by comparing the relative positions of the respective nucleotides in the ternary complex structure with a matched base pair (gray, PDB ID 2FMS). The superimposed structures indicate that α-helix N is in a ‘closed’ position. The primer terminus and its templating base (n-1) of the mismatched structure are shown as well as the backbone of the templating strand of both structures. A hydrogen bond (dashed green line) may occur between N6(dAMPCPP) and O6(dG). The atoms that directly participate in catalysis are labeled (O3’, Pα, and Mn2+).
(B) The carbons of the mismatch (dC—dAMPCPP) are colored yellow and those of the primer terminus and α-helix N are light blue. In this case, a possible hydrogen bond (dashed blue line) can form between the templating cytosine(N4) and N3 of the primer terminal cytosine.
Template Misalignment Deters Misincorporation
To permit the staggered conformation of the nascent base pair in the closed polymerase conformation, the template strand in the vicinity of the active site has shifted upstream approximately 3 Å so that the coding template base vacates the active site thereby generating a an apparent abasic coding template pocket (Figure 2). The apparent abasic site refers to the non-templating nature of the DNA at the coding position rather than an intermediate commonly observed during base excision repair. A similar upstream displacement of the template strand was observed with product DNA binary complexes with terminal mispairs (Krahn et al., 2004). The magnitude of this shift in the template strand for the binary mismatch product complexes (approaching an open polymerase conformation) is not as great as that observed for the ternary mismatched complexes (closed polymerase conformation; Figure S4). The backbone of the primer strand is not displaced in the ternary mismatched structure, but the base/sugar of the primer terminus rotates as its complementary template base is repositioned (Figure 3). This rotation positions O3’ of the primer terminus 3.9 Å away from the α-phosphate of the incoming incorrect nucleotide. Furthermore, the arrangement of catalytic atoms for nucleophilic attack now exhibits a poor geometry, if attack were to occur from this position. Note that the distance between the O3’ of the primer terminus and the α-phosphate of the correct incoming nucleotide is 3.4 Å (Batra et al., 2006).
Figure 3. Primer Terminus is Re-positioned as a Consequence of Template Slippage.
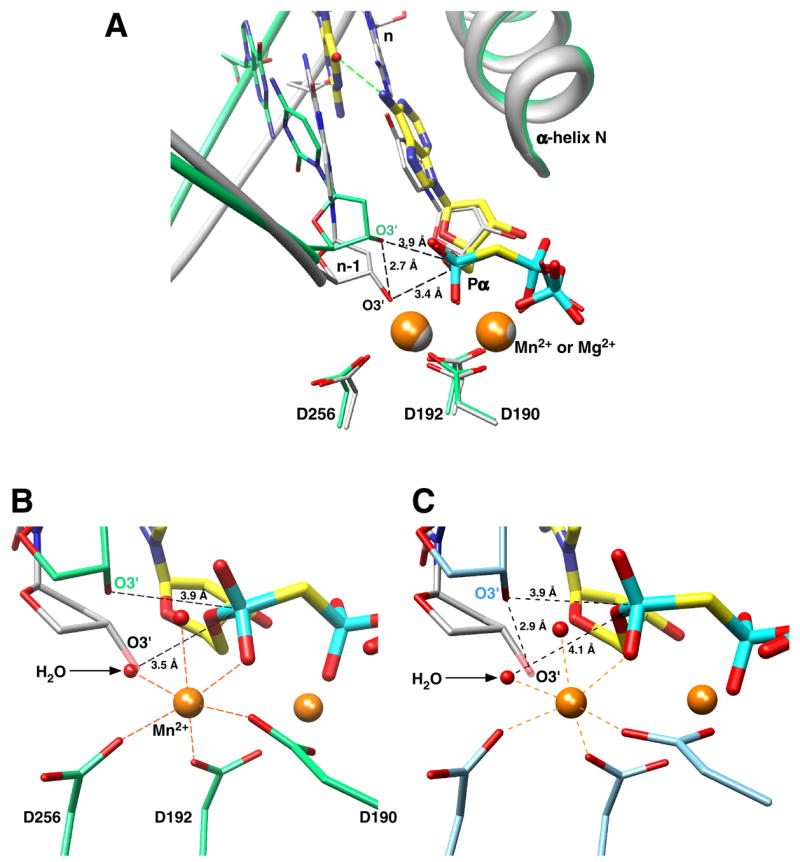
(A) The primer terminus (n-1) phosphates of the matched (gray) and dG—dAMPCPP mismatched (green) ternary complexes are in similar positions as illustrated by the ribbon representation, but the primer base and dexoxyribose of the mismatched complex are displaced from that observed in the matched ternary complex. This separates O3’ in these structures 2.7 Å, so that O3’ in the mismatch structure is now 3.9 Å from Pα of the incoming nucleotide. More importantly however, is that O3’ no longer participates in coordination of the catalytic metal and is no longer in-line with the bridging oxygen of Pα and Pβ of the incoming nucleotide. The superimposed structures also reveal that the divalent metals (Mn2+ or Mg2+, orange and grayspheres respectively) occupy identical positions in both structures. The carbons of the mismatch (dG—dAMPCPP) are colored yellow.
(B) The catalytic Mn2+ is octahedrally coordinated with oxygens from three active site aspartates (D190, D192, D256), two waters, and a non-bridging oxygen (pro-_R_P) on Pα of the incoming incorrect nucleotide. In contrast to when a correct nucleotide binds, the primer terminus O3’ is too far from the catalytic metal to participate in the inner coordination sphere. Instead, a water molecule (indicated) now occupies the position where O3’ (semi-transparent) is observed with a correct incoming nucleotide. The sugar of the primer terminus in the ternary complex structure with a correct incoming nucleotide is shown (gray).
(C) A similar view as that illustrated in panel B for the dC—dAMPCPP active site. In this case, the carbons of the active site carboxylates and primer terminus are light blue. A similarly positioned water is also observed in the mismatch structure where O3’ of a correctly positioned primer terminus is observed (gray carbons).
In contrast to the poor position of the primer terminus O3’, the sugar and triphosphate portions of the incoming correct and incorrect nucleotides superimpose nicely even though these ligands were not used to superimpose the structures (Figure 3A). Accordingly, the conformations of the incoming correct/incorrect nucleotides are nearly identical. In contrast, the position of the mismatched primer terminus of a product complex (i.e., situated in the dNTP binding pocket) is dramatically different to that of a correct/incorrect incoming dNTP due to the open-like polymerase conformation of the binary complexes (Figure S4) (Krahn et al., 2004). The two coordinating metals (Mn2+) and active site aspartate side chains (residues 190, 192, and 256) are in identical positions in the ternary complexes with correct or incorrect incoming nucleotides. The nucleotide binding metal exhibits the same inner octahedral coordination sphere as that observed with Mg2+ bound to the correct incoming nucleotide. The average coordination distance is approximately 0.1 Å longer than that observed with Mg2+ consistent with observations for other high-resolution crystal structures that bind these divalent metals (Harding, 2006). A structure of a Pol β ternary substrate complex with a correct nascent base pair (dA– dUMPNPP) and Mn2+ exhibits similar coordination distances and tertiary structure. The catalytic Mn2+ in the mismatch structures is also octahedrally coordinated similar to that observed with Mg2+ bound in the catalytic metal site with a correct incoming nucleotide (Figure 3B and C). However in the mismatch structures, O3’ of the primer terminus, a coordinating ligand for the catalytic metal with a correct incoming nucleotide, is no longer in the vicinity of the catalytic metal. Water molecules are now observed at this position and coordinate the catalytic metal. These water molecules are 3.5 and 4.1 Å from Pα of the incoming incorrect nucleotide in the G—A and C—A mismatch structures, respectively.
Local Polymerase Adjustments that Impede DNA Synthesis
There are significant differences in the position of several key residue(s) that alter their interactions when transitioning from open and closed complexes upon binding a correct nucleotide (Sawaya et al., 1997). This critical subdomain movement occurs through α-helix M (residues 262–273) rotation that brings α-helix N (residues 276–289) in contact with the nascent base pair and involves altered interactions between several key residues. In the open binary complex, Tyr271 can form a hydrogen bond with the templating base. Upon binding the correct nucleotide, closing of the N-subdomain (residues 260–335; equivalent to fingers subdomain of polymerases that utilize a right-hand nomenclature) repositions the hydroxyl of residue 271 within hydrogen bonding distance of the minor groove edge (O2 of pyrimidines or N3 of purines) of the primer terminus base. Since the primer terminal base has moved subtly upstream and into the DNA major groove upon forming the closed complex with an incorrect incoming nucleotide, Tyr271 is too distant to interact with the primer terminus, but may form a weak hydrogen bond with the minor groove edge of the incoming nucleotide in the C—A mismatch structure (Figure 4). Additionally, Arg258 forms a salt bridge with one of the active site aspartate metal ligands (Asp192) in the open ‘inactive’ binary complex, but forms hydrogen bonds with Glu295 and Tyr296 in the closed complex with a correct incoming nucleotide. Arg258 does not interact with Asp192 in the closed complex with the incorrect nascent base pairs examined here, but is observed to be in an intermediate position so that it does not interact with any protein side chains. Similar intermediate conformations of Arg258 have been observed in open binary complex structures with a primer terminus mismatch (Batra et al., 2005) or with a product complex with a mismatch in the active site (Krahn et al., 2004). Arg283 provides crucial interactions with the template strand in the closed polymerase conformation of the ternary complex with a correct incoming nucleotide. Loss of these interactions through alanine substitution dramatically reduce catalytic efficiency of correct, but not incorrect insertion (Beard et al., 1996; Beard et al., 2002a). In the mismatch structure with a templating guanine, Arg283 forms hydrogen bonds with N3 of the minor groove edge and the phosphate backbone of the templating base thereby stabilizing the upstream position of the coding base. In the case of the C—A mismatch structure, only the hydrogen bond with the phosphate backbone of the templating base is suggested. In the A—C terminal mismatch binary product complex, the open-like nature of the polymerase domain permits hydrogen bonding of the Watson-Crick edge of the mismatched cytosine at the primer terminus with Arg283 (Figure S4) (Krahn et al., 2004).
Figure 4. Position of Key Protein Residues in Closed Polymerase Complexes with Correct or Incorrect Incoming Nucleotides.

(A) Two views (major groove view, top; –90° rotation of the top view, bottom) of the nascent base pair of the mismatch structure (dG—dAMPCPP; green carbons) superimposed (see Figure 1) with that for a correctly matched base pair (gray carbons). The templating base is omitted from the correctly matched overlay for clarity. The structure illustrates the position of key protein side chains that can influence catalytic behavior. For catalytic activation with the correct incoming nucleotide, N–subdomain closing is associated with the loss of a salt bridge between Arg258 (R258) and Asp192 (D192), that coordinate both active site metals (M2+), and the formation of hydrogen bonds (black dashed lines) with Glu295 (E295) and Tyr296 (Y296). Phe272 (F272) is repositioned in the closed complex to insulate Asp192 from Arg258. Arg283 (R283) that is situated in the N–subdomain interacts with the minor groove edge of the templating strand (not shown). With an incorrect incoming nucleotide, R258 and R283 are in conformations that preclude catalytic activation. Arg283 is observed to hydrogen bond with the minor groove edge and phosphate backbone of the templating base. Other key residues (Asp192, Asn279, Phe272) are observed in similar positions as that found with a correct incoming nucleotide.
(B) Major groove view of the nascent base pair of the dG/dC—dAMPCPP superimposed mismatch structures (dC template, light blue carbons; dG template, green).
In contrast to these altered interactions, several key interactions are observed in both the matched and mismatched ternary substrate complexes. Since the correct and incorrect incoming nucleotides interact with the N–subdomain in a similar fashion, Asn279 forms a hydrogen bond with the minor groove edge (O2, dUMPNPP; N3, dAMPCPP). Likewise, Phe272 is in an identical position in these three structures. This side chain is positioned between Arg258 and the metal-coordinating Asp192 in the closed polymerase conformation.
DISCUSSION
Replicative DNA polymerases, as well as Pol β, exhibit large subdomain motions associated with binding of the incoming dNTP (Doublié et al., 1999). These motions however are generally rapid and not rate limiting (Bakhtina et al., 2005; Rothwell et al., 2005; Vande Berg et al., 2001). A number of more subtle conformational changes/adjustments have also been noted for Pol β, but their role in nucleotide insertion and/or discrimination is less certain (Batra et al., 2006; Bose-Basu et al., 2004; Vande Berg et al., 2001). Crystallographic structures of binary DNA complexes of A-family DNA polymerases indicate that an aromatic side chain stacks with the templating base immediately upstream of the nascent base pair forcing the templating coding base to reside outside of the DNA helix in a “pre-insertion” site (Kiefer et al., 1998; Li et al., 2004; Li et al., 1998). The molecular events that lead to re-positioning of the coding template base into the “insertion” site remain to be explored. For many DNA polymerases, a conformational change has been proposed to be rate limiting for correct nucleotide insertion, but the precise nature of this conformational change is unknown. DNA polymerases from divergent families and sources exhibit fidelities that correlate with the efficiency by which they insert the correct nucleotide (Beard et al., 2002a). In other words, high fidelity DNA synthesis is achieved through efficient insertion of the correct nucleotide, whereas low fidelity DNA synthesis is accompanied by poor correct nucleotide insertion efficiencies. This is kinetically due to weak binding and slow insertion of the incorrect dNTP. It is generally accepted that some aspect of chemistry is rate limiting for incorrect nucleotide insertion (Joyce and Benkovic, 2004).
The ternary substrate structures of the Pol β active site mismatches provide insight into the molecular strategy employed by polymerases generally to avert misinsertion. The poor position (i.e., distance and geometry with Pα of the incoming dAMPCPP) of the primer O3’ is the simplest explanation for the loss of insertion. A water molecule has replaced the displaced primer O3’ and completes the octahedral coordination of the catalytic metal (Figure 3). This position and coordination puts the incoming nucleotide at risk for hydrolytic cleavage. A very slow rate of hydrolysis of the incoming nucleotide can be easily measured and requires polymerase, DNA, and metal (unpublished data). Although this hydrolytic reaction is too slow to be physiologically relevant, it is an obstacle to structure determination. Employing non-hydrolysable dNTP analogs has solved this difficulty. Based on a comparison of the active site arrangement of catalytic atoms of Mycobacterium tuberculosis dUTPase and Pol β, a similar water mediated hydrolysis of dUTP has been suggested (Chan et al., 2004).
Surprisingly, the subdomain conformation of the ternary complex with the mismatch indicates a closed polymerase conformation. At the same time, the templating (coding) nucleotide is re-positioned upstream thereby vacating the template binding pocket resulting in an apparent abasic site. Consequently, although the polymerase subdomain position can be described as closed, the templating site does not contain a base (i.e., abasic) and could reasonably be described as open. Pol λ, a co-member of the X-family of polymerases, exhibits a closed protein subdomain conformation in the crystallographic structure of the binary DNA complex (Garcia-Diaz et al., 2005). However, the template strand has shifted upstream to ‘open’ the nascent base pair binding pocket. Binding a correct nucleotide induces a shift in the template strand to effectively close the binding site. Figure 5 illustrates that a superposition of Pol β and Pol λ in various liganded states indicates that the template strand in the Pol λ binary DNA complex is positioned like that observed in the Pol β mismatch (dG—dAMPCPP) structure. Likewise, Arg283 (Pol λ Arg517) stacks with the templating purine. These observations indicate that the closed polymerase conformation can exist with an open template-binding pocket in the absence of a correct incoming nucleotide. As noted above, crystallographic structures of open binary DNA complexes of members of A-family DNA polymerases indicate that they insert an aromatic protein side chain in the templating position and sequester the templating base in a pre-insertion site outside of the growing DNA duplex helix (Kiefer et al., 1998; Li et al., 2004; Li et al., 1998). This steric exclusion of a templating nucleotide prevents the fingers subdomain from providing critical interactions necessary for catalytic activation. Likewise, B-family polymerases may utilize a similar mechanism where the Gly568 backbone may sterically compete with template base binding (Freisinger et al., 2004). Accordingly, the side chain occupying the template-binding pocket would be expected to influence specificity (i.e., nucleotide choice).
Figure 5. Comparison of the Position of the Coding Templating Nucleotide in Various Polymerase Liganded States.
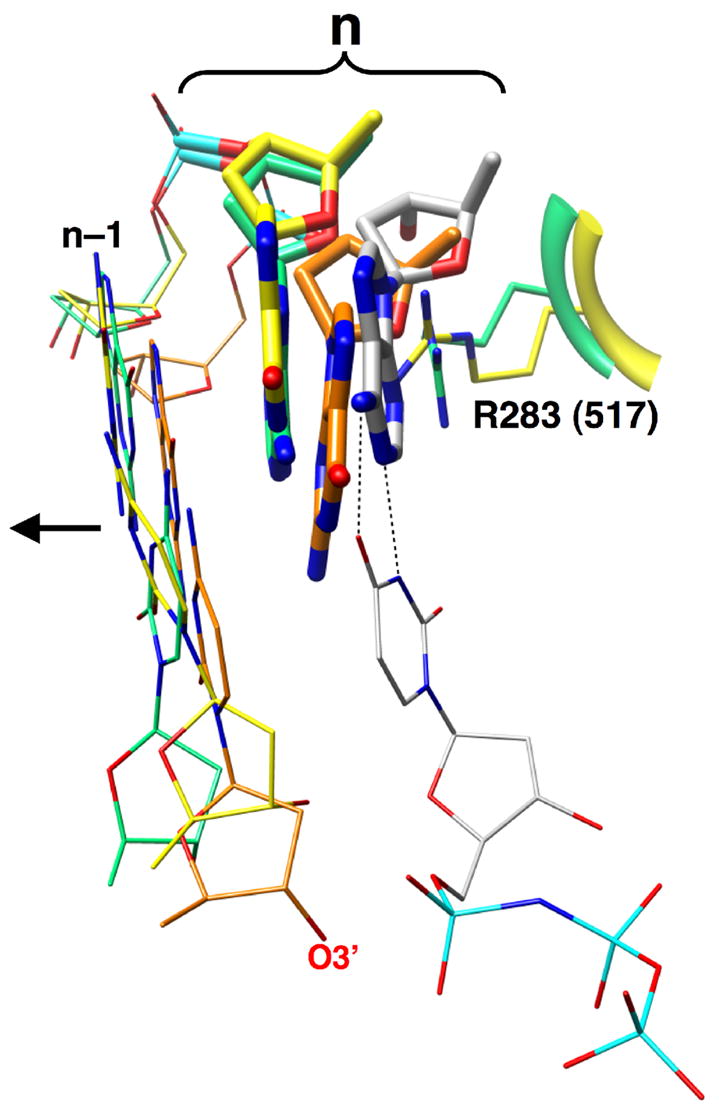
Ternary complex structures of Pol β with a Watson-Crick base pair (gray, 2FMS) and dG— dAMPCPP mismatch (yellow) where superimposed with single-nucleotide gapped binary complexes of Pol β (orange) and Pol λ (green). The templating base (n) in each case is shown in a thick stick representation and the primer terminal base pair (n–1) is illustrated as thin sticks. The arrow indicates the upstream duplex. The correct incoming nucleotide is also shown base pairing (black dashed lines) with the templating adenine. Arg283 of Pol β (R283) fills the templating base binding pocket in the mismatch structure where the templating base has moved upstream producing an apparent abasic site. A similar position of the templating nucleotide and the conserved arginine (R517) in a Pol λ binary DNA complex is observed indicating that template slippage may be a general mechanism for active site de-activation.
Through site-directed mutagenesis of Pol β, Arg283 has been shown to be essential for efficient and faithful DNA synthesis (Beard et al., 1996). In the closed ternary substrate complex, Arg283 interacts with the minor groove edge of the templating nucleotide and its 3′-neighbor. Alanine substitution results in a dramatic decrease in correct nucleotide insertion (Beard et al., 1996; Beard et al., 2002a), base substitution fidelity (Osheroff et al., 1999) and frameshift fidelity (Osheroff et al., 2000). The lower fidelity for dGTP insertion “opposite” dA or dT, could be rescued through a lysine substitution. It was postulated that the arginine or lysine side chains prevented misinsertion through an electrostatic/steric conflict with N2 of dGTP (Osheroff et al., 1999). In contrast, the ternary complex structure of Rev1 (i.e., deoxycytidyl transferase), demonstrates how an arginine residue can facilitate nucleotide selection (Nair et al., 2005). Rev1, a Y-family polymerase, exhibits a strong preference for insertion of dCTP (Nelson et al., 1996). In this case, Arg324 displaces the templating base outside of the DNA helix and forms two hydrogen bonds with the incoming dCTP. Consequently, Arg324 would discriminate against other incoming nucleotides, and the extra-helical position of the coding base would preclude template selectivity. In this context, Arg283 is observed to hydrogen bond with the Watson-Crick edge of a misinserted dCMP (templating adenine) in a product mismatched binary complex (Figure S4) (Krahn et al., 2004).
The structural characterization of mismatched base pairs in the polymerase active site has been hampered by 1) the observation that many polymerases bind the incorrect nucleotide very weakly and 2) the use of a dideoxy-terminated primer to capture abortive ternary complexes does not include a key catalytic atom. The presence of O3’ at the primer terminus is a prerequisite for the binding of catalytic divalent cation and for the assembly of catalytically complete all-atom pre-catalytic complex (Batra et al., 2006). The poor binding affinity for the incorrect incoming nucleotide is at least in part due to the open template binding pocket reducing van der Waals, π-stacking, and/or hydrogen bonding interactions. A comparison of ternary substrate complexes with a correct base pair (dUMPNPP—dA) with either Mg2+ (PDB 2FMS) or Mn2+ indicate that the overall fold is nearly identical (rmsd = 0.24 Å for 326 Cα). In addition, all side-chains are observed to be in nearly identical positions. Accordingly, the observed 10–30-fold increase in binding affinity for both correct (Table S1) and incorrect nucleotides (Table 1) appears to reflect an intrinsic property of Mn2+. Substituting Mn2+ for Mg2+ commonly lowers the fidelity of DNA synthesis (Sirover and Loeb, 1976). Since fidelity is roughly the ratio of catalytic efficiencies for right and wrong nucleotide insertion, an altered fidelity represents a differential effect of some factor (e.g., identity of the catalytic metal) on efficiency. The catalytic efficiencies of both correct and incorrect nucleotide insertion are increased in the presence of Mn2+. Since this effect is greater for incorrect insertion, fidelity is decreased. In the presence of Mn2+, the insertion of dATP opposite dC or dG is poor, but nearly identical (Table 1).
In contrast to binary DNA product complex structures with mismatches at the primer terminus (i.e., misinsertion followed by translocation) of the thermophilic Bacillus DNA polymerase I fragment (Bf) (Johnson and Beese, 2004), the mismatched nascent base pairs in the ternary substrate complexes reported here are not co-planar and do not exhibit conformations or hydrogen bonding patterns observed when these mismatches are positioned in duplex DNA. In duplex DNA, the nucleotides of the G—A mismatch are co-planar, with one or the other nucleotides in a _syn_-conformation (Brown et al., 1986; Leonard et al., 1990) or with both purines in an _anti_-conformation (Gao and Patel, 1988). Situated at the Bf primer terminus, the _anti_-anti conformation was observed coupled with a severe buckle to accommodate the G—A (template– primer) mispair forming two hydrogen bonds. In contrast, the C—A mismatch is typically observed in a Wobble conformation in duplex DNA (Hunter et al., 1986) but was disordered when situated at the Bf primer terminus.
A dT—dGTP/Ca2+ mismatch in the nascent base pair binding pocket of the Y–family DNA DNA polymerase Dpo4 has also been reported (Vaisman et al., 2005). The crystallographic asymmetric unit has two polymerase molecules. In one molecule, the mismatch is co-planar and exhibited a Wobble hydrogen-bonding pattern like that observed with this mismatch in duplex DNA. However, the sugar and triphosphate moieties are distorted with the γ-phosphate of the incorrect incoming nucleotide binding at the β-phosphate binding site. In contrast, the mismatch in the other molecule is not co-planar, but the template thymidine was positioned upstream, albeit to a lesser extent than observed in the structures reported here. Y-family DNA polymerases do not exhibit the large subdomain motions typically observed with many members of the A- and B-family polymerases, HIV-1 reverse transcriptase, and Pol β, but may involve more subtle subdomain and/or DNA conformational changes. A ternary complex structure of Dpo4 DNA polymerase with a frameshift intermediate demonstrates the flexibility inherent in the active site of this Y-family DNA polymerase. The specific contributions of the mismatch and the large ionic radius of Ca2+ need to be determined.
The structures reported here indicate that binding of a correct or incorrect nucleotide to a Pol β/DNA complex leads to a closed polymerase conformation. Whereas a tight nascent base pair binding pocket results from closing of the N-subdomain when a correct nucleotide binds, the closing motion results in an upstream shift in the template strand when an incorrect nucleotide binds producing an apparent abasic-site intermediate. Thus, the correct base pair exhibits “good geometry” and hydrogen bonding, whereas the incorrect base pair displays “poor geometry” and hydrogen bonding. The good geometry of the proper base pair positions key atoms (primer O3’, Pα of the incoming nucleotide, and catalytic metal) to facilitate nucleotide insertion. In contrast, the incorrect incoming nucleotide lacks optimum interactions with the template strand reducing binding affinity and the primer terminus is re-positioned in response to the upstream shift in the template strand resulting in poor catalytic activation. Nucleophilic attack of the primer O3’ from the observed position in the mismatch structure seems unlikely due to the increased distance, poor geometry for an in-line attack, and loss of catalytic metal coordination. Accordingly, a significant conformational change must also occur to promote significant (mis)insertion. An opening of the N-subdomain may permit the coding template base opposite the primer terminus to shift downstream thereby repositioning the sugar of the primer terminus to permit O3’ to participate in catalytic metal coordination and catalyze inefficient incorporation (Figure S4). Molecular modeling has suggested previously that ‘fine-tuning’ the catalytic metal coordination is a critical step prior to chemistry (Radhakrishnan et al., 2006). This ‘pre-chemistry’ step(s) represents an opportunity for the enzyme to induce an optimal (correct nucleotide binding) or sub-optimal metal coordination (incorrect nucleotide binding). It remains to be determined how induced active site dynamics (rapid protein/substrate segmental motions) that are observed upon binding substrates promote/inhibit this fine-tuning and therefore specificity (Bose-Basu et al., 2004; Kim et al., 2003). It will be of interest to compare mismatch structures generated with an incoming pyrimidine triphosphate, since differences in the contribution to catalytic efficiency of purine and pyrimidine deoxynucleoside triphosphate interactions with Pol β have been noted (Beard et al., 2002b) indicating that differences in the conformation of the incoming nucleotide may be expected.
Finally, the results presented here suggest that a transient abasic site intermediate could exist during misincorporation. The temporal order of template strand slippage (i.e., template base removal from the coding position generating an apparent-abasic site) and arginine occupation of the template base binding pocket is unknown; accordingly, the degree to which the template binding pocket influences insertion specificity through an empty binding pocket, polymerase side chains and/or DNA remains to be determined. Interestingly, an examination of misinserton specificities for Pols α, β, and γ indicate that purines are preferentially misinserted, and that adenine is often favored (Kunkel and Alexander, 1986). These results are consistent with the A-rule observed for many polymerases where dATP is preferentially inserted opposite a non-instructional lesion such as an abasic site (Strauss, 2002).
EXPERIMENTAL PROCEDURES
Crystallization of Pol β Ternary Substrate Complexes
Human Pol β was over-expressed in E. coli and purified (Beard and Wilson, 1995). The DNA substrate consisted of a 16-mer template, a complementary 10-mer primer strand, and a 5-mer downstream oligonucleotide. The annealed 10-mer primer creates a one-nucleotide gap with a templating A, C, or G residue. The sequence of the downstream oligonucleotide was 5′– GTCGG–3′ with a phosphorylated 5′-terminus. The template sequence was 5′–CCGAC(A, C or G)GCGCATCAGC–3′ and the primer sequence was 5′–GCTGATGCGC–3′. Oligonucleotides were dissolved in 20 mM MgCl2, 100 mM Tris/HCl, pH 7.5. Each set of template, primer, and downstream-oligonucleotides was mixed in a 1:1:1 ratio and annealed using a PCR thermocycler by heating 10 min at 90 °C and cooling to 4 °C (1 °C min−1) resulting in a 1 mM mixture of gapped duplex DNA. This solution was then mixed with an equal volume of Pol β at 4 °C, the mixture warmed to 35 °C and gradually cooled to 4 °C.
Pol β-DNA complexes were crystallized by sitting-drop vapor diffusion. The crystallization buffer was 16% PEG-3350, 350 mM sodium acetate, and 50 mM imidazole, pH 7.5. Drops were incubated at 18 °C and streak seeded after 1 day. Crystals grew in approximately 2—4 days after seeding. The ternary complexes were obtained by soaking the crystals of the binary one-nucleotide gapped DNA complex in artificial mother liquor with 50 mM MnCl2, 2 mM dAMPCPP or dUMPNPP (Jena Bioscience), 20% PEG-3350, and 12% ethylene glycol, and then flash-frozen to 100 K in a nitrogen stream. All crystals belong to the space group _P_21.
Data Collection and Structure Determination
Data were collected on a Saturn 92 CCD detector system mounted on a MicroMax-007HF (Rigaku Corporation) rotating anode generator. Data were integrated and reduced for structure refinement with HKL2000 software (Otwinowski and Minor, 1997).
Structures were determined by molecular replacement with a previously determined structure of Pol β complexed with one-nucleotide gapped DNA and a complementary incoming ddCTP, PDB ID 2FMP. The crystal structures have similar lattices and are sufficiently isomorphous to determine the molecular-replacement model position solely by rigid body refinement. The parameters and topology for dAMPCPP were based on CNS libraries with parameters for the P–C–P fragments derived from analysis of several well-characterized small molecule structures (Cambridge Structure Database). Refinement statistics are given in Table 2 and Fo–Fc simulated annealing electron density omit maps showing electron density corresponding to the mismatches and primer terminus are provided in Figure S2. The figures were prepared in Chimera (Pettersen et al., 2004).
Kinetic Assays
DNA synthesis was assayed on a single-nucleotide gapped DNA substrate where the templating base in the gap was adenine or guanine. The DNA sequence was as described previously (Beard et al., 2004) with the core sequence identical to that used for crystallization. Enzyme activities were determined using a reaction mixture containing 50 mM Tris-HCl, pH 7.4, 5 mM MnCl2, 100 mM KCl, 200 nM single-nucleotide gapped DNA, and the concentration of the incoming nucleotide varied. Reactions were incubated at room temperature and stopped with EDTA mixed with formamide dye. The substrates and products were separated on 12 or 15% denaturing polyacrylamide gels and quantified in the dried gels by phosphorimagery. Steady-state kinetic parameters were determined by fitting the rate data to the Michaelis equation.
To directly measure the rate of the first insertion (_k_pol) and the equilibrium nucleotide dissociation constant (_K_d), single-turnover kinetic assays (enzyme/DNA = 10) were performed as outlined previously (Beard et al., 2002b) employing a KinTek Model RQF-3 chemical quench-flow apparatus. Typically, a solution of Pol β (1 _μ_M) was pre-incubated with single-nucleotide gapped DNA (100 nM). This solution was rapidly mixed (two-fold dilution) with various concentrations of dNTP/Mg2+ or Mn2+. Final conditions (pH, temperature) and salt concentrations were like that described for the steady-state assay. After various time periods, the reactions were stopped with 0.25 M EDTA and the quenched samples mixed with an equal volume of formamide dye. Products were separated and quantified as described above. Under these conditions, the first-order rate constant of the exponential time-courses was dependent on the concentration of dNTP. A secondary plot of the concentration dependence of _k_obs was hyperbolic and fitted by a non-linear least-squares method to Equation 1 where _k_pol is the intrinsic rate constant for the step limiting the first insertion.
| kobs=kpol[dNTP]/(Kd+[dNTP]) | (Eq. 1) |
|---|
Supplementary Material
01
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences and was in association with the National Institutes of Health Grant 1U19CA105010.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
ACCESSION NUMBERS
Coordinates and structure factors have been deposited in the RSCB Protein Data Bank with accession codes 3C2K, 3C2L, and 3C2M for the dA–dUMPNPP, dC—dAMPCPP, and dG— dAMPCPP structures, respectively.
References
- Bakhtina M, Lee S, Wang Y, Dunlap C, Lamarche B, Tsai MD. Use of viscogens, dNTPαS, and rhodium(III) as probes in stopped-flow experiments to obtain new evidence for the mechanism of catalysis by DNA polymerase β. Biochemistry. 2005;44:5177–5187. doi: 10.1021/bi047664w. [DOI] [PubMed] [Google Scholar]
- Batra VK, Beard WA, Shock DD, Krahn JM, Pedersen LC, Wilson SH. Magnesium induced assembly of a complete DNA polymerase catalytic complex. Structure (Camb) 2006;14:757–766. doi: 10.1016/j.str.2006.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra VK, Beard WA, Shock DD, Pedersen LC, Wilson SH. Nucleotide-induced DNA polymerase active site motions accommodating a mutagenic DNA intermediate. Structure (Camb) 2005;13:1225–1233. doi: 10.1016/j.str.2005.05.010. [DOI] [PubMed] [Google Scholar]
- Beard WA, Osheroff WP, Prasad R, Sawaya MR, Jaju M, Wood TG, Kraut J, Kunkel TA, Wilson SH. Enzyme-DNA interactions required for efficient nucleotide incorporation and discrimination in human DNA polymerase β. J Biol Chem. 1996;271:12141–12144. doi: 10.1074/jbc.271.21.12141. [DOI] [PubMed] [Google Scholar]
- Beard WA, Shock DD, Vande Berg BJ, Wilson SH. Efficiency of correct nucleotide insertion governs DNA polymerase fidelity. J Biol Chem. 2002a;277:47393–47398. doi: 10.1074/jbc.M210036200. [DOI] [PubMed] [Google Scholar]
- Beard WA, Shock DD, Wilson SH. Influence of DNA structure on DNA polymerase β active site function: Extension of mutagenic DNA intermediates. J Biol Chem. 2004;279:31921–31929. doi: 10.1074/jbc.M404016200. [DOI] [PubMed] [Google Scholar]
- Beard WA, Shock DD, Yang XP, DeLauder SF, Wilson SH. Loss of DNA polymerase β stacking interactions with templating purines, but not pyrimidines, alters catalytic efficiency and fidelity. J Biol Chem. 2002b;277:8235–8242. doi: 10.1074/jbc.M107286200. [DOI] [PubMed] [Google Scholar]
- Beard WA, Wilson SH. Purification and domain-mapping of mammalian DNA polymerase β. Methods Enzymol. 1995;262:98–107. doi: 10.1016/0076-6879(95)62013-3. [DOI] [PubMed] [Google Scholar]
- Beard WA, Wilson SH. Structural insights into the origins of DNA polymerase fidelity. Structure (Camb) 2003;11:489–496. doi: 10.1016/s0969-2126(03)00051-0. [DOI] [PubMed] [Google Scholar]
- Beard WA, Wilson SH. Structure and mechanism of DNA polymerase β. Chem Rev. 2006;106:361–382. doi: 10.1021/cr0404904. [DOI] [PubMed] [Google Scholar]
- Bose-Basu B, DeRose EF, Kirby TW, Mueller GA, Beard WA, Wilson SH, London RE. Dynamic characterization of a DNA repair enzyme: NMR studies of [methyl-13C]methionine-labeled DNA polymerase β. Biochemistry. 2004;43:8911–8922. doi: 10.1021/bi049641n. [DOI] [PubMed] [Google Scholar]
- Brown T, Hunter WN, Kneale G, Kennard O. Molecular structure of the G•A base pair in DNA and its implications for the mechanism of transversion mutations. Proc Natl Acad Sci USA. 1986;83:2402–2406. doi: 10.1073/pnas.83.8.2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Segelke B, Lekin T, Krupka H, Cho US, Kim M-y, So M, Kim CY, Naranjo CM, Rogers YC, et al. Crystal structure of the Mycobacterium tuberculosis dUTPase: Insights into the catalytic mechanism. J Mol Biol. 2004;341:503–517. doi: 10.1016/j.jmb.2004.06.028. [DOI] [PubMed] [Google Scholar]
- Doublié S, Sawaya MR, Ellenberger T. An open and closed case for all polymerases. Structure (Camb) 1999;7:R31–R35. doi: 10.1016/S0969-2126(99)80017-3. [DOI] [PubMed] [Google Scholar]
- Freisinger E, Grollman AP, Miller H, Kisker C. Lesion (in)tolerance reveals insights into DNA replication fidelity. EMBO J. 2004;23:1494–1505. doi: 10.1038/sj.emboj.7600158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Patel DJ. G(syn)•A(anti) mismatch formation in DNA dodecamers at acidic pH: pH-dependent conformational transition of G•A mispairs detected by proton NMR. J Am Chem Soc. 1988;110:5178–5182. [Google Scholar]
- Garcia-Diaz M, Bebenek K, Krahn JM, Kunkel TA, Pedersen LC. A closed conformation for the Pol λ catalytic cycle. Nat Struct Mol Biol. 2005;12:97–98. doi: 10.1038/nsmb876. [DOI] [PubMed] [Google Scholar]
- Harding MM. Small revisions to predicted distances around metal sites in proteins. Acta Crystallogr D Biol Crystallogr. 2006;62:678–682. doi: 10.1107/S0907444906014594. [DOI] [PubMed] [Google Scholar]
- Hunter WN, Brown T, Anand NN, Kennard O. Structure of an adenine-cytosine base pair in DNA and its implications for mismatch repair. Nature. 1986;320:552–555. doi: 10.1038/320552a0. [DOI] [PubMed] [Google Scholar]
- Johnson SJ, Beese LS. Structures of mismatch replication errors observed in a DNA polymerase. Cell. 2004;116:803–816. doi: 10.1016/s0092-8674(04)00252-1. [DOI] [PubMed] [Google Scholar]
- Joyce CM, Benkovic SJ. DNA polymerase fidelity: Kinetics, structure, and checkpoints. Biochemistry. 2004;43:14317–14324. doi: 10.1021/bi048422z. [DOI] [PubMed] [Google Scholar]
- Kiefer JR, Mao C, Braman JC, Beese LS. Visualizing DNA replication in a catalytically active Bacillus DNA polymerase crystal. Nature. 1998;391:304–307. doi: 10.1038/34693. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Beard WA, Harvey J, Shock DD, Knutson JR, Wilson SH. Rapid segmental and subdomain motions of DNA polymerase β. J Biol Chem. 2003;278:5072–5081. doi: 10.1074/jbc.M208472200. [DOI] [PubMed] [Google Scholar]
- Krahn JM, Beard WA, Miller H, Grollman AP, Wilson SH. Structure of DNA polymerase β with the mutagenic DNA lesion 8-oxodeoxyguanine reveals structural insights into its coding potential. Structure (Camb) 2003;11:121–127. doi: 10.1016/s0969-2126(02)00930-9. [DOI] [PubMed] [Google Scholar]
- Krahn JM, Beard WA, Wilson SH. Structural insights into DNA polymerase deterrents for misincorportion support an induced-fit mechanism for fidelity. Structure (Camb) 2004;12:1823–1832. doi: 10.1016/j.str.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Alexander PS. The base substitution fidelity of eucaryotic DNA polymerases: Mispairing frequencies, site preferences, insertion preferences, and base substitution by dislocation. J Biol Chem. 1986;261:160–166. [PubMed] [Google Scholar]
- Kunkel TA, Bebenek K. DNA replication fidelity. Annu Rev Biochem. 2000;69:497–529. doi: 10.1146/annurev.biochem.69.1.497. [DOI] [PubMed] [Google Scholar]
- Leonard GA, Booth ED, Brown T. Structural and thermodynamic studies on the adenine.guanine mismatch in B-DNA. Nucleic Acids Res. 1990;18:5617–5623. doi: 10.1093/nar/18.19.5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Dutta S, Doublié S, Bdour HMd, Taylor JS, Ellenberger T. Nucleotide insertion opposite a cis-syn thymine dimer by a replicative DNA polymerase from bacteriophage T7. Nat Struct Mol Biol. 2004;11:784–790. doi: 10.1038/nsmb792. [DOI] [PubMed] [Google Scholar]
- Li Y, Korolev S, Waksman G. Crystal structures of open and closed forms of binary and ternary complexes of the large fragment of Thermus aquaticus DNA polymerase I: structural basis for nucleotide incorporation. EMBO J. 1998;17:7514–7525. doi: 10.1093/emboj/17.24.7514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeb LA, Springgate CF, Battula N. Errors in DNA replication as a basis of malignant changes. Cancer Res. 1974;34:2311–2321. [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB, III, de Bakker PIW, Word JM, Prisant MG, Richardson JS, Richardson DC. Structure validation by Ca geometry: φ,ψ and Cβ deviation. Proteins. 2003;50:437–450. doi: 10.1002/prot.10286. [DOI] [PubMed] [Google Scholar]
- Nair DT, Johnson RE, Prakash L, Prakash S, Aggarwal AK. Rev1 employs a novel mechanism of DNA synthesis using a protein template. Science. 2005;309:2219–2222. doi: 10.1126/science.1116336. [DOI] [PubMed] [Google Scholar]
- Nelson JR, Lawrence CW, Hinkle DC. Deoxycytidyl transferase activity of yeast REV1 protein. Nature. 1996;382:729–731. doi: 10.1038/382729a0. [DOI] [PubMed] [Google Scholar]
- Osheroff WP, Beard WA, Wilson SH, Kunkel TA. Base substitution specificity of DNA polymerase β depends on interactions in the DNA minor groove. J Biol Chem. 1999;274:20749–20752. doi: 10.1074/jbc.274.30.20749. [DOI] [PubMed] [Google Scholar]
- Osheroff WP, Beard WA, Yin S, Wilson SH, Kunkel TA. Minor groove interactions at the DNA polymerase β active site modulate single-base deletion error rates. J Biol Chem. 2000;275:28033–28038. doi: 10.1074/jbc.M003462200. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processsing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan R, Arora K, Wang Y, Beard WA, Wilson SH, Schlick T. Regulation of DNA repair fidelity by molecular checkpoints: “Gates” in DNA polymerase β’s substrate selection. Biochemistry. 2006;45:15142–15156. doi: 10.1021/bi061353z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PJ, Mitaksov V, Waksman G. Motions of the fingers subdomain of Klentaq1 are fast and not rate limiting: Implications for the molecular basis of fidelity in DNA polymerases. Mol Cell. 2005;19:345–355. doi: 10.1016/j.molcel.2005.06.032. [DOI] [PubMed] [Google Scholar]
- Sawaya MR, Prasad P, Wilson SH, Kraut J, Pelletier H. Crystal structures of human DNA polymerase β complexed with gapped and nicked DNA: Evidence for an induced fit mechanism. Biochemistry. 1997;36:11205–11215. doi: 10.1021/bi9703812. [DOI] [PubMed] [Google Scholar]
- Sirover MA, Loeb LA. Infidelity of DNA synthesis in vitro: Screening for potential metal mutagens or carcinogens. Science. 1976;194:1434–1436. doi: 10.1126/science.1006310. [DOI] [PubMed] [Google Scholar]
- Steitz TA, Smerdon SJ, Jager J, Joyce CM. A unified polymerase mechanism for nonhomologous DNA and RNA polymerases. Science. 1994;266:2022–2025. doi: 10.1126/science.7528445. [DOI] [PubMed] [Google Scholar]
- Strauss B. The “A” rule revisited: Polymerases as determinants of mutational specificity. DNA Repair. 2002;1:125–135. doi: 10.1016/s1568-7864(01)00014-3. [DOI] [PubMed] [Google Scholar]
- Vaisman A, Ling H, Woodgate R, Yang W. Fidelity of Dpo4: Effect of metal ions, nucleotide selection and pyrophosphorolysis. EMBO J. 2005;24:2957–2967. doi: 10.1038/sj.emboj.7600786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vande Berg BJ, Beard WA, Wilson SH. DNA structure and aspartate 276 influence nucleotide binding to human DNA polymerase β: Implication for the identity of the rate-limiting conformational change. J Biol Chem. 2001;276:3408–3416. doi: 10.1074/jbc.M002884200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
01