Mutations in CHD7, Encoding a Chromatin-Remodeling Protein, Cause Idiopathic Hypogonadotropic Hypogonadism and Kallmann Syndrome (original) (raw)
Abstract
CHARGE syndrome and Kallmann syndrome (KS) are two distinct developmental disorders sharing overlapping features of impaired olfaction and hypogonadism. KS is a genetically heterogeneous disorder consisting of idiopathic hypogonadotropic hypogonadism (IHH) and anosmia, and is most commonly due to KAL1 or FGFR1 mutations. CHARGE syndrome, a multisystem autosomal-dominant disorder, is caused by CHD7 mutations. We hypothesized that CHD7 would be involved in the pathogenesis of IHH and KS (IHH/KS) without the CHARGE phenotype and that IHH/KS represents a milder allelic variant of CHARGE syndrome. Mutation screening of the 37 protein-coding exons of CHD7 was performed in 101 IHH/KS patients without a CHARGE phenotype. In an additional 96 IHH/KS patients, exons 6–10, encoding the conserved chromodomains, were sequenced. RT-PCR, SIFT, protein-structure analysis, and in situ hybridization were performed for additional supportive evidence. Seven heterozygous mutations, two splice and five missense, which were absent in ≥ 180 controls, were identified in three sporadic KS and four sporadic normosmic IHH patients. Three mutations affect chromodomains critical for proper CHD7 function in chromatin remodeling and transcriptional regulation, whereas the other four affect conserved residues, suggesting that they are deleterious. CHD7's role is further corroborated by specific expression in IHH/KS-relevant tissues and appropriate developmental expression. Sporadic CHD7 mutations occur in 6% of IHH/KS patients. CHD7 represents the first identified chromatin-remodeling protein with a role in human puberty and the second gene to cause both normosmic IHH and KS in humans. Our findings indicate that both normosmic IHH and KS are mild allelic variants of CHARGE syndrome and are caused by CHD7 mutations.
Main Text
Idiopathic hypogonadotropic hypogonadism (IHH, MIM 146110), one of the most commonly inherited forms of hypogonadism, results from deficient hypothalamic GnRH release or action.1 IHH patients present with absent or impaired sexual development due to sex-steroid-hormone deficiency, low serum levels of the pituitary gonadotropins follicle-stimulating hormone (FSH) and luteinizing hormone (LH), and infertility.1 Kallmann syndrome (KS, MIM 308700, 147950, 244200, 610628), which couples IHH with the inability to smell (anosmia), is due to impairment of the normal embryologic migration of GnRH and olfactory neurons from the olfactory placode region into the hypothalamus.1,2 KS patients may have additional phenotypic abnormalities including cleft lip and palate, unilateral renal agenesis, dental agenesis, and neurologic abnormalities such as synkinesia and cerebellar dysfunction.2 The molecular basis of IHH and KS (IHH/KS) has been identified for approximately 25%–30% of patients, with mutations in the KAL1 (MIM 308700),3,4 FGFR1 (MIM 136350),5–7 and GNRHR (MIM 138850)8,9 genes being most common. A variety of other genes also cause IHH/KS in some patients, including GPR54 (MIM 604161),10 NR0B1 (MIM 300473),11 PROKR2 (MIM 607123),12 PROK2 (MIM 607002),12 LEP (MIM 164160),2 and LEPR (MIM 601007).2 A digenic inheritance pattern has been reported in two cases.13 To date, KAL1 mutations cause only KS,2 whereas GNRHR mutations are restricted to normosmic IHH.14 Only mutations in FGFR1 are known to cause both normosmic IHH and KS.
Heterozygous CHD7 (chromodomain helicase DNA-binding protein 7, MIM 608892) mutations have been identified in ∼60%–70% of patients with CHARGE syndrome (MIM 214800), a multisystem autosomal-dominant or sporadic disorder consisting of eye _c_oloboma, _h_eart defects, choanal _a_tresia, _r_etardation of growth and development, _g_enito-urinary anomalies, and _e_ar abnormalities (vestibular and auditory).15 The large 2997 amino acid (AA) CHD7 protein contains two chromodomains at its N terminus, followed by centrally located SNF2 and helicase domains; three conserved region (CR) domains; a switching-defective protein 3, adaptor 2, nuclear receptor corepressor, transcription factor IIIB (SANT) domain; two Brahma and Kismet (BRK) domains; and, at the C terminus, a leucine-zipper domain, which we identified by using PSORTII prediction software (Figure 1A). The leucine-zipper domain is located in AA 2888–2909 (LAFNPFLLSTMAPGLFYPSMFL). CHD7 belongs to a family of nine CHD proteins that have in common the ability to utilize ATP hydrolysis to alter nucleosome structure.16 Chromodomains have been thought to mediate chromatin interactions and have been found to interact with DNA, RNA, and histone targets.17
Figure 1.
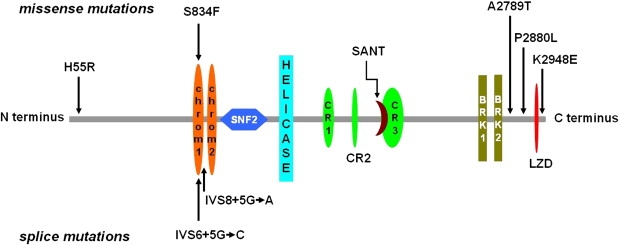
CHD7 Domains and Positions of Mutations
CHD7 structure with functional domains and the positions of five missense mutations and two splice-donor site mutations identified in IHH and KS patients are shown. The following abbreviations are used: chrom1, chromodomain 1; chrom2, chromodomain 2; SANT, SANT DNA binding domain; and LZD, leucine zipper domain. The function of CR1-CR3 and BRK domains are unknown. Three mutations affect chromodomains. Relative sizes and locations of domains are to scale.
CHD7 is expressed in the disease-associated organs of CHARGE syndrome, but also in KS-relevant tissues including the olfactory epithelium18 and pituitary19 in mice, as well as the olfactory nerve and bulb, hypothalamus, and pituitary in humans.20 Prior to the identification of CHD7, hypogonadism was reported to be associated with CHARGE syndrome, including some patients old enough to be diagnosed with IHH, as defined below.21 Several other studies implicate a possible connection between the KS phenotype and CHARGE syndrome, although it is important to emphasize that these patients with CHARGE syndrome are not yet of pubertal age. Anosmia or hyposmia has been identified in children with CHARGE syndrome,22 as well abnormal olfactory bulbs by magnetic resonance imaging (MRI).23 Most boys studied had micropenis and/or cryptorchidism suggestive of IHH, although all subjects were prepubertal.23 None of seven females under 12 years of age began puberty spontaneously, and hormonal data suggested that IHH could ensue as they got older.23 In fact, recently one female with CHARGE syndrome due to a CHD7 mutation and with some features of KS has been reported.24 However, there has been no systematic evaluation of IHH/KS patients without a diagnosis of CHARGE syndrome for the presence of CHD7 mutations. We hypothesized that IHH/KS could be a milder allelic variant of CHARGE syndrome.
We first looked at Chd7 mRNA expression in three different mouse GnRH neuronal cell lines, two migratory (GN11 and NLT)25 and one postmigratory (GT1-7).26 _Chd7_-mRNA expression was confirmed by cloning and sequencing of the PCR products (not shown). Next, RT-PCR on RNA extracted from KS-relevant rat tissues demonstrated expression in the olfactory bulb, medial basal hypothalamus, and pituitary (Figure S1 available online). Therefore, we undertook an extensive mutation analysis in IHH/KS patients without clinical features of CHARGE syndrome.
IHH was defined as the absence of puberty in females ≥ 17 yr old and in males ≥ 18 yr old with low serum gonadotropins as described previously.1 Males had low serum testosterone (<100 ng/dL, with normal being 300–1100), and females had hypoestrogenic amenorrhea, and remaining pituitary function and CNS imaging by MRI or CT were normal.1 KS was defined as IHH and anosmia or hyposmia, as detailed by the University of Pennsylvania Smell Identification Test when possible or by patient history. This study was approved by the Human Assurance Committee (Medical College of Georgia), and signed written consent was obtained from all participants. Previously, mutation screening was performed in most patients for KAL1, GNRHR, and FGFR1 genes.14,27–30
DNA sequencing of the entire 37 protein-coding exons and splice junctions of the CHD7 gene from 101 affected individuals (50 with KS and 51 with normosmic IHH) revealed six heterozygous mutations—one splice-donor site alteration and five missense mutations for a prevalence of 6% (Table 1; Figures 1 and 2; Figure S2). Another heterozygous splice-donor site mutation was identified in one of 96 additional IHH/KS patients screened for mutations only in exons 6–10. This gene region was selected for a focused mutation screen because these exons encode the highly conserved and important functional chromodomains (Figure 1). All cases with mutations were sporadic, and none of the probands had any affected family members or relatives. No identical nucleotide changes were identified in 180 controls or listed in the single-nucleotide polymorphism (SNP) database. In addition, the two CHD7 mutations identified in probands of Turkish descent were also absent in 96 Turkish controls. Six new SNPs (Table S1, submitted into the SNP database in NCBI) in the CHD7 were also detected. None of the patients with a CHD7 mutation were known to possess a mutation in another gene involved in IHH/KS.
Table 1.
CHD7 Mutations in Sporadic Patients with IHH/KS
| Patient | Gender and Phenotype | Geographic Origin | Exon or Intron | Nucleotide Change | Amino Acid Change | Confirmatory Method |
|---|---|---|---|---|---|---|
| TT20 | Male, IHH; no other anomalies | Turkey | Intron 8 | IVS8+5G→A | Premature termination | 0/180 controls & 0/96 Turkish controls |
| C187 | Female, KS, cleft lip and palate, hearing loss | USA | Intron 6 | IVS6+5G→C | 22 amino acid deletion | De novo, 0/180 controls, and 0/96 Turkish controls |
| C59 | Male, IHH and cleft lip; cryptorchidism | USA | Exon 8 | c.2501C→T | Ser834Phe | 0/180 controls |
| C148 | Male, KS; no other anomalies | USA | Exon 2 | c.164A→G | His55Arg | 0/180 controls |
| T47 | Male, IHH, myopia; no other anomalies | Turkey | Exon 38 | c.8365G→A | Ala2789Thr | 0/180 controls and 0/96 Turkish controls |
| C137 | Male, IHH; cryptorchidism | USA | Exon 38 | c.8639C→T | Pro2880Leu | 0/180 controls and 0/96 Turkish controls |
| C26 | Male, KS; no other anomalies | USA | Exon 38 | c.8842A→G | Lys2948Glu | 0/180 controls |
Figure 2.
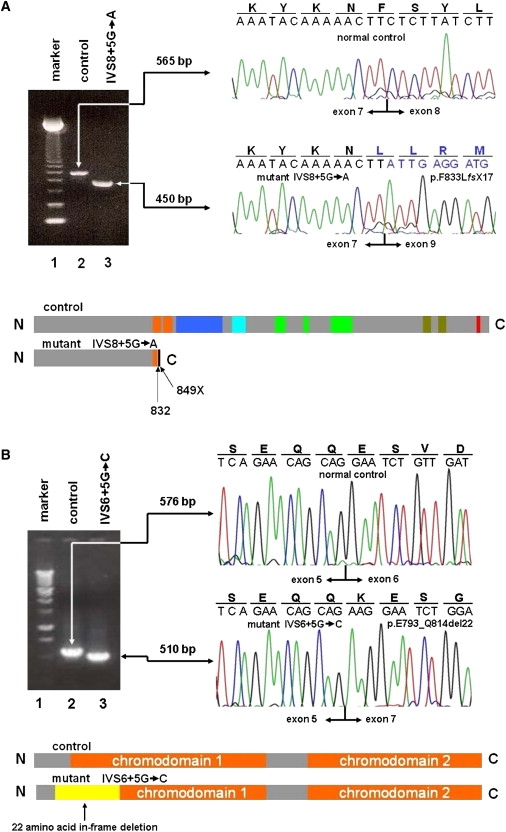
CHD7 Exon Skipping in Two Patients
(A) RT-PCR analysis of CHD7 exon-skipping event in a patient with IVS8+5G→A RT-PCR analysis confirms that the IVS8+5G→A mutation of CHD7 causes aberrant exon 8 skipping in an IHH patient. Splicing patterns are compared between a normal control and the mutant by cloned cDNA sequencing. An expected CHD7 product of 565 bp consisting of exons 4–10 is observed in the control (lane 2), whereas an abnormal product of 450 bp skipping exon 8 (115 bp) is observed in a patient with IVS8+5G→A (lane 3). Exon 9 nucleotide and out-of-frame AA sequence are depicted in blue. A 123 bp DNA marker is shown in the first lane.
The aberrant exclusion of exon 8 (115 bp) in a patient with IVS8+5G→A is predicted to introduce a frameshift in the coding region and a subsequent premature termination codon at 16 AAs downstream from exon 8 skipping. In the truncated CHD7, 16 out-of-frame AA residues generated by the frameshift are depicted as a black bar at the end. The functional domains from N terminus to C terminus are depicted in color as follows: orange, chromodomains 1 and 2; navy blue, SNF2; sky blue, helicase; green, CR1, CR2, and CR3; deep green, BRK1 and BRK2; and red, leucine zipper domain. The SANT domain within CR3 was not depicted here.
(B) RT-PCR analysis of CHD7 exon skipping event in a patient with IVS6+5G→C RT-PCR analysis shows that mutation IVS6+5G→C of CHD7 causes aberrant exon 6 skipping in a KS patient. Splicing patterns are compared between a normal control and the mutant by cloned cDNA sequencing. An expected CHD7 product of 576 bp consisting of exons 4–9 is observed in the control (lane 2), whereas an abnormal product of 510 bp skipping exon 6 (66 bp) is observed in a patient with IVS6+5G→C (lane 3). A 1 kb DNA marker is shown in the first lane.
The abnormal exon 6 (66 bp) skipping in a patient with IVS6+5G→C causes a 66 bp in-frame deletion of 22 AAs. From chromodomain 1 comprising 66 AAs, 16 residues are deleted. Protein structure of the deleted region is depicted in yellow and chromodomains 1 and 2 in orange.
Both intronic mutations impaired mRNA splicing in lymphoblasts of affected individuals and are predicted to interfere with protein function. An IVS8+5G→A mutation identified in a normosmic IHH male without any other anomalies resulted in exon 8 skipping, as determined by RT-PCR of CHD7 exons 4–10 from patient and control lymphoblastoid RNA, subsequent subcloning, and DNA sequencing. This mutant introduces 16 aberrant out-of-frame AA residues, causing a frameshift and subsequent premature termination codon at residue 849 located 49 bp downstream of the junction of exons 7 and 9. This results in a CHD7 protein predicted to be truncated more than 70% of the C terminus (Figure 2A). This transcript is not a normal splice variant because it was absent in four control lymphoblastoid cell lines, and nor was the same G→A transition identified in 180 normal controls. Importantly, this mutation removes about half of the first and all of the second chromodomains, as well as other important domains of the protein, thereby predicting a nonfunctional protein.
In a KS female with mild sensorineural deafness and cleft lip and palate, a de novo heterozygous intronic transversion, IVS6+5G→C, was identified. The mutation was absent in both parents. For direct examination of whether this variant affects splicing, CHD7 exons 4–9 were similarly analyzed by RT-PCR. In addition to the normally spliced transcript, a transcript with reduced size demonstrated exon 6 skipping not observed in four controls (Figure 2B). This results in an in-frame deletion of 22 of 66 AAs of chromodomain 1. Chromodomains are evolutionarily conserved16 and known to interact with histone tails.31,32 Interestingly, this mutation has also been reported in a patient with CHARGE syndrome.33 The patient with CHARGE syndrome has additional severe phenotypic findings, including the absence of earlobes, triangular concha, vestibular disturbance, autism-spectrum disorder, and mental retardation,33 which are absent in our KS patient.
The five missense mutations affect highly conserved AA residues when compared with known CHD7 orthologs (Figure 3A). Four of five point mutations were predicted to be deleterious by SIFT34 (Ser834Phe, Lys2948Glu, Pro2880Leu, and His55Arg), whereas Ala2789Thr was tolerated. Ser834Phe, located in DNA-binding chromodomain 1 (UniProtKB/Swiss-Prot entry Q9P2D1; INTERPRO IPR000953),35,36 a highly conserved sequence motif observed in a variety of animal and plant species, had the highest confidence measure. Further supportive evidence of the deleterious effect of Ser834Phe comes from the report of the same mutation in three patients from a family with a severe CHARGE syndrome phenotype.37
Figure 3.
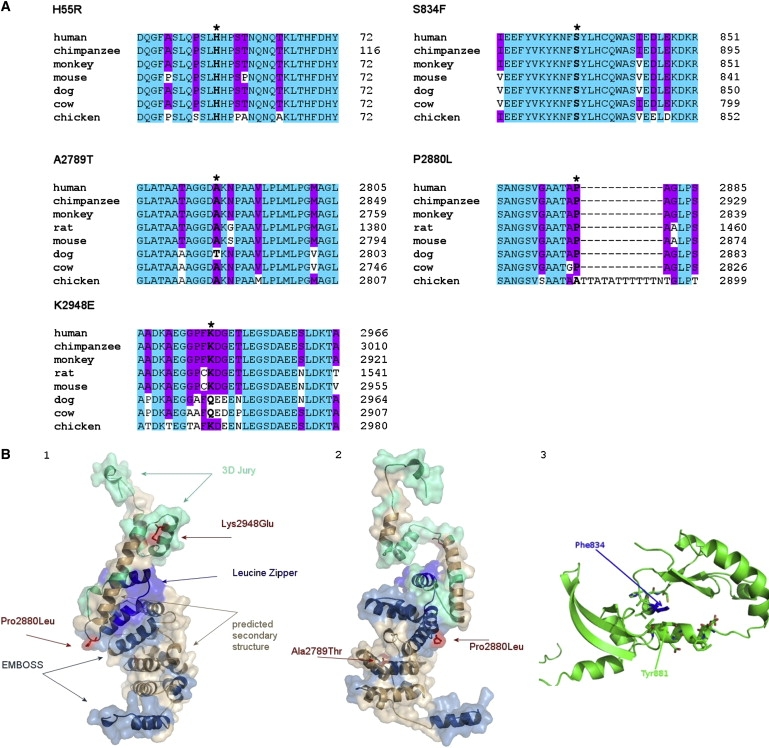
Protein Sequence Alignment and Structure Modeling of CHD7
(A) Multiple protein-sequence alignment of CHD7 with its orthologs. The positions of residues affected by missense mutations in IHH/KS patients are marked by asterisks and bold letters in available CHD7 animal orthologs. H55 and S834 are evolutionarily fully conserved, whereas A2789 and P2880 are highly conserved. K2948 shows relative conservation. Human CHD7 N-terminal residues 1–1423 are missing in the predicted rat Chd7 protein (NP_001101376). Blue shading represents the invariant residues that match the consensus exactly, and pink shading shows partial matching.
(B) CHD7 structure modeling.
(B1 and B2) Shown are alternate views of the model of the 300 AA C-terminal region of CHD7 based on the 3DJURY model, which results from the alignment of this region with that of mdia1 gbd-fh3 in complex with rhoc (1z2c/B). Regions of the model that were predicted solely on the basis of homology in 3DJURY are shown in green, but additional information was used to substantiate the model. There is an additional long consecutive region where EMBOSS found substantial sequence similarity to the CHD7 sequence—these regions are marked in dark blue. Regions that, in addition to the alignment, agree in their secondary structure with the consensus prediction of PHYRE are shown in light brown. Finally, we found a region that, in addition to the alignment and secondary structure, exhibits a motif commensurate with the leucine zipper (bright blue). Sites and side chains of the three mutations Lys2948Glu, Pro2880Leu, and Ala2789Thr are shown in red for better visibility. All mutation sites lie in highly flexible loop regions. (B1) and (B2) show the same model rotated 180° around the vertical axis though the center of the molecule.
(B3) Shown is the model of the 155 AA region around Ser834Phe (AA 795–950). The side chain of Phe834 is highlighted in dark blue for better visibility. In addition to the secondary and tertiary structure, side chains of the AA in a 6 Å radius of Phe834 are shown in the standard color coding (carbon green, oxygen red, sulfur yellow). Of particular importance is the strong stacking interaction of Phe834 (blue) with the ring of Tyr881 (green) directly underneath.
Structural models for the relevant domains suggest that Ala2789Thr and Pro2880Leu, located in the spacer sequence between the BRK2 and leucine-zipper regions, as well as Lys2948Glu, are also detrimental. All three AA residues are located in loop regions so that mutations of these residues will most likely affect structural and binding properties of the domains to their interaction partners (Figures 3B1 and 3B2). The local secondary structure of the region around Pro2880 is a random coil, and the Pro2880Leu mutation induces helix formation in this region, predicting its deleterious effect (data not shown). The Ser834 residue is also located in a loop region, coordinating strongly with adjacent residues in the neighboring helix (Tyr881, context PDYV), so Phe834 will therefore strongly affect protein stability (Figure 3B3). The Ala2789T residue is conserved in seven of eight orthologs, and Lys2948 is relatively conserved (Figure 3A).
SIFT was also used to characterize the functional significance of a 22 amino acid (ESVDAEGPVVEKIMSSRSVKKQ) in-frame deletion of exon 6 (IVS6+5G→C mutant) by predicting deleterious effects for almost all possible AA substitutions in this region (data not shown). The splice mutant IVS8+5G→A truncates the functional part of the protein starting at AA 810, including the region from AAs 920–1490. The latter is highly homologous to the SWI2/SNF2 chromatin-remodeling domain of eukaryotic Rad54 (PDB code: 1Z3I)38 and therefore an essential DNA-binding domain of the wild-type. Collectively, involvement of the conserved chromodomain by three mutations, two of which have been identified in patients with CHARGE syndrome,33,37 as well as SIFT AA conservation and protein structural analysis, indicates that these nucleotide alterations are pathogenic mutations.
To investigate developmental expression of Chd7 further in the mouse embryo, in situ hybridization analysis was performed from embryonic day 10.5 (E10.5) to postnatal day 0 (P0). Already by E10.5 and E11, high Chd7 expression was preferentially observed in the developing nervous system and its derivatives. The entire neuroepithelium was strongly labeled at that stage (Figures 4A–4C). At E10.5 and E11, high levels of expression were particularly noteworthy in the olfactory placode (Figures 4A and 4C), which gives rise to olfactory and GnRH neurons. With progressing differentiation, the label intensity declined and the signal became restricted to specific locations in the developing central and peripheral nervous systems. At E14, high signal intensity was seen in the olfactory epithelium, cochlea, anterior pituitary, and the spinal cord (Figures 4D and 4E). This pattern of expression is consistent with involvement of Chd7 in the development of the olfactory pathway and the GnRH-positive neurons.39 In the adult brain, intense Chd7 expression was restricted to the granule cell layer of the cerebellum (Figure 4H), hippocampal formation (Figure 4I), and hypothalamus (Figures 4F and 4G). Signals were absent when the sense control was used.
Figure 4.
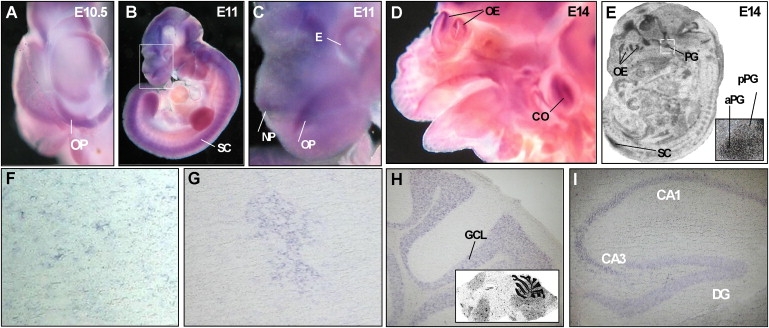
Chd7 Expression during Murine Development
The following abbreviations are used: OP, olfactory placode; GCL, granule cell layer of the cerebellum; OE, olfactory epithelium; aPG, anterior pituitary gland; pPG, posterior pituitary gland; SC, spinal cord; CA1, CA1 region of the hippocampus; CA3, CA3 region of the hippocampus; DG, dentate gyrus; CO, cochlea; E, eye; and NP, nasal pit.
(A) DIG-labeled whole-mount in situ hybridization using a Chd7 antisense probe shows high expression in the olfactory placode at E10.5.
(B and C) Strong labeling of neuroepithelial structures is seen at E11, shown in higher magnification in (C).
(D and E) Expression of Chd7 in the olfactory epithelium, developing cortex, cochlea, spinal cord, and anterior pituitary gland at E14. [35S]-UTP-labeled in situ hybridizations are shown in (E). Inset shows magnification of the pituitary gland.
(F–I) Expression of Chd7 in the adult brain: Singular DIG-positive cells are found scattered within hypothalamic nuclei of the preoptic area (F). One cluster of cells within the medial preoptic area consistently stained positively for Chd7 (G). DIG-labeled in situ hybridizations on 12 μm cryosections show expression in the cerebellum (H). The inset in (H) shows an overview of the adult brain with a [35S]-UTP labeled Chd7 antisense probe. The hippocampal region also stains positive for Chd7 (I).
The molecular basis for 70%–75% of IHH/KS patients remains unknown.2 To date, only FGFR1 mutations have been reported to cause either normosmic IHH families or KS families.7 Although a homozygous PROKR2 deletion was seen in a single family comprising both normosmic and anosmic patients, this represents variable expressivity within the same family.40 In our case, we have three unrelated probands with KS and four unrelated probands with IHH with CHD7 mutations, demonstrating that CHD7 is involved in either IHH or KS. We present new evidence for a role of CHD7 in the pathophysiology of both normosmic IHH and KS patients without a CHARGE phenotype. We first demonstrate Chd7 mRNA expression in both migratory and postmigratory GnRH neuronal cell lines. We also document mRNA expression in the hypothalamus, pituitary, and olfactory bulb in the rat—all of which are IHH/KS-relevant organs. By in situ hybridization, we confirm mRNA expression at the appropriate time when GnRH and olfactory neurons migrate from the olfactory placode region to the hypothalamus. This begins on E10–E11 in the mouse and is virtually completed by E18.5.39 In humans, GnRH migration begins at E5.5–E6.5 weeks and finishes 6–8 weeks postnatally.39 Whether or not CHD7 affects this important developmental neuron migration requires future study.
Furthermore, we demonstrate sporadic heterozygous CHD7 mutations, which were not present in ≥ 180 controls or the SNP database, in humans with both normosmic IHH and anosmic IHH (KS). Additionally, two mutations in Turkish patients were also absent in 96 Turkish controls (for a total of 276 controls). The prevalence of CHD7 mutations of ∼6% is similar to that of apparently sporadic KAL1 mutations2 and somewhat less than the 10% reported rate of FGFR1 mutations in IHH/KS.7 Two of our CHD7 mutations alter mRNA splicing in lymphoblast RNA and predict deletions of chromodomains, whereas another missense mutation within chromodomain 1 affects a highly conserved Ser residue. The disruption of these important evolutionarily conserved chromodomains is highly likely to result in deleterious consequences,16 because chromodomains are known to interact with histone tails.31,32 Chromodomain deletion has also been reported to impair nucleosome binding and remodeling by CHD proteins,41 indicating that their disruption will be detrimental to CHD7 function.
Further supportive evidence that our CHD7 mutations are deleterious comes from SIFT analysis, which indicates that four of the five missense mutations (Ser834Phe, Lys2948Glu, Pro2880Leu, and His55Arg) involve highly conserved AA residues among known species and, therefore, are not likely to be tolerated by their observed substitutions.34 These findings were also corroborated by protein structural analysis of the AA variants Ala2789Thr, Pro2880Leu, and Lys2948Glu, which were predicted to alter structural and binding properties of the domains. Taken together, both AA conservation and protein structural modeling provide additional support that these missense substitutions are highly likely to be deleterious mutations.
Importantly, our one IHH and one KS patient, who both lack the CHARGE phenotype, possess the same mutations (Ser834Phe and IVS6+5G→C) reported previously in patients with CHARGE syndrome,33,37 further demonstrating the allelic relationship of both syndromes. The KS patient with the IVS6+5G→C mutation does not fulfill Blake's criteria for CHARGE syndrome,42 although she does have hearing impairment and cleft lip and palate. This also indicates that the effects of modifying genes may determine whether the patient has the more severe CHARGE phenotype rather than the milder IHH/KS phenotype. Interestingly, our mutations have been localized to regions around four exons—2, 6, 8, and 38—suggesting the possibility of hotspots for IHH/KS mutation. Because of these findings and the absence of nonsense mutations, which often occur in CHARGE syndrome,15 we provide the first convincing evidence that IHH/KS represents a milder allelic variant of CHARGE syndrome. Although its precise function is uncertain, CHD7 appears to be important in GnRH and olfactory neuron migration to their embryologic destination in the hypothalamus. CHD7 is the first chromatin-remodeling protein involved in normal puberty in humans and is the second gene (after FGFR1) identified that results in both normosmic IHH and KS.
Acknowledgments
We thank all affected individuals and their families for their cooperation. The authors also thank I. Hermans-Borgmeyer for unrestricted support with the in situ hybridizations and interpretation of the data; B. Dierkes for excellent technical assistance; G. Schnitzler for helpful discussions; E. Moutevelis for leucine-zipper-domain structure analysis; the Computational Science Center, KIST (Seoul, Korea), and the volunteers from POEM@HOME for computational resources; Y. Shen for RT-MLPA; S. West for zinc-finger prediction analysis; L. Chorich for her efforts in supervising and running the laboratory; and E. Shah for RT-PCR analysis.
We also acknowledge support to L.C.L. from National Institutes of Health (NIH) grants HD33004 and HD040287, as well as the Medical College of Georgia Research Institute (MCGRI), Dean D.D. Miller, Institute of Molecular Medicine and Genetics (IMMAG) director R. Yu, and ob/gyn chair A.A. Murphy at MCG. J.F.G. has support from NIH grant GM061354, W.W. from Deutsche Forschungsgemeinschaft (DFG) grant WE1863/10-2 and the DFG Center for Functional Nanostructures, and I.M. from the EU Leonardo program “Unipharm Graduates.”
Supplemental Data
Document S1. Two Figures and Two Tables
Web Resources
The URLs for data presented herein are as follows:
- dbSNP, http://www.ncbi.nlm.nih.gov/SNP/
- NCBI and GenBank, http://www.ncbi.nlm.nih.gov/
- Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/
- PSORTII, http://psort.hgc.jp/form2.html
- SIFT, http://blocks.fhcrc.org/sift/SIFT.html
References
- 1.Bhagavath B., Podolsky R.H., Ozata M., Bolu E., Bick D.P., Kulharya A., Sherins R.J., Layman L.C. Clinical and molecular characterization of a large sample of patients with hypogonadotropic hypogonadism. Fertil. Steril. 2006;85:706–713. doi: 10.1016/j.fertnstert.2005.08.044. [DOI] [PubMed] [Google Scholar]
- 2.Kim H.G., Bhagavath B., Layman L.C. Clinical Manifestations of Impaired GnRH Neuron Development and Function. Neurosignals. 2008;16:165–182. doi: 10.1159/000111561. [DOI] [PubMed] [Google Scholar]
- 3.Franco B., Guioli S., Pragliola A., Incerti B., Bardoni B., Tonlorenzi R., Carrozzo R., Maestrini E., Pieretti M., Taillon-Miller P. A gene deleted in Kallmann's syndrome shares homology with neural cell adhesion and axonal path-finding molecules. Nature. 1991;353:529–536. doi: 10.1038/353529a0. [DOI] [PubMed] [Google Scholar]
- 4.Legouis R., Hardelin J.P., Levilliers J., Claverie J.M., Compain S., Wunderle V., Millasseau P., Le Paslier D., Cohen D., Caterina D. The candidate gene for the X-linked Kallmann syndrome encodes a protein related to adhesion molecules. Cell. 1991;67:423–435. doi: 10.1016/0092-8674(91)90193-3. [DOI] [PubMed] [Google Scholar]
- 5.Dode C., Levilliers J., Dupont J.M., De Paepe A., Le Du N., Soussi-Yanicostas N., Coimbra R.S., Delmaghani S., Compain-Nouaille S., Baverel F. Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet. 2003;33:463–465. doi: 10.1038/ng1122. [DOI] [PubMed] [Google Scholar]
- 6.Kim H.G., Herrick S.R., Lemyre E., Kishikawa S., Salisz J.A., Seminara S., MacDonald M.E., Bruns G.A., Morton C.C., Quade B.J. Hypogonadotropic hypogonadism and cleft lip and palate caused by a balanced translocation producing haploinsufficiency for FGFR1. J. Med. Genet. 2005;42:666–672. doi: 10.1136/jmg.2004.026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pitteloud N., Acierno J.S., Jr., Meysing A., Eliseenkova A.V., Ma J., Ibrahimi O.A., Metzger D.L., Hayes F.J., Dwyer A.A., Hughes V.A. Mutations in fibroblast growth factor receptor 1 cause both Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA. 2006;103:6281–6286. doi: 10.1073/pnas.0600962103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Roux N., Young J., Misrahi M., Genet R., Chanson P., Schaison G., Milgrom E. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N. Engl. J. Med. 1997;337:1597–1602. doi: 10.1056/NEJM199711273372205. [DOI] [PubMed] [Google Scholar]
- 9.Layman L.C., Cohen D.P., Jin M., Xie J., Li Z., Reindollar R.H., Bolbolan S., Bick D.P., Sherins R.J., Duck L.W. Mutations in the gonadotropin-releasing hormone receptor gene cause hypogonadotropic hypogonadism. Nat. Genet. 1998;18:14–15. doi: 10.1038/ng0198-14. [DOI] [PubMed] [Google Scholar]
- 10.Seminara S.B., Messager S., Chatzidaki E.E., Thresher R.R., Acierno J.S., Jr., Shagoury J.K., Bo-Abbas Y., Kuohung W., Schwinof K.M., Hendrick A.G. The GPR54 gene as a regulator of puberty. N. Engl. J. Med. 2003;349:1614–1627. doi: 10.1056/NEJMoa035322. [DOI] [PubMed] [Google Scholar]
- 11.Muscatelli F., Strom T.M., Walker A.P., Zanaria E., Recan D., Meindl A., Bardoni B., Guioli S., Zehetner G., Rabl W. Mutations in the DAX-1 gene give rise to both X-linked adrenal hypoplasia congenita and hypogonadotropic hypogonadism. Nature. 1994;372:672–676. doi: 10.1038/372672a0. [DOI] [PubMed] [Google Scholar]
- 12.Dode C., Teixeira L., Levilliers J., Fouveaut C., Bouchard P., Kottler M.L., Lespinasse J., Lienhardt-Roussie A., Mathieu M., Moerman A. Kallmann syndrome: Mutations in the genes encoding prokineticin-2 and prokineticin receptor-2. PLoS Genet. 2006;2:e175. doi: 10.1371/journal.pgen.0020175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pitteloud N., Quinton R., Pearce S., Raivio T., Acierno J., Dwyer A., Plummer L., Hughes V., Seminara S., Cheng Y.Z. Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J. Clin. Invest. 2007;117:457–463. doi: 10.1172/JCI29884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bhagavath B., Ozata M., Ozdemir I.C., Bolu E., Bick D.P., Sherins R.J., Layman L.C. The prevalence of gonadotropin-releasing hormone receptor mutations in a large cohort of patients with hypogonadotropic hypogonadism. Fertil. Steril. 2005;84:951–957. doi: 10.1016/j.fertnstert.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 15.Vissers L.E., van Ravenswaaij C.M., Admiraal R., Hurst J.A., de Vries B.B., Janssen I.M., van der Vliet W.A., Huys E.H., de Jong P.J., Hamel B.C. Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 2004;36:955–957. doi: 10.1038/ng1407. [DOI] [PubMed] [Google Scholar]
- 16.Marfella C.G., Imbalzano A.N. The Chd family of chromatin remodelers. Mutat. Res. 2007;618:30–40. doi: 10.1016/j.mrfmmm.2006.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brehm A., Tufteland K.R., Aasland R., Becker P.B. The many colours of chromodomains. Bioessays. 2004;26:133–140. doi: 10.1002/bies.10392. [DOI] [PubMed] [Google Scholar]
- 18.Bosman E.A., Penn A.C., Ambrose J.C., Kettleborough R., Stemple D.L., Steel K.P. Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum. Mol. Genet. 2005;14:3463–3476. doi: 10.1093/hmg/ddi375. [DOI] [PubMed] [Google Scholar]
- 19.Hurd E.A., Capers P.L., Blauwkamp M.N., Adams M.E., Raphael Y., Poucher H.K., Martin D.M. Loss of Chd7 function in gene-trapped reporter mice is embryonic lethal and associated with severe defects in multiple developing tissues. Mamm. Genome. 2007;18:94–104. doi: 10.1007/s00335-006-0107-6. [DOI] [PubMed] [Google Scholar]
- 20.Sanlaville D., Etchevers H.C., Gonzales M., Martinovic J., Clement-Ziza M., Delezoide A.L., Aubry M.C., Pelet A., Chemouny S., Cruaud C. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J. Med. Genet. 2006;43:211–217. doi: 10.1136/jmg.2005.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wheeler P.G., Quigley C.A., Sadeghi-Nejad A., Weaver D.D. Hypogonadism and CHARGE association. Am. J. Med. Genet. 2000;94:228–231. doi: 10.1002/1096-8628(20000918)94:3<228::aid-ajmg8>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 22.Chalouhi C., Faulcon P., Le Bihan C., Hertz-Pannier L., Bonfils P., Abadie V. Olfactory evaluation in children: Application to the CHARGE syndrome. Pediatrics. 2005;116:e81–e88. doi: 10.1542/peds.2004-1970. [DOI] [PubMed] [Google Scholar]
- 23.Pinto G., Abadie V., Mesnage R., Blustajn J., Cabrol S., Amiel J., Hertz-Pannier L., Bertrand A.M., Lyonnet S., Rappaport R. CHARGE syndrome includes hypogonadotropic hypogonadism and abnormal olfactory bulb development. J. Clin. Endocrinol. Metab. 2005;90:5621–5626. doi: 10.1210/jc.2004-2474. [DOI] [PubMed] [Google Scholar]
- 24.Ogata T., Fujiwara I., Ogawa E., Sato N., Udaka T., Kosaki K. Kallmann syndrome phenotype in a female patient with CHARGE syndrome and CHD7 mutation. Endocr. J. 2006;53:741–743. doi: 10.1507/endocrj.k06-099. [DOI] [PubMed] [Google Scholar]
- 25.Radovick S., Wray S., Lee E., Nicols D.K., Nakayama Y., Weintraub B.D., Westphal H., Cutler G.B., Jr., Wondisford F.E. Migratory arrest of gonadotropin-releasing hormone neurons in transgenic mice. Proc. Natl. Acad. Sci. USA. 1991;88:3402–3406. doi: 10.1073/pnas.88.8.3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mellon P.L., Windle J.J., Goldsmith P.C., Padula C.A., Roberts J.L., Weiner R.I. Immortalization of hypothalamic GnRH neurons by genetically targeted tumorigenesis. Neuron. 1990;5:1–10. doi: 10.1016/0896-6273(90)90028-e. [DOI] [PubMed] [Google Scholar]
- 27.Bhagavath B., Xu N., Ozata M., Rosenfield R.L., Bick D.P., Sherins R.J., Layman L.C. KAL1 mutations are not a common cause of idiopathic hypogonadotrophic hypogonadism in humans. Mol. Hum. Reprod. 2007;13:165–170. doi: 10.1093/molehr/gal108. [DOI] [PubMed] [Google Scholar]
- 28.Pedersen-White J.R., Chorich L.P., Bick D.P., Sherins R.J., Layman L.C. The prevalence of intragenic deletions in patients with idiopathic hypogonadotropic hypogonadism and Kallmann syndrome. Mol. Hum. Reprod. 2008;14:367–370. doi: 10.1093/molehr/gan027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu N., Podolsky R.H., Chudgar P., Chorich L.P., Liu C., McDonough P.G., Warrington J.A., Layman L.C. Screening candidate genes for mutations in patients with hypogonadotropic hypogonadism using custom genome resequencing microarrays. Am. J. Obstet. Gynecol. 2005;192:1274–1282. doi: 10.1016/j.ajog.2004.12.066. [DOI] [PubMed] [Google Scholar]
- 30.Xu N., Qin Y., Reindollar R.H., Tho S.P., McDonough P.G., Layman L.C. A mutation in the fibroblast growth factor receptor 1 gene causes fully penetrant normosmic isolated hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab. 2007;92:1155–1158. doi: 10.1210/jc.2006-1183. [DOI] [PubMed] [Google Scholar]
- 31.Bannister A.J., Zegerman P., Partridge J.F., Miska E.A., Thomas J.O., Allshire R.C., Kouzarides T. Selective recognition of methylated lysine 9 on histone H3 by the HP1 chromo domain. Nature. 2001;410:120–124. doi: 10.1038/35065138. [DOI] [PubMed] [Google Scholar]
- 32.Flanagan J.F., Mi L.Z., Chruszcz M., Cymborowski M., Clines K.L., Kim Y., Minor W., Rastinejad F., Khorasanizadeh S. Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature. 2005;438:1181–1185. doi: 10.1038/nature04290. [DOI] [PubMed] [Google Scholar]
- 33.Jongmans M.C., Hoefsloot L.H., van der Donk K.P., Admiraal R.J., Magee A., van de Laar I., Hendriks Y., Verheij J.B., Walpole I., Brunner H.G. Familial CHARGE syndrome and the CHD7 gene: A recurrent missense mutation, intrafamilial recurrence and variability. Am. J. Med. Genet. A. 2008;146A:43–50. doi: 10.1002/ajmg.a.31921. [DOI] [PubMed] [Google Scholar]
- 34.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mulder N.J., Apweiler R., Attwood T.K., Bairoch A., Bateman A., Binns D., Bradley P., Bork P., Bucher P., Cerutti L. InterPro, progress and status in 2005. Nucleic Acids Res. 2005;33:D201–D205. doi: 10.1093/nar/gki106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu C.H., Apweiler R., Bairoch A., Natale D.A., Barker W.C., Boeckmann B., Ferro S., Gasteiger E., Huang H., Lopez R. The Universal Protein Resource (UniProt): An expanding universe of protein information. Nucleic Acids Res. 2006;34:D187–D191. doi: 10.1093/nar/gkj161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delahaye A., Sznajer Y., Lyonnet S., Elmaleh-Berges M., Delpierre I., Audollent S., Wiener-Vacher S., Mansbach A.L., Amiel J., Baumann C. Familial CHARGE syndrome because of CHD7 mutation: Clinical intra- and interfamilial variability. Clin. Genet. 2007;72:112–121. doi: 10.1111/j.1399-0004.2007.00821.x. [DOI] [PubMed] [Google Scholar]
- 38.Thoma N.H., Czyzewski B.K., Alexeev A.A., Mazin A.V., Kowalczykowski S.C., Pavletich N.P. Structure of the SWI2/SNF2 chromatin-remodeling domain of eukaryotic Rad54. Nat. Struct. Mol. Biol. 2005;12:350–356. doi: 10.1038/nsmb919. [DOI] [PubMed] [Google Scholar]
- 39.Schwarting G.A., Wierman M.E., Tobet S.A. Gonadotropin-releasing hormone neuronal migration. Semin. Reprod. Med. 2007;25:305–312. doi: 10.1055/s-2007-984736. [DOI] [PubMed] [Google Scholar]
- 40.Pitteloud N., Zhang C., Pignatelli D., Li J.D., Raivio T., Cole L.W., Plummer L., Jacobson-Dickman E.E., Mellon P.L., Zhou Q.Y. Loss-of-function mutation in the prokineticin 2 gene causes Kallmann syndrome and normosmic idiopathic hypogonadotropic hypogonadism. Proc. Natl. Acad. Sci. USA. 2007;104:17447–17452. doi: 10.1073/pnas.0707173104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bouazoune K., Mitterweger A., Langst G., Imhof A., Akhtar A., Becker P.B., Brehm A. The dMi-2 chromodomains are DNA binding modules important for ATP-dependent nucleosome mobilization. EMBO J. 2002;21:2430–2440. doi: 10.1093/emboj/21.10.2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Blake K.D., Salem-Hartshorne N., Daoud M.A., Gradstein J. Adolescent and adult issues in CHARGE syndrome. Clin. Pediatr. (Phila.) 2005;44:151–159. doi: 10.1177/000992280504400207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Two Figures and Two Tables