Wild-type LRRK2 but not its mutant attenuates stress-induced cell death via ERK pathway (original) (raw)
. Author manuscript; available in PMC: 2009 Oct 1.
Published in final edited form as: Neurobiol Dis. 2008 Jul 8;32(1):116–124. doi: 10.1016/j.nbd.2008.06.016
Abstract
Leucine-rich repeat kinase 2 (LRRK2) is a recently identified gene that, when mutated at specific locations, results in the onset of parkinsonian symptoms with clinical features indistinguishable from idiopathic Parkinson’s disease. Based on structural and domain analysis, LRRK2 is predicted to function as a stress responsive protein scaffold mediating the regulation of mitogen activating protein kinase (MAPK) pathways. Consistent to this notion, our results supported the notion that expression of wild-type LRRK2 but not Y1699C or G2019S mutants enhanced the tolerance of HEK293 and SH-SY5Y cells towards H2O2-induced oxidative stress. This increase in stress tolerance was dependent on the presence of the kinase domain of the LRRK2 gene and manifested through the activation of the ERK pathway. Collectively, our results indicated that cells expressing LRRK2 mutants suffer a loss of protection normally derived from wild-type LRRK2, making them more vulnerable to oxidative stress.
Introduction
Leucine-rich repeat kinase 2 (LRRK2), a member of the ROCO protein family, is a recently identified gene whose mutants have been linked to the onset of parkinsonian symptoms (Haugarvoll and Wszolek, 2006; Klein and Schlossmacher, 2006; Olanow, 2007; Whaley et al., 2006). Sequence domain analysis of this gene has revealed that the 285 kDa protein for which it encodes comprises ROC, COR, kinase, and WD40 domains, in that order, with the ROC domain beginning after the first 1000 amino acids (Jain et al., 2005; West et al., 2005). The ROC domain, with its GDP/GTP binding motif has been shown to be capable of altering the kinase activity of LRRK1, a paralog of LRRK2 (Korr et al., 2006). To date, the impact of these mutations on wild-type LRRK2 is yet unclear.
Expression of LRRK2 mutant genes, such as Y1699C and R1441C, result in a loss of basal cell viability in SHSY5Y cells (Greggio et al., 2006; West et al., 2005) demonstrating their innate toxicity. Since G2019S and I2020T mutants show higher kinase activities than wild-type LRRK2, it has been speculated that alteration of kinase activity is involved in the manifestation of parkinsonian symptoms caused by LRRK2 mutations (Bialecka et al., 2005; Tomiyama et al., 2006; West et al., 2005). However, further investigation showed that different LRRK2 mutants have different inherent kinase activity based on an auto-phosphorylation assay. Indeed, some mutants have lower kinase activity than the wild-type counterpart. For example, it has been shown that the Y1699C mutant has lower kinase activity than wild-type LRRK2 and yet has the highest propensity to elicit the formation of protein aggregates (Greggio et al., 2006). Therefore, it is uncertain whether changes in kinase activities among the LRRK2 mutants contribute to the cause of pathology.
Phylogenetic analysis of the LRRK2 kinase domain has revealed sequence similarity to the receptor interaction protein (RIP) protein family as well as mixed lineage kinases (Meylan and Tschopp, 2005; Xu et al., 2001), raising the possibility of parallel functions among these kinases. In fact, Meylan and Tschopp (2005) have termed LRRK1 and LRRK2 as RIP6 and RIP7 respectively. Both of these kinase families participate in the signaling events in response to cellular stresses caused by various stimuli (Gloeckner et al., 2006). Therefore, it is likely that LRRK2 wild-type gene product is also involved in stress-induced signaling events leading to cell death.
In this study, our main objective was to investigate whether LRRK2 is involved in a stress-induced signaling cascade mediating cell death and, if so, to identify differential responses derived from LRRK2 wild-type, Y1699C and G2019S mutant genes in these events. Our results suggest that expression of LRRK2 wild-type or Y1699C mutant genes in HEK293 cells suppressed basal levels of activated ERK but not JNK, independent of its kinase domain. Moreover, the presence of LRRK2 wild-type gene product but not Y1699C or G2019S mutant gene product conferred protection against H2O2-induced cell death through activation of the ERK pathway mediated by its kinase domain in HEK293 and SH-SY5Y cells. Therefore, based on our results, this multi-functional domain LRRK2 gene is able to effect the activation of the ERK pathway.
Materials and methods
Materials
The LRRK2 wild-type, Y1699C, and G2019S mutant genes fused with GFP on their C-terminal in pCDNA3.1 expression vector were a kind gift from Dr. Matthew Farrer of the Neurogenetics Laboratories, Mayo Clinic Jacksonville. All the chemicals were obtained from Fisher Scientific (Pittsburgh, PA, USA) and all the inhibitors were obtained from BIOMOL (Plymouth Meeting, PA, USA) unless otherwise stated.
Cell culture
HEK293 cells (ATCC, Manassas, VA, USA) were maintained in DMEM media supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA). SH-SY5Y cells, a kind gift from Dr. James P. Bennett Jr, University of Virginia were maintained in DMEM media supplemented with 10% fetal bovine serum (Hyclone, Logan, UT, USA). The cells were plated in various configurations at 80% confluence before undergoing transfection. The transfection process was performed before treatments with either inhibitors or H2O2.
Transfections
Transfection of various constructs into HEK293 cells were performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) in accordance with the manufacturer’s instructions. In brief, cells were seeded in various configurations to yield 80% confluency. In the case of HEK293 cells, cell densities were 20,000 cells per well of a 96-well plate and 1 ×106 cells per well of a 6 cm plate. As for SH-SY5Y cells, cell densities were 100,000 cells per well of a 96-well plate and 5×106 cells per well of a 6 cm plates. After optimization, we found that the optimal DNA/lipofectamine ratio for HEK293 cells and SH-SY5Y cells were 1:3 and 1:2 respectively. To instigate transfection, conditioned medium was first replaced with Opti-I media (Invitrogen, Carlsbad, CA, USA). Then, the correct amounts of DNA and Lipofectamine 2000 reagent were added to Opti-I medium and incubated at room temperature for 5 min. After a further incubation at room temperature for 15 min, the DNA-lipofectamine complex was added drop-wise evenly throughout the area with cells. Then, the cells were incubated at 37°C for 4 h before the medium was changed completely back into growth medium. The transfection efficiency was estimated by eye under fluorescence illumination to visualize GFP and compared to the total number of cells under bright field illumination for HEK293 cells typically showing at least 70% transfection efficiency. On the other hand, the transfection efficiency for SH-SY5Y cells was determined by counting GFP+ cells over total cell number indicating that the efficiency was on average about 13%.
H2O2 and inhibitor treatment
Chronic treatments of H2O2, U0126 (BIOMOL, Plymouth Meeting, PA), were carried out after transfection when Opti-I medium was replaced with growth media. Cells were pretreated with MEK1 inhibitor (U0126) for 1 h before H2O2 was introduced into the medium. Typically, total cell death was quantified by WST-1 assay and necrotic cell death as described below.
Assessment of cell survival by WST-1 assay
Cell viability of HEK293 cells was determined using the WST-1 assay kit (Roche, Penzberg, Germany) in accordance with the manufacturer’s recommended protocol. In brief, after the respective treatments, the spent medium was removed, replaced with fresh growth medium, and incubated at 37°C for 3 h. The medium was then replaced again with fresh growth medium supplemented with 10% WST-1 reagent. This was followed by an additional incubation at 37°C for 30–60 min. Changes in cell viability were reflected by changes in optical density detected at 420 nm, which was measured using a spectrophotometer microplate reader (SpectraMax 340, Molecular Devices, Sunnyvale, CA, USA). All values in the figures were calculated from at least 2–3 independent experiments with each experiment containing at least 6–8 replicates in each experimental condition.
Assessment of necrosis by LDH assay
Necrosis is characterized by early loss of cell membrane integrity and the release of cytosolic lactate dehydrogenase (LDH). Hence, LDH release was measured in the present study to quantify necrotic cell death induced by MPP+ using the LDH assay kit developed by Pointe Scientific, Inc. (Fisher Scientific, Pittsburgh, PA, USA). In brief, 30 µl of the spent medium was collected from each treatment condition and mixed with 150 µl of reagent comprised of Reagent A and Reagent B pre-mixed in a ratio of 1:6. Changes in optical density at 340 nm were measured using a spectrophotometeric microplate reader and the maximum reaction rate was calculated to reflect the LDH level in the spent medium. The percentage of LDH release under each experimental condition was then calculated against the total cellular contents of LDH in control wells. Total LDH contents were assessed by subtracting the amount of LDH in untreated cell culture medium (total viable cells) from that in cultures lysed with 1% Triton X-100 for 10 min (total release).
Assessment of Cell survival by cell count post-stained with Hoechst stain
Cell density of SH-SY5Ycells expressing LRRK2 wild-type, Y1699C or G2019S mutant were determined by direct cell count due to the presence of GFP fusion to each of the genes. Twenty four hours after treatment with H2O2, the cells were fixed with 4% para-formaldehyde, 4% sucrose in PBS for 20 minutes at room temperature. Then, the cells were permeablized with 0.1% Triton X-100 in PBS for another 20 minutes at room temperature. Afterwards, the cells were stained with Hoechst stain for 1 hour before it was replaced with PBS. Then, the cells were observed via fluorescent microscopy. For each treatment conditions, three independent frames of the cells were captured showing cells expressing LRRK2-GFP fusion. The same frames of the cells stained with Hoechst stain to reveal total number of cells was captured too. In this way, the number of cells expressing LRRK2 (wild-type and mutants) and the total of cells were determined by direct cell count. In this manner, the percentage survival of SH-SY5Y cells expressing each of the LRRK2 genes was determined. In addition, the change in percentage survival of SH-SY5Y cells in response to H2O2 insult was also determined.
Deletion mutant construction in mammalian expression vector
The deletion mutants for wild-type LRRK2 and Y1699C mutant genes were created using the same approach. In each case, the gene was cut with endonucleases Cla I (New England Biolabs, Ipswich, MA, USA) and Pac I (New England Biolabs, Ipswich, MA, USA), respectively, to remove the kinase domain. Meanwhile, two oligomers of sequences “CGAGCGGCCGCGAT” and “CGCGGCCGCT” were synthesized and annealed to form a linker with ends compatible to Cla I and Pac I sites respectively. Then, the linker was used to connect the open ends of the deleted genes giving rise to the pair of deletion mutants corresponding to LRRK2 wild-type gene and Y1699C mutant gene. The excision sites of the deletion mutants were sequenced. In addition, we retained the parental vector pCDNA3.1 and the GFP fusion at the C-terminal for the deletion mutants.
PAGE and Western blotting
Standard PAGE and Western blotting protocols recommended by Cell Signaling Technology (Danvers, MA, USA) were used in this study.
Statistical analysis
Results are reported as mean value ± SEM. The significance of difference between means was assessed by ANOVA and post hoc Fisher’s protected least significant difference (PLSD) tests, with p<0.05 considered statistically significant.
Results
In this study, we characterized functional differences between wild-type LRRK2 and its mutants (Y1699C and G2019S) in response to oxidative stress. We had selected HEK293 cells as host for expressing these genes as we consistently at least 70% transfection efficiency. In addition, we have also verified the consistency of the key results in neuronal SH-SY5Y cells. Since it has been reported that 6-OHDA and H2O2 toxicities utilized similar pathways to achieve cell death (Mazzio et al., 2004; Saito et al., 2007) and in view of its stability over 6-OHDA, we chose H2O2 as the source of oxidative stress to induce degeneration in our study.
Changes in basal cell viability and H2O2-induced cell death due to expression of wild-type LRRK2 and its mutants Y1699C and G2019S
First, we have to determine the concentration of H2O2 that will elicit 50% cell death (EC50). When we treated HEK293 cells with H2O2 between 1–300µM for 18 hours, we obtained a dose response curve indicating that 150 µM H2O2 consistently elicited 50% cell death (Figure 1A). To ascertain the mode of cell death at this condition, we then measured the corresponding percentage necrotic cell death using the LDH assay (Figure 1B). We observed that chronic treatment with 150µM of H2O2 resulted in 10% of necrotic cell death, indicating that the predominant mode of cell death at these conditions is apoptosis. Therefore, the paradigm adopted in this study was the treatment of HEK293 cells with H2O2 at 150µM for 18 hours.
Figure 1. Impact of wild-type LRRK2 and its mutants Y1699C and G2019S on basal cell viability and under the insult of H2O2 in HEK293 cells.
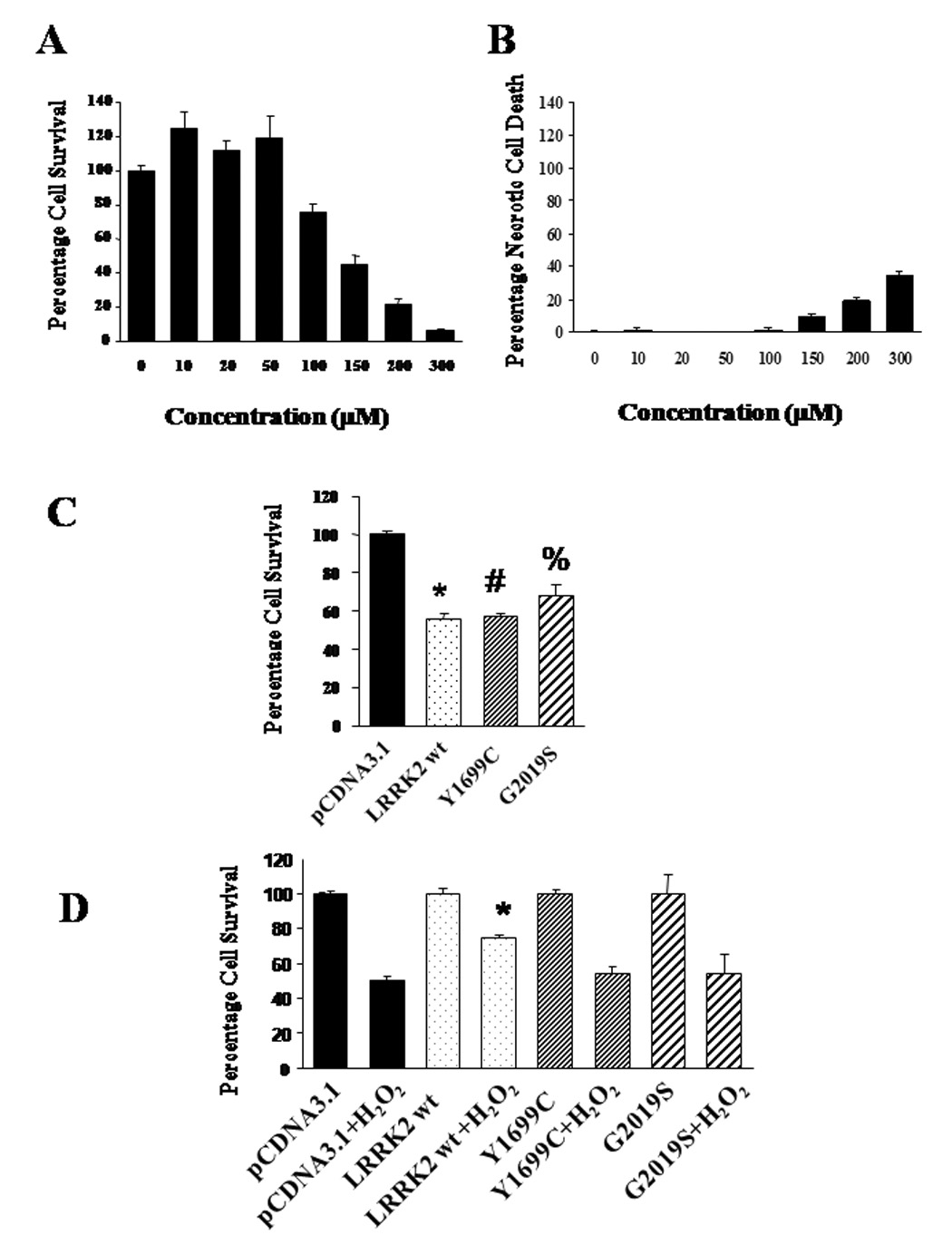
(A) Cell survival profile in response to 0–300 µM of treatment of H2O2 for 18 hours in HEK293 cells; (B) Changes in percentage cell death due to necrosis as estimated by LDH assay in response to 0–300 µM of treatment of H2O2 for 18 hours in HEK293 cells; (C) Decrease in basal cell viability in HEK293 cells expressing wild-type LRRK2 and its mutants Y1699C and G2019S. Data are means ± SEM, at least 24 readings per data point, from six independent experiments. *p < 0.01, #p < 0.01, % p< 0.01 versus viability of cells transfected with pCDNA3.1. Statistics were derived from ANOVA and post hoc Fisher’s protected least significant difference (PLSD) tests; (D) Attenuation of H2O2-induced cell death for cells expressing wild-type LRRK2 but not in cells expressing mutant Y1699C or G2019S. Percentage cell survival after H2O2 insult among cells transfected with pCDNA3.1, expressing wild-type LRRK2 or its mutants Y1699C and G2019S is normalized against untreated cells transfected with the same vector or expressing the same proteins. Data are means ± SEM, at least 24 readings per data point, from six independent experiments. * p < 0.01 versus viability of cells transfected with pCDNA3.1 after 150 µM of H2O2 treatment for 18 h. Statistics were derived from ANOVA and post hoc Fisher’s protected least significant difference (PLSD) tests.
The delivery of wild-type LRRK2, Y1699C and G2019S genes into HEK293 cells was mediated via transfection with lipofectamine 2000. After optimization, the typical transfection efficiency was at least 70% (data not shown). Comparable transfection efficiency was ensured by monitoring the expression of each LRRK2 gene with fluorescence microscopy since a GFP moiety is fused to each of their C-terminal. Twenty-four hours after transfection, we observed a consistent 30–45% drop in basal viability among cells expressing wild-type and its mutants Y1699C and G2019S with no significant difference between them (Figure 1C). However, when the cells expressing wild-type and mutant LRRK2 genes were exposed to H2O2 at 150µM for 18 hours, we observed that cells expressing wild-type LRRK2 were more tolerant to H2O2 toxicity than were those expressing each of the mutants (Figure 1D). In comparison, cells expressing wild-type LRRK2 experienced 50% attenuation in H2O2-induced cell death relative to those cells expressing Y1699C or G2019S mutants. On the other hand, there was no significant difference in the percentage cell death among cells expressing either of the mutants as well as those transfected with the parental vector pCDNA3.1. Therefore, the results suggest that Y1699C and G2019S mutants did not further exacerbate cell death in response to H2O2 toxicity. Since the impact on basal cell viability and in response to H2O2 toxicity are the same in cells expressing Y1699C and G2019S mutants, we performed more detailed investigation on the mechanistic difference between wild-type LRRK2 and Y1699C mutants in subsequent experiments.
The impact of wild-type LRRK2 and Y1699C mutants on ERK1/2
Based on structural and domain analysis, the physiological role of wild-type of LRRK2 is predicted to be a stress-responsive protein scaffold mediating the regulation of mitogen activated protein kinase (MAPK) pathways (Mata et al., 2006). Under this notion, LRRK2 must crosstalk with the proteins involving the MAP kinase cascade. Hence, we first examined changes in basal levels of phospho-ERK1/2(pERK) and phospho-JNK(pJNK) due to expression of wild-type LRRK2 or Y1699C mutant in HEK293 cells which may suggest crosstalk between LRRK2 and the MAP kinase pathways. The lysate from cells expressing wild-type LRRK2 or Y1699C mutant as well as those transfected with parental vector pCDNA3.1 were prepared and the changes in basal levels of pERK and pJNK were determined by immunoblot analysis (Figure 2A). The presence of wild-type LRRK2 and Y1699C mutant in HEK293 cells was also visualized by probing immunoblots with an antibody recognizing GFP (Figure 2A, top panel).
Figure 2. Impact of expressing wild-type LRRK2 and Y1699C mutant on basal level of phospho-ERK (pERK) and phospho-JNK (pJNK).
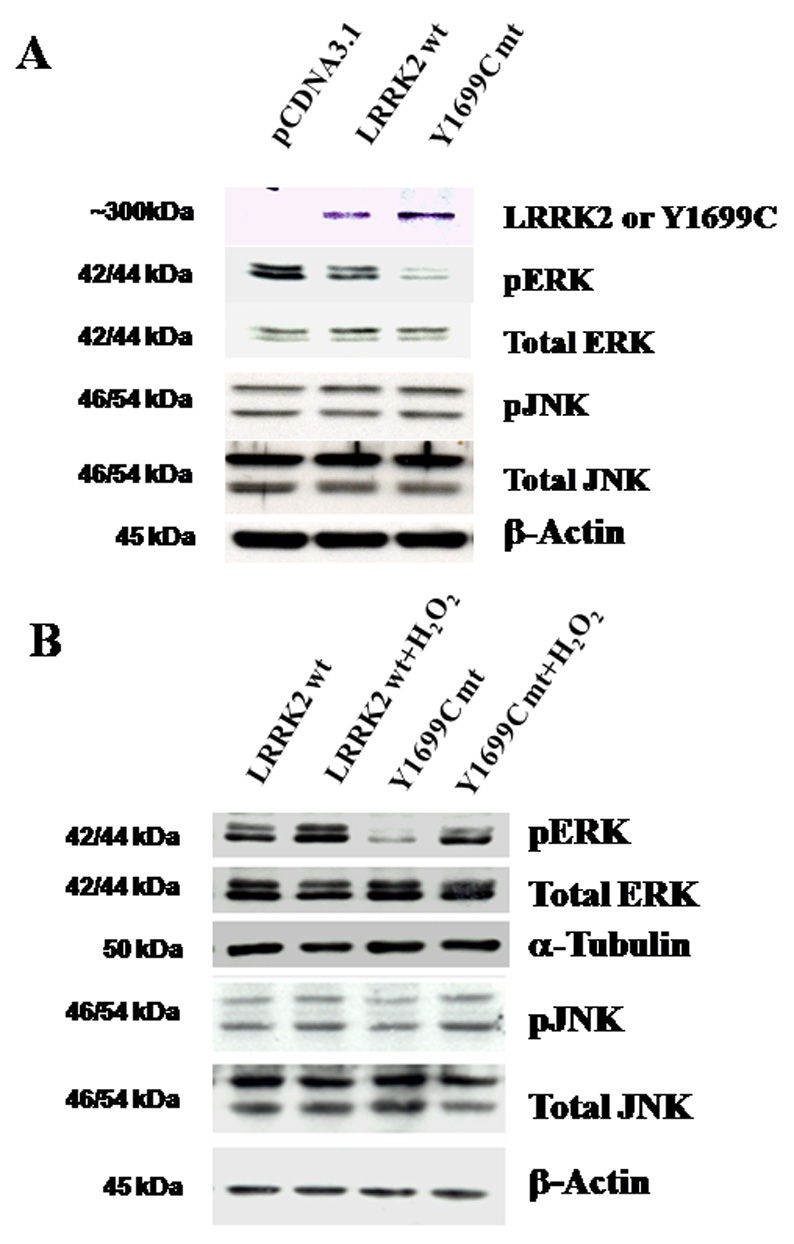
(A) Changes in basal level of pERK and pJNK in HEK293 cells expressing wild-type LRRK2 and Y1699C mutant as compared to those transfected with pCDNA3.1. In the top panel, an anti-GFP antibody was used to examine expression of wild-type LRRK2 and Y1699C mutant gene in HEK293 cells via immunoblotting. The corresponding change in basal level of pERK, total ERK, pJNK, and total JNK are also shown and β-actin was used as a loading control; (B) Corresponding changes in pERK, total ERK, pJNK, and total JNK in HEK293 cells expressing wild-type LRRK2 or Y1699C mutant with and without chronic treatment with 150 µM of H2O2 for 18 hours. β-Actin was used as loading control.
No band was detected in lysate derived from cells transfected with pCDNA3.1 vector. We also failed to detect endogenous wild-type LRRK2 in HEK293 cells with several commercially available antibodies (data not shown). By comparison, the endogenous wild-type LRRK2 was significantly lower than those being over-expressed and hence functional changes attributed to the over-expressed LRRK2 (wild-type and mutant) would receive minimal interference from endogenous wild-type LRRK2. In comparison, the basal level of pERK was decreased in cells expressing wild-type LRRK2 and Y1699C mutant with no change to the level of total ERK1/2 (tERK) (Figure 2A). Moreover, the decrease in the basal level of pERK in cells expressing Y1699C mutant was higher in cells expressing wild-type LRRK2. In contrast, there was no significant change in the basal level of pJNK or total JNK (tJNK) in cells expressing wild-type LRRK2 and Y1699C mutant to those transfected with pCDNA3.1. On the other hand, after 18 hours of treatment with H2O2 at EC50 (150µM), cells expressing wild-type LRRK2 consistently elicited a higher level of pERK than those expressing Y1699C mutant with no significant change in the level of tERK (Figure 2B). As to pJNK and tJNK, there was no significant difference in their level changes in response H2O2 toxicity between cells expressing wild-type LRRK2 and Y1699C mutant. Collectively, these results suggest the potential crosstalk between LRRK2 and the ERK1/2 pathway.
Wild-type LRRK2 activated the ERK1/2 pathway to protect against H2O2 toxicity
In view of the higher pERK level in cells expressing wild-type LRRK2 and the corresponded decrease in H2O2-induced cell death, we speculated the possibility that the activation of ERK1/2 pathway had been employed by wild-type LRRK2 to confer protection against cell death induced by H2O2 toxicity. To test this hypothesis, we cloned a dominant negative version of the ERK2 gene (dnERK2) into the same parental vector pcDNA3.1. The dominant negative version of the ERK2 gene is one with its Thr202 and Tyr204 replaced with Alanine. Over-expression of dnERK2 will saturate the activated MEK1 inhibiting the proper phosphorylation/activation of ERK1/2. As expected, upon expression of dnERK2, the activation of ERK1/2 in response to H2O2 was significantly suppressed (Figure 3A). In addition, the successful expression of the dnERK2 was confirmed indicated by the stronger upper band of the rightmost column of the second panel in Figure 3A.
Figure 3. Effect of co-expression of wild-type LRRK2, Y1699C mutant with dnERK2 respectively on basal cell viability and cell death in response to H2O2 toxicity.
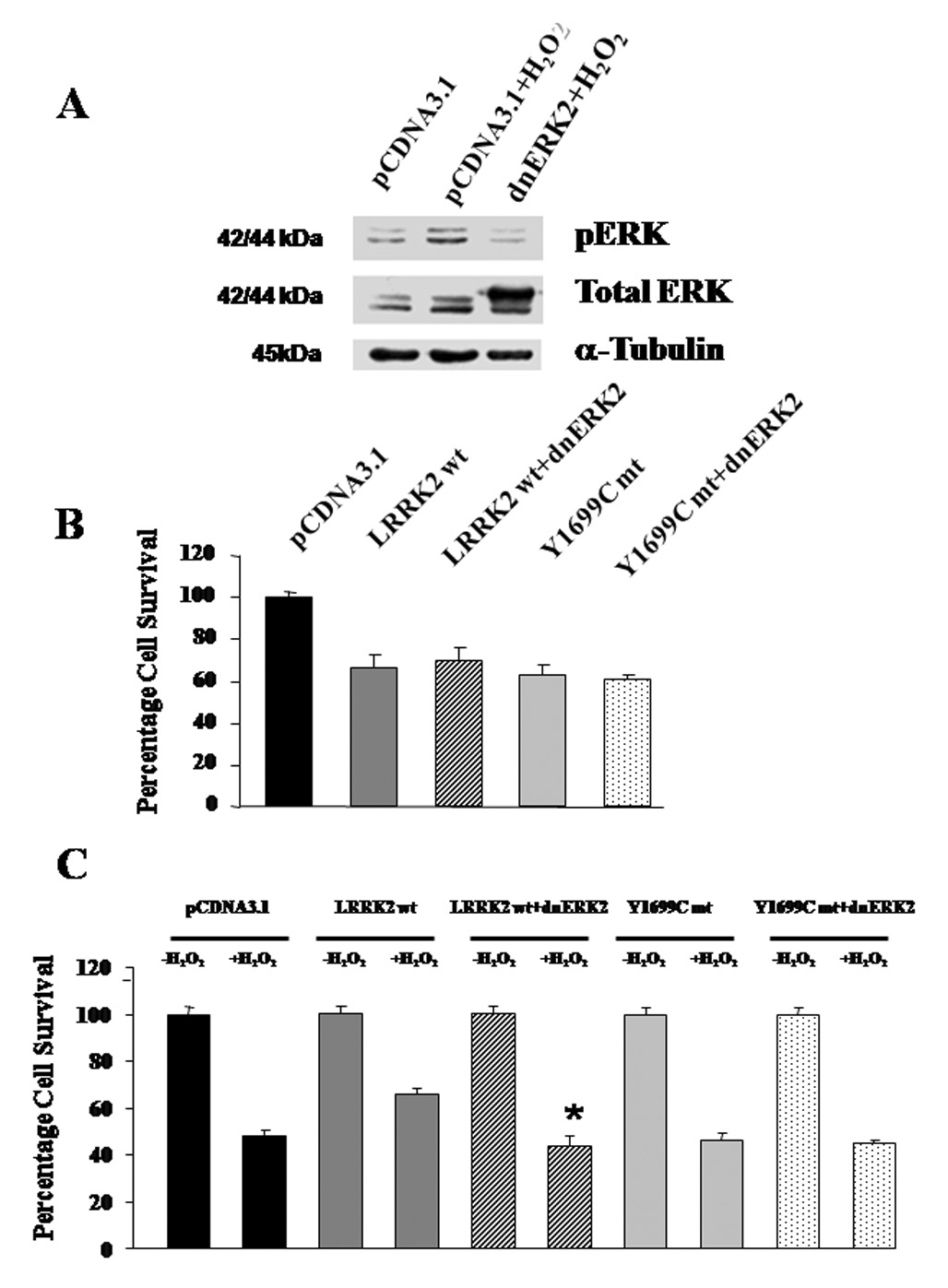
(A) Demonstration of the efficacy of using dnERK2 to silence ERK activation. In the top panel, cells expressing pCDNA3.1 showed activation in response to H2O2 (middle lane); on the other hand, cells expressing dnERK2 gene showed suppression of ERK activation under the same stimulus (right lane). The second panel indicates expression of the dnERK2 gene. α–Tubulin was used as loading control. (B) Co-expression of dnERK2 with LRRK2 wild-type gene or Y1699C mutant gene did not significantly alter the decrease in basal cell viability due to the expression of LRRK2 genes or its mutants. (C) Co-expression of dnERK2 with wild-type LRRK2 abrogated the protection conferred by the latter gene against cell death induced by H2O2 in HEK293 cells. No significant changes in cell death induced by H2O2 toxicity were detected when dnERK2 was co-expressed with Y1699C mutant in HEK293 cells. Percentage cell death induced by H2O2 among cells transfected with pCDNA3.1, expressing LRRK2 wt or Y1699C mt gene in the absence or presence of dnERK2 are normalized against untreated cells transfected with the same vector or expressing the same proteins. Data are means ± SEM, at least 12 readings per data point, from three independent experiments. * p < 0.05 versus cell viability of cells expressing LRRK2 wt without dnERK2 after chronic treatment with H2O2 for 18 hours. Statistics were derived from ANOVA and post hoc Fisher’s protected least significant difference (PLSD) tests.
Using the dnERK2 construct to suppress the activation of ERK1/2, we could examine the impact of ERK1/2 activation on basal viability and cell death induced by H2O2 toxicity. Co-expression of dnERK2 and wild-type LRRK2 and Y1699C mutant did not alter the decrease in basal viability of HEK293 cells elicited by expressing wild-type LRRK2 and Y1699C mutant alone (Figure 3B), suggesting that the decrease in basal pERK was not the cause of this drop in basal viability. On the other hand, under the persistent insult of H2O2 at 150 µM, cells co-expressing wild-type LRRK2 and dnERK2 abolished the protection against H2O2-induced cell death conferred by wild-type LRRK2 (Figure 3C). This loss of protection has brought the percentage cell death to a level comparable to cells transfected with pCDNA3.1 or expressing Y1699C mutant. Furthermore, co-expression of Y1699C mutant and dnERK2 did not elicit any further decrease in H2O2-induced cell death. Similarly, pre-treatment with the MEK inhibitor U0126 (10µM) suppressed the activation of the ERK1/2 pathway and also abrogated the protection conferred by wild-type LRRK2, without any significant impact on the H2O2-induced cell death in cells expressing Y1699C mutant (data not shown). Collectively, these results suggest the notion that wild-type LRRK2 but not Y1699C mutant activated the ERK1/2 pathway and thereby protected against H2O2-induced cell death.
Differential cell death in neuronal cells expressing LRRK2 wild-type and mutants (Y1699C or G2019S) in response to H2O2 toxicity
Thus far, we have observed that HEK293 cells expressing LRRK2 wild-type are more resistant to H2O2 toxicity as compared to cells expressing mutants (Y1699C or G2019S) and this difference can be attributed to the capacity of LRRK2 wild-type to activate the ERK1/2 pathway. At this juncture, we are interested to determine whether this differential functional impact from LRRK2 wild-type and mutants (Y1699C or G2019S) against oxidative stress can be extended to neuronal cells. To address this, we transfected the same set of LRRK2 wild-type and mutant genes into human dopaminergic neuronal SH-SY5Y cells via lipofectamine 2000, followed by determining their influence on cell viability in response to H2O2 toxicity (60 µM, 18 hours). Due to the low transfection efficiency (~13%), the percentage cell survival was determined by direct cell count of cells exhibiting GFP fluorescence against total cell number visualized by Hoechst stain.
First, the impact from these LRRK2 gene products on basal cell viability of SH-SY5Y cells was determined (Figure 4A). In comparison, we observed a decrease in basal viability of 20% in cells expressing Y1699C and nearly 30% in cells G2019S than those cells expressing LRRK2 wild-type. Consistent to reported studies, both of the LRRK2 mutants exhibited higher innate toxicity than LRRK2 wild-type (Gloeckner et al., 2006; Smith et al., 2006).
Figure 4. Impact of LRRK2 wild-type and mutants on basal viability and cell viability in response to H2O2 insult in SH-SY5Y cells.
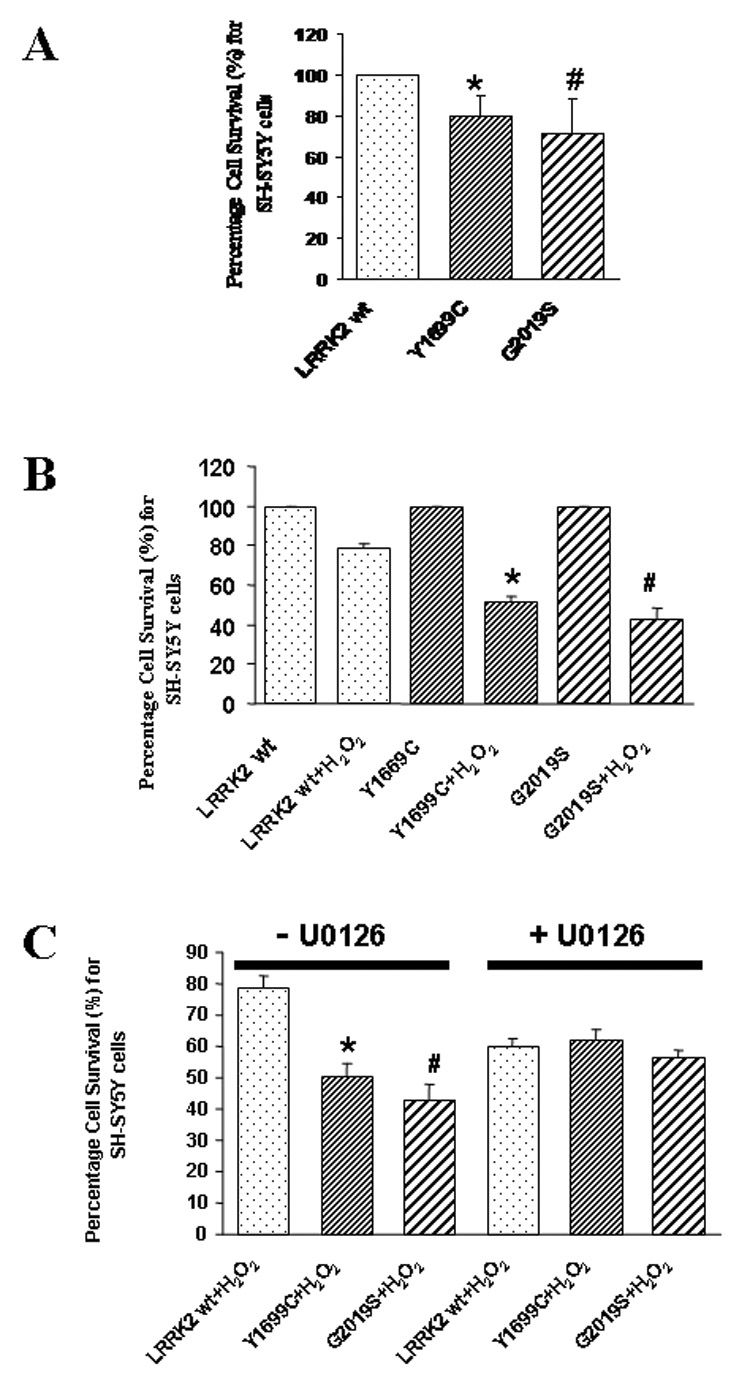
(A) Change in basal viability due to expression of LRRK2 wild-type and mutants (Y1699C and G2019S). Data are means ± SEM, 3 readings per data point, from at least free independent experiments. *p <0.05 versus basal cell viability of cells expressing LRRK2 wild-type; #p <0.05 versus basal cell viability of cells expressing LRRK2 wild-type. Statistics were derived from ANOVA and post hoc Fisher’s protected least significant difference (PLSD) tests. (B) Change in percentage cell survival between cells expressing LRRK2 wild-type and mutants (Y1699C or G2019S) in response to H2O2 insult. Data are means ± SEM, 3 readings per data point, from at least free independent experiments. *p <0.01 versus percentage cell viability of cells expressing LRRK2 wild-type after H2O2 insult; #p <0.01 versus basal cell viability of cells expressing LRRK2 wild-type after H2O2 insult. Statistics were derived from ANOVA and post hoc Fisher’s protected least significant difference (PLSD) tests. (C) Change in percentage cell survival between cells expressing LRRK2 wild-type and mutants (Y1699C or G2019S) in the absence and presence of 10µM U0126 in response to H2O2 insult. Each of the percentage cell survival of cells expressing LRRK2 wild-type and mutants (Y1699C or G2019S) after H2O2 insult was normalized from corresponding cells expressing the same LRRK2 gene without H2O2 insult. Data are means ± SEM, 3 readings per data point, from at least free independent experiments. *p <0.01 versus percentage cell viability of cells expressing LRRK2 wild-type after H2O2 insult; #p <0.01 versus basal cell viability of cells expressing LRRK2 wild-type after H2O2 insult. Statistics were derived from ANOVA and post hoc Fisher’s protected least significant difference tests.
Then, we investigated the differential change in percentage cell survival among cells expressing LRRK2 wild-type and mutants in response to H2O2 toxicity in SH-SY5Y cells. Similar to that observed in HEK293 cells, cells expressing LRRK2 wild-type experienced a 27–36% lowering in cell death as compared to cells expressing Y1699C or G2019S respectively (Figure 4B). Moreover, in the presence of 10µM U0126 pre-treated 1 hour before the treatment of H2O2, the difference in percentage cell survival between cells expressing LRRK2 wild-type and its mutants was abolished (Figure 4C) suggesting that the difference is mostly likely due to the activation of the ERK1/2 pathway mirroring that observed in HEK293 cells.
The protection conferred by wild-type LRRK2 is dependent on its kinase domain
To date, the limited biochemical characterization of LRRK2 has indicated a change in the kinase activity due to the identified mutations based on in vitro cell free kinase assay (Greggio et al., 2006; Ito et al., 2007; West et al., 2005). However, it is yet uncertain which protein along the ERK signaling cascade can interact with wild-type LRRK2 protein directly. Thus, it is the common belief that the kinase activity of LRRK2 will dictate its physiological function. Therefore, we were interested to determine whether the kinase domain is involved in the protective phenotype of wild-type LRRK2 observed in our paradigm. Since all the reported studies related to the change in kinase activity of LRRK2 due to specific mutations (Greggio et al., 2006; Ito et al., 2007; West et al., 2005) or the development of kinase dead mutations (Burke, 2007; Greggio et al., 2006; Lu and Tan, 2008) were based on cell free in vitro kinase assay using auto-phosphorylation or myelin basic protein (MBP) as substrates, it is uncertain the same specific change in kinase activity for Y1699C and G2019S can be extended to the protein that activates the ERK1/2 pathway in view of the unique protein-protein interaction between the kinase and each of its substrates. The same mutations would elicit unique impact on the activity of the kinase towards each of its substrates. Hence, under these circumstances, we decided to delete the kinase domain in lieu of mutating specific residues to create a kinase-dead version of wild-type LRRK2 and Y1699C mutant. In constructing the deletion mutants, we utilized unique endonuclease cut-sites of Cla I and Pac I flanking the kinase domain of the LRRK2 gene. Using these sites, the kinase domain was excised from the LRRK2 wild-type and Y1699C mutant gene. After the excision, the ends were joined by an in-frame short DNA linker created by annealing two complementary primers. The sequences are shown in the Material and Methods section.
Using the deletion mutants in conjunction with their corresponding complete gene, we could determine whether their differential response towards oxidative stress mandate the presence of the kinase domain. With this strategy, we observed that removal of the kinase domain from the wild-type LRRK2 further decrease the basal level of pERK to a comparable level elicited by full length Y1699C mutant (Figure 5A). On the other hand, removal of the kinase domain from Y1699C mutant did not further decrease the basal level of pERK, suggesting that the kinase domain was not involved in the suppression of basal level of pERK. Nevertheless, these results supported the notion that wild-type LRRK2 but not Y1699C mutant has the capacity to activate the ERK pathway. However, the removal of the kinase domain from full length wild-type LRRK2 and Y1699C mutant mitigated the drop in basal viability by 20% when expressed in HEK293 cells, which was 20% less than the cells expressing their full length counterpart (Figure 5B).
Figure 5. Characterization of the deletion mutants.
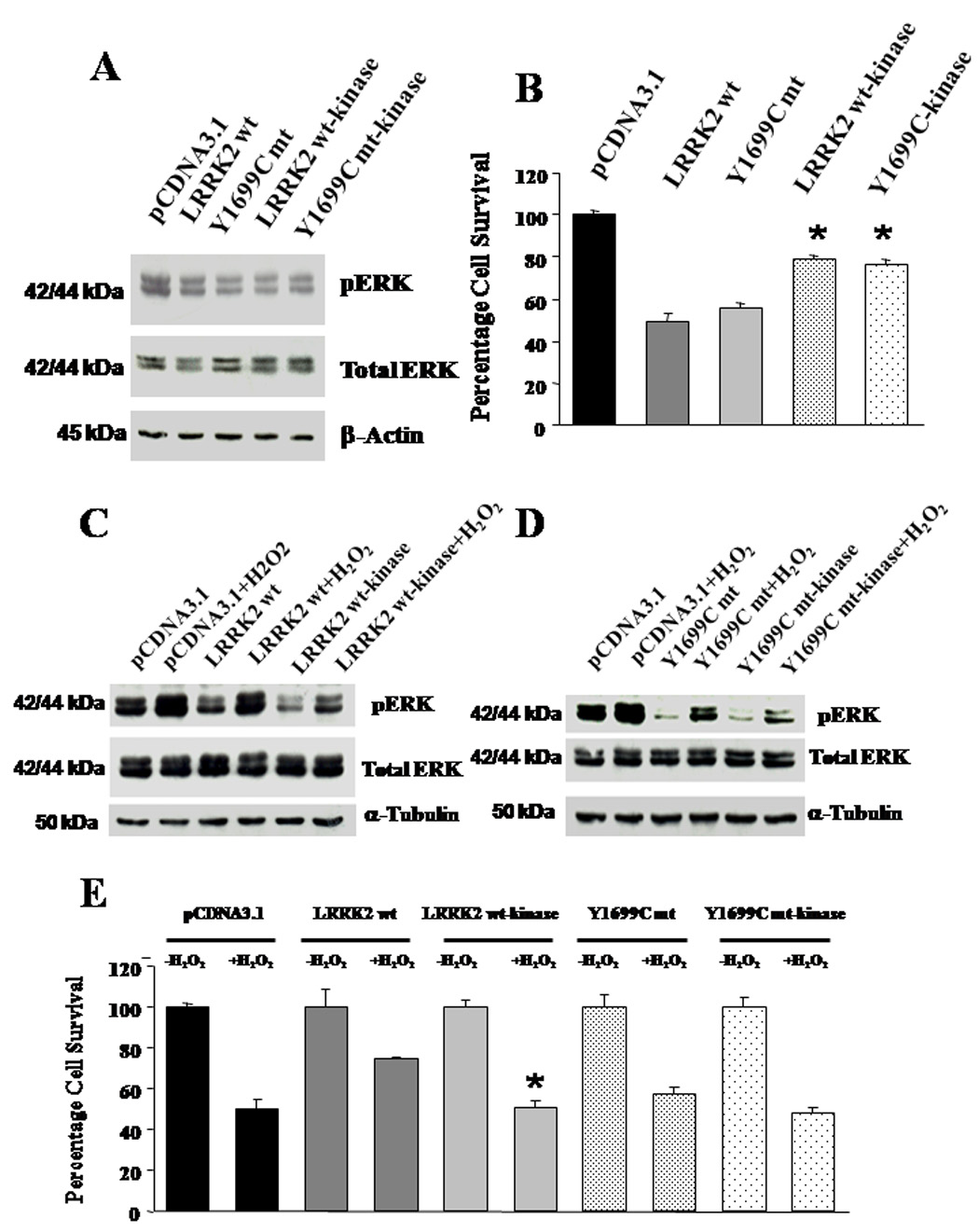
(A) Change in basal pERK in HEK293 cells expressing wild-type LRRK2, Y1699C mutant, and their respective deletion mutants. (B) Expression of deletion mutants resulted in significantly less decrease in basal cell viability as compared to their complete gene counterpart. Data are means ± SEM, at least 12 readings per data point, from three independent experiments. * p < 0.05 versus basal cell viability of cells expressing the corresponding complete gene. Statistics were derived from ANOVA and post hoc Fisher’s protected least significant difference (PLSD) tests. (C) Impact of expressing LRRK2 wild-type-kinase (deletion mutant) on ERK response in HEK293 cells towards H2O2 toxicity. The changes in pERK and total ERK with and without H2O2 treatment in cells transfected with pCDNA3.1, expressing wild-type LRRK2 (LRRK2 wt), and LRRK2 wild-type-kinase (LRRK2 wt-kinase) were visualized by immunoblotting probed with antibodies recognizing pERK, total ERK and β-actin respectively. β-actin was used as loading control. (D) Impact of expressing Y1699C mutant-kinase (deletion mutant) on ERK response in HEK293 cells towards H2O2 toxicity. The changes in pERK and total ERK with and without H2O2 treatment in cells transfected with pCDNA3.1, expresssing Y1699C mutant (Y1699C mt) and Y1699C mutant-kinase (Y1699C mt-kinase) were visualized by immunoblotting probed with antibodies recognizing pERK, total ERK and β-actin respectively. β-actin was used as loading control. (E) Comparative impact on cell death induced by treatment with 150 µM of H2O2 in cells expressing pCDNA3.1, wild-type LRRK2, LRRK2 wt-kinase, Y1699C mutant, and Y1699C mt-kinase. Percentage cell death induced by H2O2 among cells transfected with pCDNA3.1, expressing wild-type LRRK2, Y1699C mutant, or their corresponding deletion mutant are normalized against untreated cells transfected with the same vector or expressing the same proteins. Data are means ± SEM, at least 12 readings per data point, from three independent experiments. * p < 0.05 versus cell viability of cells expressing LRRK2 wt after chronic treatment with H2O2 for 18 h. Statistics were derived from ANOVA and post hoc Fisher’s protected least significant difference (PLSD) tests.
Under the chronic insult of H2O2, we found that cells expressing the deletion mutant of wild-type LRRK2 cannot elicit an increase in pERK level as observed in cells expressing full length wild-type LRRK2 (Figure 5C). In the case of Y1699C mutant, cells expressing the full length or deletion mutant version of the gene do not exhibit significant difference in their capacity to increase pERK level (Figure 5D). Correspondingly, cells expressing the deletion mutant of wild-type LRRK2 lost the protective phenotype conferred by full length wild-type LRRK2 against H2O2-induced cell death (Figure 5E), suggesting that the protective phenotype of wild-type LRRK2 is dependent on the kinase domain and hence kinase activity. Moreover, deletion of the kinase domain from Y1699C mutant did not increase the H2O2-induced cell death and was comparable to cells transfected with pCDNA3.1. Collectively, the results suggest that wild-type LRRK2 but not Y1699C mutant is capable of activating the ERK pathway to confer protection against H2O2-induced cell death.
Discussion
Studies on the biochemical characterization of functional differences between wild-type LRRK2 and its mutants are limited. The few reported biochemical studies have focused on the impact of the LRRK2 mutant kinase activity on intracellular physiological functions, including the formation of inclusion bodies and cell death (Gloeckner et al., 2006; Greggio et al., 2006; Smith et al., 2006; West et al., 2005). Overall, it has been speculated that LRRK2 mutant could elicit a “toxic gain of function” that leads to degenerative consequences within the cell. The objective of the present study was to identify functional differences between wild-type LRRK2 and the mutants Y1699C and G2019S in response to oxidative stress. Collectively, our results indicated that wild-type LRRK2 but not Y1699C or G2019S mutants attenuated H2O2–induced cell death in HEK293 cells and SH-SY5Y cells. Further mechanistic determination indicated that the mutation such as Y1699C lost the inherent protective capacity of wild-type LRRK2 against oxidative stress in its inability to activate the ERK1/2 pathway. Nevertheless, the results from this study cannot rule out the possibility that specific mutations in the LRRK2 gene abolished protective capacity of LRRK2 against oxidative stress in conjunction with a gain of toxic function as suggested in other reported studies.
Impact of wild-type LRRK2 and its mutants on basal viability
In non-neuronal HEK293 cells, expressing wild-type LRRK2, Y1699C or G2019S mutants experienced a 30–40% decrease in basal cell viability showing no significant difference in their individual toxic impact. In contrast, expression of Y1699C or G2019S mutants in neuronal SH-SY5Y cells resulted in at least 20% higher drop in basal viability than its wild-type counter-part suggesting greater toxicity for the mutants consistent to reported studies (Greggio et al., 2006; Smith et al., 2006). This discrepancy may be due to the differential endogenous protein profiles in HEK293 cells and SH-SY5Y cells. It is speculated that the LRRK2 mutants interact with neuronal specific proteins which are absence in HEK293 cells, actualizing their inherent higher toxicity than LRRK2 wild-type. On the other hand, the activation of the ERK pathway by LRRK2 wild-type in response to H2O2 toxicity was via protein/s present in HEK293 and SH-SY5Y cells and hence account for the consistent observation in both cell lines. Therefore, despite the high transfection efficiency for HEK293 cells, one must be careful in data interpretation as it is not always possible to generalize results obtained in non-neuronal cells to neuronal cells.
To date, several reported studies have suggested the toxicity of LRRK2 mutants resides solely on their change in kinase activity from LRRK2 wild-type (Greggio et al., 2006; Smith et al., 2006; West et al., 2005; West et al., 2007). If this is the case, expression of the deletion mutants should not decrease the basal viability by 20% higher than cells that have undergone transfection with pCDNA3.1 or expressing GFP alone (data not shown), albeit their corresponding impact on basal viability were 15–20% lower than cells expressing full length LRRK2 wild-type or mutants (Figure 5B). This result suggested two components for the inherent toxicity of LRRK2 wild-type and mutants. First, cells expressing the kinase deletion mutants experienced a 15–20% increase in basal viability when compared to cells expressing their full length counterpart does suggest a kinase dependent inherent toxic component of LRRK2. However, the 20% drop in basal viability in cells expressing the kinase deletion mutants when compared to cells transfected with pCDNA3.1 or expressing GFP also suggest a component of the inherent toxicity for LRRK2 that is independent of its kinase activity.
Can a kinase independent component of the inherent toxicity for LRRK2 possible? Sequence and functional domain analysis of LRRK2 led to the prediction of its function to be a protein scaffold that enables the formation of a multi-protein signaling complex similar to that of the protein kinase suppressor of Ras (Ksr)(Gloeckner et al., 2006; Kolch, 2005; Mata et al., 2006). Potential cross-talk between LRRK2 and the ERK pathway was first suggested by the decrease in basal level of pERK in cells expressing LRRK2 wild-type, its mutant and their corresponding deletion mutants which were consistent to its predicted role as a protein scaffold facilitating the activation of the ERK1/2 pathway. This is because as a protein scaffold facilitating the activation of the ERK1/2 pathway, over-expressing LRRK2 wild-type or mutants would inevitably suppress the activation of the ERK1/2 pathway which may come with detrimental consequences to cell growth and maintenance of cell viability. In parallel, it has been reported that over-expressing the scaffolding protein, JIP1, which results in the suppression of JNK activation through sequestration of MEK or JNK despite the fact that physiological function of JIP1 is to facilitate JNK activation (Dickens et al., 1997; Harding et al., 2001; Li et al., 2005; Mooney and Whitmarsh, 2004). This may account for the kinase independent component of the inherent toxicity of this protein.
Impact of LRRK2 wild-type and mutants on the cell death in response to oxidative stress
Collectively, our results indicated that LRRK2 wild-type but not its mutants has the capacity to attenuate H2O2-induced cell death via activation of the ERK1/2 pathway in both HEK293 and SH-SY5Y cells. This change in tolerance towards oxidative stress is likely to be attributed to their differential capacity to activate the ERK1/2 pathway. Our evidence further indicates that the activation of the ERK1/2 pathway by wild-type LRRK2 is kinase domain dependent. However, it is yet uncertain which protein along the ERK signaling cascade can interact with wild-type LRRK2 protein directly.
Although our study suggests a cellular function for wild-type LRRK2 in attenuating oxidative stress, it is likely that this protein has other physiological functions yet to be identified. In summary, our results suggested that mutations on LRRK2 can alter its capacity to activate the ERK1/2 signaling cascade, which significantly affect cell death in response to oxidative stress. This observation is consistent with the observation that the G2019S mutation decreases the phosphorylation of MAP kinase (White et al., 2007). However whether LRRK2 functions as an intracellular scaffold facilitating the activation of other MAP kinase signaling cascade merits further investigation.
Acknowledgements
We thank Mr. Leonard Kotveski for his technical assistance towards the completion of this project. This work is supported by NINDS (NS019608) and the U.S. Army (ERMS#03281022).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bialecka M, et al. Analysis of LRRK 2 G 2019 S and I 2020 T mutations in Parkinson's disease. Neurosci Lett. 2005;390:1–3. doi: 10.1016/j.neulet.2005.07.045. [DOI] [PubMed] [Google Scholar]
- Burke RE. Inhibition of mitogen-activated protein kinase and stimulation of Akt kinase signaling pathways: Two approaches with therapeutic potential in the treatment of neurodegenerative disease. Pharmacol Ther. 2007;114:261–277. doi: 10.1016/j.pharmthera.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickens M, et al. A cytoplasmic inhibitor of the JNK signal transduction pathway. Science. 1997;277:693–696. doi: 10.1126/science.277.5326.693. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, et al. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15:223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- Greggio E, et al. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Harding TC, et al. Inhibition of JNK by overexpression of the JNL binding domain of JIP-1 prevents apoptosis in sympathetic neurons. J Biol Chem. 2001;276:4531–4534. doi: 10.1074/jbc.C000815200. [DOI] [PubMed] [Google Scholar]
- Haugarvoll K, Wszolek ZK. PARK8 LRRK2 parkinsonism. Curr Neurol Neurosci Rep. 2006;6:287–294. doi: 10.1007/s11910-006-0020-0. [DOI] [PubMed] [Google Scholar]
- Ito G, et al. GTP binding is essential to the protein kinase activity of LRRK2, a causative gene product for familial Parkinson's disease. Biochemistry. 2007;46:1380–1388. doi: 10.1021/bi061960m. [DOI] [PubMed] [Google Scholar]
- Jain S, et al. Molecular genetic pathways in Parkinson's disease: a review. Clin Sci (Lond) 2005;109:355–364. doi: 10.1042/CS20050106. [DOI] [PubMed] [Google Scholar]
- Klein C, Schlossmacher MG. The genetics of Parkinson disease: Implications for neurological care. Nat Clin Pract Neurol. 2006;2:136–146. doi: 10.1038/ncpneuro0126. [DOI] [PubMed] [Google Scholar]
- Kolch W. Coordinating ERK/MAPK signalling through scaffolds and inhibitors. Nat Rev Mol Cell Biol. 2005;6:827–837. doi: 10.1038/nrm1743. [DOI] [PubMed] [Google Scholar]
- Korr D, et al. LRRK1 protein kinase activity is stimulated upon binding of GTP to its Roc domain. Cell Signal. 2006;18:910–920. doi: 10.1016/j.cellsig.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Li CH, et al. Activated mitogen-activated protein kinase kinase 7 redistributes to the cytosol and binds to Jun N-terminal kinase-interacting protein 1 involving oxidative stress during early reperfusion in rat hippocampal CA1 region. J Neurochem. 2005;93:290–298. doi: 10.1111/j.1471-4159.2005.03086.x. [DOI] [PubMed] [Google Scholar]
- Lu YW, Tan EK. Molecular biology changes associated with LRRK2 mutations in Parkinson's disease. J Neurosci Res. 2008 doi: 10.1002/jnr.21656. [DOI] [PubMed] [Google Scholar]
- Mata IF, et al. LRRK2 in Parkinson's disease: protein domains and functional insights. Trends Neurosci. 2006;29:286–293. doi: 10.1016/j.tins.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Mazzio EA, et al. The role of oxidative stress, impaired glycolysis and mitochondrial respiratory redox failure in the cytotoxic effects of 6-hydroxydopamine in vitro. Brain Res. 2004;1004:29–44. doi: 10.1016/j.brainres.2003.12.034. [DOI] [PubMed] [Google Scholar]
- Meylan E, Tschopp J. The RIP kinases: crucial integrators of cellular stress. Trends Biochem Sci. 2005;30:151–159. doi: 10.1016/j.tibs.2005.01.003. [DOI] [PubMed] [Google Scholar]
- Mooney LM, Whitmarsh AJ. Docking interactions in the c-Jun N-terminal kinase pathway. J Biol Chem. 2004;279:11843–11852. doi: 10.1074/jbc.M311841200. [DOI] [PubMed] [Google Scholar]
- Olanow CW. The pathogenesis of cell death in Parkinson's disease--2007. Mov Disord. 2007;22 Suppl 17:S335–S342. doi: 10.1002/mds.21675. [DOI] [PubMed] [Google Scholar]
- Saito Y, et al. Molecular mechanisms of 6-hydroxydopamine-induced cytotoxicity in PC12 cells: involvement of hydrogen peroxide-dependent and -independent action. Free Radic Biol Med. 2007;42:675–685. doi: 10.1016/j.freeradbiomed.2006.12.004. [DOI] [PubMed] [Google Scholar]
- Smith WW, et al. Kinase activity of mutant LRRK2 mediates neuronal toxicity. Nat Neurosci. 2006;9:1231–1233. doi: 10.1038/nn1776. [DOI] [PubMed] [Google Scholar]
- Tomiyama H, et al. Clinicogenetic study of mutations in LRRK2 exon 41 in Parkinson's disease patients from 18 countries. Mov Disord. 2006;21:1102–1108. doi: 10.1002/mds.20886. [DOI] [PubMed] [Google Scholar]
- West AB, et al. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, et al. Parkinson's disease-associated mutations in LRRK2 link enhanced GTP-binding and kinase activities to neuronal toxicity. Hum Mol Genet. 2007;16:223–232. doi: 10.1093/hmg/ddl471. [DOI] [PubMed] [Google Scholar]
- Whaley NR, et al. Clinical and pathologic features of families with LRRK2-associated Parkinson's disease. J Neural Transm Suppl. 2006:221–229. doi: 10.1007/978-3-211-45295-0_34. [DOI] [PubMed] [Google Scholar]
- White LR, et al. MAPK-pathway activity, Lrrk2 G2019S, and Parkinson's disease. J Neurosci Res. 2007 doi: 10.1002/jnr.21240. [DOI] [PubMed] [Google Scholar]
- Xu Z, et al. The MLK family mediates c-Jun N-terminal kinase activation in neuronal apoptosis. Mol Cell Biol. 2001;21:4713–4724. doi: 10.1128/MCB.21.14.4713-4724.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]