Role of Heparan Sulfate in Attachment to and Infection of the Murine Female Genital Tract by Human Papillomavirus (original) (raw)
Abstract
The host factors required for in vivo infection have not been investigated for any papillomavirus. Using a recently developed murine cervicovaginal challenge model, we evaluated the importance of heparan sulfate proteoglycans (HSPGs) in human papillomavirus (HPV) infection of the murine female genital tract. We examined HPV type 16 (HPV16) as well as HPV31 and HPV5, for which some evidence suggests that they may differ from HPV16 in their utilization of HSPGs as their primary attachment factor in vitro. Luciferase-expressing pseudovirus of all three types infected the mouse genital tract, although HPV5, which normally infects nongenital epidermis, was less efficient. Heparinase III treatment of the genital tract significantly inhibited infection of all three types by greater than 90% and clearly inhibited virion attachment to the basement membrane and cell surfaces, establishing that HSPGs are the primary attachment factors for these three viruses in vivo. However, the pseudoviruses differed in their responses to treatment with various forms of heparin, a soluble analog of heparan sulfate. HPV16 and HPV31 infections were effectively inhibited by a highly sulfated form of heparin, but HPV5 was not, although it bound the compound. In contrast, a N-desulfated and N-acylated variant preferentially inhibited HPV5. Inhibition of infection paralleled the relative ability of the variants to inhibit basement membrane and cell surface binding. We speculate that cutaneous HPVs, such as HPV5, and genital mucosal HPVs, such as HPV16 and -31, may have evolved to recognize different forms of HSPGs to enable them to preferentially infect keratinocytes at different anatomical sites.
Papillomaviruses (PVs) are icosahedral DNA viruses that have evolved into numerous genotypes that productively infect diverse vertebrates in a species-specific manner. These viruses also display strict tissue specificity, productively infecting only epithelial cells in the skin and mucosa. These features have inhibited viral propagation in vitro and retarded the development of in vivo models for infection. The human PVs (HPVs) belonging to the alpha genus preferentially infect the genital mucosa, and a subset of this genus include the types (e.g., HPV16, -18, -31, -33, and -45) that are the causative agents of cervical carcinoma. HPV types belonging to the beta genus generally cause asymptomatic skin infections, but certain beta types (e.g., HPV5 and -8) are associated with cutaneous squamous cell carcinomas in individuals with the rare genetic disorder epidermodysplasia verruciformis.
As with other viruses, virion attachment to the host cell is required for successful PV infection. In vitro studies have implicated cell surface heparan sulfate (HS) proteoglycans (HSPGs) as the primary attachment factors for most HPV types (13, 15). HSPGs are composed of a core protein with covalently attached repeating disaccharide units known as glycosaminoglycans. Posttranslational modification of the glycosaminoglycans by acetylation and sulfation leads to substantial heterogeneity that varies across cell type and growth conditions (20, 23). HSPGs are nearly ubiquitously expressed on mammalian cell surfaces, where they are involved in diverse biological processes, including organogenesis, growth factor and cytokine binding, and wound healing. They are also integral components of the basement membrane (BM), the specialized extracellular matrix (ECM) that surrounds most tissues. In this locale, their putative functions include regulation of BM permeability, binding of growth factors, and a role in cellular adhesion (reviewed in reference 10).
HSPGs can also help mediate infection by acting as receptors/coreceptors for some bacterial and viral pathogens (reviewed in reference 12). It is well established that HPV16 utilizes attachment to HSPGs for efficient infection in vitro. However, in vitro studies investigating other HPV types, such as HPV31 and HPV5, have described possible differences. Infection with HPV31 has been reported to be HSPG independent in keratinocyte lines such as HaCaT, although not in other, more transformed lines (17). Also, heparin, which shares the same disaccharide units with HS but is more homogeneous and has a higher level of sulfation, did not inhibit HPV5 infection at doses that efficiently blocked HPV16 infection in vitro (3).
In addition to binding cell surfaces, PVs also bind strongly to the ECM deposited by epithelial cells in vitro and onto the BM in vivo (5, 9, 18). Laminin 5 appears to be the primary molecule mediating in vitro ECM binding (6). However, interaction with an HS moiety on the ECM may be critical for transfer of infectious virions to the cell surface (21). PV cell surface binding in vitro may arise independently of ECM binding; however, the kinetics of in vivo infection suggest that virion binding to the BM may be essential. It is therefore possible that this aspect of in vivo infection could differ from what has been seen in vitro.
It is unclear if HSPGs play any role in PV infection in vivo, as the cellular factors and processes involved in PV infection of epithelial tissues in vivo have not been examined previously. There is a clear precedent of in vitro HSPG dependence for infection of cell lines that does not reflect an in vivo function. For instance, HSPGs facilitate human immunodeficiency virus infection of certain permissive lymphoid cell lines in vitro, yet they play no role in the infection of primary blood lymphocytes (14).
In this study, we utilized our recently developed murine cervicovaginal challenge model (18), which is useful to examine establishment of HPV infection in vivo, to investigate the HSPG dependency of HPV infection, examining both binding and infection of HPV16 pseudovirions in the presence of agents that either compete for HS binding or remove HS from cell surfaces. Because of the published data suggesting possible differences from HPV16 in HSPG dependency for in vitro infection, we also evaluated HPV5 and HPV31 pseudovirions.
MATERIALS AND METHODS
Antibodies and reagents.
The following were obtained from Sigma: heparin (H4784), _N_-acetylheparin (A8036), _N_-acetyl-de-_O_-sulfated heparin (A6039), de-_N_-sulfated heparin (D4776), chondroitin-6-sulfate (C4384), and heparinase III (H8891). Prior to use, heparinase was resuspended in 20 mM Tris, 50 mM NaCl, 4 mM CaCl2, 0.01% bovine serum albumin. The rabbit polyclonal antisera raised against HPV16 L1 capsids and HPV31 L1 capsids have been previously described (19). The same rabbit immunization protocol was used here to raise polyclonal antiserum to HPV5 L1 capsids. These sera recognize both conformational and linear epitopes. The rabbit polyclonal antiserum recognizing laminin 5 was purchased from Abcam (ab14509). The anti-HPV16 antibody Camvir-1 was purchased from Abcam (ab69).
Cell lines and pseudovirus production.
HaCaT cells and 293TT cells were grown in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Pseudovirus stocks were produced by the production method available on our laboratory website (http://ccr.cancer.gov/staff/links.asp?profileid=5637). Nucleotide maps of the HPV16, HPV31, and HPV5 expression plasmids, p16Shell, p31Shell, and p5Shell, and the reporter plasmids for luciferase (pCLucf) and red fluorescent protein (RFP) (pRwb) are also available at this site. To purify capsids, the clarified and matured cell lysates were ultracentrifuged through Optiprep gradients, as previously described (2).
Pseudovirus characterization.
Pseudovirus stocks were characterized for encapsidated DNA, L1 content, and infectivity. Encapsidated DNA was released from the L1/L2 capsids by incubation with proteinase K (10 μl pseudovirus preparation plus 10 μl of 2% proteinase K solution, incubated for 10 min at 55°C), and the capsids were resolved by electrophoresis and quantified. The L1 content of the pseudovirus stocks was determined by comparison to bovine serum albumin standards after sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The titer of the pseudovirus stocks that encapsidated pRwB was determined through flow cytometric analysis of 293TT cells at 48 h following infection. The titers of pseudovirions with encapsidated pCLucf were determined by quantification of luciferase activity using the BriteLite Plus method (Perkin-Elmer) on a BMG Polarstar Optima microplate reader.
In vivo infection of murine genital tracts.
The protocol previously described (18) was followed, with the exception of the additional use of the Whitten effect to induce hormonal synchronization of the mice. For this modification, bedding from the cages of male BALB/c mice, harvested after at least 4 days of habitation, was added to cages containing female mice 8 days prior to instillation of pseudovirus (7). Depo-Provera treatment according to the published protocol occurred on the fourth day after bedding addition, 4 days prior to pseudovirion instillation.
Pseudovirus stocks encapsidating pCLucf and pRwB were coinstilled after premixing with either the inhibitor in question or an equal volume of diluent with 4% carboxymethylcellulose (CMC) and brought to a final volume of 30 μl. The volume of inhibitor (from a 100-mg/ml heparin stock) added to the inocula was in excess by at least 1,000-fold compared to the amount of L1 (ng/μl) determined to be present in the pseudovirus stock, typically 500 μg of heparin. At 48 h following the introduction of pseudovirus, the mice were each given an intravaginal instillation of 20 μl of luciferin (0.3 mg) and imaged with a Xenogen IVIS 100 (Caliper Life Sciences). Images were taken at 3 min postinstallation of luciferin at medium binning with a 30-s exposure. Images were then analyzed by drawing an equally sized region of interest for each mouse and measuring total flux (photons/second). Statistical analysis was done with GraphPad Prism software, in which a one-tailed unpaired t test was used to determine P values.
Following imaging, the mice were sacrificed, and their genital tracts were excised, washed with phosphate-buffered saline (PBS), and frozen in tissue freezing medium (Triangle Biomedical Sciences). Following cryostat sectioning, slides were fixed in 2% paraformaldehyde in PBS for 15 min and subsequently soaked in 100 mM glycine with 0.1% sodium azide in PBS overnight. When indicated, tissue sections were overlaid with DAPI (4′,6′-diamidino-2-phenylindole)-containing mounting medium (Invitrogen), and infectious events, indicated by RFP signal, were measured. To monitor in vivo binding, mice were sacrificed 4 hours after genital challenge, and their genital tissues were excised, frozen, cryosectioned, and subjected to immunofluorescent staining, using the appropriate rabbit anticapsid polyclonal antiserum.
For the in vivo heparinase treatment, mice were hormonally prepared for genital challenge as described above. Prior to pseudovirus instillation, mice were treated with a 4% nonoxonyl-9 solution in 4% CMC spiked with either heparinase buffer or heparinase III (1.7 units/mouse). At 5 hours following pretreatment, the mice were challenged with HPV16-RFP, combined with an additional treatment of heparinase III (3.3 units/mouse, for the mice pretreated with heparinase III) or heparinase buffer (diluted into 4% CMC for a total volume of 30 μl, for mice pretreated with buffer). Mice were sacrificed 4 hours later, and their genital tissues were excised and analyzed for capsid binding by immunofluorescent staining. All microscopy was performed on a Zeiss LSM 510 system. Additionally, mice were prepared as described above and challenged with HPV16-pCLucf. Luciferase expression was measured after 24, 48, and 72 h to determine in vivo inhibition.
Immunofluorescent staining.
Prior to staining, tissue sections were blocked with 10% donkey serum in PBS with 0.1% Brij 58 for 30 min at room temperature. Antisera recognizing the HPV capsids were diluted 1:1,000. The anti-laminin 5 serum was diluted 1:100. Bound antibody was detected with Alexa Fluor 488-conjugated donkey anti-rabbit serum (Invitrogen). Following staining, sections were mounted with DAPI-containing mounting solution (Invitrogen).
In vitro infection.
Pseudovirus was added to HaCaT cells that had been grown overnight at a plating density of 1 × 105 cells per well in 24-well plates. When heparin (H4784) was used, it was serially diluted from 200 μg/ml to 0.2 μg/ml and added immediately following addition of pseudovirus. The percentage of RFP-transduced cells was determined by flow cytometric analysis after a 72-h infection. Each condition was performed in triplicate.
Heparin binding column.
Heparin binding capacity was assessed through the use of the HiTrap Heparin HP (Amersham Biosciences) system. HiTrap heparin columns (1 ml) were equilibrated with 10 to 15 ml of binding buffer (10 mM sodium phosphate buffer [pH 7.0], 0.36 M NaCl). When indicated, heparin (H4784) was preincubated with the pseudovirus stock (∼2 μg of L1) at a concentration of 50 mg/ml in a 200-μl final volume for 90 min on ice. Pseudovirus samples were then diluted 1:10 with binding buffer and applied to the column, and flowthrough fractions were collected. The column was then washed with 10 to 15 ml of binding buffer prior to elution with 10 mM sodium phosphate buffer (pH 7.0)-0.8 M NaCl. After elution, fractions were collected, the regeneration buffer (10 mM sodium phosphate buffer [pH 7.0], 2.0 M NaCl) was added to the column, and the remaining fractions were collected. Fractions collected following addition of the sample, the elution buffer, and the regeneration buffer were assayed for protein content in a bicinchoninic acid assay (Pierce, microplate procedure). Western blot analysis was performed to specifically determine which fractions contained L1 protein. HPV16 L1 was detected with an anti-L1 monoclonal antibody (Camvir1; Abcam), whereas HPV31 and HPV5 L1 species were each detected with their respective rabbit polyclonal antisera. Imaging was performed with a Fujifilm LAS-4000 chemiluminescent imaging system.
RESULTS
Effect of heparin on in vivo HPV infection.
HPV16 virions are known to bind heparin in vitro, and this binding inhibits in vitro infection in association with the inhibition of virion adsorption to cell surface (9, 13, 15). We sought to determine whether heparin treatment would inhibit in vivo infection by HPV16, HPV31, and HPV5 with a similar degree of efficiency. To enable us to conduct a valid comparison of the in vivo infectivities of the three pseudoviruses, we first determined the L1 content and encapsidated marker plasmid DNA content of the pseudovirus preparations. For in vivo infection, we normalized the amount of the three pseudovirion stocks for inoculation based on the amount of the encapsidated marker plasmid. For assays that employed heparin, we adjusted the amount of heparin per L1 (by weight) for inclusion in the infection and binding inhibition assays.
Mice were prepared for intravaginal pseudovirus instillation by progesterone and nonoxonol-9 treatment, according to the protocol previously established in the laboratory (18). However, while pseudovirus infection had previously been detected with an RFP-encoding marker plasmid, for this study, the marker plasmid pCLucF, encoding luciferase, was encapsidated and transduced, to conveniently enable the quantitative measurement of pseudovirus infection in living animals over time. The ability to measure infection at more than one time point made it possible to readily determine if infection had been prevented, or merely temporally retarded, by various treatments.
When the relative infectivities of the three HPV types were compared, luciferase activities resulting from HPV16 and HPV31 infection fell within a similar range (representative mice are shown in Fig. 1). However, despite normalization of the encapsidated DNA delivered, HPV5 infected less efficiently than HPV16 or HPV31 and is therefore shown in Fig. 1 with a different scale of flux.
FIG. 1.

Visualization of luciferase-expressing pseudovirus infection in vivo. Quantification of luciferase expression was performed on an IVIS 100 imaging system at 48 h postinfection. Representative animals are shown for the three PV types. The color scale represents expression levels. Note that the scales vary among the three virus types, indicating different luciferase expression levels.
To determine the degree to which heparin could affect cervicovaginal infection, we instilled the same normalized amount of each virus after it had been preincubated with a heparin stock. For this experiment, we used the highly sulfated heparin (Sigma H-4784) that had previously found to be the most inhibitory variant for HPV33 infection in vitro (22). This treatment significantly inhibited HPV16 and HPV31 infection (Fig. 2). HPV16 was inhibited by 95% (P < 0.0001), and HPV31 infection was inhibited by greater than 99% (P = 0.0043). In contrast, HPV5 was not significantly inhibited by this dose of heparin (P = 0.0991), although there was a trend toward inhibition, as the luciferase activity was about one-half of that obtained in the absence of heparin. It is notable that for HPV31, which was the HPV type most inhibited by heparin, the luciferase activity in the presence of heparin was significantly greater than that in the negative control (P = 0.0022), implying that even its inhibition by heparin was not complete. We found no evidence that heparin treatment delayed the kinetics of pseudovirus infection. Mice were imaged daily for 5 days following infection (data not shown).
FIG. 2.

Inhibition of in vivo infection with heparin. The luciferase signal following infection with the pseudoviruses, either untreated or with heparin coinstillation, is shown at 48 h postinfection. Each group was composed of 10 animals. The percent inhibition is indicated above the bars. The significance is noted when relevant. Error bars indicate standard errors.
Inhibition of in vitro infection with heparin.
To evaluate in parallel the heparin inhibition of the three HPV types in vitro, we performed infections of HaCaT cells using RFP-expressing pseudovirus. Both HPV16 and HPV31 infections of HaCaT cells were similarly diminished by heparin H4784 in a dose-dependent manner, with 50% inhibitory concentrations of 2.3 μg/ml and 2.8 μg/ml, respectively (Fig. 3). However, HPV5 infection was not substantially inhibited by heparin H4784. In fact, a slight enhancement of infection was observed at the lower doses in multiple experiments. This in vitro observation is consistent with our previously published observations (3). Thus, as with the in vivo results, HPV16 and HPV31 are sensitive to heparin H4783 in vitro, while HPV5 is resistant.
FIG. 3.

Inhibition of in vitro infection with heparin. The effect of heparin (H4784) on pseudovirus infection was determined on HaCaT cells. Pseudovirions that contained an encapsidated RFP-expressing reporter plasmid were utilized. The heparin was serially diluted from 256 μg/ml to 1 μg/ml, and infection was compared to samples with no exogenous heparin. All infections were performed in triplicate. Each of the untreated pseudovirus inocula infected approximately 20% of the cells. Infection was evaluated at 48 h. Error bars indicate standard errors .
Inhibition of in vivo infection by other forms of heparin.
As in vitro studies have demonstrated a clear hierarchy of inhibitory potentials among various formulations of heparin and other sulfated polysaccharides (3, 22), we evaluated a panel of heparins and chondroitin-6-sulfate in the genital challenge model (Fig. 4). Luminescence values compared to the positive control suggested that that _N_-acetylheparin (A8036) inhibits infection with HPV16 and HPV31 approximately as well as H4784 and that de-_N_-heparin (D4776) may be more inhibitory against HPV5 than the H4784 formulation. The other heparin variants, as well as chondroitin-6-sulfate (C4384), did not substantially inhibit cervicovaginal infection by HPV16, HPV31, or HPV5. Taken together, these results suggest a role for the N sulfation of HS in enabling efficient infection of HPV16 and HPV31. However, there was considerable variation within each sample set, the sample size was small (n = 3), and none of the differences between the positive control and the experimental conditions reached statistical significance. Thus, this conclusion must be considered tentative.
FIG. 4.

Inhibition of in vivo infection with polysaccharides. Infection, as measured by luciferase expression, was determined following coinstillation of pseudoviruses with chondroitin-6 sulfate or different heparin formulations as indicated. Each group was composed of three mice. Mice were examined at 48 h postinfection. Error bars indicate standard errors.
Microscopic examination of tissues.
In addition to luminescence analysis, the cellular distribution within the infected tissue in the presence of the inhibitors was evaluated by confocal microscopy for all three HPV types. For this analysis, we coinfected the genital epithelium with pseudovirions containing a plasmid encoding RFP and with the pseudovirions containing the luciferase plasmid. Thus, we are able to analyze infection by two different methods: quantitatively, by measuring luminescence detected in the tissue, and histologically, by microscopically analyzing fluorescence in specific cells of the excised cervicovaginal tissue. Coinstillation of pseudovirus stocks of the same HPV type with different encapsidated marker plasmids did not affect the magnitude or spectrum of infected cells compared to instillation of the individual stocks (data not shown). As previously reported for HPV16-RFP pseudovirus (18), we found that HPV31 and HPV5 pseudovirions also specifically infected epithelial cells. All layers of the N-9-disrupted epithelium, from the basal keratinocytes to the superficial cells, contained infected cells at 3 days postinoculation (data not shown). None of the inhibitors altered the pattern of infection, although some, as described below, clearly affected the relative number of infected cells, in parallel with the relative reduction in magnitude of the intravaginal luminescence.
Effect of heparin on capsid binding in the female genital tract.
To evaluate what aspect(s) of the infectious process might be inhibited by heparin, we compared the tissue distribution pattern and relative intensity of pseudovirion binding within the N-9-disrupted cervicovaginal tissue. As previously reported for HPV16 (18), untreated pseudovirions of all three HPV types bound strongly to the BM at an early time point after inoculation (4 h). However, in the presence of heparin, the BM association of both HPV16 and HPV31 was dramatically diminished to a signal indistinguishable from background (Fig. 5B and D). There was also no indication of binding to cells under these conditions by immunohistological assessment. The heparin-treated HPV5 showed more variable BM binding than that observed with the untreated capsids. HPV5 binding to the BM was generally decreased in the presence of heparin, but a strong association, indistinguishable from that of the untreated virus, was observed in some regions of the tissue (Fig. 5F). For all three HPV types, we did not find any indication of cellular binding in conjunction with the heparin treatment. However, as previously shown, the majority of untreated capsids are associated with the BM at this early time point.
FIG. 5.

Pseudovirion binding in the presence of heparin. Genital tissue was excised 4 hours after instillation of the pseudoviruses to evaluate the effect of heparin on tissue binding. Pseudovirions were detected with rabbit polyclonal antisera and Alexa Fluor 488-conjugated donkey anti-rabbit secondary antiserum. HPV16 is shown in panels A and B. HPV31 is shown in panels C and D. HPV5 is shown in panels E and F. The leftmost image in each case (A, C, and E) shows the detection of untreated virus. The rightmost image (B, D, and F) shows detection of virus in the presence of heparin.
We also examined the pattern of capsid binding with the panel of polysaccharides utilized in the infectivity analyses. Among the heparin variants used in this study, A8036, which has diminished N sulfation, prevented BM association most dramatically. Notably, this heparin variant not only inhibited binding of HPV16 and HPV31 but additionally affected HPV5 binding. In contrast, D4776, which completely lacks N sulfation, displayed no inhibitory effect on the binding of HPV16 and HPV31, but it did substantially prevent binding of HPV5. BM binding of all three virus preparations in the presence of _N_-acetyl-de-_O_-heparin (A6039) or chondroitin-6-sulfate (C4384) was indistinguishable from that of the positive controls (data not shown). Relative BM binding under the various conditions is indicated in Table 1. In general, the inhibition of BM association correlated with the ability of a particular heparin derivative to prevent infection in vivo.
TABLE 1.
Relative BM binding of HPV16, HPV31, and HPV5 following coinstillation with different polysaccharide preparations
| Polysaccharide prepn | Relative BM binding ofa: | ||
|---|---|---|---|
| HPV16 | HPV31 | HPV5 | |
| None (untreated) | +++ | +++ | +++ |
| H4784 | − | − | +++ |
| D4776 | +++ | +++ | + |
| A6039 | ++ | +++ | ++ |
| A8036 | + | + | ++ |
| C4384 | +++ | ++ | +++ |
Heparin binding ability of PV capsids.
Given the observation that unmodified heparin did not substantially inhibit HPV5 infection in vitro or in vivo, we wanted to determine if these capsids might interact less efficiently with heparin. We therefore compared the abilities of the three different HPV types to bind to a Hi-Trap heparin column (the heparin in the column is similar, but not identical, to heparin H4784). In this assay, the virus preparation was applied to the column, unbound virus was detected in the flowthrough, and bound virus was detected after elution with 0.8 M salt. The relevant fractions were screened for L1 content by Western blot analysis (Fig. 6). For both HPV16 and HPV31, L1 was not detected in the flowthrough but was clearly detected after elution by salt addition. With HPV5, some capsids were evident in the flowthrough, although the majority were associated with the column until salt elution. The amount of HPV5 capsids in the flowthrough fractions was somewhat variable but was observed in multiple independent experiments.
FIG. 6.
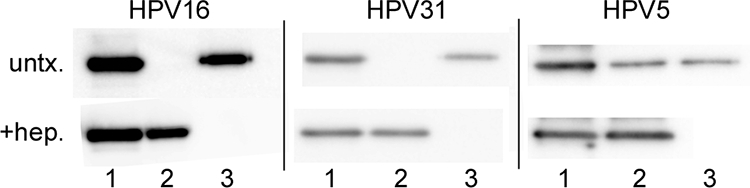
Interaction of pseudovirions with heparin. The ability of pseudovirions to bind to a heparin HiTrap column was evaluated by Western analyses. Lanes 1, input pseudovirions; lanes 2, pseudovirions present in the flowthrough fraction; lanes 3, pseudovirions present following 0.8 M NaCl elution. untx., profile of untreated pseudovirions; +hep., profile of pseudovirions following incubation with heparin (H4784).
To determine if the heparin formulation (H4784) used in this study was specifically able to bind to HPV5, we preincubated each HPV pseudovirus with this heparin and added it to the column. When preincubated with H4784, all three virus preparations were detected in the flowthrough fractions, indicating an interaction of the particles, including HPV5, with this particular heparin variant.
These results indicate that the inability of heparin to significantly inhibit HPV5 infection is not attributable to an inability of heparin to bind the capsids. However, they do not rule the possibility that heparin may bind to HPV5 capsids with a lower affinity than to either HPV16 or HPV31 capsids.
Effect of heparinase treatment of pseudovirus infection.
The heparin inhibition studies outlined above suggest that HSPGs function as the primary attachment factors for HPV16 and HPV31 in vivo. However, they do not rule out the possibility that heparin binding prevents binding and infection by occluding sites on the capsid involved in the interaction with an HSPG-independent receptor or that HPV5 can bind to some modifications of HSPGs even when the capsids are complexed with heparin. To more directly address the role of the HSPGs in virus attachment and infection, we evaluated these parameters after in vivo treatment of the cervicovaginal tract with heparinase III, which is a heparin-degrading lyase that recognizes HS as its primary substrate (11). Pretreatment with heparinase III inhibited infection by all three pseudoviruses by at least 89% at days 1, 2, and 3 postinoculation (Fig. 7 shows day 2 data), and the differences were significant for all three viruses (P values using an unpaired t test were >0.05 for HPV16 and HPV31 and >0.005 for HPV5 for the day 2 results). There was no suggestion that HPV5 or HPV31 was less HSPG dependent than HPV16 as evaluated by this technique. It is noteworthy that infection by HPV16 and -31 in heparinase III-pretreated mice was above background, as we had also observed with heparin treatment. Incomplete HSPG cleavage or HPSG-independent infection by a fraction of the capsids could account for this residual infection.
FIG. 7.

Heparinase inhibition of in vivo infection. The luciferase signal at 48 h following infection with the pseudoviruses either of untreated animals or following heparinase treatment (hepx) is shown. Each group was composed of five animals. The percent inhibition is indicated above the bars. Error bars indicate standard errors.
BM binding of HPV16 was also examined following enzymatic digestion in vivo with heparinase III. We found that this treatment caused a substantial diminution of BM association compared to that observed in the untreated samples (Fig. 8, compare panels A and B). There was essentially no capsid binding to the BM in two of the treated mice, whereas there was detectable, but greatly diminished, binding to the BM in the third treated mouse. Heparinase treatment also effectively prevented cell surface binding. The distribution and intensity of laminin 5 were unaffected by the heparinase treatment as determined by immunostaining (Fig. 8C and D).
FIG. 8.

Detection of HPV16 binding and laminin 5 following heparinase treatment. Genital tissue was excised 4 hours after instillation of HPV16 pseudovirus to evaluate the effect of heparinase treatment on tissue binding. Detection of HPV16 binding is shown in panel A (untreated) and panel B (heparinase treated). Laminin 5 staining is shown in panel C (untreated) and panel D (heparinase treated).
DISCUSSION
The results of this study strongly support the conclusion that HSPGs are the primary attachment factors utilized by HPVs when they infect the genital tract. It was evident that binding to both the BM and the cell surface is dependent upon HSPGs in vivo. Because we found that the treatments employed equally disrupted viral interactions with the BM and cells, our results cannot distinguish whether HSPG-mediated binding of the BM, cell surface, or both is required for efficient infection of the female genital tract. We recently reported that capsids can efficiently infect cultured cells lacking HS in the presence of exogenous furin (8), raising the possibility that cell surface HSPGs may not play an obligatory role in vivo if a sufficient level of furin is present in the female genital tract, either in the extracellular space or in the BM. Furin has been reported to have a naturally truncated, secreted form which exhibits functional activity (24) and therefore could reasonably be present in the extracellular space in vivo. It is also possible to envision that capsid binding to HSPG present on the BM is followed by a conformational change in the capsid that allows furin cleavage and subsequent transfer to a non-HSPG cell surface receptor. The residual infection observed after heparin or heparinase treatment may indicate that a subset of pseudovirions are in a conformation that can be more readily be cleaved by furin and thus bind to the cell surface without the contribution of HSPGs. Alternatively, it may reflect the existence of a relatively inefficient HS-independent pathway of HPV infection in the female genital tract.
Our results do not support a role of laminin 5 in BM binding, as previously proposed for ECM binding in vitro (6), as heparinase treatment greatly inhibited capsid binding but did not detectably affect the laminin 5 content of the BM. This finding suggests that the ECM deposited by monolayer culture of keratinocytes and the genital tract BM are not equivalent structures with respect to PV binding. Another recent study suggested that although the binding to ECM in vitro was not largely due to HSPG, PV interaction with HSPG was important for infectious transfer from the ECM to cells (21). This observation supports the idea that viral interaction with the HSPG on the BM and ECM may be mechanistically similar. Although our data clearly show that initial BM association is not laminin 5 dependent, subsequent interaction cannot be eliminated.
We found no evidence that HPV16 and HPV31 differ in any substantive way in their interaction with HSs or their dependency on HSPGs for infection. The inhibition of in vivo infection exhibited by various forms of heparin was indistinguishable for the two viruses, as were the effects of heparinase treatment. At present it is unclear why our findings for HPV31 differ from those in a previous study which concluded that infection of cultured keratinocytes by HPV31 was HSPG independent (17). It is unlikely to simply be a difference between the requirements for in vivo and in vitro infection. Using the same keratinocyte line, HaCaT, employed in the previous study, we found that the 50% inhibitory concentrations for heparin were virtually identical for HPV16 and HPV31. It is possible that there may be differences in particle maturation and furin accessibility during the harvest of raft-derived viruses utilized in the previous study and culture-derived pseudoviruses employed here.
The HSPG dependency for in vivo HPV infection was also assessed for HPV5, a representative of the beta genus. This was the first evaluation of genital tract infection by a beta type, which are normally detected on nongenital skin surfaces. Relatively robust infection of HPV5 was detected, albeit at lower levels than that seen with an equivalent number of pseudogenomes encapsidated by HPV16 or HPV31 genomes. Therefore, it appears that tissue tropism, as with species specificity, is determined primarily by events after the initial establishment of virus infection. Consistent with this conclusion, HPV16 pseudovirions were recently shown to infect mouse nongenital epidermis (1).
The results of the heparinase studies clearly indicate that HSPGs are also involved in the BM attachment and infection of HPV5. This was a somewhat unexpected result, as heparin treatment did not significantly block HPV5 infection in vivo or in vitro, despite having the capacity to bind to the capsid. The most likely explanation for this discrepancy is that HPV5 has several distinct HS binding sites and that a site that does not efficiently interact with soluble heparin mediates BM/cell surface attachment. The presence of at least two distinct HS binding sites on alpha types was previously postulated (16). Another consideration is that the basic amino acids, arginine and lysine, which are essential for heparin binding, have been shown to be less important for HS interaction. The more heterogeneous HS can also accommodate a wider range of structure, and nonionic interactions could be more relevant in this binding (4). Therefore, we must also conclude that results obtained using soluble analogs of attachment factors in inhibitor studies do not invariably predict the biological significance of the factor.
The importance of particular N- and O-sulfation patterns of heparin for inhibition of in vitro infection of alpha types was previously documented (22). Although equally dependent on HSPGs for in vivo infection, HPV5 may preferentially bind HS chains with a subset of modifications different from those with which HPV16 and HPV31 interact. For example, our preliminary data suggest that de-N-sulfated heparin and _N_-acetylheparin are more inhibitory than the highly sulfated H4784 variant for in vivo HPV5 infection but less inhibitory for HPV16 and HPV31. Differences in the hierarchy of HS affinities between the alpha and beta genera may reflect the adaptation to preferentially infect keratinocytes at different anatomic sites, which might be distinguished by subtle differences in HSPG sulfation patterns.
Acknowledgments
This research was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research.
Footnotes
▿
Published ahead of print on 10 December 2008.
REFERENCES
- 1.Alphs, H. H., R. Gambhira, B. Karanam, J. N. Roberts, S. Jagu, J. T. Schiller, W. Zeng, D. C. Jackson, and R. B. Roden. 2008. Protection against heterologous human papillomavirus challenge by a synthetic lipopeptide vaccine containing a broadly cross-neutralizing epitope of L2. Proc. Natl. Acad. Sci. USA 1055850-5855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Buck, C. B., D. V. Pastrana, D. R. Lowy, and J. T. Schiller. 2004. Efficient intracellular assembly of papillomaviral vectors. J. Virol. 78751-757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buck, C. B., C. D. Thompson, J. N. Roberts, M. Muller, D. R. Lowy, and J. T. Schiller. 2006. Carrageenan is a potent inhibitor of papillomavirus infection. PLoS Pathog. 2e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caldwell, E. E., V. D. Nadkarni, J. R. Fromm, R. J. Linhardt, and J. M. Weiler. 1996. Importance of specific amino acids in protein binding sites for heparin and heparan sulfate. Int. J. Biochem. Cell. Biol. 28203-216. [DOI] [PubMed] [Google Scholar]
- 5.Culp, T. D., L. R. Budgeon, and N. D. Christensen. 2006. Human papillomaviruses bind a basal extracellular matrix component secreted by keratinocytes which is distinct from a membrane-associated receptor. Virology 347147-159. [DOI] [PubMed] [Google Scholar]
- 6.Culp, T. D., L. R. Budgeon, M. P. Marinkovich, G. Meneguzzi, and N. D. Christensen. 2006. Keratinocyte-secreted laminin 5 can function as a transient receptor for human papillomaviruses by binding virions and transferring them to adjacent cells. J. Virol. 808940-8950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dalal, S. J., J. S. Estep, I. E. Valentin-Bon, and A. E. Jerse. 2001. Standardization of the Whitten effect to induce susceptibility to Neisseria gonorrhoeae in female mice. Contemp Top. Lab Anim. Sci. 4013-17. [PubMed] [Google Scholar]
- 8.Day, P. M., D. R. Lowy, and J. T. Schiller. 2008. Heparan sulfate-independent cell binding and infection with furin-precleaved papillomavirus capsids. J. Virol. 8212565-12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Day, P. M., C. D. Thompson, C. B. Buck, Y. Y. Pang, D. R. Lowy, and J. T. Schiller. 2007. Neutralization of human papillomavirus with monoclonal antibodies reveals different mechanisms of inhibition. J. Virol. 818784-8792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erickson, A. C., and J. R. Couchman. 2000. Still more complexity in mammalian basement membranes. J. Histochem Cytochem. 481291-1306. [DOI] [PubMed] [Google Scholar]
- 11.Ernst, S., R. Langer, C. L. Cooney, and R. Sasisekharan. 1995. Enzymatic degradation of glycosaminoglycans. Crit. Rev. Biochem. Mol. Biol. 30387-444. [DOI] [PubMed] [Google Scholar]
- 12.Fears, C. Y., and A. Woods. 2006. The role of syndecans in disease and wound healing. Matrix Biol. 25443-456. [DOI] [PubMed] [Google Scholar]
- 13.Giroglou, T., L. Florin, F. Schafer, R. E. Streeck, and M. Sapp. 2001. Human papillomavirus infection requires cell surface heparan sulfate. J. Virol. 751565-1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ibrahim, J., P. Griffin, D. R. Coombe, C. C. Rider, and W. James. 1999. Cell-surface heparan sulfate facilitates human immunodeficiency virus type 1 entry into some cell lines but not primary lymphocytes. Virus Res. 60159-169. [DOI] [PubMed] [Google Scholar]
- 15.Joyce, J. G., J. S. Tung, C. T. Przysiecki, J. C. Cook, E. D. Lehman, J. A. Sands, K. U. Jansen, and P. M. Keller. 1999. The L1 major capsid protein of human papillomavirus type 11 recombinant virus-like particles interacts with heparin and cell-surface glycosaminoglycans on human keratinocytes. J. Biol. Chem. 2745810-5822. [DOI] [PubMed] [Google Scholar]
- 16.Knappe, M., S. Bodevin, H. C. Selinka, D. Spillmann, R. E. Streeck, X. S. Chen, U. Lindahl, and M. Sapp. 2007. Surface-exposed amino acid residues of HPV16 L1 protein mediating interaction with cell surface heparan sulfate. J. Biol. Chem. 28227913-27922. [DOI] [PubMed] [Google Scholar]
- 17.Patterson, N. A., J. L. Smith, and M. A. Ozbun. 2005. Human papillomavirus type 31b infection of human keratinocytes does not require heparan sulfate. J. Virol. 796838-6847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts, J. N., C. B. Buck, C. D. Thompson, R. Kines, M. Bernardo, P. L. Choyke, D. R. Lowy, and J. T. Schiller. 2007. Genital transmission of HPV in a mouse model is potentiated by nonoxynol-9 and inhibited by carrageenan. Nat. Med. 13857-861. [DOI] [PubMed] [Google Scholar]
- 19.Roden, R. B., H. L. Greenstone, R. Kirnbauer, F. P. Booy, J. Jessie, D. R. Lowy, and J. T. Schiller. 1996. In vitro generation and type-specific neutralization of a human papillomavirus type 16 virion pseudotype. J. Virol. 705875-5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt, A., A. Skaletz-Rorowski, and E. Buddecke. 1995. Basic fibroblast growth factor controls the expression and molecular structure of heparan sulfate in corneal endothelial cells. Eur. J. Biochem. 234479-484. [DOI] [PubMed] [Google Scholar]
- 21.Selinka, H. C., L. Florin, H. D. Patel, K. Freitag, M. Schmidtke, V. A. Makarov, and M. Sapp. 2007. Inhibition of transfer to secondary receptors by heparan sulfate-binding drug or antibody induces noninfectious uptake of human papillomavirus. J. Virol. 8110970-10980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selinka, H. C., T. Giroglou, T. Nowak, N. D. Christensen, and M. Sapp. 2003. Further evidence that papillomavirus capsids exist in two distinct conformations. J. Virol. 7712961-12967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turnbull, J., A. Powell, and S. Guimond. 2001. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 1175-82. [DOI] [PubMed] [Google Scholar]
- 24.Vey, M., W. Schafer, S. Berghofer, H. D. Klenk, and W. Garten. 1994. Maturation of the trans-Golgi network protease furin: compartmentalization of propeptide removal, substrate cleavage, and COOH-terminal truncation. J. Cell Biol. 1271829-1842. [DOI] [PMC free article] [PubMed] [Google Scholar]