Rapid rheological screening to identify conditions of biomaterial hydrogelation (original) (raw)
. Author manuscript; available in PMC: 2010 Jan 1.
Published in final edited form as: Soft Matter. 2009 Jan 1;5(4):740–742. doi: 10.1039/b818178k
Abstract
Hydrogels engineered for biomedical applications consist of numerous components, each of which can affect the material assembly and final mechanical properties. We present methods that rapidly generate rheological libraries to identify regimes of hydrogel assembly in a large composition parameter space. This method conserves both material and time, and leads to critical insight into assembly mechanisms and mechanics, which can then be used for further materials development and optimization.
The assembly of precursor molecules and macromolecules into hydrogels, and the resulting material mechanical properties, are principal design factors in the engineering of many biologically active and therapeutic materials.1 For instance, the gel microstructure and modulus are known to affect the viability and migration of cells2 and the differentiation of stem cells.3 Hydrogels intended for such biological applications are typically composed of numerous structural compounds, in addition to soluble and attached epitopes and markers,2,4,5 each of which can influence, by design or unintentional consequence, the assembly, final material properties, and ultimate material performance. In order to facilitate the design and optimization of therapeutic hydrogel materials over a large parameter space, we apply methods of high-throughput rheological characterization using multiple particle tracking microrheology. This simple, yet powerful approach enables us to rapidly assay compositions for hydrogel formation in a relatively short period of time, while consuming only small quantities of material.
In this study, the hydrogelator materials consist of maleimide-functionalized high molecular weight heparin (HMWH, MW = 15 000).6 Covalent cross-linking occurs by the addition of dithiolized poly(ethylene glycol) (PEG, MW = 10 000).6 Heparin has the unique ability to sequester and stabilize soluble proteins, including growth factors that promote cellular proliferation and differentiation.7 Because of this, heparin hydrogels have recently been engineered to stimulate communication between cells and a surrounding gel matrix,2 behave as stimuli responsive materials,4,8 and serve as a three-dimensional matrix for stem cell culture.5 In general, the high charge and conformational stiffness of many biomolecules, including heparin, may alter the anticipated gelation conditions and rheology. Thus, rapid evaluation of gelation in biomolecular gels provides a valuable tool for the synthesis of materials that mimic the chemical, structural and rheological characteristics of native biological matrices.
We systematically vary the number of maleimide sites available for cross-linking on the heparin backbone. The average numbers of maleimide functional groups per heparin, determined by NMR spectroscopy, are f = 4.2, 6.5, 8.3 and 11.6 Samples are prepared by varying the concentration of both the backbone and cross-linker molecules, and repeated for each heparin functionality. Each sample is made separately by mixing HMWH and PEG from 4 wt% stock solutions. Both polymers are suspended in phosphate buffered saline (1X PBS, pH 7.4). Each sample is then loaded into a capillary tube (0.2 × 2.0 × 50 mm), and sealed with fast-curing UV epoxy (Norland Optics, NOA81). Arrays of eight capillaries are fixed to one microscope slide, as illustrated in Fig. 1. This method increases the efficiency and accuracy of the manual data collection. Specifically, this is an optimal number of samples to allow the sample vessels to be imaged with common microscope equipment while simultaneously minimizing the time between sample loading and sealing, thus limiting possible contamination or evaporation. Samples equilibrate for at least two hours before measurements are taken.
Fig. 1.
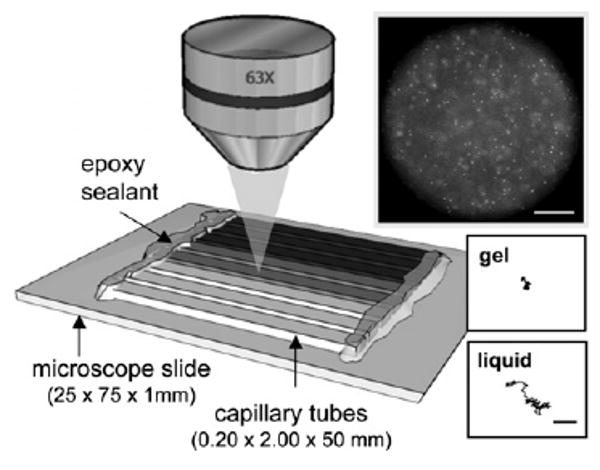
Eight individual samples are prepared in capillaries and attached to a single microscope slide. Particle trajectories are captured at 63 × magnification, as shown by the sample video frame (scale bar 50 μm). Insets show example trajectories of particles in a liquid and arrested in a gel (scale bar 2 μm).
Rheological libraries are created using multiple particle tracking microrheology.9–12 To perform particle tracking, monodisperse fluorescent polystyrene probe particles (diameter 2_a_ = 1.05 ± 0.01 (μm) are introduced into the material during mixing at a volume fraction φ ≈ 1 × 10−2. After equilibration, ensembles of particle trajectories are collected using video microscopy. The movement of the embedded tracer particles identifies the rheological state of each equilibrated sample, as illustrated in Fig. 1. The material viscoelastic properties are related to the ensemble average mean-squared displacement (MSD) by the generalized Stokes–Einstein relationship (GSER), J(τ) = π_a_〈Δ_r_2(τ) 〉/k_B_T, where J(τ) is the creep compliance and k_B_T is the thermal energy.10 Samples in which probes exhibit diffusive trajectories (〈_r_2(τ) 〉 ∼ τ) are indicative of a liquid with viscosity η = τ/J(τ). In contrast, restricted movement and corresponding sub-diffusive logarithmic slope of the MSD indicate that the tracer particles experience a viscoelastic network. At the liquid–solid transition, the MSD exhibits a power-law dependence over all lag times, 〈Δ r_2(τ) 〉 ∼ τ_n, where n ≈ 0.45 is the critical relaxation exponent of the gel network.13–15
A rheological library consisting of 73 samples requires only 48 mg of total polymer in our experiments due to the small sample volumes (approximately 20–40 (μL). Because the samples are prepared and equilibrated in parallel, the total time required to make rheological measurements for the entire library (approximately one day) is significantly shorter than traditional rheological characterization methods, which typically require serial sample preparations and are limited to 5–10 measurements per day.11 Excluding the equilibration time, each sample takes 15 min for preparation, data acquisition and multiple particle tracking analysis. To ensure the statistical accuracy of the data, 800 frames are collected with a field of view covering 65 000 (μm2. Although this sample preparation is simple in execution, the uniqueness and impact of this technique is exhibited in the creation of the gelation libraries.
The critical relaxation exponent divides the samples into their respective rheological states; samples with logarithmic slopes greater than n = 0.45 are viscous liquids, while those with n < 0.45 are viscoelastic solids.13–15 Fig. 2 shows MSDs for f = 6.5 HMWH. Each curve represents a single equilibrated sample with a unique composition. Data for the entire sample library is compiled in Fig. 3a across the parameter space consisting of the HMWH and PEG polymer concentrations and HMWH maleimide functionality. The symbol colors indicate the value of the MSD logarithmic slope, providing an immediate identification of compositions that lead to gel formation. As the backbone functionality increases, the area of this “gelation envelope” increases. We also find that a lower concentration limit of the gelation envelope occurs for all HMWH maleimide functionalities in the range of 3–4 wt% total polymer, including heparin backbone and PEG cross-linker.
Fig. 2.
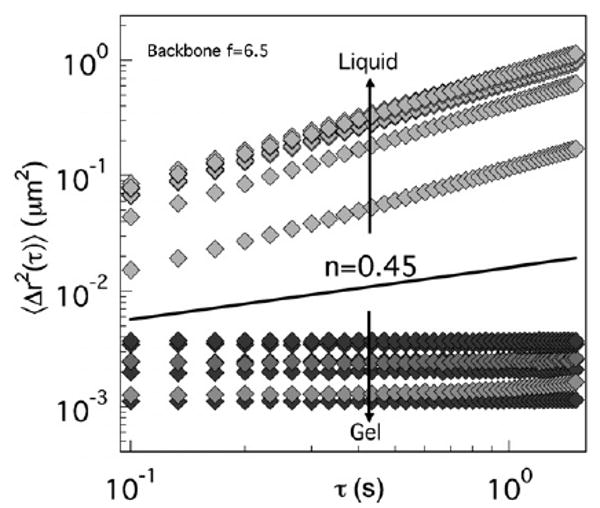
Probe particle mean-squared displacement (MSD) for equilibrated samples, each with a unique composition. The solid line represents the critical relaxation exponent, which distinguishes the rheological state of the sample as either a solid (gel) or a liquid (non-gel).
Fig. 3.
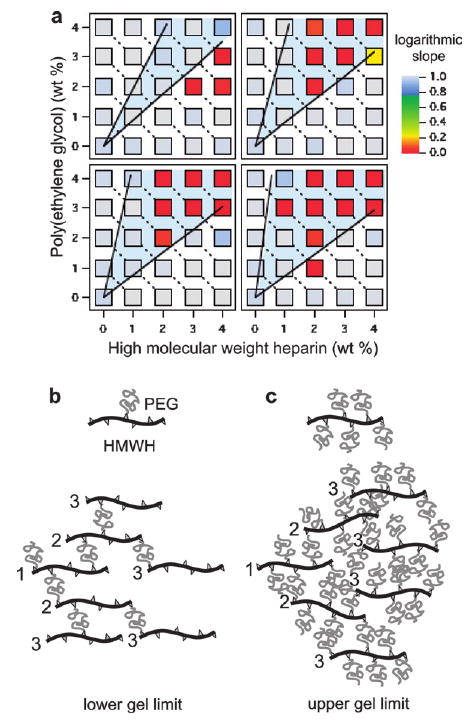
Rheological libraries of hydrogel assembly. (a) Libraries for four backbone functionalities. They are f = 4.2 (top left), 6.5 (top right), 8.3 (bottom left), and 11 (bottom right). Each square represents a discrete sample condition. The symbol colors correspond to the MSD logarithmic slope. Solid black lines represent the Flory–Stockmayer lower (b) and upper (c) limits of gelation. The numbers in each network diagram indicate the generation from an arbitrary reference molecule. The minimum condition for gelation occurs when children cross-link to the same number of molecules as their parent.
Gelation limits based on Flory–Stockmayer theory16 are in good agreement with the experimental gelation envelope for functionalities 6.5, 8.3 and 11 at concentrations greater than the measured lower functionalities gelation concentration, as shown by the solid lines and shaded regions in Fig. 3a. The lower line in each plot represents the minimum concentration of PEG necessary for gelation, corresponding to approximately one bifunctional PEG molecule for each heparin backbone _n_PEG = f/(f − 1)_n_HMWH, where f is the functionality of the HMWH molecules and _n_PEG and _n_HMWH are the moles of PEG and HMWH, respectively. This minimum amount of cross-linker ensures that each HMWH molecule in the network has the same number of cross-links as their parents, which represents the critical condition for growth of an infinite cluster (percolation), as shown in Fig. 3b. The upper gelation limit represents conditions when at least one unreacted maleimide group on each HMWH remains available for cross-linking by a dithiolized PEG, given by _n_PEG = (f − 1)_n_HMWH and illustrated in Fig. 3c. In essence, the effective cross-linkers in this case are the unreacted maleimide sites. Again, as long as the functionality of the heparin is greater than three, an infinite, percolated network can form. The general agreement of the microrheology experiments with the Flory–Stockmayer theory demonstrates that the PEG thiol groups are not being consumed by dithiol formation, but are participating in the cross-linking reaction with the HMWH maleimide functional groups.
Interestingly, for the lowest functionality we tested (f = 4.2), the gel region shifts to higher HMWH concentrations than predicted by Flory–Stockmayer theory. This indicates that, although the average maleimide functionality is 4.2 in these samples, heparin molecules with lower functionalities are also present in sufficient quantity to terminate the network cluster growth before percolation occurs, necessitating higher HMWH concentrations. Other potential deviations from the expected Flory–Stockmayer behavior are likely. For instance, at high PEG concentrations (near the upper gelation limit), significant steric constraints caused by the large number of PEG molecules attached to each heparin backbone may limit the accessibility of the unreacted maleimide site, especially for higher functionalities. Furthermore, PEG molecules could react with functional sites on the same heparin backbone. These potential complications highlight the power and importance of empirically confirming the gelation conditions.
In all, we demonstrated that multiple particle tracking microrheology provides a rapid, powerful and straightforward method for generating rheological libraries of hydrogelation conditions over a large composition parameter space. The applications of this method include the identification of hydrogel assembly and efficient optimization of biomaterial rheology for therapeutic applications. Using this method, we have identified the limits of gelation for a multi-component hydrogel system. The identification of gelation envelopes furthers our understanding of hydrogel assembly using functionalized heparin, which in turn will enable networks to be engineered that will closely mimic the mechanical and signaling microenvironments of biological materials. The simplicity of both the experiment and analysis will enable researchers investigating novel biomaterial gelators to perform rapid rheological screening without a tremendous investment in time, resources or materials.
The authors acknowledge the financial support of the National Institutes of Health (1 R01 EB003172-01), a National Science Foundation Graduate Research Fellowship (K.M.S.), and the National Science Foundation IGERT (DGE-0221651).
Notes and References
- 1.Langer R, Tirrell DA. Nature. 2004;428:487. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 2.Raeber GP, Lutolf MP, Hubbell JA. Biophys J. 2005;89:1374. doi: 10.1529/biophysj.104.050682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engler AJ, Sen S, Lee Sweeney H, Discher DE. Cell. 2006;126:677. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 4.Seal B, Panitch A. Macromolecules. 2006;39:2268. [Google Scholar]
- 5.Benoit DSW, Durney AR, Anseth KS. Biomaterials. 2007;28:66. doi: 10.1016/j.biomaterials.2006.08.033. [DOI] [PubMed] [Google Scholar]
- 6.Nie T, Baldwin A, Yamaguchi N, Kiick KL. J Controlled Release. 2007;122:287. doi: 10.1016/j.jconrel.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee KY, Peters MC, Anderson KW, Mooney DJ. Nature. 2000;408:998. doi: 10.1038/35050141. [DOI] [PubMed] [Google Scholar]
- 8.Yamaguchi N, Zhang L, Chae BS, Palla CS, Furst EM, Kiick KL. J Am Chem Soc. 2007;129:3040. doi: 10.1021/ja0680358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mason TG, Weitz DA. Phys Rev Lett. 1995;74:1250. doi: 10.1103/PhysRevLett.74.1250. [DOI] [PubMed] [Google Scholar]
- 10.Crocker JC, Grier DG. J Colloid Interface Sci. 1996;179:298. [Google Scholar]
- 11.Breedveld V, Pine DJ. J Mater Sci. 2003;38:4461. [Google Scholar]
- 12.Savin T, Doyle PS. Biophys J. 2005;88:623. doi: 10.1529/biophysj.104.042457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Veerman C, Rajagopal K, Palla CS, Pochan DJ, Schneider JP, Furst EM. Macromolecules. 2006;39:6608. [Google Scholar]
- 14.Larsen TH, Schultz KM, Furst EM. Korea-Aust Rheol J. 2008;20:165. [Google Scholar]
- 15.Larsen TH, Furst EM. Phys Rev Lett. 2008;100:146001. doi: 10.1103/PhysRevLett.100.146001. [DOI] [PubMed] [Google Scholar]
- 16.Flory PJ. J Am Chem Soc. 1941;63:3083. [Google Scholar]