Estrogen Regulation of Thrombospondin-1 in Human Breast Cancer Cells (original) (raw)
. Author manuscript; available in PMC: 2010 Sep 1.
Published in final edited form as: Int J Cancer. 2009 Sep 1;125(5):1045–1053. doi: 10.1002/ijc.24373
Abstract
Expression of thrombospondin-1 (TSP-1), a large extracellular matrix protein, has been associated with modulation of angiogenesis and tumor growth. Both pro- and anti-angiogenic properties of TSP-1 have been described, and the role of TSP-1 expression in the growth and progression of human breast cancer is not clear. Because estrogens cause progression of many breast cancers, and estradiol (E2) downregulates a TSP-1 receptor, we examined whether TSP-1 is regulated by estrogen and involved in tumor progression. E2 induced TSP-1 expression in T47-D and MCF-7 breast cancer cells in vitro within 3-6 h; the induction was blocked by the anti-estrogen ICI 182,780, indicating that estrogen receptors (ER) are necessary for this effect. Furthermore, E2 caused the production of TSP-1 protein from tumor cells in an ER-alpha–dependent manner. The E2-mediated TSP-1 RNA induction was dose-dependent and blocked by actinomycin D, indicating that the response to E2 was at least partly transcriptional. Transfection studies with deletion constructs of the TSP-1 promoter identified an estrogen-responsive region in the human TSP-1 promoter, located between -2200 and -1792 bp upstream of the transcription start site. An antibody against TSP-1 restricted the proliferation of E2-dependent MCF-7 cells in vitro and in vivo. A panel of breast cancer cells proliferated in the presence of low concentrations of exogenous TSP-1, whereas higher concentrations inhibited proliferation. A real-time PCR analysis showed that E2 also induced TSP-1 mRNA in the normal mammary glands of immature ovariectomized mice in an ER-dependent manner. In summary, we report the novel observation that TSP-1 production is directly controlled by estrogens in ER-positive breast cancer cells, and the released protein has pro-growth regulatory functions. Consequently, we propose that TSP-1 could be a therapeutic target for anti-tumor therapy in early-stage tumors.
Keywords: Estrogen, TSP-1, Breast Cancer
Introduction
Thrombospondin-1 (TSP-1) is an extracellular matrix protein that was the first natural inhibitor of angiogenesis to be identified (1, 2). However, the exclusive role of TSP-1 as an anti-angiogenic protein has recently been questioned, and TSP-1 is now believed to play a proliferative role in breast cancer (3-8). Indeed, its role appears to vary with cell type, concentration, and context (2, 9). The in vivo anti-angiogenic properties of TSP-1 have been demonstrated primarily in animal model systems under nonphysiological conditions, for example in tumor-prone TSP-1 knockout mice (10) and in TSP-1 transgenic animals that overexpress TSP-1 from the MMTV promoter and have reduced tumor burden (11). In contrast, when synthesis of endogenously produced TSP-1 is blocked, progression of breast tumors is reduced (4), a finding that is more consistent with a pro-angiogenic, or proliferative, rather than anti-angiogenic role. TSP-1 is present at a higher level in malignant and invasive human breast tumors than in noninvasive tumors or normal human breast tissue (12-14), supporting the notion that TSP-1 is in fact pro-angiogenic or pro-proliferative in its effect on breast tumor development. Other studies have shown pro-angiogenic effects of TSP-1 that are dose-dependent, including an ability to stimulate cell survival and endothelial cell migration (9, 16, 17). As is becoming increasingly apparent, the role of TSP-1 is complex and likely to vary depending on cellular context, cell type, hormonal milieu, and TSP-1 receptor–dependent signaling (2, 9, 16, 17). Furthermore, individual regions of the TSP-1 protein may independently stimulate or inhibit angiogenesis (2, 18). This raises the interesting possibility that breast tumors may produce a pro-angiogenic environment in which the estradiol (E2)-induced TSP-1 protein either interacts with other proteins or that TSP-1 is cleaved generating angiogenic regions, leading ultimately to tumor cell proliferation and tumor progression.
Many breast cancer cells express steroid hormone receptors (19), including estrogen receptor (ER) and (E2) affects the proliferation of many hormone receptor–positive breast cancer cells. E2 can function via both ER-alpha and ER-beta, the two isoforms known for ER, though most proliferative functions of E2 are associated with ER-alpha and ER-beta is thought to prevent such proliferation (20). We recently observed that E2 downregulates CD36, a TSP-1 receptor in human breast epithelial cells (21). Here we sought to determine whether TSP-1 expression is regulated by E2 in such cells, and found that E2 directly controls TSP-1 production in human breast cancer cells via the alpha type of ER (ER-alpha). Furthermore, E2-induced TSP-1 stimulated breast tumor cell proliferation, both in vitro and in vivo in nude mice.
Materials and Methods
Cell Culture, Cell Treatments, and RNA collection
Cells of the T47-D, MCF-7, and MDA-MB-231 breast cancer cell lines (ATCC, Manassas, VA) were grown in phenol red–free DMEM/F12 medium (Invitrogen Corporation & Life Technologies, Grand Island, NY) supplemented with 10% fetal bovine serum (FBS; JRH Biosciences, Lenexa, KS). When the cells were 60%-70% confluent, they were washed once with PBS and incubated for 18-24 h in 5 mL phenol red–free DMEM/F12 medium supplemented with 5% charcoal-stripped serum (22). Fresh medium (phenol red–free DMEM/F12 medium supplemented with 5% charcoal-stripped serum) was added with the indicated concentrations of ligands and antihormones, and cells were incubated for a further 6 h (unless indicated otherwise), after which they were washed with PBS and harvested for total RNA preparation as described previously (23 24). For real-time quantitative PCR assays RNA was prepared using the Ultraspec RNA Isolation System (Biotecx Laboratories, Inc., Houston, TX). Estradiol, actinomycin-D, puromycin, and TSP-1 protein were purchased from Sigma (St. Louis, MO). Faslodex (ICI 182,780); 4, 4′, 4″-(4-propyl-[1H]-pyrazole-1,3,5-triyl) trisphenol (PPT, an ER-alpha–specific ligand); and 2,3-bis-(4-hydroxyphenyl)-propionitrile (DPN, an ER-beta–specific ligand) were purchased from Tocris Cookson Inc. (Ellisville, MO).
Northern Blot Analysis
RNA (20 μg) was denatured for 30 min in 15 mM methyl mercuric hydroxide (Alfa, Salt Lake City, UT) and separated in a 1% agarose gel containing 6% (v/v) formaldehyde. After electrophoresis, gels were stained with ethidium bromide to verify equal loading and RNA was transferred to a Duralon membrane (Stratagene, La Jolla, CA) by electroblotting. Blots were probed for 28S RNA, stripped, and reprobed for TSP-1 (23, 24). The TSP-1 probe was prepared by nick translation of a plasmid containing the human TSP-1 gene, kindly provided by Dr. Jack Lawler (Beth Israel Deaconess Medical Center, Harvard Medical School, Boston). Blots were hybridized, washed, and visualized as described previously (23, 24). Results were quantified by densitometric analysis where indicated.
Real-Time PCR Assays
The Ultraspec RNA Isolation System (Biotecx Laboratories, Inc., Houston, TX) was used to extract total RNA from the mammary glands of ovariectomized Balb/c mice (21 days old) that were treated with E2 (40 μg/kg) ± ICI 182,780 (3 mg/kg), as well as from vehicle-treated controls. RNA was pretreated with DNase I (Invitrogen, Carlsbad, CA), and SuperScript (Invitrogen) was used to synthesize cDNA according to the manufacturer's recommended conditions. The human TSP-1 primers and probe were designed by Lark Technologies, Inc. (Houston, TX) and are proprietary. Real-time quantitative PCR was performed using an ABI PRISM 7700™ Sequence Detection System (Applied Biosystems, Foster City, CA). The experiment was conducted in triplicate and each sample was analyzed in duplicate, using ribosomal 18S RNA as internal control. After amplification, the relative differences in amounts of RNA were calculated based on the 2-Δ ΔCT method (Applied Biosystems Prism Sequence Detection System User Bulletin #2).
Western Blot Analysis
T47-D cells were grown in DMEM/F12 until they were 70% confluent. Cells were then washed with PBS and incubated for 24 h in DMEM/F12 supplemented with 5% dextran-coated charcoal, after which they were washed twice with PBS and media replaced with serum-free DMEM/F12. Cells were incubated with 10 nM Estradiol (± 1 uM ICI 182,780) for 18 hours. One group of cells was treated with ICI 182,780 alone, while controls were treated with vehicle alone. Cell culture supernatant (conditioned medium) from each group was collected and protease cocktail (Active Motif, Carlsbad, CA) added. Media was then concentrated at 3000g for 15 min using a Centricon-50 filter (Millipore Corporation, Billerica, MA). 20μg of protein from concentrated cell culture supernatants were separated by NuPAGE 10% Bis-Tris Gel (Invitrogen, Carlsbad, CA). Electrophoresis was performed at 120 V for 1.5 hr using NuPAGE MES-SDS running buffer. Separated proteins were transferred to polyvinylidene difluoride membranes (Bio-Rad Laboratories, Hercules, CA) at 35 V for 1.5 h and blots subsequently blocked for I hr in 5% non-fat dry milk in TBS overnight. Blots were then incubated with TSP-1 antibody (Ab-11, 1:200 diluted, from Thermo Scientific, Fremont, CA) for 2 hr at RT, washed three times with TBS containing 0.1% Tween 20 (TBS-T) at RT, and then incubated with secondary antibody for 1 h at RT. Blots were finally washed seven times with TBS-T and immunoreactive bands visualized using an ECL plus detection kit (Amersham, Pharmacia Biotech, Arlington Heights, IL).
Cell Viability Assay
Viable cells were quantitated using the sulforhodamine B (SRB) assay as described previously (25, 26). This cell protein dye-binding assay is based on the measurement of protein content of surviving cells as an index to determine cell growth and cell viability. Briefly, 100 μl aliquots of culture medium containing 6-8×103 cells were seeded into each well of a 96 well plate and incubated overnight at 37°C under conditions of 5% CO2. Cells were then washed with DMEM/F12 medium once and treated with various concentrations of recombinant TSP-1 (R & D Systems, Minneapolis, MN) for 24 h to replicate the time point in Fig 5A. Surviving or adherent cells were fixed in situ by adding 100 μl 50% cold trichloroacetic acid (TCA), then incubated at 4°C for 1 h. Cells were washed 5 times with ice-cold water, dried and stained in 50 ul of 4% SRB for 8 min at room temperature (RT). Unbound dye was washed 5 times with cold 1% acetic acid, the plate dried at RT, and bound stain was solubilized with 150 μl 10 mM Tris. Sample absorbance at 520nm was measured using a SpecTRA MAX 190 microplate reader (Sunnyvale, CA). Six to twelve wells per concentration were analyzed and each experiment was performed in either duplicate or triplicate.
Figure 5. Effect of TSP-1 on proliferation of breast cancer cells.
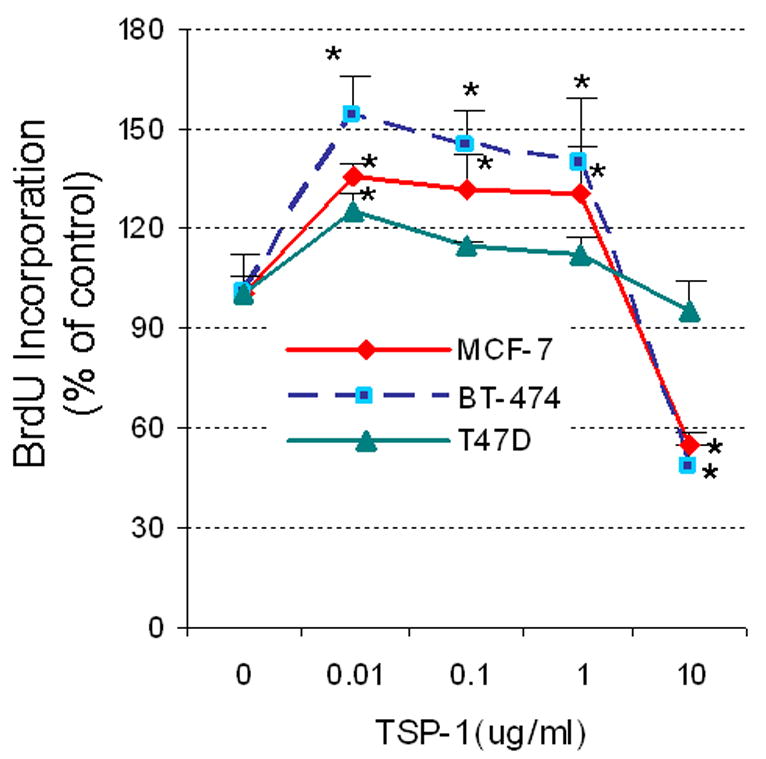
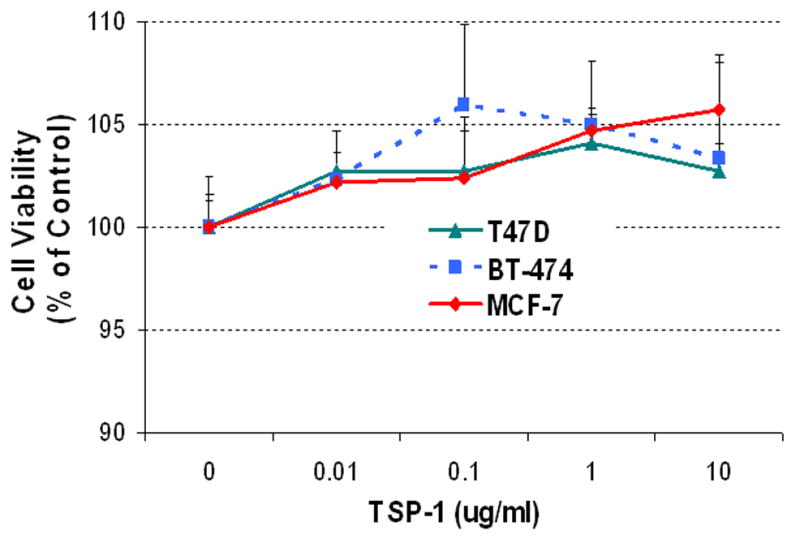
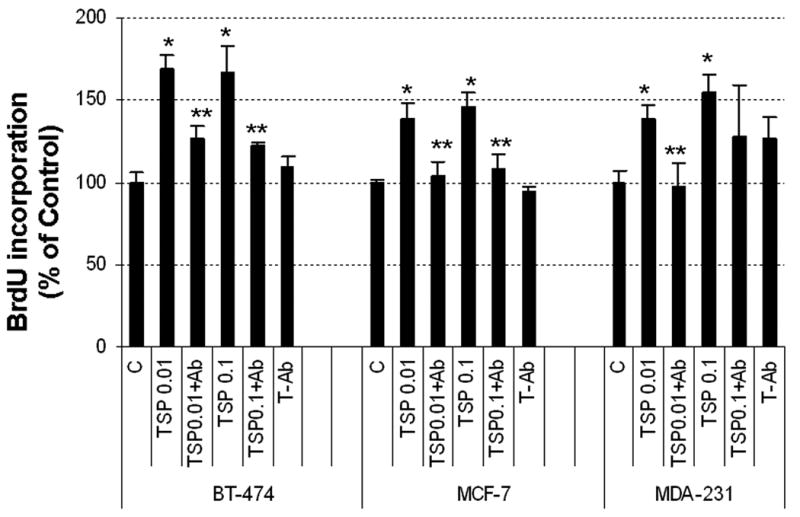
(A) Cells were serum-starved and then incubated in fresh medium without serum with the indicated concentrations of exogenous TSP-1 for 15 h. BrdU was added to the media, and the cells were incubated for an additional 3 h. The cells were harvested, and BrdU was quantified by ELISA. (B) The effect of TSP-1 on breast cancer cell viability. Cells were seeded into 96-well plates overnight as described in Methods. The medium was removed and cells were washed once with DMEM/F12, and treated with identical concentrations shown in (A) in serum free DMEM/F12 medium for 24 hrs. Cell growth and viability were determined by SRB assay as described in Methods. (B) Specificity of TSP-1 effects on breast cancer cells. Cells were cultured as above and treated with either 0.01 or 0.1 μg/ml TSP-1 ± an antibody against TSP-1 (Ab) for 18 h. BrdU was added to the media 3 h prior to termination of the experiment (at 15h). Cells were harvested and BrdU was quantified by ELISA. * Significantly different from control; ** significantly different from hormone treatment in the absence of Ab (p < 0.05, ANOVA).
TSP-1 ELISA Assay
T47-D and MCF-7 breast cancer cells were plated in DME/F12 + 10% FBS overnight, washed with PBS, and incubated in DMEM/F12 with 5% charcoal stripped serum for 24 h. Cells were then washed with PBS and placed in serum free DMEM/F12 for 24 h. Cells were then incubated for a further 18 h in the presence of E2 and various ligands as indicated in the figure legends in serum free DMEM/F12 (T47-D) or DMEM/F12 with 1% charcoal stripped serum (MCF-7 cells do not survive in serum free media for extended periods of time). The media were collected and TSP-1 quantified using a Quantakine kit from R&D Systems (Minneapolis, MN) according to the manufacturer's protocol. TSP-1 values were calculated by plotting absorbance at 450 and 540 nm. Experimental values were compared to standard values. All values were normalized to total cellular protein, which was determined using a BCA assay. For this assay, cell pellets were washed once with cold PBS and resuspended in 0.2-0.3 mL lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, and 1% NP-40). The cell suspension was vortexed for 10 s, incubated on ice for 30 min, and centrifuged at 14,000 rpm for 15 min at 4°C. Supernatants were transferred to a fresh tube and protein levels quantified.
Transient Transfection Assay
The TSP-1 promoter and its deletion constructs were kindly provided by Drs. Hong and Kang from the Catholic University of Korea. Cells were seeded into 6-well plates at 3 × 105 cells/well, grown to 60%-70% confluence, washed with PBS, and then incubated with DMEM/F12 medium supplemented with 5% dextran-coated charcoal-stripped serum for 18-24 h. Cells were washed and transfected with the indicated plasmids using Superfect reagent (Qiagen, Valencia, CA) according to the manufacturer's guidelines. Next, cells were washed and treated with hormone in medium with 5% charcoal stripped serum for 18-24 h and then harvested. Luciferase activity was quantified using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI) and a Sirius luminometer (Berthold Detection Systems, GmbH). Renilla luciferase was expressed from pRL-CMV (Promega E2261). This plasmid was cotransfected with a consensus estrogen responsive element (ERE)-Luc at a ratio of 1:10.
Autocrine Effects of Estrogen-induced TSP-1 on Tumor Cell Proliferation
MCF-7 cells were grown in 100-mm tissue culture dishes in medium containing 10% FBS and allowed to reach approximately 60%-70% confluence. Cells were washed twice with PBS and treated with 5% charcoal stripped FBS in DME/F12 for 24 h. Cells were washed and medium replaced with serum-free DME/F12 containing 10 nM E2 ± TSP-1 antibody (Clone A4.1, Neomarkers, Fremont, CA), after which cells were incubated for 24 h. Three hours prior to the end of the experiment, 100 μM bromodeoxyuridine (BrdU) was added to each well, and BrdU incorporation was measured with an ELISA kit (Roche Diagnostics Corporation, Indianapolis, IN).
Animal Studies
Female athymic nude (nu/nu) mice, 5 to 6 weeks old and weighing 18 to 22 g, were purchased from Harlan Sprague Dawley, Inc (Indianapolis, IN). The mice were housed in a laminar air-flow cabinet under specific pathogen–free conditions. All facilities were accredited by the American Association for Accreditation of Laboratory Animal Care, Inc. in accordance with the current regulations and standards of the United States Department of Agriculture and the Public Health Service.
Nude mice were inoculated on the back with 17-β-estradiol pellets (1.7 mg/pellet, 60-day release) or placebo pellets (both from Innovative Research of America, Sarasota, FL) 24-48 h prior to inoculation with tumor cells. Cultured breast cancer cells were harvested by trypsinization with 1× 0.05% trypsin-EDTA (Invitrogen/Life Technologies) and washed twice with DMEM/F12 medium. MCF-7 cells (5 × 106) were resuspended in 0.15 ml Matrigel (BD Biosciences, Bedford, MA):DMEM/F12 medium; 1:4 [v/v]). Cells were then injected subcutaneously (sc) into both flanks of each mouse (0.15 ml at each site). Tumor size was measured every 2 or 3 days with a digital caliper and tumor volumes were calculated by the formula (L × W × H) × π/6 (27). Intraperitoneal (ip) injections with a TSP-1–specific antibody or a matched IgM control (Neomarkers) were started when tumor volumes reached approximately 80 mm3. Following the first injection on day 9, tumors began to shrink. Treatment was then continued on days 11-13 with single daily ip injections. Animals were weighed throughout the experiment to monitor any toxic effects and were sacrificed on day 19.
Statistics
Values are reported as mean ± SEM. Statistical analysis was carried out using ANOVA and values were considered significant at P < 0.05. Student's _t_-test was used to compare mean values for transient transfection assays and dose-dependent production of TSP-1 message in T47-D and MCF-7 cells.
Results
E2 induces TSP-1 mRNA in breast cancer cells
We treated T47-D cells with 10 nM E2 and collected RNA at various points to determine a time course for the expression of TSP-1 mRNA. E2 increased the levels of TSP-1 message over time up to 12 h (Fig. 1A). Further studies showed that TSP-1 levels remained at a high level even after 24 h exposure to E2 (not shown). Actinomycin-D completely blocked the induction of TSP-1 by E2, indicating that E2 induces TSP-1 transcription; however, administration of puromycin, a protein synthesis inhibitor, had no such effect (Fig 1B), indicating that TSP-1 induction was independent of new protein synthesis. Furthermore, the induction of TSP-1 was specific and likely mediated via ER, because a 100-fold excess of ICI-182,780, an E2 antagonist that competitively binds ER, completely blocked the E2-mediated TSP-1 induction (Fig 1C). E2 also induced TSP-1 in a dose-dependent manner in the ER-positive MCF-7 cell line (Fig 1D), although to a much lower extent than in T47-D cells. No E2-dependent induction of TSP-1 was observed in breast cancer cell lines that lack ER-alpha, such as MDA-MB-231 (data not shown).
Figure 1. E2 induces TSP-1 message and protein in breast cancer cells.
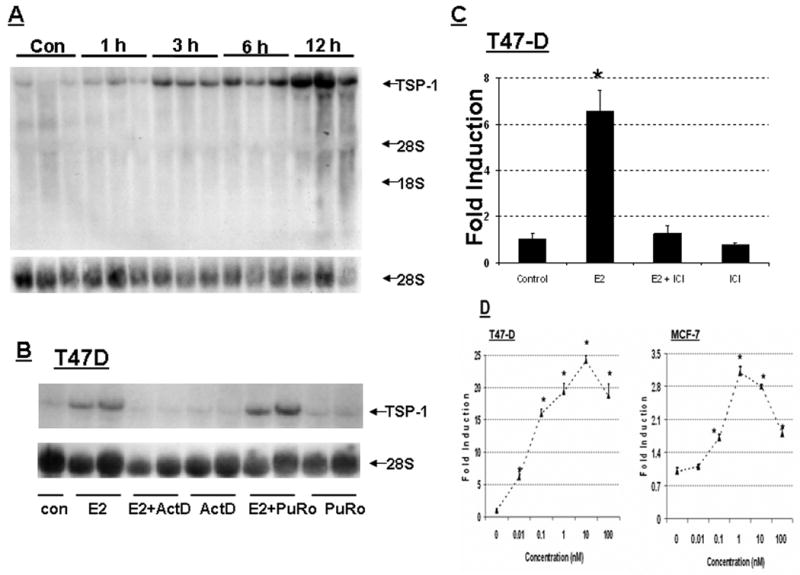
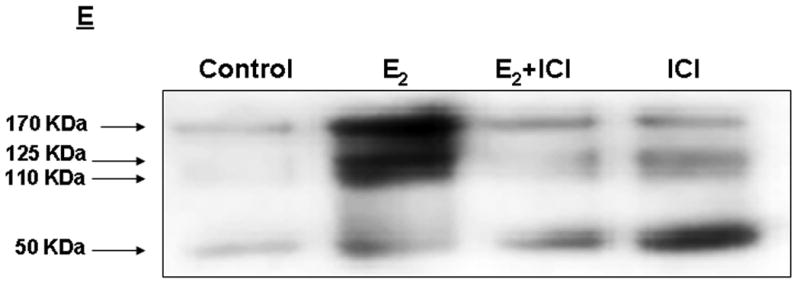
T47-D and MCF-7 cells were cultured and total RNA prepared and assessed for the expression of TSP-1 mRNA by Northern blot analysis. The blots were then stripped and probed for 28S RNA as a loading control. (A) T47-D cells were treated with 10 nM E2 for 1, 3, 6, or 12 h, as indicated. The TSP-1 band is marked with an arrow, and the locations of 28S and 18S ribosomal RNA are shown as reference points. (B) Cells were treated with 10 nM E2 for 6 h in the presence of actinomycin D (Act D; 2 μg/ml) or puromycin (PuRo; 10 μg/ml). (C) Cells were treated for 6 h with 10 nM E2, 10 nM E2 in the presence of 1 μM ICI 182,780 (E2 + ICI), or 1 μM ICI 182,780 alone (ICI). (D) Cells were incubated for 6 h with the indicated dose of E2, and TSP-1 message was visualized as shown in panel B. Autoradiograms were scanned and the densitometric values for the TSP-1 bands were normalized to the densitometric values for 28S in each lane. Values are mean + SEM (n = 3 per dose). * Significantly different from control (p < 0.05; Student t-test). (E) Western blot analysis of serum free media collected after treating T47-D cells with estradiol (E2) in the absence (control) and presence or absence of ICI 182,780 (ICI). Treatment of media prior to western blot analysis is described in Methods.
To ensure that induced TSP-1 message led to synthesis and secretion of estrogen dependent TSP-1 from breast cancer cells, we performed a Western blot analysis of media collected following an overnight incubation of cells with the hormone. As shown in Fig I E, estradiol induced a 5-6 fold induction of TSP-1 in media (170 kDa). Induction of two additional lower molecular weight fragments (125- and 110- kDa) was also observed. A 50-KDa band was also detected, though this fragment was unaffected by estradiol treatment. Further quantitation of TSP-1 in response to various ligands was conducted using an ELISA kit as described below.
The human TSP-1 promoter contains an estrogen-responsive region
To investigate how E2, acting via the ER, induces TSP-1 at the transcriptional level, we examined whether the promoter region for TSP-1 contains an estrogen-responsive region. As shown in Fig 2, T47-D cells transfected with the full-length TSP-1 promoter region, containing 2200 bp upstream of the transcription start site, were responsive to E2, and the response was blocked in the presence of 100-fold excess of ICI 182,780. The response was smaller than that observed with the consensus estrogen response element present in the vitellogenin gene (20-fold compared with 50- to 60-fold). Transfection of a truncated promoter (-1732/+754) completely abolished the response to E2 (Fig 2). Further deletions up to the -35/+754 region did not restore the response observed with the full-length -2200 promoter, suggesting that an estrogen-responsive element is present in the human TSP-1 gene promoter in the region between -2200 and -1734 bp upstream of the transcription start site. Similar results were obtained with MCF-7 cells (data not shown).
Figure 2. The TSP-1 promoter contains an estrogen-responsive region.
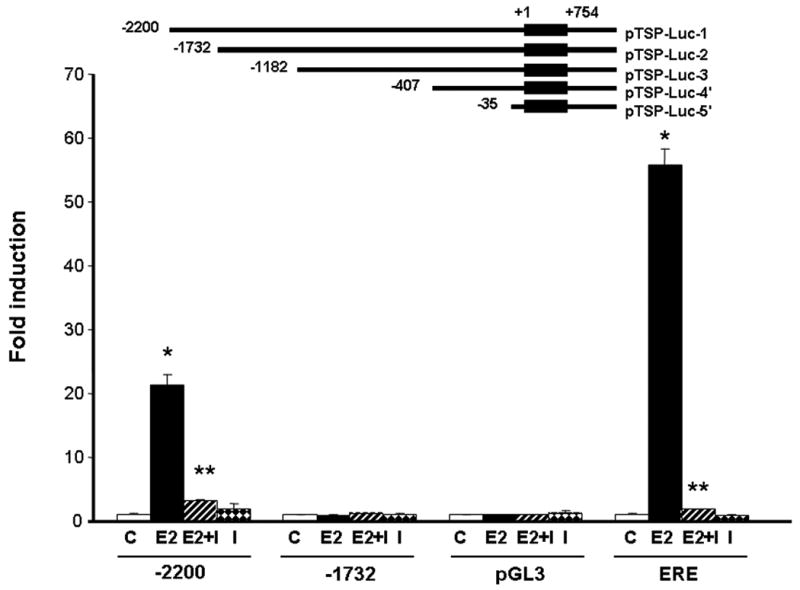
Reporter gene constructs are shown schematically in the upper part of the figure. The DNA sequence coordinates of the human TSP-1 promoter are indicated; +1 indicates the transcription start site of the TSP-1 gene. Cells were treated with 10 nM E2 ± 1 μM ICI 182,780 (I) for 18 h. Cells were harvested and luciferase activity was quantified. pGL3, empty vector; ERE, estrogen response element from the vitellogenin gene; * significantly different from control; ** Significantly different than E2 treatment in the absence of ICI 182,780 (p < 0.05, Student _t_-test, n = 3 per treatment).
E2 promotes the release of TSP-1 from human breast cancer cells in an ER-alpha–dependent manner
We next performed studies to determine whether induction of TSP-1 mRNA by E2 in breast tumor cells leads to the release of TSP-1 protein in a hormone-dependent manner, and whether such release is dependent on the presence of ER-alpha or ER-beta. T47-D and MCF-7 cells were treated with 10 nM E2, which binds to both forms of the receptor, together with an agonist specific for either ER-alpha (PPT) or ER-beta (DPN). Treatment of both T47-D and MCF-7 cells with 10 nM E2 and PPT resulted in the release of TSP-1 protein (Fig. 3). In contrast, DPN did not cause the production of TSP-1 from either cell type, demonstrating that TSP-1 release is controlled via ER-alpha in these human breast cancer cells.
Figure 3. E2 causes release of TSP-1 protein from breast cancer cells.
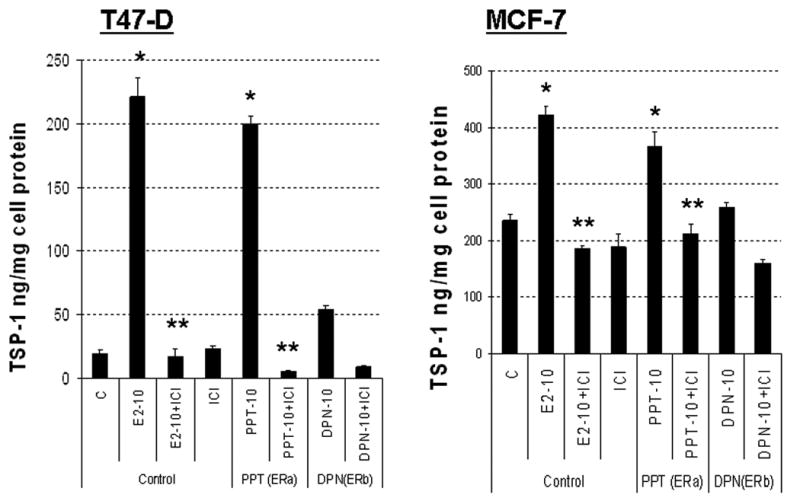
T47-D or MCF-7 cells were incubated overnight in DMEM/F12 + 5% charcoal stripped FBS. Fresh serum-free media was added and cells were incubated for 24 h. The media was then replaced with either serum free DMEM/F12 (T47-D) or DMEM/F12 with 1% charcoal stripped serum (MCF-7) and cells treated with 10 nM E2 (E2-10), an ER-alpha–specific ligand (PPT; 10 nM), or an ER-beta–specific ligand (DPN; 10 nM) ± 1 μM ICI 182,780 (ICI) for 18 h as indicated. The media were collected, and TSP-1 was quantified by ELISA. * significantly different from control; ** significantly different from hormone treatment in the absence of ICI 182,780 (p < 0.05 ANOVA).
E2-dependent proliferation of breast cancer cells is suppressed by a TSP-1 antibody, both in vivo and in vitro
Using the well-characterized MCF-7 cell-line, we examined whether E2-induced TSP-1 is involved in the in vitro proliferation of human breast cancer cells and whether we could replicate the same results in vivo. To ascertain whether E2-induced TSP-1 directly affects the proliferation of breast cancer cells, we incubated cultured MCF-7 cells with E2 and with or without a TSP-1–specific antibody. As determined by measuring BrdU incorporation, exposure to E2 caused the proliferation of breast cancer cells in vitro (Fig 4A). The TSP-1–specific antibody prevented this proliferation, indicating that the effect can be attributed to the induction of TSP-1 by E2.
Figure 4. TSP-1-induced proliferation of breast cancer cells.
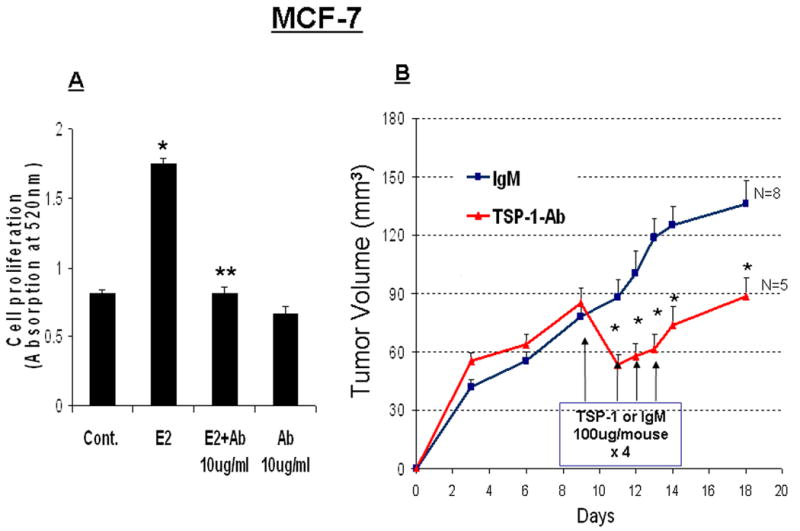
(A) MCF-7 cells were cultured in DME/F12 with 5% DCC serum. The medium was changed to serum-free medium, and the cells were treated for 24 h in the presence of E2 alone, E2 + an antibody against TSP-1 (E2 + Ab), or Ab alone. BrdU was added to the media, and the cells were incubated for an additional 3 h. Cells were then harvested and BrdU was quantified by ELISA. * significantly different from control; ** significantly different from E2 treated group (p < 0.05 ANOVA). (B) MCF-7 xenografts were grown in intact nude mice supplemented with an E2 pellet as described in the Methods. On day 9, the mice were treated with an antibody against TSP-1 or an IgM control. Additional antibody injections were given on days 11, 12, and 13. * Significantly different from IgM-treated control animals (p < 0.05, student t-test).
In order to determine whether the E2-induced TSP-1 activity is also involved in the progression of breast tumors, we established xenografts from MCF-7 cells in nude mice. We implanted the mice with E2 pellets before injecting the tumor cells because MCF-7 tumors do not grow efficiently without E2. The tumor-bearing mice were given either a TSP-1 antibody or an IgM antibody (as a control). Following the first injection of TSP-1 antibody, we observed a dramatic reduction in the growth of tumor xenografts (Fig 4B). Tumor growth remained low until injections of TSP-1 antibody ceased (Fig 4B). The IgM control antibody did not block tumor progression.
To assess whether TSP-1 has a direct effect on proliferation of breast cancer cells, we performed a dose-response study with exogenous TSP-1 and 3 commonly used cell lines, MCF-7, BT-474, and T47-D. Low concentrations (0.01-1.0 μg/ml) caused tumor cells to proliferate, whereas a higher concentration (10 μg/ml) actually inhibited tumor cell proliferation (Fig 5A). The extent of induction was most pronounced in BT-474 cells and was minimal in T47-D cells, with an intermediate response in MCF-7 cells, suggesting that each cell type responds differently to TSP-1. To establish whether the effect of TSP-1 on various breast cancer cell lines was specific, we suppressed TSP-1 activity with a specific antibody. To ensure that higher doses of TSP-1 (10 ug/ml) did not induce apoptosis and consequent loss of cells, we performed a cell viability assay as described previously (28). As illustrated in Fig 5B no cell loss occurred during 24 h exposure to TSP-1, confirming that incorporation of BrdU represented new DNA synthesis and that reduced proliferation values occurring as a consequence of exposure to higher doses of TSP-1 were not due to lower cell numbers. Because T47-D cells responded minimally to TSP-1, we introduced an additional cell line (MDA-MB-231) into our studies, along with BT-474 and MCF-7 cells. All three cell lines tested showed increased proliferation in response to low concentrations of TSP-1 (Fig 5C); this response was inhibited in all cell lines by a TSP-1 antibody, demonstrating the specificity of the response.
E2 induces TSP-1 in normal mouse mammary glands
We next examined whether E2-mediated TSP-1 induction in the mammary gland was conserved between mouse and human. Ovariectomized mice were given E2, and RNA was prepared from the mammary gland collected at different time points to assess TSP-1 mRNA induction. Due to potentially low levels of TSP-1 message in the normal mammary gland, we used real-time quantitative PCR for this analysis. A single injection of E2 transiently upregulated TSP-1 message, with maximal induction occurring 3 h after E2 administration (Fig 6A). The induction of TSP-1 mRNA was transient, declining to basal levels about 12 h after the E2 injection. This was in contrast to the effect in T47-D cells, which continued to accumulate TSP-1 mRNA until about 12 h after E2 administration. To determine whether the in vivo TSP-1 induction was ER-dependent, we injected animals with the ER antagonist ICI 182,780 30 min before administering E2 and collected RNA 3 h after the E2 injection. Real-time PCR assays showed that ICI 182,780 completely blocked the production of E2-induced TSP-1 in mouse mammary gland, indicating that, as in human breast cancer cells, induction of TSP-1 was ER-dependent in mouse mammary glands (Fig 6B).
Figure 6. E2-induced TSP-1 message in normal mouse mammary gland.
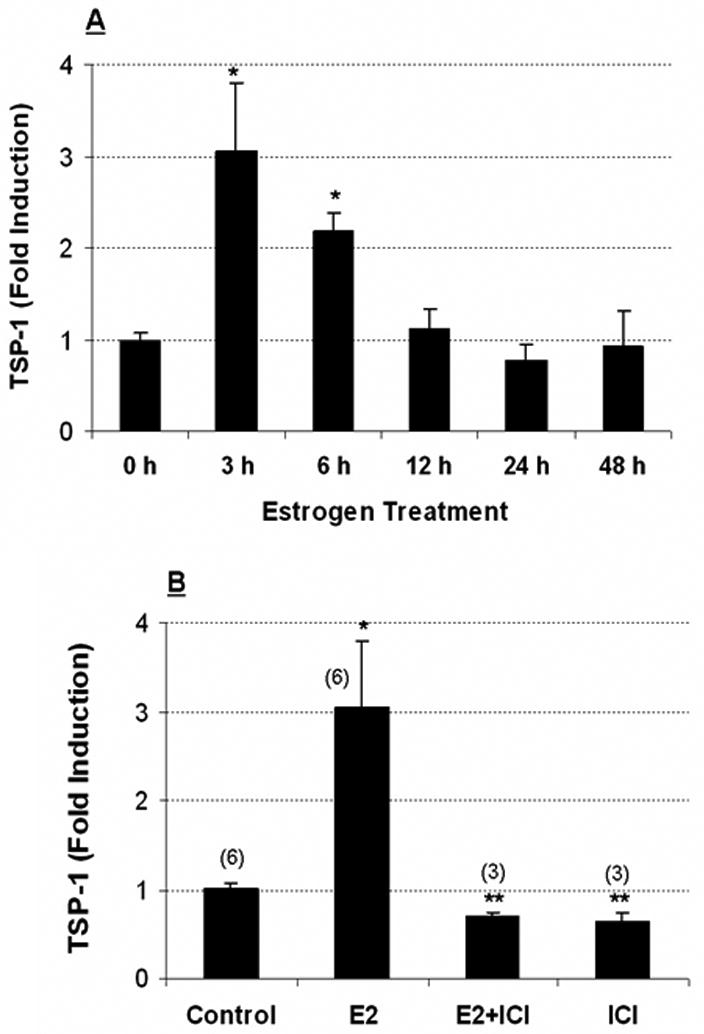
(A) Ovariectomized Balb/c mice (n = 3 per time point) were injected with 40 μg/kg E2 and sacrificed at the times shown. RNA was prepared as described in Methods. TSP-1 message was quantified using real-time PCR with 18S as an internal control. (B) ICI 182,780 (ICI) inhibits estradiol-induced TSP-1 in mouse mammary gland. Ovariectomized mice were injected with ICI (3 mg/kg) 30 min prior to the administration of E2. The mice were sacrificed 3 h later, and RNA was prepared using a Qiagen RNeasy kit. TSP-1 message was quantified using real-time PCR analysis with 18S as an internal control. Numbers in parentheses refer to the number of animals used. * Significantly different from control (p < 0.05; ANOVA).
Discussion
TSP-1 is an extracellular protein that has several biological functions, including roles in cell proliferation, wound healing, adhesion, and metastasis (29-31). TSP-1 has also been considered to be anti-angiogenic, because it is believed to promote endothelial cell apoptosis (32). However, its anti-angiogenic function may depend on the microenvironment, and several studies have raised serious doubts about its role exclusively as an anti-angiogenic protein (3-8, 33, 34). Indeed, a recent commentary on TSP-1 stated that “thrombospondin is downregulated in many but by no means all human neoplasms, invasive ductal breast carcinoma being a conspicuous counter example…. perhaps the famed anti-angiogenic behavior of TSP-1 requires some serious prodding.” (8). This statement is reinforced by our observation that TSP-1 may play a direct proliferative role in breast cancer cells, and there are reports that TSP-1 plays a role in tumor progression, metastasis, and tumor cell adhesion in breast cancer (3-8). However, TSP-1 fragments have been reported to possess both pro- and anti-angiogenic functions (35). Although there is evidence that breast epithelial cells produce TSP-1, and the protein is elevated in malignant or aggressive breast cancer cells (3, 14,15), the effects of E2 on the induction of TSP-1 message and protein have not been studied in detail in breast cancer cells. Furthermore, there are no reports describing the effects of TSP-1 produced by tumor cells on the cells' proliferation.
We recently observed that E2, a hormone that drives the progression of many human breast cancers, downregulates CD36, a TSP-1 receptor in human breast epithelial cancer cells (21). CD36 has been chiefly characterized in endothelial cells, where it plays a role in TSP-1–mediated apoptosis (32). Because E2 is intimately involved in the progression of breast disease, we undertook this series of studies to investigate whether E2 might also regulate TSP-1 in breast tumor cells. Surprisingly, we found that upon exposure to E2, certain tumor cells expressed TSP-1 mRNA and produced TSP-1 protein. Because TSP-1 has also been implicated in cell proliferation, we examined the role of E2 and its receptor in mediating TSP-1 production by human breast cancer cells and investigated the mechanism and consequences of this phenomenon. E2-induced proliferation of mammary cells requires the formation of an extracellular matrix, and TSP-1 is known to affect cell-cell adhesion, angiogenesis, and expansion (36). We therefore also sought to determine whether E2 might induce TSP-1 in normal mammary cells.
Our findings clearly show that TSP-1 is under direct E2 regulation in both breast cancer cells and normal mammary gland and, furthermore, that these effects are mediated via ER-alpha in human tumor cells. In this respect it is interesting to note that ER-alpha but not ER-beta is linked with proliferation of breast cancers (20). In fact ER-beta is believed to prevent the proliferative effect promoted via ER-alpha (37). Because women are exposed to estrogens both physiologically and pharmacologically, it is important that we understand the potential effects of estrogen-selective ligands on TSP-1 production. Although many breast tumors are under the influence of steroid hormones, including synthetic hormones consumed by millions of women worldwide, surprisingly little attention has been paid to the effects of these pharmacologically administered compounds on TSP-1 production. The few studies addressing steroid hormone control of TSP-1 have focused almost exclusively on the effects of estrogens and progestins in the uterus and on uterine cells in culture (38-40). We therefore felt it was important to examine the role estrogen might play in TSP-1 induction in normal breast tissue as well as breast cancers. Our findings clearly demonstrate that TSP-1 is under hormonal regulation in both normal and neoplastic breast cells.
The time course study conducted using T47-D breast cancer cells showed that unlike many mRNA species, which undergo transient E2 induction (including TSP-1 production in normal mammary gland, Fig 6A), TSP-1 mRNA was continuously induced for up to 24 h in vitro in the presence of E2. The kinetics of E2-mediated TSP-1 induction therefore appear to be quite different in human tumor cells than in normal mouse mammary gland, although it is possible that species differences could play a role. It is possible that effects of E2 on TSP-1 production could vary in vivo compared with in vitro situations. Induction of TSP-1 message was blocked by actinomycin-D, indicating that ER is directly involved in TSP-1 induction in breast cancer cells, likely through an estrogen-responsive region in the TSP-1 promoter located between -2200 and -1732 bp upstream of the transcription start site of the gene. The direct effect of E2 was further confirmed by the complete arrest of TSP-1 induction by the antiestrogen ICI 182,780 in both breast cancer cells and normal mouse mammary gland. These findings lead us to conclude that the TSP-1 gene must be considered a bona fide estrogen-responsive gene in human breast cancer cells, though it remains to determined whether ER physically contacts the estrogen responsive region or whether ER functions through other indirect mechanisms including activation of extranuclear signal transduction pathways and/or indirectly by interacting with other transcription factors such as AP-1 (41, 42).
Using receptor-specific agonists we determined that E2 induction of TSP-1 was dependent upon its binding to ER-alpha in breast cancer cells, and, furthermore, that tumor cell proliferation occurred via an autocrine mechanism (Fig 4A). Antibodies to TSP-1 also prevented E2-dependent tumor cell progression in vivo. Although use of the antibody did not lead to complete tumor loss, our antibody treatment was short-lived; consequently, it is possible that over a prolonged period of exposure to antibody, greater tumor loss might be observed. This notion is strengthened by the observation that immediately after the last injection of TSP-1 AB, there was a dramatic increase in the rate of progression of tumor growth approaching that seen under control conditions. There is a report that tumors are able to overcome the inhibitory effects of TSP-1, though the authors did not examine the role of TSP-1 in proliferation (43). In our in vivo and in vitro studies, E2 might induce factors that override the previously reported anti-angiogenic effects of TSP-1, thereby facilitating tumor cell survival and proliferation. Such factors may include VEGF, a potent angiogenic protein that some reports have shown to be under E2 control (44, 45). On the other hand breast cancer cells may produce fragments of TSP-1 with angiogenic potential that directly induce proliferation of breast cancer cells, a possibility that has previously been discussed (35). Also, using Western blot analysis we detected a TSP-1 related increase in fragments that were smaller than the full length protein (170 KDa) in conditioned media prepared from estrogen treated breast cancer cells. Whether these bands represent an angiogenic fragment of TSP-1, or whether full length TSP-1 can also be angiogenic in a context-dependent manner, will be subjects of a future study.
Interestingly, TSP-1 also seemed to increase breast cancer cell proliferation in a paracrine manner following the administration of exogenous TSP-1 protein. Proliferation only occurred when cells were exposed to lower concentrations of TSP-1; raising the TSP-1 concentration attenuated cell proliferation. A dose-dependent effect of TSP-1 has previously been reported in endothelial cells (17). Other studies support a pro-proliferative role for TSP-1 in wound healing (30), vascular smooth muscle cells (31), pancreatic cancer (46), and hepatocellular carcinoma (33). It appears therefore that TSP-1 may play a direct proliferative role in many different types of tumors. In addition, its effects on cell adhesion might also promote an environment in the extracellular compartment conducive for the proliferation of both tumor and endothelial cells.
The growing number of studies seeking to further examine a proliferative role for TSP-1 in both physiological (7, 18, 30, 31) and pathological situations (47-49) underscores the importance of our studies exploring the relationship between sex steroids and TSP-1 during the progression of breast disease. On the basis of our findings we suggest that attempts to elevate endogenous TSP-1 levels in an effort to exploit its anti-angiogenic functions should be considered with caution, because breast tumors may respond differently to this protein depending on their microenvironment. This would be especially true if a tumor were developing in response to E2. We further propose that any TSP-1–mediated anti-angiogenic therapy should be used in combination with other anti-cancer or anti-hormonal agents. Conversely, however, targeting TSP-1 in developing tumor tissues could provide a therapeutic means by which we might prevent further tumor progression. Most importantly, gaining a deeper understanding of how estrogens affect TSP-1 production by breast cancer cells may further the potential for using anti-angiogenic agents or antihormones for treating breast disease.
Acknowledgments
Supported by Susan G Komen for the Cure grant BCTR0600704, a COR award from the University of Missouri-Columbia, and in part by NIH grant CA-86916.
We would like to thank Dr. Jack Lawler, Harvard University for providing the human TSP- 1 probe, as well as Drs. Hong and Kang from the Catholic University of Korea for the provision of human TSP-1 luciferase promoter constructs. We acknowledge the excellent technical help provided by Zhifang Zhu, Constance Chiappetta, and Vanessa Welbern during the course of this study. SMH also wishes to express his gratitude to Dr. George Stancel from the University of Texas Health Sciences Center in Houston for initial consultation and advice on this project. SMH is the Zalk Missouri Professor of Tumor Angiogenesis.
References
- 1.Good DJ, Polverini PJ, Rastinejad F, Le Beau MM, Lemons RS, Frazier WA, Bouck NP. A tumor suppressor-dependent inhibitor of angiogenesis is immunologically and functionally indistinguishable from a fragment of thrombospondin. Proc Natl Acad Sci. 1990;87:6624–8. doi: 10.1073/pnas.87.17.6624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sid B, Sartelet H, Bellon G, El Btaouri H, Rath G, Delorme N, Haye B, Martiny L. Thrombospondin 1: a multifunctional protein implicated in the regulation of tumor growth. Crit Rev Oncol Hematol. 2004;49:245–8. doi: 10.1016/j.critrevonc.2003.09.009. [DOI] [PubMed] [Google Scholar]
- 3.Wang-Rodriguez J, Urquidi V, Rivard A, Goodison S. Elevated osteopontin and thrombospondin expression identifies malignant human breast carcinoma but is not indicative of metastatic status. Breast Cancer Res. 2003;5:R136–43. doi: 10.1186/bcr620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang TN, Qian XH, Granick MS, Solomon MP, Rothman VL, Berger DH, Tuszynski GP. Inhibition of breast cancer progression by an antibody to a thrombospondin-1 receptor. Surgery. 1996;120:449–54. doi: 10.1016/s0039-6060(96)80322-9. [DOI] [PubMed] [Google Scholar]
- 5.Wang TN, Qian X, Granick MS, Solomon MP, Rothman VL, Berger DH, Tuszynski GP. Thrombospondin-1 (TSP-1) promotes the invasive properties of human breast cancer. J Surg Res. 1996;63:39–43. doi: 10.1006/jsre.1996.0219. [DOI] [PubMed] [Google Scholar]
- 6.Wong SY, Purdie AT, Han P. Thrombospondin and other possible related matrix proteins in malignant and benign breast disease. An immunohistochemical study. Am J Pathol. 1992;140:1473–82. [PMC free article] [PubMed] [Google Scholar]
- 7.Ichii T, Koyama H, Tanaka S, Shioi A, Okuno Y, Otani S, Nishizawa Y. Thrombospondin-1 mediates smooth muscle cell proliferation induced by interaction with human platelets. Arterioscler Thromb Vasc Biol. 2002;22:1286–92. doi: 10.1161/01.atv.0000024684.67566.45. [DOI] [PubMed] [Google Scholar]
- 8.Thomas-Tikhonenko A, Iruela-Arispe ML. Whence Thrombospondin. Cancer Biol & Therapy. 2004;3:e60–e61. doi: 10.4161/cbt.3.4.737. [DOI] [PubMed] [Google Scholar]
- 9.Motegi K, Harada K, Pazouki S, Baillie R, Schor AM. Evidence of a bi-phasic effect of thrombospondin-1 on angiogenesis. Histochem J. 2002;34:411–21. doi: 10.1023/a:1023687505139. [DOI] [PubMed] [Google Scholar]
- 10.Lawler J, Miao WM, Duquette M, Bouck N, Bronson RT, Hynes RO. Thrombospondin-1 gene expression affects survival and tumor spectrum of p53-deficient mice. Am J Pathol. 2001;159:1949–56. doi: 10.1016/S0002-9440(10)63042-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodriguez-Manzaneque JC, Lane TF, Ortega MA, Hynes RO, Lawler J, Iruela-Arispe ML. Thrombospondin-1 suppresses spontaneous tumor growth and inhibits activation of matrix metalloproteinase-9 and mobilization of vascular endothelial growth factor. Proc Natl Acad Sci. 2001;98:12485–90. doi: 10.1073/pnas.171460498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown LF, Guidi AJ, Schnitt SJ, Van De Water L, Iruela-Arispe ML, Yeo TK, Tognazzi K, Dvorak HF. Vascular stroma formation in carcinoma in situ, invasive carcinoma, and metastatic carcinoma of the breast. Clin Cancer Res. 1999;5:1041–56. [PubMed] [Google Scholar]
- 13.Tuszynski GP, Nicosia RF. Localization of thrombospondin and its cysteine-serine-valine-threonine-cysteine- glycine-specific receptor in human breast carcinoma. Lab Invest. 1994;70:228–33. [PubMed] [Google Scholar]
- 14.Bertin N, Clezardin P, Kubiak R, Frappart L. Thrombospondin-1 and -2 messenger RNA expression in normal, benign, and neoplastic human breast tissues: correlation with prognostic factors, tumor angiogenesis, and fibroblastic desmoplasia. Cancer Res. 1997;57:396–9. [PubMed] [Google Scholar]
- 15.Tuszynski GP, Smith M, Rothman VL, Capuzzi DM, Joseph RR, Katz J, Besa EC, Treat J, Switalska HI. Thrombospondin levels in patients with malignancy. Thromb Haemost. 1992;67:607–611. [PubMed] [Google Scholar]
- 16.RayChaudhury A, Frazier WA, D'Amore PA. Comparison of normal and tumorigenic endothelial cells: differences in thrombospondin production and responses to transforming growth factor-beta. J Cell Sci. 1994;107:39–46. doi: 10.1242/jcs.107.1.39. [DOI] [PubMed] [Google Scholar]
- 17.Pazouki S, Pendleton N, Heerkens E, Smither RL, Moore JV, Schor AM. Biphasic effect of thrombospondin-1 (TSP-1) in the regulation of angiogenesis in human breast carcinoma. Biochem Soc Trans. 1996;24:368S. doi: 10.1042/bst024368s. [DOI] [PubMed] [Google Scholar]
- 18.Taraboletti G, Morbidelli L, Donnini S, Parenti A, Granger HJ, Giavazzi R, Ziche M. The heparin binding 25 kDa fragment of thrombospondin-1 promotes angiogenesis and modulates gelatinase and TIMP-2 production in endothelial cells. FASEB J. 2000;14:1674–6. doi: 10.1096/fj.99-0931fje. [DOI] [PubMed] [Google Scholar]
- 19.Osborne CK. Steroid hormone receptors in breast cancer management. Breast Cancer Res Treat. 1998;51:227–38. doi: 10.1023/a:1006132427948. [DOI] [PubMed] [Google Scholar]
- 20.Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC. Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res. 2004;64:423–8. doi: 10.1158/0008-5472.can-03-2446. [DOI] [PubMed] [Google Scholar]
- 21.Uray I, Liang Y, Hyder SM. Estradiol down-regulates CD36 expression in human breast cancer cells. Cancer Lett. 2004;20:101–107. doi: 10.1016/j.canlet.2003.10.021. [DOI] [PubMed] [Google Scholar]
- 22.Hyder SM, Murthy L, Stancel GM. Progestin regulation of vascular endothelial growth factor in human breast cancer. Cancer Res. 1998;58:392–395. [PubMed] [Google Scholar]
- 23.Hyder SM, Chiappetta C, Stancel GM. Pharmacological and endogenous progestins induce vascular endothelial growth factor expression in human breast cancer cells. Int J Cancer. 2001;92:469–473. doi: 10.1002/ijc.1236. [DOI] [PubMed] [Google Scholar]
- 24.Hyder SM, Stancel GM, Chiappetta C, Murthy L, Boettger-Tong HL, Makela S. Uterine expression of vascular endothelial growth factor is increased by estradiol as well as tamoxifen. Cancer Res. 1996;56:3954–60. [PubMed] [Google Scholar]
- 25.Rubinstein LV, Shoemaker RH, Paull KD, Simon RM, Tosini S, Skehan P, Scudiero DA, Monks A, Boyd MR. Comparison of in vitro anticancer–drug- screening data generated with a tetrazolium assay versus a protein assay against a diverse panel of human tumor cell lines. J Natl Cancer Inst. 1990;82:1113–1118. doi: 10.1093/jnci/82.13.1113. [DOI] [PubMed] [Google Scholar]
- 26.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. New colorimetric cytotoxicity assay for anti-cancer-drug screening. J Natl Cancer Inst. 1990;82:1107–1112. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 27.El Etreby MF, Liang Y. Effect of antiprogestins and tamoxifen on growth inhibition of MCF-7 human breast cancer cells in nude mice. Breast Cancer Res Treat. 1998;49:109–19. doi: 10.1023/a:1006098910000. how to measure tumors. [DOI] [PubMed] [Google Scholar]
- 28.Liang Y, Hyder SM. Proliferation of Endothelial and Tumor Epithelial Cells by Progestin-Induced VEGF from Human Breast Cancer Cells: Paracrine and Autocrine Effects. Endocrinology. 2005;146:3632–41. doi: 10.1210/en.2005-0103. [DOI] [PubMed] [Google Scholar]
- 29.Incardona F, Lawler J, Cataldo D, Panet A, Legrand Y, Foidart JM, Legrand C. Heparin-binding domain, type 1 and type 2 repeats of thrombospondin mediate its interaction with human breast cancer cells. J Cell Biochem. 1996;2:431–42. doi: 10.1002/(sici)1097-4644(19960915)62:4<431::aid-jcb1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 30.DiPietro LA, Nissen NN, Gamelli RL, Koch AE, Pyle JM, Polverini PJ. Thrombospondin 1 synthesis and function in wound repair. Am J Pathol. 1996;148:1851–60. [PMC free article] [PubMed] [Google Scholar]
- 31.Patel MK, Lymn JS, Clunn GF, Hughes AD. Thrombospondin-1 is a potent mitogen and chemoattractant for human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1997;17:2107–14. doi: 10.1161/01.atv.17.10.2107. [DOI] [PubMed] [Google Scholar]
- 32.Reiher FK, Volpert OV, Jimenez B, Crawford SE, Dinney CP, Henkin J, Haviv F, Bouck NP, Campbell SC. Inhibition of tumor growth by systemic treatment with thrombospondin-1 peptide mimetics. Int J Cancer. 2002;98:682–9. doi: 10.1002/ijc.10247. [DOI] [PubMed] [Google Scholar]
- 33.Poon RT, Chung KK, Cheung ST, Lau CP, Tong SW, Leung KL, Yu WC, Tuszynski GP, Fan ST. Clinical significance of thrombospondin 1 expression in hepatocellular carcinoma. Clin Cancer Res. 2004;10:4150–7. doi: 10.1158/1078-0432.CCR-03-0435. [DOI] [PubMed] [Google Scholar]
- 34.de Fraipont F, Keramidas M, El Atifi M, Chambaz EM, Berger F, Feige JJ. Expression of the thrombospondin-1 fragment 167-569 in C6 glioma cells stimulates tumorigenicity despite reduced neovascularization. Oncogene. 2004;23:3642–9. doi: 10.1038/sj.onc.1207438. [DOI] [PubMed] [Google Scholar]
- 35.Sargiannidou I, Zhou J, Tuszynski GP. The role of thrombospondin-1 in tumor progression. Exp Biol Med. 2001;226:726–33. doi: 10.1177/153537020222600803. [DOI] [PubMed] [Google Scholar]
- 36.Esemuede N, Lee T, Peirre-Paul D, Sumpio BE, Gahtan V. The role of thrombospondin-1 in human disease. J Surg Res. 2004;122:135–142. doi: 10.1016/j.jss.2004.05.015. [DOI] [PubMed] [Google Scholar]
- 37.Lindberg MK, Movérare S, Skrtic S, Gao H, Dahlman-Wright K, Gustafsson JA, Ohlsson C. Estrogen receptor (ER)-beta reduces ERalpha-regulated gene transcription, supporting a “ying yang” relationship between ER-alpha and ERbeta in mice. Mol Endocrinol. 2003;17:203–8. doi: 10.1210/me.2002-0206. [DOI] [PubMed] [Google Scholar]
- 38.Iruela-Arispe ML, Porter P, Bornstein P, Sage HE. Thrombospondin-1, an Inhibitor of Angiogenesis, is Regulated by Progesterone in the Human Endometrium. J Clin Invest. 1996;97:403–12. doi: 10.1172/JCI118429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mirkin S, Archer DF. Effects of levonorgestrel, medroxyprogesterone acetate, norethindrone, progesterone, and 17beta-estradiol on thrombospondin-1 mRNA in Ishikawa cells. Fertil Steril. 2004;82:220–2. doi: 10.1016/j.fertnstert.2004.02.100. [DOI] [PubMed] [Google Scholar]
- 40.Kapiteijn K, Koolwijk P, Van Der Weiden R, Helmerhorst F, Kooistra T, Van Hinsbergh VW. Steroids and cytokines in endometrial angiogenesis. Anticancer Res. 2001;21:4231–42. [PubMed] [Google Scholar]
- 41.Song RX, Zhang Z, Santen RJ. Estrogen rapid action via protein complex formation involving ERalpha and Src. Trends Endocrinol Metab. 2005;16:347–53. doi: 10.1016/j.tem.2005.06.010. [DOI] [PubMed] [Google Scholar]
- 42.Marino M, Galluzzo P, Ascenzi P. Estrogen signaling multiple pathways to impact gene transcription. Curr Genomics. 2006;7:497–508. doi: 10.2174/138920206779315737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fontana A, Filleur S, Guglielmi J, Frappart L, Bruno-Bossio G, Boissier S, Cabon F, Clézardin P. Human breast tumors override the antiangiogenic effect of stromal thrombospondin-1 in vivo. Int J Cancer. 2005;116:686–691. doi: 10.1002/ijc.20584. [DOI] [PubMed] [Google Scholar]
- 44.Nakamura J, Savinov A, Lu Q, Brodie A. Estrogen regulates vascular endothelial growth/permeability factor expression in 7,12 dimethylbenz(a)anthracene induced rat mammary tumors. Endocrinology. 1996;137:5589–96. doi: 10.1210/endo.137.12.8940388. [DOI] [PubMed] [Google Scholar]
- 45.Hyder SM, Stancel GM, Chiappetta C, Murthy L, Boettgertong HL, Makela S. Uterine expression of vascular endothelial growth factor is increased by estradiol and tamoxifen. Cancer Res. 1996;56:3954–60. [PubMed] [Google Scholar]
- 46.Tobita K, Kijima H, Dowaki S, Oida Y, Kashiwagi H, Ishii M, Sugio Y, Sekka T, Ohtani Y, Tanaka M, Inokuchi S, Makuuchi H. Thrombospondin-1 expression as a prognostic predictor of pancreatic ductal carcinoma. Int J Oncol. 2002;21:1189–95. [PubMed] [Google Scholar]
- 47.Yee KO, Connolly CM, Duquette M, Kazerounian S, Washington R, Lawler J. The effect of thrombospondin- 1 on breast cancer metastasis. Breast Cancer Research Treatment. 2008 doi: 10.1007/s10549-008-9992-6. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pratt DA, Miller WR, Dawes J. Thrombospondin in malignant and non-malignant breast tissue. Eur J Cancer Clin Oncol. 1989;25:343–50. doi: 10.1016/0277-5379(89)90028-x. [DOI] [PubMed] [Google Scholar]
- 49.Byrne GJ, Hayden KE, McDowell G, Lang H, Kirwan CC, Tetlow L, Kumar S, Bundred NJ. Angiogenic characteristics of circulating and tumoural thrombospondin-1 in breast cancer. Int J Oncol. 2007;31:1127–32. doi: 10.3892/ijo.31.5.1127. [DOI] [PubMed] [Google Scholar]