Linking Cell Cycle to Asymmetric Division: Aurora-A Phosphorylates the Par Complex to Regulate Numb Localization (original) (raw)
. Author manuscript; available in PMC: 2010 Nov 22.
SUMMARY
Drosophila neural precursor cells divide asymmetrically by segregating the Numb protein into one of the two daughter cells. Numb is uniformly cortical in interphase but assumes a polarized localization in mitosis. Here, we show that a phosphorylation cascade triggered by the activation of Aurora-A is responsible for the asymmetric localization of Numb in mitosis. Aurora-A phosphorylates Par-6, a regulatory subunit of atypical protein kinase C (aPKC). This activates aPKC, which initially phosphorylates Lethal (2) giant larvae (Lgl), a cytoskeletal protein that binds and inhibits aPKC during interphase. Phosphorylated Lgl is released from aPKC and thereby allows the PDZ domain protein Bazooka to enter the complex. This changes substrate specificity and allows aPKC to phosphorylate Numb and release the protein from one side of the cell cortex. Our data reveal a molecular mechanism for the asymmetric localization of Numb and show how cell polarity can be coupled to cell-cycle progression.
INTRODUCTION
Many different cell types are the products of asymmetric cell divisions, in which a cell divides into two daughters of distinct size, gene expression, or developmental potential (Doe, 2008; Gonczy, 2008; Knoblich, 2008). Sensory organ precursor (SOP) cells of the Drosophila peripheral nervous system divide asymmetrically into an anterior daughter, programmed to produce the internal cells of the sensory organ, and a posterior daughter, giving rise to the external cells. Similarly, in the Drosophila central nervous system, neuroblasts divide asymmetrically into a basal daughter, destined for differentiation, and an apical daughter, which retains neuroblast identity. To divide asymmetrically, these cells segregate protein determinants into one of the two daughter cells during mitosis.
The Drosophila protein Numb is the prototype of a segregating determinant. Numb is a membrane-associated protein that is uniformly distributed on the cell cortex and in the cytoplasm in interphase. In late prophase, Numb concentrates on the anterior cortex of SOP cells and on the basal cortex of neuroblasts, resulting in its segregation into only one of the two daughter cells (Rhyu et al., 1994). Because Numb acts as a repressor of Notch signaling, this causes unequal levels of Notch activity, which results in the establishment of distinct cell fates (Guo et al., 1996).
The molecular events leading to the asymmetric localization of cell fate determinants in mitosis are poorly understood. The Par complex, comprising the PDZ domain proteins Bazooka (Baz; Par-3 in other species) and Par-6 as well as atypical protein kinase C (aPKC), is localized to the posterior cell cortex of SOP cells and to the apical cortex of neuroblasts (Gonczy, 2008). A critical substrate for the Par complex is the cytoskeletal protein Lethal (2) giant larvae (Lgl) (Betschinger et al., 2003), but how the phosphorylation of Lgl by aPKC on one side of the cortex leads to the localization of Numb to the opposite side is unclear (Wirtz-Peitz and Knoblich, 2006).
Whereas the Par complex provides the spatial cue for Numb localization, it is unknown what triggers Numb to localize asymmetrically only in mitosis. A candidate for this is the mitotic kinase Aurora-A (AurA) since it is activated at the onset of mitosis and is required for Numb asymmetry (Berdnik and Knoblich, 2002; Lee et al., 2006a; Wang et al., 2006). AurA localizes to centrosomes and promotes centrosome maturation (Glover et al., 1995), but neither functional centrosomes nor intact microtubules are necessary for Numb localization (Berdnik and Knoblich, 2002). Therefore, the well-studied role of AurA at spindle poles cannot account for its function in cortical polarity, suggesting that novel substrates remain to be identified.
Here, we elucidate a molecular mechanism for the asymmetric localization of Numb in mitosis. We identify Par-6 as a cortical substrate for AurA and show that its phosphorylation triggers an exchange of Lgl for Baz in the Par complex. The remodeled complex binds and phosphorylates Numb on one side of the cell cortex. Since phosphorylated Numb is released from the cortex (Nishimura and Kaibuchi, 2007; Smith et al., 2007), these events restrict Numb into a cortical crescent on the opposite side.
RESULTS
AurA Activates aPKC by Phosphorylating Par-6
Numb is localized asymmetrically in mitosis in an AurA-dependent manner (Berdnik and Knoblich, 2002). As in aurA mutants, Numb is mislocalized around the cell cortex upon expression of Lgl3A, a nonphosphorylatable mutant in which the three aPKC phosphorylation sites are mutated to Ala (Betschinger et al., 2003). To test whether a failure to phosphorylate Lgl is the underlying defect in aurA mutants, we probed extracts from aurA mutant larval brains for phosphorylated Lgl (p-Lgl). Indeed, p-Lgl levels were reduced in brains homozygous for the hypomorphic allele _aurA_37 and were even lower in the more severe heteroallelic combination _aurA_37/Ac-3 (Figure 1A). Since aPKC is the Lgl kinase (Betschinger et al., 2003) (Figure S1A available online), we probed these mutants for levels of autophosphorylated aPKC (p-aPKC), which is a direct readout of kinase activity (Hirai and Chida, 2003). Whereas total aPKC levels were unchanged, p-aPKC levels were diminished (Figure 2D), demonstrating that aPKC activity is reduced in aurA mutants. We conclude that AurA activates aPKC, leading to the phosphorylation of Lgl.
Figure 1. AurA Phosphorylates the Par Complex to Activate aPKC.

(A) Lgl is underphosphorylated in aurA mutants. Larval brain extracts of the indicated genotypes were analyzed.
(B) AurA induces the phosphorylation of Lgl by activation of aPKC. Immunoprecipitate (IP) from _aurA_37/Ac-3 brains was incubated with ATP and recombinant AurA as indicated.
(C) ClustalW alignment of the AurA phosphorylation site (red) in Par-6 orthologs. Residues identical or similar to the Drosophila protein are colored magenta and blue, respectively. The bottom line shows the AurA consensus (Ferrari et al., 2005); 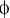 denotes any hydrophobic residue.
denotes any hydrophobic residue.
(D) Constructs used in the kinase assay.
(E) AurA phosphorylates Par-6 on Ser34 in vitro. Recombinant proteins were incubated with [32P]ATP and recombinant AurA as indicated. The gel was Coomassie-stained (CBB), followed by autoradiography (32P). The arrowhead indicates autophosphorylated AurA. Autoradiographs were quantified by summing the signal of the full-length bands and normalizing for the signal of Par-6WT (set to 1). Averages and standard deviations are shown (n = 4).
(F) AurA phosphorylates Par-6 on Ser34 in vivo. Phosphospecific antibodies directed against Ser34 were used to immunoprecipitate p-Par-6 from brains of the indicated genotypes. A second round (#2) of immunoprecipitation from the supernatant confirms the depletion of p-Par-6. To avoid the overlapping IgG signal and to control for the specificity of the antibodies, extracts from wild-type animals and from _par-6_Δ226 mutants complemented by a genomic rescue construct expressing Par-6-GFP (_par-6_GFP) were used.
Figure 2. Phosphorylation of Par-6 on Ser34 Regulates aPKC Activity and Numb Localization.

(A–C) Phosphorylation of Par-6 on Ser34 regulates the interaction of Par-6 with aPKC. Recombinant proteins were used to precipitate aPKC from larval brains. (A) The baits were preincubated with recombinant AurA and ATP as indicated. (B) Phosphomimetic forms of Par-6 (S34D/E) and a control mutant (K23A) defective for binding to aPKC (Noda et al., 2003) were used in the pull-down assays. (C) Par-6 interacts with aPKC through dimerization of the PB1 domains.
(D) Phosphorylation on Ser34 is required for full activity of aPKC toward Lgl. Brain extracts from par-6 mutants complemented by genomic rescue constructs expressing either Par-6WT or Par-6S34A were analyzed; aurA mutants were included as controls. (#1) and (#2) refer to independent insertions of the rescue construct. Western blots were quantified and normalized for Par-6WT (set to 1). Averages and standard deviations are shown (n = 3). Differences in p-aPKC and p-Lgl levels between _aurA_37/37 and _par-6_S34A brains are insignificant (p > 0.45 and p > 0.81, respectively).
(E–N) Phosphorylation on Ser34 is required for timely Numb localization. Larval neuroblasts of the indicated genotypes were stained for p-H3 to label DNA in mitotic cells and for Numb. Apical is up. Percentages shown for prometaphase cells include late prophase cells.
To ask whether AurA regulates aPKC in a direct or indirect manner, we examined the effect of AurA on the purified Par complex. We isolated the Par complex by immunoprecipitation of Par-6 from _aurA_37/Ac-3 brains and measured its activity by probing for p-Lgl after incubation with ATP. The Par complex from aurA mutants showed only marginal activity toward Lgl (Figure 1B). This was dramatically enhanced by the addition of recombinant AurA to the purified complex, although AurA did not itself phosphorylate the site recognized by p-Lgl antibodies (Figure S1B). Consistent with this, levels of p-aPKC were strongly increased after adding AurA (Figure 1B). Thus, direct phosphorylation of the Par complex by AurA stimulates aPKC activation.
To identify the AurA substrate in the Par complex, we performed in vitro kinase assays on recombinant Par-6 and aPKC. Both Par-6 and the catalytic domain of aPKC were phosphorylated by recombinant AurA (Figure S2A). Deletion analysis of aPKC mapped the phosphorylated region to a C-terminal fragment containing no conserved AurA phosphorylation sites (data not shown), suggesting that Par-6 rather than aPKC is the relevant target of AurA. We mapped the phosphorylated site in Par-6 to the PB1 domain (Figure S2B). This region contains one putative AurA phosphorylation site at Ser34, which is highly conserved (Figure 1C). Ala substitution of Ser34 suppressed phosphorylation in a manner similar to deletion of the PB1 domain (Figures 1D, 1E, and S2C), demonstrating that Ser34 is the main phosphorylation site for AurA in this domain of Par-6. To investigate Par-6 phosphorylation in vivo, we raised phosphospecific antibodies against Ser34. These antibodies recognized recombinant Par-6 in western blots and immunoprecipitations only after phosphorylation by recombinant AurA (Figure S1C). Phosphorylated Par-6 (p-Par-6) was only weakly detectable in total lysate and in total Par-6 immunoprecipitates (data not shown), suggesting that in vivo phosphorylation levels are low. Nevertheless, probing p-Par-6 immunoprecipitates for total Par-6 revealed that Par-6 phosphorylation was abolished in _aurA_37/Ac-3 mutants (Figure 1F). We conclude that Par-6 is phosphorylated on Ser34 in vivo in an AurA-dependent manner.
The PB1 domain of Par-6 dimerizes with the PB1 domain of aPKC (Noda et al., 2003)(Figure 2C). To test if Ser34 phosphorylation regulates this interaction, we performed pull-down assays from brain lysate with recombinant Par-6. Indeed, prior incubation of the Par-6 bait with recombinant AurA (Figure 2A) as well as phosphomimetic mutations of Ser34 (Figure 2B) suppressed the binding to endogenous aPKC. We conclude that AurA-dependent phosphorylation of Par-6 on Ser34 negatively regulates its physical interaction with aPKC.
Par-6 directly inhibits aPKC activity (Yamanaka et al., 2001), raising the possibility that AurA activates aPKC by dissociating it from Par-6. To test this, we analyzed par-6 mutant brains complemented by genomic rescue constructs expressing either wild-type (_par-6_WT) or nonphosphorylatable Par-6 in which Ser34 was substituted for Ala (_par-6_S34A). Indeed, levels of p-aPKC and p-Lgl were reduced in _par-6_S34A brains, comparable to levels seen in _aurA_37/37 brains (Figure 2D), demonstrating that phosphorylation of Ser34 accounts for most of the activity of AurA on aPKC. Consistent with this, Numb was mislocalized around the cell cortex in _par-6_S34A neuroblasts (Figures 2E–2J; Table S1), comparable to the pattern seen in _aurA_37/37 brains (Figures 2K–2N) and similar to the pattern seen upon expression of Lgl3A (Figure S5N). We conclude that phosphorylation of Par-6 on Ser34 is responsible for the timely localization of Numb in mitosis. Although Par-6 is a key phosphorylation target of AurA, the stronger phenotype of _aurA_37/Ac-3 mutants (Figures 2D and S5A; Table S1) suggests additional substrates for AurA.
aPKC Releases Lgl from the Cortex in Mitosis
The activation of aPKC by AurA implies that Lgl is phosphorylated in mitosis. Since Lgl is insoluble when unphosphorylated, but soluble when phosphorylated (Betschinger et al., 2003), the subcellular distribution of Lgl might reflect its phosphorylation status. We therefore imaged a functional Lgl-GFP fusion in SOP cells of living Drosophila pupae (Bellaiche et al., 2001a). Lgl-GFP was strongly localized to the cell cortex in interphase (data not shown) and at the onset of mitosis (Figure 3A; Movie S1). In prophase, Lgl-GFP was first released from the posterior cortex, giving rise to a transient cortical asymmetry at mid-prophase (Figure 3A, arrowheads). Cortical release continued until Lgl-GFP was completely cytoplasmic by nuclear envelope break-down (NEBD). Since aPKC is localized to the posterior cortex (Bellaiche et al., 2001b), the asymmetric localization of Lgl-GFP to the anterior suggested that aPKC phosphorylates Lgl, thereby releasing the protein from the posterior cortex. To test this, we coexpressed aPKCΔN, a constitutively active form of aPKC (Betschinger et al., 2003), which led to redistribution of Lgl-GFP into the cytoplasm throughout the cell cycle (Figure 3B; Movie S2). Conversely, the nonphosphorylatable Lgl3A-GFP remained strongly localized to the cell cortex throughout mitosis (Figure 3C; Movie S3). Finally, when the Par complex was relocalized to the anterior cortex by overexpression of Inscuteable (Bellaiche et al., 2001b), the cortical asymmetry of Lgl was inverted (Figure 3D, arrowheads; Movie S4). We conclude that aPKC phosphorylation first leads to a cortical asymmetry of Lgl, and then to the complete removal of Lgl from the cell cortex.
Figure 3. aPKC Releases Lgl from the Cortex in Mitosis.

(A–I) Lgl-GFP and Histone-RFP were coexpressed in pupal SOP cells. NEBD is t = 0. Anterior is oriented toward the left. (A) Lgl-GFP is released from the cortex in prophase, is asymmetrically localized (arrowheads) at mid-prophase, and returns to the cortex after mitosis. (B) Expression of aPKCDN redistributes Lgl-GFP from the cortex into the cytoplasm independent of the cell cycle. (C) Lgl3A-GFP remains cortical throughout mitosis. (D) Overexpression of Insc inverts Lgl-GFP asymmetry (arrowheads). (E) In _aurA_37/37 mutants, cortical release of Lgl-GFP is delayed. (F) Expression of aPKCΔN restores cortical release of Lgl-GFP in _aurA_37/37 mutants. (G) Lgl-GFP is released from the cortex prematurely when AurA is overexpressed. (H) Lgl3A-GFP is unaffected by AurA overexpression. (I) Lgl-GFP is released from the cortex in prophase in _baz_Xi106 clones.
To confirm that Lgl phosphorylation in prophase is initiated by AurA, we examined the localization of Lgl-GFP in aurA mutants. Indeed, in hypomorphic _aurA_37/37 mutants, Lgl-GFP remained on the cortex at NEBD and was fully released only in late prometaphase (Figure 3E; Movie S5). Conversely, overexpression of AurA caused a premature release of Lgl-GFP from the cortex (Figure 3G; Movie S6). Since expression of aPKCΔN in aurA mutants depleted the excess levels of cortical Lgl (Figure 3F), underphosphorylation of Lgl by aPKC is responsible for the delay in the cortical release of Lgl. Moreover, overexpression of AurA had no effect on Lgl3A-GFP (Figure 3H), demonstrating that AurA induces the cortical release of Lgl in a phosphorylation-dependent manner. We conclude that AurA is both necessary and sufficient to induce the cortical release of Lgl in prophase. It is likely that the late cortical release in aurA mutants is due to residual activity of the _aurA_37 allele.
AurA Regulates the Subunit Composition of the Par Complex
Our data reveal that AurA triggers the cortical release of Lgl, but how this leads to the asymmetric localization of Numb is unknown. In addition to AurA and the Lgl/Par-6/aPKC complex, Numb asymmetry requires Baz (Bellaiche et al., 2001b; Schober et al., 1999). In baz mutant SOP cells, Lgl-GFP was completely released from the cortex in prophase (Figure 3I), suggesting that Baz acts downstream of Lgl. To investigate this further, we examined the localization of Baz and Lgl by two-color live imaging. At the onset of mitosis, a functional Baz-GFP fusion protein (Benton and St Johnston, 2003) was localized to the posterior cortex at the level of the apical adherens junctions (Figure S3B) (Bellaiche et al., 2001b). In prophase, Baz-GFP redistributed to the posterior lateral cortex as Lgl-RFP was released from this side (Figure 4A). In aurA mutants, however, Baz-GFP failed to localize to the lateral cortex (Figure 4B) and instead was trapped at the level of the adherens junctions (Figure S3D). Since aurA mutants fail to release Lgl from the lateral cortex in prophase (Figure 3E), this suggests that Lgl inhibits the cortical localization of Baz.
Figure 4. AurA Regulates the Subunit Composition of the Par Complex.

(A and B) Cortical release of Lgl regulates the localization of Baz to the posterior lateral cortex. Baz-GFP was coexpressed with either Lgl-RFP or His-RFP in pupal SOP cells. NEBD is t = 0. Anterior is oriented toward the left. (A) In prophase, Baz-GFP localizes to the posterior lateral cortex, as Lgl-RFP is released from this side. (B) Posterior lateral localization of Baz-GFP fails in _aurA_37/37 mutants.
(C and D) AurA promotes and Lgl inhibits the assembly of the Baz complex. Immunoprecipitates (IP) from larval brains expressing Baz-GFP were analyzed. (C’) Quantification of (C). The IP signal was adjusted to the corresponding input signal and normalized for wild-type (WT) (set to 1). Averages and standard deviations are shown (n = 5). Differences to WT are significant (p < 0.05). (D) Immunoprecipitates from brains expressing Baz-GFP alone (control) or together with either LglWT-myc or Lgl3A-myc were analyzed. (D’) Quantification of (D). The IP signal was adjusted to the corresponding input signal and normalized for control (set to 1). Averages and standard deviations are shown (n = 3 for Baz; n = 6 for Lgl). Baz levels are significantly different from the control, and Lgl levels are significantly different from each other (p < 0.05).
In epithelial cells, Lgl competes with Baz for interaction with Par-6 and aPKC (Yamanaka et al., 2003, 2006). In Drosophila, distinct Lgl/Par-6/aPKC and Baz/Par-6/aPKC complexes (here-after referred to as Lgl and Baz complexes, respectively) are assembled in vivo (Figure 6A). To test whether the interaction of Baz with Par-6 and aPKC is inhibited by Lgl, we analyzed Par-6 immunoprecipitates from larval brains expressing Baz-GFP. Par-6 immunoprecipitates from wild-type brains contained both Lgl and Baz (Figure 4C). However, an excess of Baz was coimmunoprecipitated with Par-6 from lgl mutant brains (Figure 4C), whereas overexpression of Lgl reduces the amounts of coimmunoprecipitated Baz (Figure 4D). Interestingly, Par-6 immunoprecipitates from aurA mutants contained an excess of Lgl at the expense of Baz (Figure 4C). This was phenocopied by expression of Lgl3A (Figure 4D), demonstrating that entry of Baz into the Par complex requires AurA to initiate the phosphorylation-dependent release of Lgl from the cell cortex. Thus, AurA triggers a remodeling of the Par complex from the Lgl configuration into the Baz configuration by activating aPKC at the onset of mitosis.
Figure 6. Baz Changes the Substrate Specificity of aPKC.

(A) Baz and Lgl assemble into distinct Par complexes with differential activity toward Numb. Baz and Lgl complexes were isolated from _baz_CC01941 embryos expressing Baz-GFP by immunoprecipitation (IP) of GFP and Lgl, respectively. The immunoprecipitates were incubated with recombinant Numb, ATP, and aPKC inhibitor where indicated.
(B) Numb is in the Baz complex, but not in the Lgl complex. Recombinant proteins were used in pull-down assays from embryos expressing Baz-GFP.
(C) NumbS52F is not in the Baz complex. Recombinant proteins were used in pull-down assays from larval brains expressing Baz-GFP.
(D) Proposed mechanism whereby the activation of AurA leads to the phosphorylation of Numb.
(E) Lgl has higher cortical mobility than Numb. Lgl3A-GFP and Numb-GFP were photobleached on the anterior cortex of pupal SOP cells in metaphase (see Figures S6B and S6C). and the recovery of fluorescence was recorded. The values were normalized to prebleach intensity after correction for background variation and fluorescence loss. Averages are plotted, and standard deviations for the individual time points are shown as gray bars (n = 33 for Lgl3A-GFP; n = 26 for Numb-GFP).
(F) Numb is dephosphorylated more rapidly than Lgl. myctagged Numb and Lgl were immunoprecipitated from embryos, in vitro phosphorylated by PKCz, and subjected to dephosphorylation by brain extract for the indicated durations. Addition of phosphatase inhibitors suppressed the decay of phosphospecific signal. (F’) Quantification of (F). Averages are plotted, and standard deviations for the individual time points are shown (n = 2).
Numb Localization Requires Appropriate Levels of Baz Complex
If a failure to assemble the Baz complex is responsible for the mislocalization of Numb in aurA mutants, then restoring the levels of Baz complex should rescue aurA mutants. To test this, we constructed a Baz-Par-6 fusion protein to force Baz into the Par complex. Indeed, expression of this construct in aurA mutant SOP cells rescued the asymmetric localization of the Numb reporter GFP-Pon (Lu et al., 1999) (Figures 5A–5C; Movies S7–S9). Overexpression of Baz alone failed to rescue GFP-Pon asymmetry in aurA mutant SOP cells (Figure 5D; Movie S10), although it affected the apical-basal distribution of GFP-Pon (Figure S4D’). We conclude that AurA induces the asymmetric localization of Numb by promoting the interaction of Baz with Par-6.
Figure 5. Numb Localization Requires Appropriate Levels of Baz Complex.

(A–D) AurA functions in Numb localization by promoting the interaction of Baz with Par-6. GFP-Pon and Histone-RFP were expressed in pupal SOP cells. NEBD is t = 0. The axis of Pon asymmetry (if any) is left to right. Transparent z-projections are shown. (A) In wild-type cells, GFP-Pon is localized asymmetrically from NEBD onward. (B) In aurA mutants, GFP-Pon remains uniformly cortical, but becomes partially polarized in metaphase. (C) Expression of Baz-Par-6 in aurA mutant cells restores the asymmetric localization of GFP-Pon, but does not rescue the delay in mitotic progression. (D) Overexpression of Baz fails to rescue GFP-Pon asymmetry.
(E–M) Larval neuroblasts of the indicated genotypes were stained for p-H3 to label DNA in mitotic cells and for Numb. Apical is up. (E–G) Knockdown of Baz rescues lgl mutants. (H–J) A temperature-sensitive heteroallelic combination of lgl rescues the strong _aurA_37/Ac-3 mutant at the restrictive temperature. (K–M) Lgl overexpression enhances the hypomorphic _aurA_37/37 mutant.
We show that lgl mutants contain an excess amount of Baz complex. If this is responsible for Numb mislocalization in these mutants, they should be rescued by lowering Baz levels. Indeed, moderate knockdown of Baz (Figures S5AC–S5AE) rescued the asymmetric localization of Numb (Figures 5E–5G) in lgl mutant neuroblasts. We conclude that the role of Lgl in the asymmetric localization of Numb is to inhibit the assembly of the Baz complex.
Excess or insufficient levels of Baz complex both disrupt the asymmetric localization of Numb. Since AurA and Lgl act in an antagonistic manner on the formation of the Baz complex, we analyzed Numb localization in lgl aurA double mutants. As predicted, a weak heteroallelic combination of lgl partially rescued Numb asymmetry in _aurA_37/Ac-3 neuroblasts (Figures 5H–5J). However, overexpression of Lgl in hypomorphic _aurA_37/37 mutants enhanced the penetrance of the Numb mislocalization phenotype (Figures 5K–5M). These results confirm the antagonistic roles of AurA and Lgl in regulating the localization of Numb.
Baz Changes the Substrate Specificity of aPKC
Our data suggest that the exchange of Lgl for Baz converts the Par complex into its active configuration. What is the activity of the Baz complex? Recently, it was shown that Numb is a substrate for aPKC, and that phosphorylation releases Numb from the cortex into the cytoplasm (Nishimura and Kaibuchi, 2007; Smith et al., 2007). Importantly, the nonphosphorylatable Numb5A localizes symmetrically in SOP cells (Smith et al., 2007). We hypothesized that the Baz complex phosphorylates Numb, whereas the Lgl complex does not. To test this, we isolated the two complexes from embryos bearing a GFP trap in the baz gene (Buszczak et al., 2007) by immunoprecipitation of GFP and Lgl, respectively. We incubated these immunoprecipitates with ATP and recombinant Numb and assayed for activity by using antibodies specific for phosphorylated Numb (p-Numb) (Figures S1D and S1E). Although the immunoprecipitates contained comparable amounts of aPKC, only the Baz complex phosphorylated Numb, and this effect was negated by the addition of an aPKC inhibitor (Figure 6A). We conclude that the exchange of Lgl for Baz enables the Par complex to phosphorylate Numb.
What is the molecular basis for the difference in activity between the two complexes? Since Numb can directly bind to Par-3 in mammalian cells (Nishimura and Kaibuchi, 2007), Baz might recruit Numb into the Par complex. To test this, we performed pull-down assays from embryos (Figure 6B) or larval brains (data not shown) expressing Baz-GFP. Recombinant Numb precipitated Baz, Par-6, and aPKC, but not Lgl. We conclude that the Baz complex recruits Numb, whereas the Lgl complex does not.
The functional significance of the Numb-Baz interaction is supported by the point-mutated allele _numb_S52F. Its protein product localizes symmetrically in SOP cells (Bhalerao et al., 2005), although it can still be phosphorylated by aPKC in vitro (Smith et al., 2007). Our findings raise the possibility that NumbS52F is defective for binding to Baz, as this would prevent it from being phosphorylated by aPKC in vivo. To test this, we performed pull-down assays from brains expressing Baz-GFP. Recombinant NumbS52F failed to precipitate Baz and bound only minor amounts of Par-6 and aPKC, although its interaction with the downstream effector α-adaptin was unimpaired (Figure 6C). This suggests that the interaction of Numb with the Baz complex is essential for its asymmetric localization.
Par-3 is itself phosphorylated by aPKC (Lin et al., 2000; Nagai-Tamai et al., 2002), raising the possibility that Baz acts as a competitive substrate with Numb. However, overexpression of Par-3 in cultured cells stimulated, rather than inhibited, Numb phosphorylation (Figure S7D).
We conclude from these data that Baz confers novel substrate specificity on aPKC by recruiting Numb into proximity to aPKC. Thus, the exchange of Lgl for Baz in the Par complex leads to the phosphorylation of Numb by aPKC on one side of the cell cortex (Figure 6D). p-Numb is released from the cortex (Nishimura and Kaibuchi, 2007; Smith et al., 2007), restricting the protein into a cortical crescent on the opposite side.
Although Numb and Lgl are both substrates for aPKC, phosphorylation of Lgl results in uniform cytoplasmic localization, whereas phosphorylation of Numb results in asymmetric cortical localization. How might this be explained? Fluorescence recovery after photobleaching (FRAP) of Lgl3A-GFP demonstrates that, in prophase, unphosphorylated Lgl on the anterior cortex of SOP cells diffuses to the posterior side, where it may be phosphorylated by aPKC (Figure S6A; Movie S11). Low cortical mobility, however, would protect a substrate on the anterior cortex from phosphorylation by aPKC. Indeed, quantitative FRAP of Numb-GFP and Lgl3A-GFP reveals that Numb has lower cortical mobility than Lgl (Figures 6E, S6B, and S6C). Moreover, we expect an unidentified phosphatase to offset the aPKC-dependent release from the cell cortex. To investigate this, we measured the relative dephosphorylation rates of p-Numb and p-Lgl in brain extract. This revealed that Numb is indeed dephosphorylated more rapidly than Lgl (Figure 6F). Together, these data suggest that the striking difference in how Numb and Lgl localize in response to phosphorylation by aPKC derives, at least in part, from differences in the cortical mobilities and dephosphorylation rates of these two substrates.
We elucidated a mechanism for the asymmetric localization of Numb, but the adaptor proteins Pon and Miranda (Mira) are localized asymmetrically in an Lgl-dependent manner as well (Langevin et al., 2005; Ohshiro et al., 2000; Peng et al., 2000). To investigate if these are localized by a similar mechanism, we assessed their physical interaction with members of the Par complex in pull-down assays from larval brains. Both recombinant Pon and Mira precipitated Baz, aPKC, and Par-6, but neither bait precipitated Lgl in amounts suggestive of a direct interaction (Figure S7A). In addition, both Pon and Mira were phosphorylated by aPKC in vitro (Figure S7B). These data suggest that the mechanism of Numb localization is used for Pon and Mira as well, although redundancy can be inferred from the fact that Mira is unaffected in aurA mutants (Lee et al., 2006a; Wang et al., 2006).
Our data link AurA to the aPKC-dependent phosphorylation of Numb. To test if this pathway is conserved in mammals, we transfected AurA or one of its activators into HeLa cells and probed for p-Numb. Indeed, Numb phosphorylation was enhanced by overexpression or activation of AurA, and these effects were negated by cotransfection of dominant-negative aPKC (Figure S7C). We conclude that the role of AurA in regulating the aPKC-dependent phosphorylation of Numb is conserved.
AurA and Lgl Regulate Neuroblast Proliferation through the Baz-Dependent Phosphorylation of Numb by aPKC
In the larval brain, asymmetric cell division balances differentiation with self-renewal, giving rise to one differentiating daughter cell and one proliferating neuroblast at each neuroblast division. numb mutant neuroblasts divide symmetrically into two neuroblasts, resulting in a neuroblast tumor (Lee et al., 2006a; Wang et al., 2006). Interestingly, expression of aPKCCAAX, a constitutively active form of aPKC, causes a similar phenotype (Lee et al., 2006b). To test if Numb is a relevant target of aPKC in regulating neuroblast proliferation, we tried to offset the excess activity of aPKCCAAX by overexpressing Numb. Indeed, overexpression of Numb completely suppressed the tumor phenotype induced by aPKCCAAX (Figures 7A–7C and S8A–S8D), although overexpression of Numb alone had no effect on neuroblast numbers (data not shown; Lee et al., 2006a; Wang et al., 2006). Moreover, the aPKCCAAX phenotype was enhanced by the null allele _numb_15 but weakly suppressed by the _numb_S52F allele (Figures 7D–7G). Therefore, the aPKCCAAX phenotype is sensitive to the dosage of Numb and at least partially depends on the interaction of Numb with the Baz complex.
Figure 7. AurA and Lgl Regulate Neuroblast Proliferation through the Baz-Dependent Phosphorylation of Numb by aPKC.

(A–T) Third-instar larval brains were stained for Miranda and p-H3 (both green) to label neuroblasts and for Prospero (red) to label neurons. Surface views of (A–C) whole brains and (D–T) posterior surface views of single lobes are shown, except in (I), in which both posterior and anterior sides are exposed. Dotted lines demarcate the central brain (CB) (right) from the optic lobe (left). Scale bars are 100 mm. Note that scale varies among the images. (A and D) In the wild-type CB, a defined number of neuroblasts (green) give rise to chains of differentiating progeny (red). (A–G) aPKC regulates neuroblast proliferation through Numb. (B and C) Expression of aPKCCAAX at 29°C induces strong overproliferation of neuroblasts at the expense of neurons, which is suppressed to wild-type by coexpression of Numb. (E–G) Expression of aPKCCAAX at 18°C induces modest overproliferation of neuroblasts at the expense of neurons, which is enhanced by heterozygosity for _numb_15, but weakly suppressed by heterozygosity for _numb_S52F. (H–O) Lgl reg-ulates neuroblast proliferation through Baz and Numb. (H–L) Neuroblast overproliferation in lgl mutants is enhanced by overexpression of Baz-GFP, but suppressed to wild-type by knockdown of Baz. Neither has any effect on neuroblast numbers in the wild-type. (M–O) At the early third-instar stage, lgl mutant neuroblasts weakly overproliferate, which is enhanced by heterozygosity for _numb_15, but weakly suppressed by heterozygosity for _numb_S52F. (P–T) AurA regulates neuroblast proliferation through subunit exchange in the Par complex. (P–R) Lgl overexpression has no effect on neuroblast numbers in the wild-type, but induces neuroblast overproliferation in _aurA_37/37 mutants, which are otherwise free of tumors. (S and T) Baz-GFP overexpression suppresses neuroblast overproliferation in _aurA_37/Ac-3 mutants.
lgl mutants develop neuroblast tumors similar to numb mutants. Our model proposes that lgl mutants assemble an excess of Baz complex, leading to ectopic Numb phosphorylation. To test if this is responsible for tumor formation in the lgl mutant, we attempted to modify this phenotype by altering the levels of Baz. Indeed, overexpression of Baz led to a further increase in neuroblast numbers, whereas knockdown of Baz rescued the lgl mutant to wild-type, although neither had an effect on neuroblast numbers in wild-type brains (Figures 7H–7L). Analogous to the aPKCCAAX phenotype, the lgl phenotype was enhanced by _numb_15, weakly suppressed by _numb_S52F, and rescued to wild-type by overexpression of Numb (Figures 7M–7O and S8E–S8K). Together, these genetic interactions suggest that Lgl acts as a tumor suppressor, at least in part, by blocking the Baz-dependent phosphorylation of Numb by aPKC.
aPKC is mislocalized in lgl mutants (Figure S5W), and this has been proposed as the root cause of tumor formation (Lee et al., 2006b). However, aPKC mislocalization in lgl mutants was unaffected by either knockdown or overexpression of Baz (Figures S5W, S5X, and S5Z; Table S2), suggesting additional constraints on tumor formation. Moreover, we found that _numb_S52F mutant neuroblasts, which missegregate Numb into both daughter cells, did not overproliferate (Figure S9; Table S3). Therefore, the localization of Numb and aPKC in neuroblasts did not correlate with tumor formation in lgl mutants. A plausible explanation is that differentiation requires a threshold amount of unphosphorylated Numb, which is influenced by its asymmetric segregation but is primarily determined by the overall level of Numb phosphorylation in the neuroblast and in the differentiating daughter cell (Figure S8M; see Discussion).
Neuroblast tumors have also been reported for the aurA mutant. To test if a failure of the Par complex to undergo subunit exchange is responsible for tumor formation in aurA mutant brains, we attempted to modify this phenotype by altering the levels of Lgl and Baz. Overexpression of Lgl did not cause a phenotype in wild-type brains but induced neuroblast overproliferation in aurA hypomorphs that were otherwise free of tumors (Figures 7P–7R). However, overexpression of Baz suppressed tumor formation in the stronger aurA heteroallelic combination (Figures 7S and 7T). Consistent with this, the mislocalization of Numb and aPKC in aurA mutants was suppressed by overexpression of Baz but was enhanced by overexpression of Lgl (Figures S5A, S5D–S5H, S5O, and S5R–S5V; Tables S1 and S2). In contrast to lgl mutants, therefore, tumor formation in the aurA mutant background correlated well with mislocalization of Numb and aPKC. These data confirm the prevailing view that AurA acts as a tumor suppressor by regulating the localization of Numb and aPKC during mitosis (Lee et al., 2006a; Wang et al., 2006) (Figure S8M).
DISCUSSION
A Molecular Mechanism for the Asymmetric Localization of Numb
Since the discovery of Numb asymmetry (Rhyu et al., 1994), several proteins required for Numb localization have been identified (Gonczy, 2008), but how they cooperate remained unclear. Here, we describe a cascade of interactions among these proteins that culminates in the asymmetric localization of Numb in mitosis (Figure 6D). In interphase, Lgl localizes to the cell cortex, where it forms a complex with Par-6 and aPKC. At the onset of mitosis, AurA phosphorylates Par-6 in this complex, thereby releasing aPKC from inhibition by Par-6. Activated aPKC phosphorylates Lgl, causing its release from the cell cortex. Since Baz competes with Lgl for entry into the Par complex, the disassembly of the Lgl/Par-6/aPKC complex allows for the assembly of the Baz/ Par-6/aPKC complex. Baz is a specificity factor that allows aPKC to phosphorylate Numb on one side of the cell cortex. Since p-Numb is released from the cortex (Nishimura and Kaibuchi, 2007; Smith et al., 2007), these events restrict Numb into a cortical crescent on the opposite side.
The Function of Lgl as a Molecular Buffer
Our data show that Lgl acts as an inhibitory subunit of the Par complex. Given that Par-6 inhibits aPKC activity until the onset of mitosis, why would an additional layer of regulation be required?
Like all phosphoproteins Numb is in a dynamic equilibrium between the phosphorylated and unphosphorylated states. Too high a rate of phosphorylation shifts this equilibrium toward the phosphorylated state, mislocalizing Numb into the cytoplasm. Too low a rate shifts it toward the unphosphorylated state, mislocalizing Numb around the cell cortex. Importantly, our data show that only the Baz complex can phosphorylate Numb. Assuming an abundance of Lgl over cortical Par-6, an increase in aPKC activity would translate into a comparatively small increase in the levels of Baz complex. This is because assembly of the Baz complex requires free subunits of Par-6 and aPKC, which become available only once the pool of cortical Lgl has been completely phosphorylated. Therefore, we propose that Lgl acts as a molecular buffer for the activity of the Par complex toward Numb. This maintains Numb phosphorylation within a range that is sufficiently high to exclude Numb from one side of the cell cortex but sufficiently low to permit the cortical localization of Numb to the other side.
What is the evidence for this model? Lgl3A, having infinite buffering capacity, induces the mislocalization of Numb around the cell cortex (Betschinger et al., 2003; Langevin et al., 2005). Conversely, in lgl mutants, having no buffering capacity, Numb is mislocalized into the cytoplasm (Langevin et al., 2005; Ohshiro et al., 2000). Moreover, our model predicts the loss of buffering capacity in the lgl mutant to be offset by an increase in the amount of substrate, as this would render the excess activity of the Par complex limiting. Indeed, overexpression of Numb in lgl mutants restores the cortical localization of Numb as well as its cortical asymmetry (Langevin et al., 2005).
Our results indicate that Lgl gain- and loss-of-function phenotypes are entirely accounted for by the role of Lgl in inhibiting the assembly of the Baz complex. Previously, however, it was thought that the asymmetric phosphorylation of Lgl by aPKC restricts an activity of Lgl to the opposite side of the cell cortex (Betschinger et al., 2003). Based on this model, it was subsequently proposed that Lgl mediates the asymmetric localization of cell fate determinants by inhibiting the cortical localization of myosin-II (Barros et al., 2003). In addition, the role of the yeast orthologs of Lgl in exocytosis led to speculation that Lgl establishes an asymmetric binding site for cell fate determinants by promoting targeted vesicle fusion (Ohshiro et al., 2000; Peng et al., 2000; Wirtz-Peitz and Knoblich, 2006). However, our data show that Lgl asymmetry is extremely transient, and that the protein is completely cytoplasmic from NEBD onward. Lgl cannot therefore interact with any cortical proteins in prometaphase or metaphase, when myosin-II was reported to localize asymmetrically (Barros et al., 2003), or establish a stable landmark for vesicle fusion. Interestingly, a recent study demonstrated that yeast Lgl inhibits the assembly of SNARE complexes by sequestering a plasma membrane SNARE (Hattendorf et al., 2007). This mechanism is reminiscent of fly Lgl sequestering Par-6 and aPKC from interaction with Baz, suggesting that the defining property of Lgl-family members is not a specific role in exocytosis, but a more generic role in regulating the assembly of protein complexes.
The Roles of AurA, Lgl, and aPKC in the Regulation of Numb Activity
Our data identify Numb as a key target of aPKC in tumor formation and suggest that Lgl acts as a tumor suppressor in the larval brain by inhibiting the aPKC-dependent phosphorylation of Numb. Although it is tempting to conclude that tumor formation in lgl mutants results from the missegregation of Numb, missegregation of Numb in _numb_S52F mutants (this study) or upon expression of Lgl3A (Lee et al., 2006b) does not cause neuroblast tumors. How might this be explained?
During mitosis, unphosphorylated cortical Numb is inherited by the differentiating daughter. At the same time, Baz and aPKC are excluded from this daughter, which limits Numb phosphorylation after exit from mitosis. In the subsequent interphase, some differentiating daughters reexpress members of the Baz complex (Bowman et al., 2008), but Numb continues to be protected from phosphorylation since cortical Lgl prevents the reassembly of the Baz complex. Thus, Lgl acts both in mitosis and interphase to maximize the amount of unphosphorylated Numb in the differentiating daughter cell (Figure S8M).
In lgl mutants, Numb phosphorylation is increased in mitosis, and less unphosphorylated Numb is segregated into the basal daughter cell. Moreover, the assembly of the Baz complex is unrestrained in the subsequent interphase, which is exacerbated by the missegregation of aPKC into both daughter cells (Lee et al., 2006b). Together, these defects minimize the amount of unphosphorylated Numb in the differentiating daughter cell.
Why is the amount of unphosphorylated Numb critical for differentiation? Recently, it was shown that aPKC-dependent phosphorylation of Numb inhibits not only its cortical localization, but also its activity, owing to the reduced affinity of p-Numb for its endocytic targets (Nishimura and Kaibuchi, 2007). Therefore, ectopic phosphorylation of Numb leads to its inactivation, transforming the basal daughter cell into a neuroblast in a manner similar to mutation of numb. Consistent with this model, studies in SOP cells have documented ectopic Notch signaling in lgl mutants (Langevin et al., 2005; Roegiers et al., 2005). Although the _numb_S52F mutant and Lgl3A overexpression also lead to missegregation of Numb, the levels of active unphosphorylated Numb are increased rather than decreased in these cases and are sufficient to support differentiation.
Our data also provide additional insight into the mechanism of tumor formation in aurA mutants. In aurA mutants, the differentiating daughter cell inherits less Numb because Numb is mislocalized around the cell cortex. At the same time, aPKC is missegregated into the differentiating daughter cell, where it promotes Numb phosphorylation in the subsequent interphase. Together, these events result in subthreshold amounts of unphosphorylated Numb in some basal daughter cells, transforming these into neuroblasts. This model explains why aurA mutants are characterized by reduced aPKC activity in mitosis, but are nonetheless suppressed by aPKC mutations (Lee et al., 2006a; Wang et al., 2006), since a lack of aPKC in the differentiating daughter cell restores threshold amounts of unphosphorylated Numb.
Our data reveal that Lgl inhibits Numb phosphorylation to maintain Numb activity, whereas AurA promotes Numb phosphorylation in mitosis to ensure its asymmetric segregation. We conclude that Lgl and AurA act on opposite ends of a regulatory network that maintains appropriate levels of Numb phosphorylation at the appropriate time in the cell cycle.
EXPERIMENTAL PROCEDURES
Drosophila Strains and Constructs
UAS constructs were expressed in SOP cells by using neur > Gal4 and in neuroblasts by using insc > Gal4, except for experiments shown in Figure S1A, in which act5C > Gal4 was used. GAL80ts was used to suppress the expression of aPKCΔN prior to pupariation. All fly stocks and constructs are described in detail in the Supplemental Experimental Procedures.
Lgl-GFP is functional because it complements _lgl_1/4 mutants in the larval brain (Figure S8L). The Par-6-GFP genomic rescue construct is functional because it rescues _par-6_Δ226 mutants to viability (data not shown).
Live Imaging
Live imaging of SOP cells was performed essentially as described (Bellaiche et al., 2001a). Images were acquired on a Zeiss LSM510 Meta confocal microscope with a Plan-Apochromat 633/1.40 oil immersion objective at 53 zoom (single cells) or 13 zoom (whole notum). For two-dimensional (2D) recordings, images were acquired at intervals of 6–12 s. For 3D recordings, a z-stack of 10 or 20 slices at 1 μm distance was acquired at intervals of 42 s or 84 s, respectively. Images were processed in LSM software (Zeiss), ImageJ (NIH), and Imaris (Bitplane).
For FRAP analysis, a Plan-Apochromat 633/1.40 oil immersion objective was used at 53 zoom with a pinhole of 2 Airy units. The bleach pulse consisted of 10 passes of a 488 nm laser at 100% power. Quantitative experiments were performed in bidirectional mode at 8 fps, and 8 prebleach frames and 480 postbleach frames were acquired. Integrated densities of the bleached area, a whole-cell, and a background region were determined for each time point by using LSM software (Zeiss). Values were corrected for background in Microsoft Excel. In addition, the double normalization method was used to correct for loss of total fluorescence due to bleach pulse and acquisition bleaching (Phair et al., 2004). Each experiment was normalized to the average prebleach fluorescence, which allowed averages and standard deviations to be calculated for the individual time points.
Antibodies and Immunohistochemistry
Phosphospecific antibodies against Ser34 of Par-6 were raised in rabbits against the peptide EFRRWpSFKRNEAE and were affinity purified. Baz antibodies were raised in rabbits against the N-terminal 318 amino acids of Baz fused to MBP.
Antibodies used in western blotting, immunoprecipitation, and/or immunohistochemistry were rabbit anti-p-Ser34-Par-6 and anti-Baz (1:200; this study); rabbit anti-Par-6 (1:200; Petronczki and Knoblich, 2001); rabbit anti-Lgl (1:100) and rabbit anti-p-Ser660-Lgl (1:200; Betschinger et al., 2003); mouse anti-cmyc (sc-40) and rabbit anti-PKCζ (1:1,000; sc-216, Santa Cruz); rabbit anti-p-Thr555-PKCζ (1:100; ab5813) and rabbit anti-GFP (1:1,000; ab6556, Abcam); mouse anti-GFP (1:100; Roche); rabbit anti-p-Ser7-Numb (1:200;Nishimura and Kaibuchi, 2007); rabbit anti-Miranda (1:200; Betschinger et al., 2006); rabbit anti-p-H3 (1:1000; Millipore); mouse anti-Prospero (1:20; MR1A, DSHB); rabbit anti-Numb (1:100; Schober et al., 1999); and rabbit anti-α-adaptin (1:200; Dornan et al., 1997). Rabbit anti-GFP (A-6455, Molecular Probes) was used for immunoprecipitation of Baz-GFP.
For immunohistochemistry, third-instar wandering larvae were dissected in PBS, and brains with attached ventral nerve cords were fixed for 10 min in 3.7% formaldehyde in PBS containing 0.2% Triton X-100 and processed as described (Betschinger et al., 2006). Images were acquired on a Zeiss LSM510 confocal microscope and were processed in Photoshop (Adobe Systems).
Kinase Assays
For in vitro kinase assays, bacterially produced proteins were incubated with either 30 ng recombinant human AurA (Millipore) for 30 min at 30°C or with 50 ng recombinant PKCζ (Calbiochem) for 10 min at 25°C in kinase buffer (25 mM Tris-HCl [pH 7.5], 25 mM NaCl, 5 mM MgCl2,10 mM β-glycerophosphate, 1 mM EGTA, 1 mM DTT, 100 mM ATP) containing 1.5 mCi [γ-32P]ATP. For IP kinase assays, the immunoprecipitates were incubated for 30 min at 30°C in kinase buffer (20 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 0.1 μM Calyculin A, 1 mM EGTA, 1 mM DTT, 0.1% Triton X-100, 100 mM ATP). A total of 100 ng recombinant AurA was used in the assay illustrated in Figure 1B. A total of 40 μg/ml of the peptide SIYRRGARRWRKL was used as a pseudosubstrate inhibitor against aPKC in the experiment illustrated in Figure 6A. Reactions were terminated by the addition of SDS sample buffer.
Immunoprecipitations and Binding Assays
For immunoprecipitations, embryos or larval brains were extracted with lysis buffer (20 mM Tris-HCl [pH 7.5], 1 mM EDTA, 100 mM NaCl, 1% Triton X-100, 10 μg/ml PMSF, Complete Protease Inhibitor Cocktail [Roche]), cleared by centrifugation at 16,000 3 g for 15 min, and incubated with antibodies for 1–2 hr at 4°C. The immunocomplexes were then precipitated by using Protein A-Sepharose 4B beads (Amersham), washed three times with lysis buffer, and eluted by boiling in SDS sample buffer. For in vitro binding assays, GST fusion proteins were immobilized onto glutathione Sepharose 4B beads (Amersham) and incubated with embryonic lysate or larval brain lysate in lysis buffer for 1–2 hr at 4°C. The beads were washed three times with lysis buffer and were eluted by boiling in SDS sample buffer.
Dephosphorylation Assay
Embryonic myc-Lgl and myc-Numb immunoprecipitates were washed three times with high-salt buffer (20 mM Tris-HCl [pH 7.5], 1 mM EDTA, 500 mM NaCl, 1% Triton X-100, 10 mg/ml PMSF) and twice with kinase buffer (25 mM Tris-HCl [pH 7.5], 100 mM NaCl, 5 mM MgCl2,10mM β-glycerophosphate, 1 mM DTT, 1 mM EGTA, 0.1% Trition X-100) at 4°C and were incubated with 100 ng recombinant PKCζ (Calbiochem) for 30 min at 30°C in kinase buffer containing 100 μM ATP. Subsequently, the samples were washed three times with high-salt buffer and twice with lysis buffer.
Phosphorylated samples were incubated with larval brain extract in phosphatase buffer (50 mM Tris-HCl [pH 7.5], 100 mM NaCl, 0.1 mM EGTA, 2 mM DTT, 1 mM MnCl2, 0.5% Trition X-100, 10 mg/ml PMSF, EDTA-free Complete Protease Inhibitor Cocktail [Roche]) with or without phosphatase inhibitors (0.1 μM Calyculin A, 20 mM β-glycerophosphate, 5 mM NaF, 1 mM sodium pyrophosphate) for the indicated durations at 20°C, terminated by the addition of chilled lysis buffer with phosphatase inhibitors, centrifuged, and eluted by boiling in SDS sample buffer.
Supplementary Material
supplementary materials and methods, figures, tables and references
ACKNOWLEDGMENTS
We thank C. Jüschke and S.K. Bowman for comments on the manuscript; Y. Bellaïche, T. Hirota, Y.N. Jan, D. St Johnston, K. Kaibuchi, F. Matsuzaki, T. Murphy, S. Ohno, H. Saya, F. Schweisguth, and the Developmental Studies Hybridoma Bank for constructs, antibodies, and fly stocks; J. Betschinger, D. Berdnik, and A. Hutterer for generating transgenic fly lines and constructs; N. Nishimura and S. Farina Lopez for technical assistance; and M.P. Postiglione, S. Goulas, and other members of our lab for discussion. F.W.P. was supported by a PhD fellowship of the Boehringer Ingelheim Fonds; T.N. is supported by a long-term fellowship of the Human Frontiers Science Program; and work in J.A.K.’s lab is supported by the Austrian Academy of Sciences, the Austrian Science Fund, the Vienna Science and Technology Fund, the European Union EUROSYSTEMS, and ONCASYM.
Footnotes
REFERENCES
- Barros CS, Phelps CB, Brand AH. Drosophila nonmuscle myosin II promotes the asymmetric segregation of cell fate determinants by cortical exclusion rather than active transport. Dev. Cell. 2003;5:829–840. doi: 10.1016/s1534-5807(03)00359-9. [DOI] [PubMed] [Google Scholar]
- Bellaiche Y, Gho M, Kaltschmidt JA, Brand AH, Schweisguth F. Frizzled regulates localization of cell-fate determinants and mitotic spindle rotation during asymmetric cell division. Nat. Cell Biol. 2001a;3:50–57. doi: 10.1038/35050558. [DOI] [PubMed] [Google Scholar]
- Bellaiche Y, Radovic A, Woods DF, Hough CD, Parmentier ML, O’Kane CJ, Bryant PJ, Schweisguth F. The Partner of Inscuteable/Discs-large complex is required to establish planar polarity during asymmetric cell division in Drosophila. Cell. 2001b;106:355–366. doi: 10.1016/s0092-8674(01)00444-5. [DOI] [PubMed] [Google Scholar]
- Benton R, St Johnston D. Drosophila PAR-1 and 14–3-3 inhibit Bazooka/PAR-3 to establish complementary cortical domains in polarized cells. Cell. 2003;115:691–704. doi: 10.1016/s0092-8674(03)00938-3. [DOI] [PubMed] [Google Scholar]
- Berdnik D, Knoblich JA. Drosophila Aurora-A is required for centrosome maturation and actin-dependent asymmetric protein localization during mitosis. Curr. Biol. 2002;12:640–647. doi: 10.1016/s0960-9822(02)00766-2. [DOI] [PubMed] [Google Scholar]
- Betschinger J, Mechtler K, Knoblich JA. The Par complex directs asymmetric cell division by phosphorylating the cytoskeletal protein Lgl. Nature. 2003;422:326–330. doi: 10.1038/nature01486. [DOI] [PubMed] [Google Scholar]
- Betschinger J, Mechtler K, Knoblich JA. Asymmetric segregation of the tumor suppressor brat regulates self-renewal in Drosophila neural stem cells. Cell. 2006;124:1241–1253. doi: 10.1016/j.cell.2006.01.038. [DOI] [PubMed] [Google Scholar]
- Bhalerao S, Berdnik D, Torok T, Knoblich JA. Localization-dependent and -independent roles of numb contribute to cell-fate specification in Drosophila. Curr. Biol. 2005;15:1583–1590. doi: 10.1016/j.cub.2005.07.061. [DOI] [PubMed] [Google Scholar]
- Bowman SK, Rolland V, Betschinger J, Kinsey KA, Emery G, Knoblich JA. The tumor suppressors Brat and Numb regulate transit-amplifying neuroblast lineages in Drosophila. Dev. Cell. 2008;14:535–546. doi: 10.1016/j.devcel.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buszczak M, Paterno S, Lighthouse D, Bachman J, Planck J, Owen S, Skora AD, Nystul TG, Ohlstein B, Allen A, et al. The carnegie protein trap library: a versatile tool for Drosophila developmental studies. Genetics. 2007;175:1505–1531. doi: 10.1534/genetics.106.065961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doe CQ. Neural stem cells: balancing self-renewal with differentiation. Development. 2008;135:1575–1587. doi: 10.1242/dev.014977. [DOI] [PubMed] [Google Scholar]
- Dornan S, Jackson AP, Gay NJ. a-adaptin, a marker for endocytosis, is expressed in complex patterns during Drosophila development. Mol. Biol. Cell. 1997;8:1391–1403. doi: 10.1091/mbc.8.8.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrari S, Marin O, Pagano MA, Meggio F, Hess D, El-Shemerly M, Krystyniak A, Pinna LA. Aurora-A site specificity: a study with synthetic peptide substrates. Biochem. J. 2005;390:293–302. doi: 10.1042/BJ20050343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover DM, Leibowitz MH, McLean DA, Parry H. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995;81:95–105. doi: 10.1016/0092-8674(95)90374-7. [DOI] [PubMed] [Google Scholar]
- Gonczy P. Mechanisms of asymmetric cell division: flies and worms pave the way. Nat. Rev. Mol. Cell Biol. 2008;9:355–366. doi: 10.1038/nrm2388. [DOI] [PubMed] [Google Scholar]
- Guo M, Jan LY, Jan YN. Control of daughter cell fates during asymmetric division: interaction of Numb and Notch. Neuron. 1996;17:27–41. doi: 10.1016/s0896-6273(00)80278-0. [DOI] [PubMed] [Google Scholar]
- Hattendorf DA, Andreeva A, Gangar A, Brennwald PJ, Weis WI. Structure of the yeast polarity protein Sro7 reveals a SNARE regulatory mechanism. Nature. 2007;446:567–571. doi: 10.1038/nature05635. [DOI] [PubMed] [Google Scholar]
- Hirai T, Chida K. Protein kinase Cz (PKCz): activation mechanisms and cellular functions. J. Biochem. 2003;133:1–7. doi: 10.1093/jb/mvg017. [DOI] [PubMed] [Google Scholar]
- Knoblich JA. Mechanisms of asymmetric stem cell division. Cell. 2008;132:583–597. doi: 10.1016/j.cell.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Langevin J, Le Borgne R, Rosenfeld F, Gho M, Schweisguth F, Bel-laiche Y. Lethal giant larvae controls the localization of notch-signaling regulators numb, neuralized, and Sanpodo in Drosophila sensory-organ precursor cells. Curr. Biol. 2005;15:955–962. doi: 10.1016/j.cub.2005.04.054. [DOI] [PubMed] [Google Scholar]
- Lee CY, Andersen RO, Cabernard C, Manning L, Tran KD, Lanskey MJ, Bashirullah A, Doe CQ. Drosophila Aurora-A kinase inhibits neuroblast self-renewal by regulating aPKC/Numb cortical polarity and spindle orientation. Genes Dev. 2006a;20:3464–3474. doi: 10.1101/gad.1489406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CY, Robinson KJ, Doe CQ. Lgl, Pins and aPKC regulate neuroblast self-renewal versus differentiation. Nature. 2006b;439:594–598. doi: 10.1038/nature04299. [DOI] [PubMed] [Google Scholar]
- Lin D, Edwards AS, Fawcett JP, Mbamalu G, Scott JD, Pawson T. A mammalian PAR-3-PAR-6 complex implicated in Cdc42/Rac1 and aPKC signalling and cell polarity. Nat. Cell Biol. 2000;2:540–547. doi: 10.1038/35019582. [DOI] [PubMed] [Google Scholar]
- Lu B, Ackerman L, Jan LY, Jan YN. Modes of protein movement that lead to the asymmetric localization of partner of Numb during Drosophila neuroblast division. Mol. Cell. 1999;4:883–891. doi: 10.1016/s1097-2765(00)80218-x. [DOI] [PubMed] [Google Scholar]
- Nagai-Tamai Y, Mizuno K, Hirose T, Suzuki A, Ohno S. Regulated protein-protein interaction between aPKC and PAR-3 plays an essential role in the polarization of epithelial cells. Genes Cells. 2002;7:1161–1171. doi: 10.1046/j.1365-2443.2002.00590.x. [DOI] [PubMed] [Google Scholar]
- Nishimura T, Kaibuchi K. Numb controls integrin endocytosis for directional cell migration with aPKC and PAR-3. Dev. Cell. 2007;13:15–28. doi: 10.1016/j.devcel.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Noda Y, Kohjima M, Izaki T, Ota K, Yoshinaga S, Inagaki F, Ito T, Sumimoto H. Molecular recognition in dimerization between PB1 domains. J. Biol. Chem. 2003;278:43516–43524. doi: 10.1074/jbc.M306330200. [DOI] [PubMed] [Google Scholar]
- Ohshiro T, Yagami T, Zhang C, Matsuzaki F. Role of cortical tumour-suppressor proteins in asymmetric division of Drosophila neuroblast. Nature. 2000;408:593–596. doi: 10.1038/35046087. [DOI] [PubMed] [Google Scholar]
- Peng CY, Manning L, Albertson R, Doe CQ. The tumour-suppressor genes lgl and dlg regulate basal protein targeting in Drosophila neuroblasts. Nature. 2000;408:596–600. doi: 10.1038/35046094. [DOI] [PubMed] [Google Scholar]
- Petronczki M, Knoblich JA. DmPAR-6 directs epithelial polarity and asymmetric cell division of neuroblasts in Drosophila. Nat. Cell Biol. 2001;3:43–49. doi: 10.1038/35050550. [DOI] [PubMed] [Google Scholar]
- Phair RD, Gorski SA, Misteli T. Measurement of dynamic protein binding to chromatin in vivo, using photobleaching microscopy. Methods Enzymol. 2004;375:393–414. doi: 10.1016/s0076-6879(03)75025-3. [DOI] [PubMed] [Google Scholar]
- Rhyu MS, Jan LY, Jan YN. Asymmetric distribution of numb protein during division of the sensory organ precursor cell confers distinct fates to daughter cells. Cell. 1994;76:477–491. doi: 10.1016/0092-8674(94)90112-0. [DOI] [PubMed] [Google Scholar]
- Roegiers F, Jan LY, Jan YN. Regulation of membrane localization of Sanpodo by lethal giant larvae and neuralized in asymmetrically dividing cells of Drosophila sensory organs. Mol. Biol. Cell. 2005;16:3480–3487. doi: 10.1091/mbc.E05-03-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schober M, Schaefer M, Knoblich JA. Bazooka recruits Inscuteable to orient asymmetric cell divisions in Drosophila neuroblasts. Nature. 1999;402:548–551. doi: 10.1038/990135. [DOI] [PubMed] [Google Scholar]
- Smith CA, Lau KM, Rahmani Z, Dho SE, Brothers G, She YM, Berry DM, Bonneil E, Thibault P, Schweisguth F, et al. aPKC-mediated phosphorylation regulates asymmetric membrane localization of the cell fate determinant Numb. EMBO J. 2007;26:468–480. doi: 10.1038/sj.emboj.7601495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Somers GW, Bashirullah A, Heberlein U, Yu F, Chia W. Aurora-A acts as a tumor suppressor and regulates self-renewal of Drosophila neuroblasts. Genes Dev. 2006;20:3453–3463. doi: 10.1101/gad.1487506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirtz-Peitz F, Knoblich JA. Lethal giant larvae take on a life of their own. Trends Cell Biol. 2006;16:234–241. doi: 10.1016/j.tcb.2006.03.006. [DOI] [PubMed] [Google Scholar]
- Yamanaka T, Horikoshi Y, Suzuki A, Sugiyama Y, Kitamura K, Maniwa R, Nagai Y, Yamashita A, Hirose T, Ishikawa H, et al. PAR-6 regulates aPKC activity in a novel way and mediates cell-cell contact-induced formation of the epithelial junctional complex. Genes Cells. 2001;6:721–731. doi: 10.1046/j.1365-2443.2001.00453.x. [DOI] [PubMed] [Google Scholar]
- Yamanaka T, Horikoshi Y, Sugiyama Y, Ishiyama C, Suzuki A, Hirose T, Iwamatsu A, Shinohara A, Ohno S. Mammalian Lgl forms a protein complex with PAR-6 and aPKC independently of PAR-3 to regulate epithelial cell polarity. Curr. Biol. 2003;13:734–743. doi: 10.1016/s0960-9822(03)00244-6. [DOI] [PubMed] [Google Scholar]
- Yamanaka T, Horikoshi Y, Izumi N, Suzuki A, Mizuno K, Ohno S. Lgl mediates apical domain disassembly by suppressing the PAR-3-aPKC-PAR-6 complex to orient apical membrane polarity. J. Cell Sci. 2006;119:2107–2118. doi: 10.1242/jcs.02938. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
supplementary materials and methods, figures, tables and references