A mechanosensitive transcriptional mechanism that controls angiogenesis (original) (raw)
. Author manuscript; available in PMC: 2009 Aug 26.
Published in final edited form as: Nature. 2009 Feb 26;457(7233):1103–1108. doi: 10.1038/nature07765
Abstract
Angiogenesis is controlled by physical interactions between cells and extracellular matrix as well as soluble angiogenic factors, such as VEGF. However, the mechanism by which mechanical signals integrate with other microenvironmental cues to regulate neovascularization remains unknown. Here we show that the Rho inhibitor, p190RhoGAP, controls capillary network formation in vitro and retinal angiogenesis in vivo by modulating the balance of activities between two antagonistic transcription factors – TFII-I and GATA2 – that govern gene expression of the VEGF receptor, VEGFR2. Moreover, this novel angiogenesis signaling pathway is sensitive to extracellular matrix elasticity as well as soluble VEGF. This is the first known functional cross-antagonism between transcription factors that controls tissue morphogenesis, and that responds to both mechanical and chemical cues.
Keywords: VEGFR2, p190RhoGAP, TFII-I, GATA2, mechanotransduction, angiogenesis, capillary endothelial cell
Deregulation of angiogenesis – the growth of blood capillaries – contributes to development of many diseases, including cancer, arthritis and blindness1, 2. FDA-approved angiogenesis inhibitors solely target the oxygen-sensitive vascular endothelial growth factor, VEGF; however, neovascularization is also controlled by other microenvironmental signals, including mechanical forces conveyed by extracellular matrix (ECM). For example, although capillary development is driven by angiogenic mitogens, cell sensitivity to these soluble cues can be modulated by physical interactions between cells and ECM that alter cell shape and cytoskeletal structure3-8. Similar changes in capillary cell shape and function can be produced by changing ECM elasticity, adhesivity or topography, applying mechanical stresses, or altering cell-generated traction forces3-8. Mechanical tension also stimulates capillary growth and vascular remodeling in vivo9, and regional variations of ECM mechanics and cell shape appear mediate how neighboring cells undergo localized differentials of growth and differentiation that drive three-dimensional (3D) tissue pattern formation10, 11. But the mechanism by which mechanical signals conveyed by ECM converge with those elicited by growth factors to control gene transcription required for angiogenic control remains unknown.
VEGF and its receptor VEGFR2 are of particular importance in angiogenesis because they are essential for normal blood vessel development12, 13, and deregulation of these factors leads to various pathological conditions1, 2, 14. We therefore investigated the role of VEGF and VEGFR2 in the mechanism by which these mechanical forces regulate capillary development. Analysis of cellular mechanotransduction during angiogenesis has revealed that the small GTPase, Rho, mediates growth control in vitro10, 15, as well as blood vessel development in vivo9, by modulating the mechanical force balance that governs cell shape. Stress-induced distortion of the capillary cell cytoskeleton regulates Rho activity by controlling its upstream inhibitor, p190RhoGAP16. p190RhoGAP was shown to bind to the transcription factor TFII-I and sequester it in the cytoplasm of fibroblasts17, and TFII-I is a multifunctional transcription factor that regulates VEGFR2 expression in large vessel endothelial cells 18 19. Although it remains unknown whether it plays a role in angiogenesis, TFII-I deletions are associated with cardiovascular defects20. These findings raise the possibility that TFII-I might also contribute to intracellular signaling mechanisms triggered by mechanical forces during vascular network formation.
p190RhoGAP and TFII-I regulate VEGFR2
To explore whether p190RhoGAP modulates vascular development by altering gene transcription, we knocked down p190RhoGAP in human microvascular endothelial (HMVE) cells using siRNA. TFII-I protein levels were approximately 1.5 times higher in the nuclear fraction of knockdown cells (Supplementary Fig. S2a), and this was confirmed by fluorescence microscopy (Supplementary Fig. S2b). As first shown in fibroblasts17, we found that p190RhoGAP co-precipitates with TFII-I (and vice versa) (Supplementary Fig. S2c), suggesting that p190RhoGAP may also bind TFII-I and sequester it in the cytoplasm of human capillary cells.
TFII-I upregulates VEGFR2 protein expression in human aortic endothelial cells by binding to the VEGFR2 promoter18. But TFII-I knockdown in our HMVE cells increased (rather than decreased) VEGFR2 mRNA and protein levels by 2- to 3-fold relative to control cells, and overexpressing TFII-I (delta isoform21) using lentiviral transduction produced the opposite effect (Fig. 1a, Supplementary Fig. S3a). This may be due to the fact that macrovascular and microvascular endothelial cells undergo distinct morphogenetic programs (produce large tubes versus branching capillaries).
Fig. 1. TFII-I and GATA2 control VEGFR2 expression via p190RhoGAP.
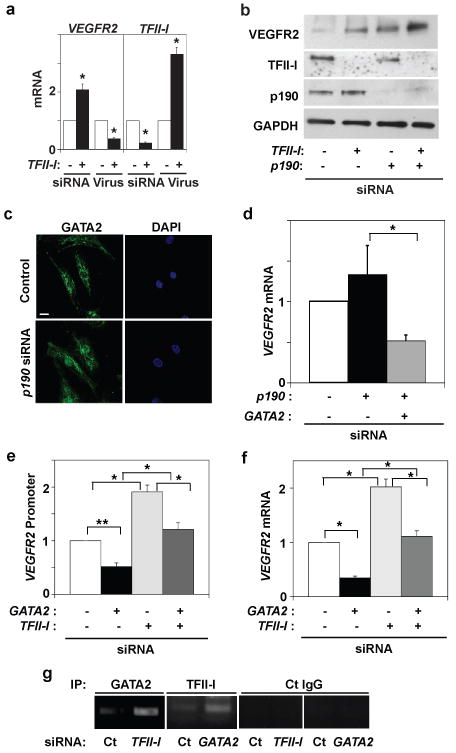
a) VEGFR2 and TFII-I mRNA levels in cells treated with TFII-I siRNA or lentiviral vectors (virus) relative to control cells (*, p < 0.05; unpaired Student's t-test is used throughout). b) Immunoblots showing VEGFR2, TFII-I, p190RhoGAP, and GAPDH protein levels in cells treated with TFII-I or p190RhoGAP siRNAs, or both. c) Immunofluorescence micrographs showing GATA2 distribution and nuclear DAPI staining in control and p190RhoGAP knockdown cells cultured with 0.3% serum (bar, 5μm). d) VEGFR2 mRNA level in cells transfected with p190RhoGAP siRNA alone or with GATA2 siRNA (*, p<0.01). VEGFR2 promoter activities (e) and mRNA levels (f) in cells transfected with GATA2 or TFII-I siRNA alone or in combination (*, p<0.01; **, p<0.05). g) ChIP analysis showing VEGFR2 promoter co-immunoprecipitating with GATA2 or TFII-I antibodies in cells transfected with TFII-I or GATA2 siRNA (Ct, control; n=3; error bars = s.e.m. throughout).
TFII-I binds to the Inr region of the VEGFR2 gene promoter, and smaller portions of this promoter (human VEGFR2: -225 to +268, -570 to +268, and -780 to +268) have similar or greater activity compared to the full length promoter (4kb)18, 22. When these various VEGFR2 promoters were characterized using a luciferase assay in human umbilical vein endothelial (HUVE) cells, TFII-I knockdown increased, and overexpression of TFII-I using DNA transfection decreased, VEGFR2 promoter activity by 1.5 to 2-fold and one-fifth normal levels, respectively (Supplementary Fig. S3b). The promoter-less pGL3 basic reporter showed no promoter activity (not shown). Moreover, mutagenesis of the VEGFR2 Inr (-780+268; 780MUT) decreased promoter activity in both control and TFII-I knockdown cells (Supplementary Fig. S3b), confirming that TFII-I decreases VEGFR2 promoter activity through the Inr region. Importantly, the specificity of these effects of TFII-I knockdown were confirmed by demonstrating that reconstitution of TFII-I can reverse these effects (Supplementary Fig. S3c).
We next asked whether p190RhoGAP stimulates VEGFR2 expression by restricting nuclear translocation of TFII-I (Supplementary Fig. S2a,b). TFII-I knockdown increased, and p190RhoGAP knockdown decreased, VEGFR2 promoter activity (-780+268) relative to control cells (Supplementary Fig. S3d). The double knockdown of p190RhoGAP and TFII-I exhibited the same stimulation as observed in the TFII-I knockdown cells (Supplementary Fig. S3d). However, p190RhoGAP knockdown did not decrease VEGFR2 mRNA and protein levels; in fact, it increased VEGFR2 protein levels (Fig. 1b, Supplementary Fig. S3d). Moreover, p190RhoGAP and TFII-I double knockdown produced similar or slightly higher levels of VEGFR2 mRNA and protein expression (Fig. 1b, Supplementary Fig. S3d) compared to either p190RhoGAP or TFII-I knockdown alone. These results raised the intriguing possibility that an antagonist of TFII-I activity might exist that also contributes to p190RhoGAP-dependent control of VEGFR2 expression.
GATA2 upregulates VEGFR2
GATA2 is another transcription factor that binds the VEGFR2 promoter and regulates its activity23, 24. Because many GATA family members antagonize the effects of other transcription factors at promoter sites25-28, we explored whether GATA2 mediates p190RhoGAP-dependent control of VEGFR2 expression by opposing TFII-I activity. When p190RhoGAP was knocked down, GATA2 levels in the nucleus increased dramatically (> 10-fold) relative to control cells even though total GATA2 levels remained the same, as shown by immunoblotting (Supplementary Fig. S3e) and fluorescence microscopy (Fig. 1c). p190RhoGAP also co-immunoprecipitated with GATA2 and vice versa (Supplementary Fig. S3f). Importantly, this increase in nuclear GATA2 was significantly higher than the 1.5-fold increase in nuclear TFII-I produced using p190RhoGAP siRNA (Supplementary Fig. S2a,b), suggesting that p190RhoGAP binds and sequesters GATA2 in the cytoplasm more efficiently than TFII-I in capillary cells. GATA2 knockdown using siRNA decreased VEGFR2 promoter activity, as well as levels of mRNA and protein (Supplementary Fig. S4a,b) as previously observed23, while GATA2 overexpression produced the opposite effects. These effects of GATA2 siRNA on VEGFR2 promoter activity and protein expression were specific as they were reversed by Gata2 reconstitution (Supplementary Fig. S3c). Interestingly, p190RhoGAP knockdown increased the expression of VEGFR2 mRNA and protein, and double knockdown with GATA2 inhibited these effects (Fig. 1d, Supplementary Fig. S4c). Thus, GATA2 appears to upregulate VEGFR2 promoter activity and mediate the effects of p190RhoGAP on VEGFR2 gene expression in capillary cells. However, knockdown of p190RhoGAP, which releases more GATA2 than TFII-I and increases VEGFR2 expression, did not increase VEGFR2 promoter activity (Supplementary Fig. S3d). This may be because cellular TFII-I levels are 5 times higher than GATA2 levels (not shown). We also measured VEGFR2 promoter activity using only a portion of its promoter, and hence, TFII-I and GATA2 also may exert regulatory activities at other promoter sites; alternatively, p190RhoGAP might elicit signals that influence mRNA stability.
We next asked whether GATA2 and TFII-I directly antagonize each other. Simultaneous knockdown of TFII-I and GATA2 abrogated each other's effects on VEGFR2 promoter activity (Fig. 1e), mRNA production (Fig. 1f) and protein expression levels (Supplementary Fig. S4e), and simultaneous overexpression of TFII-I and GATA2 produced similar effects (Supplementary Fig. S4d,e). These effects were specific for VEGFR2 as knockdown of TFII-I or GATA2 did not alter expression of VEGFR1 or VEGFR3 in HMVE cells (Supplementary Fig. S5a). Importantly, although p190RhoGAP is a Rho inhibitor, altering Rho activity with constitutively active RhoA, membrane-permeable C3 exoenzyme or siRNA directed to another Rho-inhibiting RhoGAP (p73RhoGAP) 29 did not change VEGFR2 mRNA or protein levels (Supplementary Fig. S5b,c). Thus, p190RhoGAP appears to control VEGFR2 expression solely by its ability to sequester these transcription factors.
Analysis of this mechanism of functional cross-antagonism revealed that GATA2 and TFII-I associate with each other, as GATA2 co-immunoprecipitated with TFII-I, and vice versa (Supplementary Fig. S6a). Furthermore, GATA2 even binds to TFII-I in p190RhoGAP knockdown cells and to p190RhoGAP in TFII-I knockdown cells (Supplementary Fig. S6b). These results suggest that the various heterodimeric combinations of TFII-I, GATA2, and p190RhoGAP exist in separate pools, and that these heterodimers then associate to form a larger ternary complex (Supplementary Fig. S1).
Chromatin immunoprecipitation (ChIP) analysis revealed that TFII-I knockdown cells exhibited increased recruitment of GATA2 to the GATA binding site (-150-+150) compared to control cells and vice versa (Fig. 1g), whereas control IgG did not immunoprecipitate these DNAs (Fig. 1g). p190RhoGAP knockdown decreased recruitment of TFII-I, but not GATA2, to this promoter site, which resulted in a relative net increase in GATA2 recruitment to this site (Supplementary Fig. S6c). TFII-I and GATA2 therefore compete with each other for occupancy of a common region of the VEGFR2 promoter, which is controlled by p190RhoGAP.
Mechanical control of VEGFR2
Soluble growth factors, integrin binding to ECM and mechanical distortion of the cytoskeleton all regulate p190RhoGAP activity in cells30. Cell binding to growth factors and adhesive contact formation with ECM also control VEGFR2 expression31, 32, and soluble mitogens (5% serum plus VEGF, bFGF and PDGF) increase nuclear translocation of TFII-I and GATA2 in HMVE cells (Supplementary Fig. S7a). We therefore next explored whether changes in mechanical interactions between cells and ECM regulate this pathway as well. When HMVE cells were cultured in the absence of mitogens on fibronectin-coated polyacrylamide gels with different elasticity (Young's moduli of 150 to 4000Pa), they appeared round on the soft gels, while they flattened on the stiffer gels, as previously observed33, 34, which is based on differences in their ability to physically resist cell traction forces8, 33, 34. Nuclear GATA2 levels were significantly higher in cells on the stiffer gels, whereas nuclei exhibit similar high levels of TFII-I regardless of ECM stiffness (Fig. 2a), and similar results were obtained in the presence of multiple soluble factors or VEGF alone (Supplementary Fig. S7b). Moreover, VEGFR2 mRNA and protein levels were higher in cells on the stiffer (4000 Pa) gels (Fig. 2b, Supplementary Fig. S7c). Interestingly, this relatively stiff, but still compliant, ECM gel appeared to support optimal responsiveness of this signaling pathway as VEGFR2 mRNA and protein levels were significantly lower in cells cultured on rigid glass ECM substrates (Fig. 2b, Supplementary Fig. S7c). ECM elasticity may therefore regulate VEGFR2 expression preferentially via GATA2, particularly over the stiffness range analyzed in this study.
Fig. 2. Matrix elasticity controls VEGFR2 expression via TFII-I and GATA2.
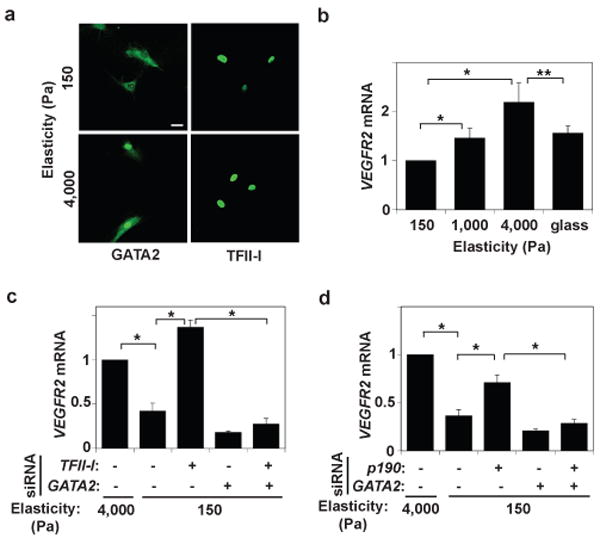
a) Immunofluorescence micrographs showing GATA2 and TFII-I distribution in HMVE cells cultured on the fibronectin-coated gels of different stiffness (Young's moduli of 150 and 4000 Pa) in EBM2 with 0.3% serum (bar, 5μm). b) VEGFR2 mRNA level in HMVE cells cultured on rigid fibronectin-coated glass or the gels of different elasticity (150, 1000 and 4000 Pa; normalized to that in cells on the softest gels; *, p<0.01, **, p<0.05). c) VEGFR2 mRNA levels in HMVE cells on soft gels (150 Pa) transfected with TFII-I or GATA2 siRNA alone, or in combination (normalized to cells on the stiffest gels; *, p<0.01). d) VEGFR2 mRNA levels in HMVE cells on soft gels transfected with p190RhoGAP or GATA2 siRNA alone, or in combination (normalized to cells on stiffest gels; *, p<0.01). Error bars represent s.e.m. of 3 replica experiments.
Further analysis revealed that TFII-I knockdown restored VEGFR2 expression in cells on soft gels to levels similar to those in cells on stiff gels, and double knockdown with GATA2 (which decreases VEGFR2 expression) abrogated TFII-I's effects (Fig. 2c, Supplementary Fig. S7d). p190RhoGAP knockdown also increased expression of VEGFR2 mRNA and protein, and double knockdown with GATA2 inhibited these effects on soft gels (Fig. 2d, Supplementary Fig. S7d). These data indicate that p190RhoGAP and the mutually antagonistic TFII-I and GATA2 transcription factors mediate the effects of ECM elasticity on VEGFR2 expression in these cells, which is preferentially shifted to GATA2 on stiffer gels.
Transcriptional control of angiogenesis
We next explored the functional relevance of this antagonism between GATA2 and TFII-I by analyzing capillary cell migration and differentiation (tube formation) in vitro. When HMVE cells were transfected with TFII-I siRNA and analyzed using the Transwell migration assay, VEGF-stimulated cell motility increased by 2-fold, whereas this induction was prevented by knocking down GATA2 simultaneously with TFII-I (Fig. 3a). The VEGFR2 kinase inhibitor SU5416 totally inhibited the migration of these cells (Fig. 3a), confirming that these effects are mediated by VEGFR2 signaling. Moreover, GATA2 overexpression increased VEGF-induced cell migration by 3-fold, and simultaneous overexpression of GATA2 and TFII-I abolished these effects (Fig. 3a). These effects were specific in that reconstitution of TFII-I or Gata2 reversed the effects of knocking down TFII-I or GATA2, respectively (Supplementary Fig. S8a).
Fig. 3. Antagonism between GATA2 and TFII-I controls capillary cell migration and tube formation in vitro.
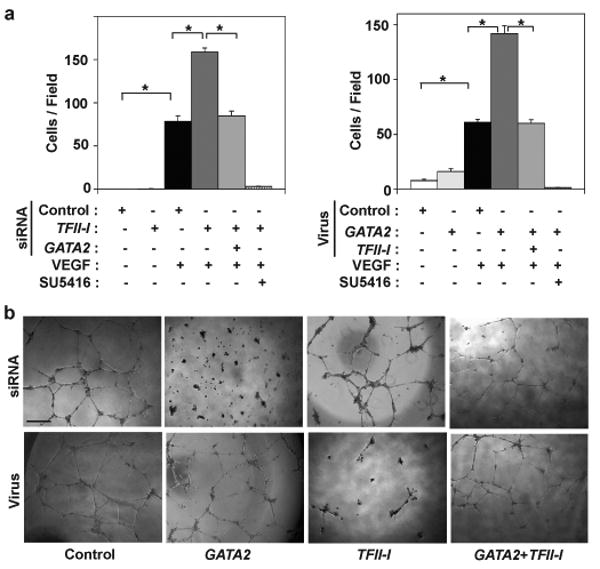
a) The motility of HMVE cells transfected with human siRNAs or transduced with lentiviral vectors encoding GATA2 or TFII-I, alone or in combination, was quantitated using the Transwell migration assay (*, p< 0.01). Where indicated, VEGF (10 ng/ml) was added to the lower chamber and SU5416 was added in both chambers. Error bars represent s.e.m. of 3 replica experiments. b) Micrographs showing in vitro tube formation induced by VEGF (10 ng/ml) in HMVE cells transfected with siRNAs or transduced with lentiviral vectors encoding GATA2 or TFII-I, alone or in combination.
We then studied capillary tube formation by HMVE cells cultured within the ECM gel, Matrigel, which supports angiogenesis in part because of its flexibility. Tube formation was stimulated by VEGF in a dose-dependent manner, whereas bFGF and PDGF were ineffective, and the effects of VEGF were inhibited by SU5416 (Supplementary Fig. S8b). GATA2 knockdown and TFII-I overexpression suppressed VEGF-stimulated capillary development, and once again either knocking down or overexpressing both transcription factors simultaneously negated these effects (Fig. 3b, Supplementary Fig. S8c). TFII-I knockdown or GATA2 overexpression alone did not produce significant effects on tube formation, apparently because it was already optimally stimulated under these conditions (Fig. 3b); knockdown or overexpression of p190RhoGAP also did not alter tube formation (Supplementary Fig. S8d). Thus, functional antagonism between GATA2 and TFII-I at the level of VEGFR2 transcription translates into biologically relevant changes in capillary cell behavior that are required for formation of 3D capillary networks.
Control of angiogenesis in vivo
We next examined whether ECM mechanics governs vessel formation in vivo using a modified Matrigel implant assay. Maximal levels of cell infiltration, capillary blood vessel formation, and VEGFR2 expression were observed in Matrigel with intermediate stiffness (800 Pa) compared to cells in more or less rigid gels (900 or 700 Pa, respectively)(Fig. 4a,c and Supplementary Fig. S9a,b). VEGFR2, GATA2, and TFII-I all localized within cells lining CD31-positive staining microvessels (Supplementary Fig. S9c). The finding that the optimal ECM stiffness required for angiogenesis in vivo (800 Pa) was different than that observed in vitro (4000 Pa) is likely due to different requirements by cells when cultured on 2D ECM versus within a 3D ECM gel; it also may relate to how the Matrigel is remodeled over time in vivo.
Fig. 4. Matrix elasticity controls vessel formation via TFII-I and GATA2 in vivo.
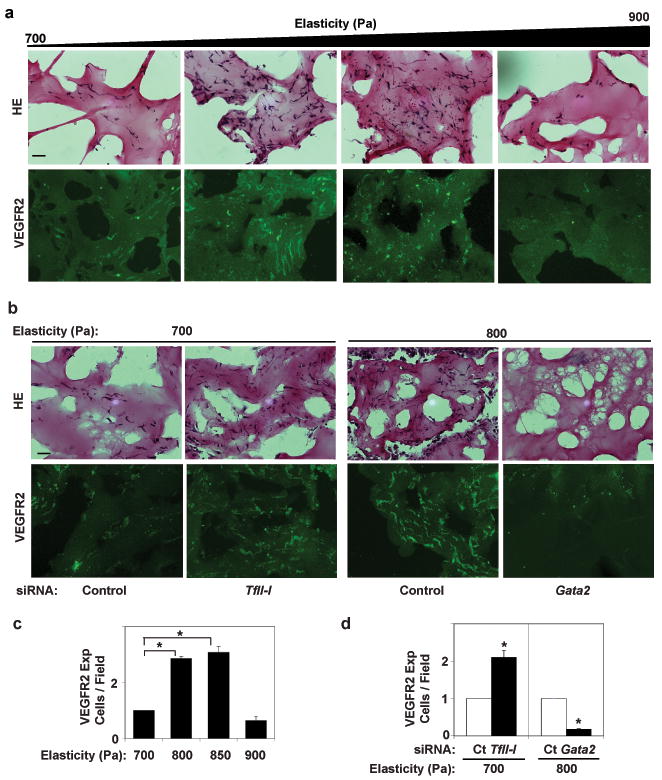
Light (H&E-stained) (a,b top), immunofluorescence micrographs (a,b bottom) and vessel densities per high power field (c,d) showing cell infiltration and VEGFR2 expression in Matrigel with different elasticity implanted in mice for 7 days without (a,c) or with TFII-I or Gata2 siRNAs (b,d) (bar, 25 μm). The number of VEGFR2-positive blood vessels was normalized to that in the 700Pa gels in (c) and to that in gels treated with control siRNA in (d) (n=6, mean ± S.E.M., *, p< 0.01).
We next performed siRNA-mediated gene knockdown in the in vivo Matrigel assay. Interestingly, TFII-I knockdown increased cell infiltration, capillary vessel formation and VEGFR2 expression by cells in the soft gels so that it mimicked the behavior of cells on the intermediate stiffness gel, whereas Gata2 knockdown produced the opposite effects (Fig. 4b,d and Supplementary Fig. S10a), and p190RhoGAP knockdown also increased the level of vessel formation in the soft gels (Supplementary Fig. S10b). These results indicate that TFII-I, GATA2, and p190RhoGAP mediate the signaling effects of ECM mechanics on vessel formation in vivo.
To unequivocally confirm the functional and clinical relevance of regulatory interactions between TFII-I, GATA2, and p190RhoGAP, we modulated their expression in the neonatal mouse retina because angiogenesis in this growing organ is tightly regulated by VEGF and its receptors14, 35, 36. TFII-I, GATA2, and VEGFR2 localized to the three layers of the retina where blood vessels are located in postnatal day 15 mice (Supplementary Fig. S11a,b). Consistent with in vitro data, knocking down TFII-I using intravitreal injection of siRNA to p14 mice resulted in increased Vegfr2 expression and the appearance of highly tortuous dilated vessels and a significant increase in vascular density in the retina, whereas Gata2 knockdown suppressed Vegfr2 expression, disrupted capillary network formation and decreased vascular density (Fig. 5a, Supplementary Fig. S12a,b). Knockdown of p190RhoGAP also increased Vegfr2 expression and vessel density, but it resulted in a slightly different capillary growth pattern, perhaps in part because of its known effects on vascular permeability37 (Fig. 5a and Supplementary Fig. S12a,b). Similar effects were observed in earlier (P5) retina that contain rapidly growing microvessels (Supplementary Fig. S13). Furthermore, overexpression of TFII-I and GATA2 produced opposite effects on vascular density in P14 retina, and simultaneous overexpression of both factors abolished these effects, confirming our in vitro findings (Fig. 5b and Supplementary Fig. S12c). Knockdown of p190RhoGAP or TFII-I did not significantly change Vegf expression levels in the retina, whereas Gata2 knockdown increased (rather than decreased) its expression (Supplementary Fig. S12d). The decrease of Vegfr2 expression appears to be sufficient to abrogate this Vegf response in Gata2 knockdown retina, and thereby inhibit angiogenesis. Hence, p190RhoGAP controls Vegfr2 expression and vascular development by modulating the balance between TFII-I and GATA2 activities such that TFII-I activity dominates (and Vegfr2 expression is suppressed) in retina.
Fig. 5. TFII-I, GATA2, and p190RhoGAP regulate retinal vessel formation in vivo.
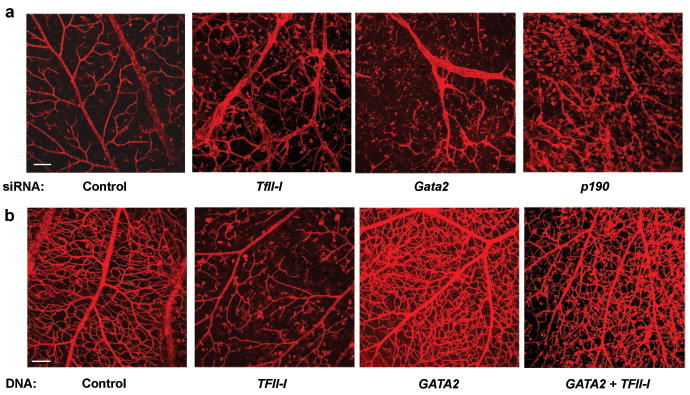
a) Confocal fluorescence micrographs showing projected images of vessels stained with Alexa 594-isolectin in the retina of a control mouse eye versus eyes transfected with TFII-I, Gata2, or p190RhoGAP siRNAs (bar = 0.1 mm). b) Confocal fluorescence micrographs showing projected images of vessels stained with Alexa 594-isolectin in the retina of a control mouse eye versus eyes overexpressed TFII-I or GATA2, alone or both in combination (bar = 0.1 mm).
Discussion
Transcription factors change their activities in a spatiotemporal manner during development, and thereby specify cell fate38, 39. Here we show that p190RhoGAP, which has been shown to be regulated by growth factors, ECM binding and cytoskeletal distortion also controls VEGFR2 expression, as well as angiogenesis in vitro and in vivo, by altering the balance between two mutually antagonistic transcription factors: TFII-I and GATA2 (Supplementary Fig. S1). Moreover, we demonstrate that p190RhoGAP and this downstream transcriptional control mechanism are controlled by mechanical signals conveyed by variations in ECM elasticity. This mechanism is analogous to other developmental mechanisms used by hematopoietic cells and mammary epithelium38-40 28; however, this is the first demonstration that transcriptional cross-antagonism can govern histodifferentiation and tissue morphogenesis, and be sensitive to mechanical as well as chemical cues.
Our data suggest that an appropriate level of ECM stiffness may be required for optimal VEGFR2 expression and vascular development in vitro and in vivo. In fact, the fates of different cell types are exquisitely sensitive to distinct ECM elasticity values that often match those exhibited by their host tissues41. Abrupt local changes in ECM mechanics also accompany the switch between active growth and quiescent differentiation of functional capillary networks in living tissues42. Cell rounding suppressed p190RhoGAP activity within 1 to 2 hours by altering its binding to the cytoskeletal protein, filamin16. A similar cytoskeleton-based effect could mediate the effect of ECM elasticity on p190RhoGAP activity at later times, and thereby control the TFII-I:GATA2 balance and VEGFR2 transcription. Since VEGFR2 is expressed in neurons as well as in endothelial cells in retina, some of the effects of gene knockdown we observed may not be specific to capillary cells. But even these alterations might be relevant for control of vascular development because neuron-vessel interactions are indispensable for normal microvasculature function and patterning43.
In summary, these data reveal a previously unknown mechanosensitive signaling pathway that controls VEGFR2 promoter activity and expression, and which represents a point of convergence for all three classes of microenvironmental signals that regulate capillary morphogenesis. Development of specific modifiers of this pathway could therefore lead to novel therapeutic approaches for various angiogenesis-dependent diseases, including proliferative retinopathy, arthritis, and cancer in the future.
Methods Summary
Expression of TFII-I, GATA2, and VEGFR2 were evaluated by qRT-PCR and immunoblotting. A luciferase reporter assay was used to measure VEGFR2 promoter activity. To test the effects of TFII-I and GATA2 on VEGFR2 expression and angiogenesis in vitro, siRNA-mediated knockdown or lentiviral transduction was performed in HMVE cells. In vitro analysis of angiogenesis was carried out using Transwell migration and Matrigel tube formation assays, and similar results were obtained using both native and growth factor-reduced forms of Matrigel. A subcutaneous Matrigel angiogenesis assay was used to analyze the effects of ECM mechanics on capillary formation in vivo. Retinal vessel formation was also studied in newborn C57BL/6 mice, and gene expression was manipulated in whole living retina by intravitreal injection of siRNA or DNA into the eye at P5 or P14.
Methods
Materials
Anti-GATA2 polyclonal antibody was from Abcam (Cambridge, MA); anti-HA monoclonal antibody from Covance (Princeton, NJ); monoclonal antibodies against TFII-I, p190RhoGAP, PECAM (CD31), and paxillin from Transduction laboratory (Lexington, KY); anti-GAPDH antibody from Chemicon (Temecula, CA); anti-lamin monoclonal antibody from Upstate (Lake Placid, NY); anti-VEGFR2 antibody from Cell Signaling (Danvers, MA); and anti-myc antibody from Santa Cruz (Santa Cruz, CA). Protein G-sepharose was from Amersham-Pharmacia (Uppsala, Sweden) and SU5416 was from Calbiochem (SanDiego, CA). VEGF-A was from NIH; bFGF and PDGF were from Roche (Basel, Switzerland) and Biovision (Mountain View, CA) respectively. Cell permeable Rho inhibitor (C3 exoenzyme) was from Cytoskeleton (Denver, CO). HMVE and HUVE cells (Cambrex, Walkersville, MD) were cultured in EBM2 medium containing 5% FBS and growth factors (VEGF, bFGF, and PDGF) for all experiments16 except the nuclear translocation assays in which we used EBM2 with 0.3% serum. Cells were plated on plastic dishes for molecular biochemical assays, and on fibronectin-coated glass coverslips for cell staining, except for experiments using flexible substrates.
Plasmid construction and gene knockdown
pGL3-_VEGFR2_-225 (-225+268), -570 (-570+268), -780 (-780+268) were constructed using the reverse transcription (RT)-PCR with genomic DNA from HUVE cells, and subcloned into pGL3 vector (Promega) at the SacI/XhoI sites. For pGL3-_VEGFR2_-780Inr-MUT, the Inr (CACT to GTGC) was point mutated using the QuickChange mutagenesis kit (Stratagene, La Jolla, CA). For lentivirus construction, human myc-TFII-I (delta isoform), HA-GATA2, and HA-p190RhoGAP were constructed by PCR using template plasmids from Open Biosystems (Huntsville, AL) and H. Sabe (OBI, Osaka, Japan). For retrovirus construction, mouse myc-TFII-I (gamma isoform) and HA-Gata2 were constructed using template plasmids from Open Biosystems and T. Nakano (Osaka University, Osaka, Japan), respectively. To generate delta isoform of mouse TFII-I, 256-274 and 294-314aa were deleted from the gamma isoform. Construction of constitutively active RhoA and generation of viral vectors were previously described 37. For gene knockdown, siRNA transfection was performed as described16. siRNA sequences are shown in Supplementary Table S1.
Biochemical Methods
For luciferase reporter assays, HUVE cells were transfected using Superfect (QIAGEN) and assayed using Dual-Luciferase reporter assay kit (Promega). Luciferase activity was measured in duplicate using a luminometer (TD20/20, Turner Designs). Cytoplasmic and nuclear cell extracts were prepared with a Nuclear Extraction Kit (Chemicon).
Molecular biological methods
Quantitative RT-PCR was performed with the Quantitect SYBR Green RT-PCR kit (QIAGEN) using ABI7300 real-time PCR system (Applied Biosystems, Foster City, CA); β2 microglobulin or cyclophilin controlled for cDNA content. The primers used are shown in Supplementary table S2. For ChIP assay, DNA from HMVE cells transfected with control or TFII-I siRNA was immunoprecipitated with the GATA2 antibody or control immunoglobulin (Jackson Immuno Research), or vice versa, according to the manufacturer instructions (Active Motif). GATA2- and TFII-I-binding region was amplified using primers, 5′-GTAAATGGGCTTGGGGAGCTG-3′ and 5′-GGCGGCTGCAGGGGCGTCT-3′.
Cell analysis methods
Flexible polyacrylamide gel culture substrates were prepared as described34, 44 and coated with fibronectin (1 μg/cm2). Substrate flexibility was controlled by varying the acrylamide (2–4%) and the bis-acrylamide (0.1–0.5%) concentration; the Young's modulus (stiffness) was determined as described45. HMVE cells were cultured for 6 hrs on the gels and immunostaining was performed and analyzed using confocal Leica SP2 microscope 15. For cell migration assay, Transwell membranes (Coster, NY) were coated with 0.5% gelatin, and cells were seeded (105 cells/100 μl) with 0.3%FBS/EBM2. Cells were stained with Giemsa solution 16 h later, and counted in 10 random fields (×400). For the in vitro angiogenesis assay, HMVE cells (104 cells/150 μl of EBM-2) were plated on MatrigelTM (BD biosciences), and incubated for 12-16 hrs in the presence of VEGF (10 ng/ml); tube formation was assessed in 10 random fields (4×).
In vivo Matrigel implantation assay
All animal studies were reviewed and approved by the animal care and use committee of Children's Hospital Boston. Matrigel plugs with different elasticity were cast in 4 × 4mm (ID×H) polydimethylsiloxane (PDMS) molds and incubated at 37°C overnight before implanting them subcutaneously on the backs of C57BL/6 mice. The stiffness of the Matrigel was modulated over a narrow range (i.e., without making it rigid) by altering ECM protein cross-linking using a microbial transglutaminase (2.5-20 U/g; Ajinomoto, Japan) 46. The storage modulus (G′) of the gels was measured with an AR-G2 rheometer (TA Instruments) using a standard 20 mm aluminum parallel plate (1Hz, 1% strain, 37°C). The Young's modulus for an equivalent polyacrylamide gel was calculated by E = 2*G′(1+ν) using an average Poisson's Ratio (ν) of 0.5. After 7 days, the PDMS molds containing the gel plugs were harvested, fixed, and cryosectioned. H&E staining and immunostaining were performed as described 15 47. Stacks of optical sections (20 μm thick) were compiled to form 3D images using Velocity4.4 (Improvision, PerkinElmer). Vessel formation was evaluated by counting the number of vessels that stained positive for fluorescein-conjugated ConA injected into the tail vein or VEGFR2 in 5 different areas (n=6), individual cell nuclei were identified by DAPI staining. Cell recovery solution (BD Biosciences) was used to collect cells from the recovered Matrigel plugs. In some studies, siRNA (7 μg) was mixed into the Matrigels and 10 μg of additional siRNA was injected into the implanted Matrigel after 3 days (n=6); the same amount of scrambled siRNA was used as a control. Gene knockdown was evaluated by counting the number of cells expressing each gene in the five different regions.
In vivo analysis for retinal vessel formation
For gene knockdown in living retina, siRNA (0.5μg) for each gene was injected intravitreally into one eye of C57BL/6 mouse at P5 or P14, and the same amount of control siRNA was injected to the other eye. To overexpress genes, the complex of DNA (0.5μg) for TFII-I and/or GATA2 and jetPEI transfection reagent (Polyplus transfection, CA) was injected to the eye at P14. Vascular network formation in the retina was assessed 2 days after injection using flat-mounted, fluorescein-conjugated isolectin-staining and immunohistochemical analysis (n=7). Retinal RNA was purified and gene expression was quantified using qRT-PCR (n=7). Quantification of vessel density was performed with Adobe photoshop.
Supplementary Material
Supple Fig S 1
Supple Fig S 10
Supple Fig S 11
Supple Fig S 12
Supple Fig S 13
Supple Fig S 2
Supple Fig S 3
Supple Fig S 4
Supple Fig S 5
Supple Fig S 6
Supple Fig S 7
Supple Fig S 8
Supple Fig S 9
SuppleTableS1
SuppleTableS2
Acknowledgments
We thank T. Polte, E. Pravda, M. de Bruijn, and K. Johnson for their technical suggestions and assistance, T. Nakano and H. Sabe for providing plasmid, NIH for providing VEGF, and D. Weitz for providing assistance with Rheometry measurements. This work was supported by funds from NIH (to D.E.I., L.E.H.S. and K.M.C.), V. Kann Rasmussen Foundation (to L.E.H.S.), Children's Hospital Mental Retardation and Developmental Disabilities Research Center (to L.E.H.S.), a Research to Prevent Blindness Lew Wasserman Merit Award (to L.E.H.S.), AHA (to A.M.), and a Children's Hospital House Officer Development Award (to A.M.); D.E.I. is a recipient of a DoD Breast Cancer Innovator Award.
Footnotes
Author Contributions: A.M. conceived of the experiments, and performed experiments, designed research and analyzed data with assistance from K.M.C., T.M., C.W.Y., D.H., C.M.A., G.M., L.E.H.S. and D.E.I. A.M. wrote the manuscript with D.E.I., with input from L.E.H.S.
References
- 1.Ferrara N, Gerber HP, LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003;9:669–76. doi: 10.1038/nm0603-669. [DOI] [PubMed] [Google Scholar]
- 2.Ferrara N, Mass RD, Campa C, Kim R. Targeting VEGF-A to treat cancer and age-related macular degeneration. Annu Rev Med. 2007;58:491–504. doi: 10.1146/annurev.med.58.061705.145635. [DOI] [PubMed] [Google Scholar]
- 3.Ingber DE, Folkman J. Mechanochemical switching between growth and differentiation during fibroblast growth factor-stimulated angiogenesis in vitro: role of extracellular matrix. J Cell Biol. 1989;109:317–30. doi: 10.1083/jcb.109.1.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen CS, Mrksich M, Huang S, Whitesides GM, Ingber DE. Geometric control of cell life and death. Science. 1997;276:1425–8. doi: 10.1126/science.276.5317.1425. [DOI] [PubMed] [Google Scholar]
- 5.Dike LE, et al. Geometric control of switching between growth, apoptosis, and differentiation during angiogenesis using micropatterned substrates. In vitro Cell Dev Biol Anim. 1999;35:441–8. doi: 10.1007/s11626-999-0050-4. [DOI] [PubMed] [Google Scholar]
- 6.Parker KK, et al. Directional control of lamellipodia extension by constraining cell shape and orienting cell tractional forces. Faseb J. 2002;16:1195–204. doi: 10.1096/fj.02-0038com. [DOI] [PubMed] [Google Scholar]
- 7.Matthews BD, Overby DR, Mannix R, Ingber DE. Cellular adaptation to mechanical stress: role of integrins, Rho, cytoskeletal tension and mechanosensitive ion channels. J Cell Sci. 2006;119:508–18. doi: 10.1242/jcs.02760. [DOI] [PubMed] [Google Scholar]
- 8.Kumar S, et al. Viscoelastic retraction of single living stress fibers and its impact on cell shape, cytoskeletal organization, and extracellular matrix mechanics. Biophys J. 2006;90:3762–73. doi: 10.1529/biophysj.105.071506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moore KA, et al. Control of basement membrane remodeling and epithelial branching morphogenesis in embryonic lung by Rho and cytoskeletal tension. Dev Dyn. 2005;232:268–81. doi: 10.1002/dvdy.20237. [DOI] [PubMed] [Google Scholar]
- 10.Huang S, Ingber DE. The structural and mechanical complexity of cell-growth control. Nat Cell Biol. 1999;1:E131–8. doi: 10.1038/13043. [DOI] [PubMed] [Google Scholar]
- 11.Folkman J, Moscona A. Role of cell shape in growth control. Nature. 1978;273:345–9. doi: 10.1038/273345a0. [DOI] [PubMed] [Google Scholar]
- 12.Folkman J, Kalluri R. Cancer without disease. Nature. 2004;427:787. doi: 10.1038/427787a. [DOI] [PubMed] [Google Scholar]
- 13.Matsumoto T, Claesson-Welsh L. VEGF receptor signal transduction. Sci STKE 2001. 2001:RE21. doi: 10.1126/stke.2001.112.re21. [DOI] [PubMed] [Google Scholar]
- 14.Wong CG, Rich KA, Liaw LH, Hsu HT, Berns MW. Intravitreal VEGF and bFGF produce florid retinal neovascularization and hemorrhage in the rabbit. Curr Eye Res. 2001;22:140–7. doi: 10.1076/ceyr.22.2.140.5528. [DOI] [PubMed] [Google Scholar]
- 15.Mammoto A, Huang S, Moore K, Oh P, Ingber DE. Role of RhoA, mDia, and ROCK in cell shape-dependent control of the Skp2-p27kip1 pathway and the G1/S transition. J Biol Chem. 2004;279:26323–30. doi: 10.1074/jbc.M402725200. [DOI] [PubMed] [Google Scholar]
- 16.Mammoto A, Huang S, Ingber DE. Filamin links cell shape and cytoskeletal structure to Rho regulation by controlling accumulation of p190RhoGAP in lipid rafts. J Cell Sci. 2007;120:456–67. doi: 10.1242/jcs.03353. [DOI] [PubMed] [Google Scholar]
- 17.Jiang W, et al. An FF domain-dependent protein interaction mediates a signaling pathway for growth factor-induced gene expression. Mol Cell. 2005;17:23–35. doi: 10.1016/j.molcel.2004.11.024. [DOI] [PubMed] [Google Scholar]
- 18.Jackson TA, Taylor HE, Sharma D, Desiderio S, Danoff SK. Vascular endothelial growth factor receptor-2: counter-regulation by the transcription factors, TFII-I and TFII-IRD1. J Biol Chem. 2005;280:29856–63. doi: 10.1074/jbc.M500335200. [DOI] [PubMed] [Google Scholar]
- 19.Roy AL. Biochemistry and biology of the inducible multifunctional transcription factor TFII-I. Gene. 2001;274:1–13. doi: 10.1016/s0378-1119(01)00625-4. [DOI] [PubMed] [Google Scholar]
- 20.Francke U. Williams-Beuren syndrome: genes and mechanisms. Hum Mol Genet. 1999;8:1947–54. doi: 10.1093/hmg/8.10.1947. [DOI] [PubMed] [Google Scholar]
- 21.Roy AL. Signal-induced functions of the transcription factor TFII-I. Biochim Biophys Acta. 2007;1769:613–21. doi: 10.1016/j.bbaexp.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patterson C, et al. Cloning and functional analysis of the promoter for KDR/flk-1, a receptor for vascular endothelial growth factor. J Biol Chem. 1995;270:23111–8. doi: 10.1074/jbc.270.39.23111. [DOI] [PubMed] [Google Scholar]
- 23.Minami T, Rosenberg RD, Aird WC. Transforming growth factor-beta 1-mediated inhibition of the flk-1/KDR gene is mediated by a 5′-untranslated region palindromic GATA site. J Biol Chem. 2001;276:5395–402. doi: 10.1074/jbc.M008798200. [DOI] [PubMed] [Google Scholar]
- 24.Minami T, et al. Interaction between hex and GATA transcription factors in vascular endothelial cells inhibits flk-1/KDR-mediated vascular endothelial growth factor signaling. J Biol Chem. 2004;279:20626–35. doi: 10.1074/jbc.M308730200. [DOI] [PubMed] [Google Scholar]
- 25.Cantor AB, Orkin SH. Hematopoietic development: a balancing act. Curr Opin Genet Dev. 2001;11:513–9. doi: 10.1016/s0959-437x(00)00226-4. [DOI] [PubMed] [Google Scholar]
- 26.Grogan JL, Locksley RM. T helper cell differentiation: on again, off again. Curr Opin Immunol. 2002;14:366–72. doi: 10.1016/s0952-7915(02)00340-0. [DOI] [PubMed] [Google Scholar]
- 27.Pai SY, Truitt ML, Ho IC. GATA-3 deficiency abrogates the development and maintenance of T helper type 2 cells. Proc Natl Acad Sci U S A. 2004;101:1993–8. doi: 10.1073/pnas.0308697100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kouros-Mehr H, Slorach EM, Sternlicht MD, Werb Z. GATA-3 maintains the differentiation of the luminal cell fate in the mammary gland. Cell. 2006;127:1041–55. doi: 10.1016/j.cell.2006.09.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su ZJ, et al. A vascular cell-restricted RhoGAP, p73RhoGAP, is a key regulator of angiogenesis. Proc Natl Acad Sci U S A. 2004;101:12212–7. doi: 10.1073/pnas.0404631101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arthur WT, Petch LA, Burridge K. Integrin engagement suppresses RhoA activity via a c-Src-dependent mechanism. Curr Biol. 2000;10:719–22. doi: 10.1016/s0960-9822(00)00537-6. [DOI] [PubMed] [Google Scholar]
- 31.Robinson CJ, Stringer SE. The splice variants of vascular endothelial growth factor (VEGF) and their receptors. J Cell Sci. 2001;114:853–65. doi: 10.1242/jcs.114.5.853. [DOI] [PubMed] [Google Scholar]
- 32.Sheibani N, Frazier WA. Down-regulation of platelet endothelial cell adhesion molecule-1 results in thrombospondin-1 expression and concerted regulation of endothelial cell phenotype. Mol Biol Cell. 1998;9:701–13. doi: 10.1091/mbc.9.4.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Numaguchi Y, et al. Caldesmon-dependent switching between capillary endothelial cell growth and apoptosis through modulation of cell shape and contractility. Angiogenesis. 2003;6:55–64. doi: 10.1023/a:1025821517679. [DOI] [PubMed] [Google Scholar]
- 34.Polte TR, Eichler GS, Wang N, Ingber DE. Extracellular matrix controls myosin light chain phosphorylation and cell contractility through modulation of cell shape and cytoskeletal prestress. Am J Physiol Cell Physiol. 2004;286:C518–28. doi: 10.1152/ajpcell.00280.2003. [DOI] [PubMed] [Google Scholar]
- 35.Pierce EA, Avery RL, Foley ED, Aiello LP, Smith LE. Vascular endothelial growth factor/vascular permeability factor expression in a mouse model of retinal neovascularization. Proc Natl Acad Sci U S A. 1995;92:905–9. doi: 10.1073/pnas.92.3.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stalmans I, et al. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J Clin Invest. 2002;109:327–36. doi: 10.1172/JCI14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mammoto T, et al. Angiopoietin-1 requires p190RhoGAP to protect against vascular leakage in vivo. J Biol Chem. 2007 doi: 10.1074/jbc.M702169200. [DOI] [PubMed] [Google Scholar]
- 38.Singh H, Medina KL, Pongubala JM. Contingent gene regulatory networks and B cell fate specification. Proc Natl Acad Sci U S A. 2005;102:4949–53. doi: 10.1073/pnas.0500480102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swiers G, Patient R, Loose M. Genetic regulatory networks programming hematopoietic stem cells and erythroid lineage specification. Dev Biol. 2006;294:525–40. doi: 10.1016/j.ydbio.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 40.Gottgens B, et al. Establishing the transcriptional programme for blood: the SCL stem cell enhancer is regulated by a multiprotein complex containing Ets and GATA factors. Embo J. 2002;21:3039–50. doi: 10.1093/emboj/cdf286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;126:677–89. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 42.Clark ER, Clark EL. Microscopic observations on the growth of blood capillaries in the living mammal. Am J Anat. 1938;64:251–301. [Google Scholar]
- 43.Carmeliet P, Tessier-Lavigne M. Common mechanisms of nerve and blood vessel wiring. Nature. 2005;436:193–200. doi: 10.1038/nature03875. [DOI] [PubMed] [Google Scholar]
- 44.Pelham RJ, Jr, Wang Y. Cell locomotion and focal adhesions are regulated by substrate flexibility. Proc Natl Acad Sci U S A. 1997;94:13661–5. doi: 10.1073/pnas.94.25.13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang N, et al. Cell prestress. I. Stiffness and prestress are closely associated in adherent contractile cells. Am J Physiol Cell Physiol. 2002;282:C606–16. doi: 10.1152/ajpcell.00269.2001. [DOI] [PubMed] [Google Scholar]
- 46.Yung CW, et al. Transglutaminase crosslinked gelatin as a tissue engineering scaffold. J Biomed Mater Res A. 2007;83:1039–46. doi: 10.1002/jbm.a.31431. [DOI] [PubMed] [Google Scholar]
- 47.Connor KM, et al. Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat Med. 2007;13:868–73. doi: 10.1038/nm1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supple Fig S 1
Supple Fig S 10
Supple Fig S 11
Supple Fig S 12
Supple Fig S 13
Supple Fig S 2
Supple Fig S 3
Supple Fig S 4
Supple Fig S 5
Supple Fig S 6
Supple Fig S 7
Supple Fig S 8
Supple Fig S 9
SuppleTableS1
SuppleTableS2