Coxiella burnetii Acid Phosphatase Inhibits the Release of Reactive Oxygen Intermediates in Polymorphonuclear Leukocytes (original) (raw)
Abstract
Coxiella burnetii, the etiological agent of Q fever, is a small, Gram-negative, obligate intracellular bacterium. Replication of C. burnetii during infection has been shown to be increased by decreasing oxidative stress using p47phox −/− and iNOS−/− mice in vivo and by pharmacologic inhibitors in vitro. Building upon this model, we investigated the role polymorphonuclear leukocytes (PMN) play in the control of infection, since NADPH oxidase-mediated release of reactive oxygen intermediates (ROI) is a primary bactericidal mechanism for these cells that is critical for early innate clearance. Earlier studies suggested that C. burnetii actively inhibited release of ROI from PMN through expression of an unidentified acid phosphatase (ACP). Recent genomic annotations identified one open reading frame (CBU0335) which may encode a Sec- and type II-dependent secreted ACP. To test this model, viable C. burnetii propagated in tissue culture host cells or axenic media, C. burnetii extracts, or purified recombinant ACP (rACP) was combined with human PMN induced with 4-phorbol 12-myristate 13-acetate (PMA). The release of ROI was inhibited when PMN were challenged with viable C. burnetii, C. burnetii extracts, or rACP but not when PMN were challenged with electron beam-inactivated C. burnetii. C. burnetii extracts and rACP were also able to inhibit PMA-induced formation of NADPH oxidase complex on PMN membranes, suggesting a molecular mechanism responsible for this inhibition. These data support a model in which C. burnetii eludes the primary ROI killing mechanism of activated PMN by secreting at least one acid phosphatase.
Coxiella burnetii, the etiological agent of Q fever, is a small, Gram-negative, obligate intracellular bacterium. Human Q fever is most commonly acquired through aerosol inhalation of soil contaminated with livestock birth products (43). There are two distinct disease progressions associated with Q fever: acute and chronic infection. The acute infection presents as a flu-like illness often including severe periorbital headache, and clinical presentations have included the neurological, gastrointestinal, endocrine, and renal systems (34, 35). Chronic Q fever has been characterized by the development of endocarditis, with less common manifestations of hepatitis and osteoarticular or vascular infections (3). C. burnetii resides within specialized phagolysosomes (27) of host cells during infection, including macrophages (22, 26) and potentially polymorphonuclear leukocytes (PMN). The ability of C. burnetii to survive and replicate within the phagolysosome requires it to either inhibit or withstand degradative enzymes and an oxidative burst from PMNs that generate reactive oxygen intermediates (ROI), such as superoxide anions. Earlier studies suggested that PMN challenged with C. burnetii Nine Mile phase I (NMI) did not stimulate an oxidative burst (1). Additionally, C. burnetii extracts have acid phosphatase activity that can serve as an inhibitor of an oxidative burst in _N_-formyl-Met-Leu-Phe (fMLP)-stimulated human PMN (4) by disrupting tyrosine phosphorylation/dephosphorylation (25).
C. burnetii isolates used in this study were C. burnetii Nine Mile RSA493 (NMI) and C. burnetii Nine Mile RSA439 (NMII). These two isolates are considered phase variants, with NMI being a virulent isolate and NMII being relatively avirulent. The phase differences are caused, in part, by a large deletion in the NMII genome (6, 17). This deletion is approximately 23,000 bp and includes genes encoding lipopolysaccharide (LPS), causing NMII to have a deep rough LPS as opposed to the smooth LPS of NMI. The phase variation phenomenon is similar to the smooth-to-rough-LPS transition of Gram-negative enteric bacteria (17, 18, 20, 21).
Zamboni and Rabinovitch (46) reported that peritoneal mouse macrophages (PMM) or bone marrow-derived macrophages (BMDM) treated with the nitric oxide (NO) donor sodium nitroprusside and challenged with NMII had lower bacterial loads over time than untreated macrophages. PMM treated with the NO synthase suppressors aminoguanidine and _N_-methyl-l-arginine had an increased bacterial load over time compared to the control. Brennan et al. (7) demonstrated that p47phox −/− and iNOS−/− mice infected with NMI were significantly reduced in the ability to control infection compared to wild-type mice. IFN-γ-treated primary macrophages and the J774.A16 cell line, which exhibits an increased NADPH expression, inhibited C. burnetii replication due, in part, to increased hydrogen peroxide (H2O2). These studies show that C. burnetii is sensitive to both ROI and reactive nitrogen intermediates (RNI) and that both ROI and RNI are required to reduce C. burnetii growth.
PMN are one of the first in a cascade of cell types that migrate to a wound or site of infection (9). The initial role of PMN in an infection is one of search and destroy. To accomplish this task, PMN have an arsenal of enzymes at their disposal in the form of granules, both azurophilic (primary) and specific (secondary), metalloproteases, and NADPH oxidase (13, 30).
NADPH oxidase is a multicomponent enzyme that is assembled on a cellular or vacuolar membrane. The assembly of NADPH oxidase on the membrane initiates the conversion of O2 to ROIs, including singlet oxygen (1O2) and superoxide anion (O2−) (37). ROIs can additionally participate in a variety of reactions (i.e., with halide-containing molecules) to produce other highly toxic compounds (23). The release of ROI initiates a charge buildup across the vacuole membrane, which is compensated when ions (such as K+ and Cl−) move across the vacuole membrane, providing an environment conducive to degranulation and enzymatic digestion (39). For the assembly of the NADPH complex to proceed, the cytoplasmic portions (i.e., p47phox, p60phox, and p40phox) are phosphorylated (12, 38). These phosphorylation events produce conformational changes and subsequent binding to the membrane-associated components (i.e., gp91phox and p22phox) (10, 38).
Previous studies suggested that C. burnetii infection (both phase I and phase II) of PMN did not stimulate production of ROI (1). This lack of response was thought to be caused, in part, by acid phosphatase activity that was detected in C. burnetii extracts (4, 25). Recently, Siemsen et al. reported that PMN challenged with opsonized NMII did not activate the translocation of p47phox or p60phox to the membrane (42). This suggested that C. burnetii actively inhibited the phosphorylation of p47phox and p60phox, although it was not determined if C. burnetii components acted directly or indirectly upon p47phox and p60phox phosphorylation. We report that C. burnetii encodes an acid phosphatase (ACP) that can be secreted. This ACP was found to be inhibited by acid phosphatase inhibitors, including sodium tartrate. The ROI response in PMN was inhibited by native and recombinant ACP and, when exogenously added to PMN, prevented the translocation of p47phox to the membrane.
MATERIALS AND METHODS
Isolation of human PMN.
Human PMN were harvested from healthy volunteers under a currently approved Institutional Review Board protocol for human subjects in research. Peripheral blood was obtained by venipuncture and combined with a 6% dextran solution at a 10:1 ratio. The erythrocytes were allowed to sediment (30 to 40 min), leaving leukocyte-rich plasma (LRP). LRP was layered on top of Nycoprep 1.077 (Axis-Shield, Oslo, Norway) at a ratio of 2:1. This gradient was centrifuged for 25 min at 600 × g. The PMN-enriched pellet was resuspended in ice-cold distilled water for 30 s to lyse contaminating erythrocytes, after which an equal volume of 1.7% NaCl was added to restore isotonic conditions. The mixture was then centrifuged for 10 min at 250 × g. The resulting PMN were resuspended in Hanks balanced salt solution (HBSS) (without Mg2+ or Ca2+) and enumerated using a hemocytometer. Purified PMN were Wright stained and evaluated for morphology and purity. They were also stained with fluorescein isothiocyanate (FITC)-conjugated CD16b antibody (Ab Direct/Serotec, Raleigh, NC), a specific marker for PMN, and analyzed by flow cytometer to evaluate purity. Preparations with greater than 95% purity were used in this study.
Oxidative burst in PMN challenged with C. burnetii.
In a 96-well plate, 106 PMN were combined at a multiplicity of infection (MOI) of 10 bacteria to 1 PMN with virulent NMI or avirulent NMII. Challenged PMN were exogenously activated with 50 ng/ml 4-phorbol 12-myristate 13-acetate (PMA) 30 min postchallenge. The plate was maintained at 37°C and 5% CO2. Superoxide anion production by PMN was monitored on a spectrophotometer (Spectromax M2; Molecular Dynamics, Sunnyvale, CA) by an increase in fluorescence (excitation, 488 nm; emission, 530 nm) after the addition of 10 μg/ml OxyBurst H2HFF green bovine serum albumin (BSA) (Invitrogen, Carlsbad, CA).
Acid phosphatase purification from C. burnetii.
Purification was done as described before by Li et al. (25). Briefly, previously purified (from tissue culture or acidified citrate cysteine medium [ACCM] propagation) and frozen NMII, approximately 1 mg, was centrifuged at 20,000 × g for 30 min, and freezing media were discarded. The pellet was resuspended in 2 ml of 10 mM HEPES (pH 7.4), 0.8% NaCl, 0.2% Triton X-100 with complete protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany) and gently shaken at 4°C for 80 min. This was followed by centrifugation at 100,000 × g for 30 min. The supernatant had a specific activity of 0.094 μM/min/mg and was stored at −20°C until use. (Throughout this paper, 1 U is equal to 1 mM/min/mg.)
Production of recombinant C. burnetii ACP.
The open reading frame (ORF) CBU0335 (NCBI, GeneID: 1208217) was PCR amplified and cloned into the pFLAG-MAC vector (Sigma-Aldrich, St. Louis, MO) by the In-fusion system (Clonetech, Mountain View, CA) using the following primers: forward primer, 5′-ATA TAA GCT TAT GAT TCT TAG CTT CAC ACC GAT G-3′; reverse primer, 5′-CAT GAG ATC TTT AAT ACA TAA AAT TAG GTA ATT TAT AAC TGT GTT CG-3′. The plasmid, designated pJH101, was sequenced to confirm the predicted sequence. Recombinant ACP (rACP) was produced and purified as follows. A 5-ml overnight culture was grown, added to 100 ml of LB broth with 50 μg/ml carbenicillin, and grown at 37°C to an optical density at 600 nm (OD600) of approximately 0.5. At this growth stage, 2 μM IPTG was added and incubation was continued at 37°C for 2 h. The culture was centrifuged at 15,000 × g for 20 min, and the supernatant was removed. The pellet was resuspended in 5 ml lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, pH 7.4) and sonicated (model S-3000; Misonix Inc., Farmingdale, NY) three times, 30 s each, at power setting 8. The sonicated solution was centrifuged for 1 h at 25,000 × g at 4°C. The supernatant was combined with FLAG-conjugated agarose beads (Sigma-Aldrich) and gently shaken for 4 h at 4°C. The FLAG beads were loaded onto a column and washed three times with 10 ml Tris-buffered saline (TBS), pH 7.2. Elution was done with 0.1 M glycine, pH 3.5, three times, 1 ml each. The eluate had a specific activity of 0.024 μM/min/mg, and the eluates were stored at −20°C until use.
Acid phosphatase activity assay.
Acid phosphatase activity was determined using the Acid Phosphatase Enzcheck system (Invitrogen) according to the manufacturer's specification. Briefly, a 50-μl reaction buffer containing 10 μM DiFMUP (6,8-difluoro-4-methyumbelliferyl phosphate) and 50 μl of sample was added to a 96-well plate. The plate was kept in the dark at room temperature for the duration of the experiment. The acid phosphatase activity was monitored with a spectrophotometer by an increase in fluorescence (excitation, 360 nm; emission, 460 nm).
PMN membrane isolation.
Human PMN were treated with either Escherichia coli, live NMII, C. burnetii extract, or rACP for 30 min or PMA (50 ng/ml) for 10 min. After treatment, the reaction was stopped by the addition of 1 ml ice-cold Tris-sucrose buffer (TSB) (100 mM Tris, 340 mM sucrose, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride [PMSF], 1× complete protease inhibitor cocktail [Roche], and 0.1× HALT protease cocktail [Pierce, Thermo Fisher Scientific, Rockford, IL]). PMN were collected by centrifugation at 1,000 × g for 1 min. PMN were resuspended in 200 μl TSB and sonicated on power setting 0.5 for 35 s. The remaining intact PMN were removed by centrifugation at 1,000 × g for 1 min. The supernatant was then centrifuged for 30 min at 100,000 × g. The resulting pellet (membrane fraction) was resuspended in 200 μl TSB. Sample buffer was then added, and all samples were boiled for 10 min. Samples were separated by SDS-PAGE and Western blotted onto nitrocellulose membranes. The membranes were then probed with anti-p47phox (Santa Cruz Biotechnology, Santa Cruz, CA) or anti-actin (Cell Signaling Technology, Danvers, MA).
Electron beam inactivation of C. burnetii.
Tissue culture-propagated NMI or NMII was suspended in 0.25 M sucrose phosphate (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.76 mM KH2PO4, 250 mM sucrose) in a 50-ml polypropylene conical tube. This was then exposed to a minimum of 10,000 grays (10 kGy) of ionizing radiation at the National Center for Electron Beam Research (Texas A&M University, College Station, TX) (31, 32). The resultant material was deemed inactivated by infecting severe combined immunodeficiency (SCID) mice intraperitoneally with 108 genome equivalents with no observable sickness or splenomegaly 14 days postinfection (2).
ACCM propagation of C. burnetii.
C. burnetii was grown as described by Anders et al. (33). Briefly, NMII was seeded in ACCM at 106 genome equivalents/ml. The cultures were gently shaken for 7 days at 37°C and 5% CO2 and 2.5% O2. C. burnetii was harvested by centrifugation at 20,000 × g for 30 min and frozen at −80°C until used.
Statistical analyses.
Results were expressed as the means ± the standard deviation (SD) and were compared using a Student's t test. Differences were considered significant at P values of <0.05.
RESULTS
PMNs challenged with viable C. burnetii were inhibited in the ability to express ROI after exogenous stimulation.
PMN are normally stimulated to release ROI when encountering bacteria. We wanted to investigate if PMN were stimulated to release ROI when encountering C. burnetii. Human PMN were challenged with viable or electron beam-inactivated C. burnetii (Fig. 1 A). There was no significant ROI response, compared to results with the control, when PMN were challenged with either viable or electron beam-inactivated C. burnetii. This was in contrast to the result with PMN that were challenged with viable E. coli, which induced a robust ROI response. The lack of ROI response to C. burnetii by PMN has been reported previously (1, 14, 15, 42), as well as results for several other organisms (5, 8, 29, 44). Since there was no significant difference of ROI release upon C. burnetii challenge compared to that with the control, we wanted to know if C. burnetii were actively inhibiting PMN ROI release or just being undetected by the PMN. We used PMA, a potent PMN stimulator (11, 41), measured its effect, and chose 50 ng/ml as the dose that initiated a robust ROI release. Higher doses did not produce an appreciably larger ROI release (data not shown). An alternate PMA stimulator of ROI release is fMLP. We did not choose this because in our hands, it did not consistently produce a robust ROI release. PMN were challenged with viable or electron beam-inactivated C. burnetii and incubated at 37°C for 30 min. They were treated with 50 ng/ml PMA, and incubation was continued at 37°C for the duration of the experiment (Fig. 1B). PMA-stimulated PMN challenged with electron beam-inactivated C. burnetii had an ROI response similar to that of the control (nonchallenged) PMN, while PMN that were challenged with viable C. burnetii inhibited the ROI response (Fig. 1B).
FIG. 1.

Coxiella burnetii does not stimulate ROI release from human PMN. (A) Human PMN (106) were challenged with live or killed NMI or NMII or live E. coli at an MOI of 10 bacteria to 1 PMN. (B) At 30 min postchallenge, 50 ng/ml PMA was added. Cells were incubated at 37°C and 5% CO2 for the duration of the experiment. Data represent mean ± SD of triplicate values from one experiment representative of a total of three. *, P < 0.05; **, P < 0.005.
Identification and testing of a C. burnetii acid phosphatase.
Acid phosphatase enzymes have been identified in other intracellular organisms that inhibit PMN ROI release (36) or reduce organism viability in the presence of PMN when deleted (28). Analysis of the C. burnetii genome identified 29 annotated phosphatase enzymes, yet, of these, only 2 were annotated as acid phosphatase enzymes, CBU0335 (ACP) and CBU1671 (survival protein E [surE]) (40). We speculated that secretion was essential for an enzyme to subvert the host ROI response by either acting directly on or interfering in the assembly of the NADPH oxidase complex. The only ORF that had a predicted signal sequence was CBU0335, which is 666 bp in length and encodes a predicted 25.43-kDa protein with the predicted signal sequence and 22.71 kDa without the predicted signal sequence (Fig. 2 A). The predicted signal sequence is a 20-amino-acid residue that is positive by SignalP 3.0 (http://www.cbs.dtu.dk/services/SignalP/) and SIG-Pred: Signal Peptide Prediction algorithms (http://bmbpcu36.leeds.ac.uk/prot_analysis/Signal.html). CBU0335 also has a high degree of similarity to acid phosphatase enzymes from other known intracellular bacteria. Francisella tularensis encodes three distinct acid phosphatase enzymes, of which CBU0335 has a high level of homology with one member (F. tularensis subsp. novicida U112, FTN1526 [identity, 31%; similarity, 54%], and F. tularensis subsp. tularensis SCHU S4, FTT0156 [identity, 31%; similarity, 54%]). Legionella pneumophilia encodes two distinct acid phosphatase enzymes, of which CBU0335 shares high homology with one member (L. pneumophila strain Lens, lpl2544 [identity, 45%; similarity, 66%], and L. pneumophila strain Paris, lpp2674 [identity, 45%; similarity, 65%]) (Fig. 2B). Interestingly, among the other genes that have similarity with CBU0335, only the L. pneumophilia ORFs encode a predicted secretion signal by SignalP 3.0.
FIG. 2.

Amino acid alignment of CBU0335 with ClustalW and predicted cleavage site. (A) The cleavage site was determined for ORF CBU0335 using the Gram-negative secretion prediction group from SignalP. n-region, h-region, and c-region, N terminus, middle, and C terminus, respectively, of the predicted signal sequence. (B) Sequences of L. pneumophilia strain Paris ORF lpp2674 (Lp_Paris), C. burnetii RSA493 ORF CBU0335 (Cb_RSA493), and F. tularensis subsp. Schu S4 ORF FTT0156 (Ft_SCHU_S4). Asterisks indicate identical amino acids; colons indicate conserved substitutions; periods indicate semiconserved substitutions.
Expression of ACP by C. burnetii.
C. burnetii must survive in the extreme conditions present in a PMN and phagolysosome and would express genes and produce proteins to help combat those environmental stresses. Microarray data showed that transcripts for CBU0335 were present and at similar levels under oxidative stress conditions in experiments comparing _C. burnetii_-infected J774.A16 cells and J774.D9 cells (K. Mertens, unpublished data). To evaluate the expression of ACP protein, an rACP containing an N-terminal FLAG epitope but with no predicted signal sequence (predicted size, 24.18 kDa) was purified under partial denaturing conditions and used to immunize mice and rabbits for antibody production. This antibody recognized only the rACP and not the native protein (data not shown). An alternative antibody was identified that was generated against a GST-fusion-derived rACP (a kind gift from Robert Heinzen, Rocky Mountain Laboratory). This antibody recognized the FLAG-Acp fusion protein and a band of equal mass in the C. burnetii extract, suggesting that rACP was identical to C. burnetii ACP (Fig. 3).
FIG. 3.
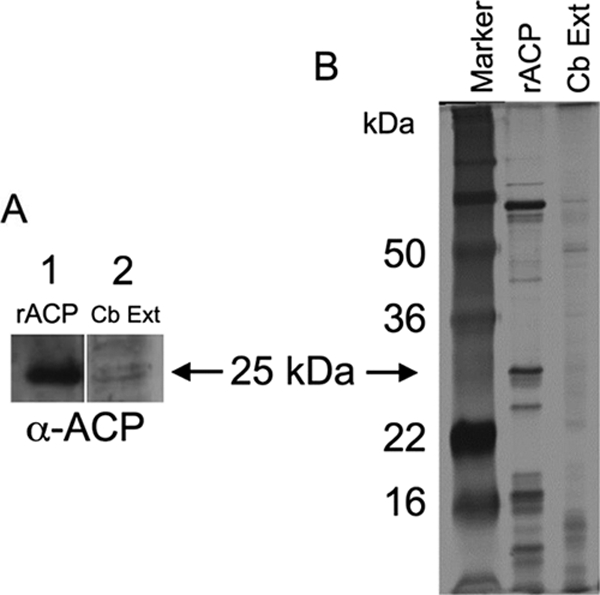
Detection of ACP. (A) Western blot using anti-rACP primary antibody of rACP (lane 1) and C. burnetii extract (lane 2). (B) Silver stain of SDS-PAGE marker (lane 1), rACP (lane 2), and C. burnetii extract (lane 3).
pH-dependent acid phosphatase activity.
Purified rACP and C. burnetii extracts were assayed for acid phosphatase and pH-dependent activities. rACP and C. burnetii extract were incubated in sodium acetate buffers at different pH values (pH 3 to 6) or Tris-buffered saline (pH 7 and 8). Both the purified rACP and C. burnetii extracts had a pH-dependent acid phosphatase activity (Fig. 4 A and B). The optimum pH of activity for these preparations differed slightly, with the peak of rACP activity at pH 7 but extending through pH 3, while the C. burnetii extract had an optimum activity at pH 6. Since known inhibitors of acid phosphatase (4) have been used in previous studies characterizing C. burnetii ACP activity, we compared several inhibitors to test the sensitivity of rACP and C. burnetii extract. The rACP or C. burnetii extract was added to reaction buffer along with 10 μM sodium molybdate (Sigma-Aldrich), 10 μM sodium orthovanadate (Sigma-Aldrich), or 10 mM sodium tartrate (Sigma-Aldrich). There was a marked inhibition of acid phosphatase activity with all three antagonists (Fig. 5). It was also noted that both the rACP and C. burnetii extract were sensitive to sodium tartrate.
FIG. 4.

Acid phosphatase activity of C. burnetii extract and rACP. C. burnetii extract (A) and rACP (B) (0.01 U and 0.00001 U, respectively) were added to 100 μl of either 1 M sodium acetate buffer (pH 3 to 6) or TBS (pH 7 to 8). DiFMUP (100 μM) was added, and the increase in fluorescence was monitored. Data represent mean ± SD of triplicate values from one experiment representative of a total of three. *, P < 0.05; **, P < 0.005.
FIG. 5.

Inhibition of acid phosphatase activity. 0.0001 U rACP or 0.08 U C. burnetii extract were combined with 10 μM sodium molybdate, 10 μM sodium orthovanadate, or 10 mM sodium tartrate in 100 μl reaction buffer. Acid phosphatase activity was detected by an increase in fluorescence as described in Materials and Methods. Percent inhibition is relative to that of the untreated control. Data represent the mean of triplicate values from one experiment representative of a total of three.
C. burnetii extracts from axenically propagated NMII and rACP inhibit the ROI response of exogenously stimulated PMN.
As reported by Li et al., acid phosphatase activity unique to C. burnetii may be one mechanism by which the ROI response is inhibited. Initial C. burnetii extracts in our study were made according to the method outlined by Li et al. (25) using tissue culture-propagated NMII. Li et al. reported that by employing a subset of heteromolybdate compounds, bacterial acid phosphatase activity was inhibited and able to be discriminated from host (mammalian) acid phosphatase activity. We were unable to obtain these compounds for our experiments; however, C. burnetii was grown outside a host cell in an artificial medium (ACCM) (33), and when this culture method was used, our ACCM-propagated C. burnetii extracts (ACE) did not contain mammalian protein contamination. As an additional control, ACCM was shown to be devoid of ACP activity (data not shown). When human PMN were treated with ACE or rACP for 10 min (Fig. 6) and subsequently exogenously stimulated by the addition of 50 ng/ml PMA, the ROI response was significantly reduced compared to that of the control. This complete lack of detectable reactive oxygen could be due either to an active inhibition of NADPH oxidase complex function or to scavenging by enzymes such as superoxide dismutase (SOD) (19) and secreted Cu/Zn SOD (R. Brennan, unpublished data). The ACE was tested for SOD activity and was found to have none detectable (data not shown). SOD activity was detected using the Sigma-Aldrich SOD determination kit based on the color change of a water-soluble formozan dye. This strongly suggests that the ROI inhibition was not due to SOD activity in ACE.
FIG. 6.
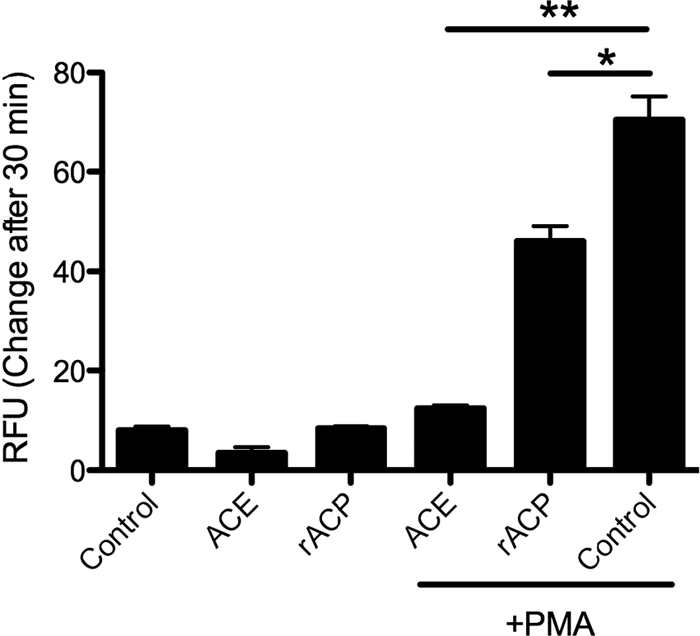
Inhibition of ROI release by rACP and ACE. Human PMN (106) were incubated with 0.0001 U rACP or 0.08 U ACE for 10 min. Cells were then treated with 50 ng/ml PMA or not treated and were incubated for 30 min at 37°C. Data represent mean ± SD of triplicate values from one experiment representative of a total of three. *, P ≤ 0.005; **, P ≤ 0.001.
ACE and rACP inhibit the assembly of the NADPH oxidase complex.
The cytosolic components of the NADPH oxidase complex need to move to the membrane to initiate ROI release. To investigate how NMII may influence the NADPH assembly, E. coli, NMII, ACE, or rACP was added to PMN that were then stimulated, or not, with 50 ng/ml PMA. The PMN were harvested and lysed. Membranes were separated from cytosolic components, and the location of p47phox was examined by Western blotting (Fig. 7). PMN treated with NMII and untreated PMN had comparable amounts of p47phox mobilized to the membrane, consistent with an inability to activate PMN. The results of NMII treatment followed by PMA stimulation were not significantly different from those of PMA treatment alone (3- to 5-fold induction). In contrast, the exogenous addition of ACE or rACP was able to completely inhibit the movement of p47phox to the membrane even after PMA stimulation.
FIG. 7.
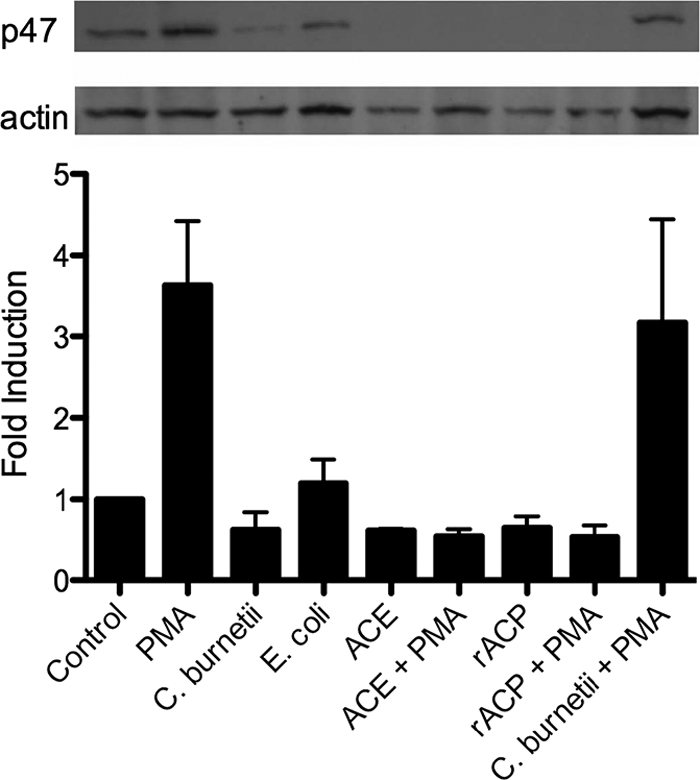
ACE and rACP inhibit the movement of p47phox to the membrane. E. coli (30 min), NMII (30 min), ACE (10 min), or rACP (10 min) was incubated with PMN and stimulated or not stimulated with 50 ng/ml PMA (10 min). Membranes were run on SDS-polyacrylamide gels, blotted to polyvinylidene difluoride (PVDF), and probed with anti-p47phox or anti-actin. Fold induction = (average pixel density of α-p47phox/average pixel density of α-actin)/(average pixel density of α-p47phox control/average pixel density of α-actin control). Data represent means ± SD of values from three experiments.
DISCUSSION
The inability of C. burnetii to be propagated outside host cells has limited the approaches available to discover and characterize virulence factors. The inclusion of host proteins in C. burnetii preparations has been problematic when describing interactions between C. burnetii and host cells. Recently, methods for C. burnetii propagation in vitro in an acidified citrate medium were published (33). This has allowed the analysis of pure C. burnetii cultures devoid of the contamination of host proteins.
C. burnetii, like other intracellular organisms, likely manipulates the host in a variety of ways to establish a replication niche. This likely requires the secretion of proteins that interact with the host. F. tularensis secretes an acid phosphatase that can inhibit the ROI response in PMN and allow for survival in a macrophage (2, 27, 31). We have shown that C. burnetii rACP, C. burnetii extract (devoid of host contamination), and viable C. burnetii inhibit the ROI response in PMA-activated PMN. This supports a role for Acp in C. burnetii intracellular survival. As noted in Results and Materials and Methods, the rACP is a fusion protein with an N-terminal FLAG epitope. This is one of three different strategies employed by our lab to clone and express an active ACP. We used a variety of denaturing conditions to purify ACP but found no detectable ACP activity. Employing renaturation steps after purification rescued some ACP activity. Finally, with the most recent FLAG epitope construct, a partially denaturing protein purification protocol was developed. With this protocol, recombinant protein with significant ACP activity was obtained. This leads us to speculate that the ACP protein is sensitive to conformational change from its native state. As described in previous studies, some phosphatase proteins will unfold and demonstrate irreversible loss of activity due to thermal or chemical stress (45, 47). This may explain the differences seen between the C. burnetii extract and rACP activity. One would expect a much higher specific activity from purified protein than from a lysate. However, if a conformational change occurs, we speculate that activity could be diminished. Ongoing work on improving native protein purification could result in further increases in enzymatic activity.
The ability of C. burnetii to inhibit NADPH complex formation has recently been demonstrated by Siemsen et al. (42). They used opsonized C. burnetii to show that with FcR-mediated phagocytosis, there was no increase in p47phox or p67phox translocation to the membrane. Unexpectedly, we showed that while C. burnetii alone did not stimulate NADPH complex formation, C. burnetii infection did not inhibit its formation in the presence of PMA stimulation. This may indicate that infection-mediated suppression of PMA-stimulated ROI release is downstream of NADPH oxidase complex formation at the membrane. Alternatively, infection-mediated suppression may only partially inhibit NADPH oxidase complex formation, as suggested by the partial inhibition of ROI release (Fig. 1). Hence, ROI release may be a more sensitive monitor of ACP-mediated activity by C. burnetii infection than membrane localization analysis. When PMN were preincubated with either C. burnetii extracts or rACP and then PMA stimulated, NAPDH oxidase formation was inhibited. One would expect that rACP or the C. burnetii extracts were taken up, by either pinocytosis or receptor-mediated endocytosis, but as described by another group (36), acid phosphatases may be agonists to extracellular signals, thereby manipulating the PMN from the outside.
We speculate that C. burnetii, due to its slow replication rate, probably does not undergo significant replication inside PMN but rather uses PMN as a trafficking mechanism to be delivered to the preferred host cell, a macrophage, as is the case for Leishmania donovani (16, 24). C. burnetii may survive in the PMN awaiting the programmed apoptosis that PMN are destined to undergo. Once the PMN undergoes apoptosis, C. burnetii may be included in an apoptotic bleb waiting for a macrophage to phagocytose the apoptotic body. The expression of ACP may be an alternate strategy, should C. burnetii be phagocytosed by a primed or activated PMN, thus allowing C. burnetii to dampen the ROI response and allow for survival.
In this paper, we show that C. burnetii encodes an ACP that can inhibit the ROI response of PMN. This ACP was molybdate and tartrate sensitive and had pH-dependent activity. Also, engagement of C. burnetii by PMN did not lead to ROI induction or NADPH oxidase assembly. Future experiments will include examination of the mechanism behind this inhibition, which could include proteins such as protein kinase C, Akt, and Erk1/2. This would be a valuable step not only toward understanding the role of the PMN in C. burnetii infection but also in forming a more complete picture of the process by which PMN undergo ROI generation and NADPH oxidase assembly.
Acknowledgments
Funding was provided by NIH grants AI037744 and AI057768 awarded to J.E.S.
ACP antibody was a kind gift from Robert Heinzen.
Footnotes
▿
Published ahead of print on 15 November 2010.
REFERENCES
- 1.Akporiaye, E., D. Stefanovich, V. Tsosie, and G. Baca. 1990. Coxiella burnetii fails to stimulate human neutrophil superoxide anion production. Acta Virol. 34**:**64-70. [PubMed] [Google Scholar]
- 2.Andoh, M., T. Naganawa, A. Hotta, T. Yamaguchi, H. Fukushi, T. Masegi, and K. Hirai. 2003. SCID mouse model for lethal Q fever. Infect. Immun. 71**:**4717-4723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Angelakis, E., and D. Raoult. 2010. Q fever. Vet. Microbiol. 140**:**297-309. [DOI] [PubMed] [Google Scholar]
- 4.Baca, O., M. Roman, R. Glew, R. Christner, J. Buhler, and A. Aragon. 1993. Acid phosphatase activity in Coxiella burnetii: a possible virulence factor. Infect. Immun. 61**:**4232-4239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barquero-Calvo, E., E. Chaves-Olarte, D. S. Weiss, C. Guzmán-Verri, C. Chacón-Díaz, A. Rucavado, I. Moriyón, and E. Moreno. 2007. Brucella abortus uses a stealthy strategy to avoid activation of the innate immune system during the onset of infection. PLoS One 2**:**e631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beare, P. A., J. E. Samuel, D. Howe, K. Virtaneva, S. F. Porcella, and R. A. Heinzen. 2006. Genetic diversity of the Q fever agent, Coxiella burnetii, assessed by microarray-based whole-genome comparisons. J. Bacteriol. 188**:**2309-2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brennan, R., K. Russell, G. Zhang, and J. Samuel. 2004. Both inducible nitric oxide synthase and NADPH oxidase contribute to the control of virulent phase I Coxiella burnetii infections. Infect. Immun. 72**:**6666-6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carlyon, J., D. Abdel-Latif, M. Pypaert, P. Lacy, and E. Fikrig. 2004. Anaplasma phagocytophilum utilizes multiple host evasion mechanisms to thwart NADPH oxidase-mediated killing during neutrophil infection. Infect. Immun. 72**:**4772-4783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cassatella, M. A. 2003. The neutrophil: an emerging regulator of inflammatory and immuune response. Karger Publishers, Basel, Switzerland.
- 10.Clark, R. A. 1999. Activation of the neutrophil respiratory burst oxidase. J. Infect. Dis. 179(Suppl. 2)**:**S309-S317. [DOI] [PubMed] [Google Scholar]
- 11.DeLeo, F. R. A. 1999. NADPH oxidase activation and assembly during phagocytosis. J. Immunol. 163**:**6732-6740. [PubMed] [Google Scholar]
- 12.El-Benna, J., P. M. Dang, M. A. Gougerot-Pocidalo, J. C. Marie, and F. Braut-Boucher. 2009. p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp. Mol. Med. 41**:**217-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Elgert, K. D. 2009. Immunology, second ed. Wiley-Blackwell, Hoboken, NJ.
- 14.Ferencik, M., S. Schramek, J. Kazar, and J. Stefanovic. 1984. Effect of Coxiella burnetii on the stimulation of hexose monophosphate shunt and on superoxide anion production in human polymorphonuclear leukocytes. Acta Virol. 28**:**246-250. [PubMed] [Google Scholar]
- 15.Fumarulo, R., D. Giordano, A. Mosca, R. Monno, and G. Miragliotta. 1989. Different priming on human neutrophil respiratory burst by lipopolysaccharides from phase I and phase II Coxiella burnetii. Microbiologica 12**:**55-60. [PubMed] [Google Scholar]
- 16.Gueirard, P., A. Laplante, C. Rondeau, G. Milon, and M. Desjardins. 2008. Trafficking of Leishmania donovani promastigotes in non-lytic compartments in neutrophils enables the subsequent transfer of parasites to macrophages. Cell. Microbiol. 10**:**100-111. [DOI] [PubMed] [Google Scholar]
- 17.Hackstadt, T. 1986. Antigenic variation in the phase I lipopolysaccharide of Coxiella burnetii isolates. Infect. Immun. 52**:**337-340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hackstadt, T., M. G. Peacock, P. J. Hitchcock, and R. L. Cole. 1985. Lipopolysaccharide variation in Coxiella burnetii: intrastrain heterogeneity in structure and antigenicity. Infect. Immun. 48**:**359-365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heinzen, R. A., M. E. Frazier, and L. P. Mallavia. 1992. Coxiella burnetii superoxide dismutase gene: cloning, sequencing, and expression in Escherichia coli. Infect. Immun. 60**:**3814-3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hitchcock, P. J., and T. M. Brown. 1983. Morphological heterogeneity among Salmonella lipopolysaccharide chemotypes in silver-stained polyacrylamide gels. J. Bacteriol. 154**:**269-277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hotta, A., M. Kawamura, H. To, M. Andoh, T. Yamaguchi, H. Fukushi, and K. Hirai. 2002. Phase variation analysis of Coxiella burnetii during serial passage in cell culture by use of monoclonal antibodies. Infect. Immun. 70**:**4747-4749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kazar, J., E. Skultetyova, and R. Brezina. 1975. Phagocytosis of Coxiella burneti[sic] by macrophages. Acta Virol. 19**:**426-431. [PubMed] [Google Scholar]
- 23.Klebanoff, S. J. 2005. Myeloperoxidase: friend and foe. J. Leukoc. Biol. 77**:**598-625. [DOI] [PubMed] [Google Scholar]
- 24.Laskay, T., G. van Zandbergen, and W. Solbach. 2008. Neutrophil granulocytes as host cells and transport vehicles for intracellular pathogens: apoptosis as infection-promoting factor. Immunobiology 213**:**183-191. [DOI] [PubMed] [Google Scholar]
- 25.Li, Y., G. Curley, M. Lopez, M. Chavez, R. Glew, A. Aragon, H. Kumar, and O. Baca. 1996. Protein-tyrosine phosphatase activity of Coxiella burnetii that inhibits human neutrophils. Acta Virol. 40**:**263-272. [PubMed] [Google Scholar]
- 26.Little, J. S., R. A. Kishimoto, and P. G. Canonico. 1983. Intracellular fate of phase I Coxiella burnetii in guinea pig peritoneal macrophages. J. Reticuloendothel. Soc. 33**:**331-341. [PubMed] [Google Scholar]
- 27.Maurin, M., A. Benoliel, P. Bongrand, and D. Raoult. 1992. Phagolysosomes of _Coxiella burnetii_-infected cell lines maintain an acidic pH during persistent infection. Infect. Immun. 60**:**5013-5016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohapatra, N. P., S. Soni, T. J. Reilly, J. Liu, K. E. Klose, and J. S. Gunn. 2008. Combined deletion of four Francisella novicida acid phosphatases attenuates virulence and macrophage vacuolar escape. Infect. Immun. 76**:**3690-3699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mott, J., and Y. Rikihisa. 2000. Human granulocytic ehrlichiosis agent inhibits superoxide anion generation by human neutrophils. Infect. Immun. 68**:**6697-6703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murphy, K. P., P. Travers, C. Janeway, and M. Walport. 2008. Janeway's immunobiology, seventh ed. Garland Science, New York, NY.
- 31.Nieto-Sandoval, J. M., L. Almela, J. A. Fernández-López, and J. A. Muñoz. 2000. Effect of electron beam irradiation on color and microbial bioburden of red paprika. J. Food Prot. 63**:**633-637. [DOI] [PubMed] [Google Scholar]
- 32.O'Bryan, C. A., P. G. Crandall, S. C. Ricke, and D. G. Olson. 2008. Impact of irradiation on the safety and quality of poultry and meat products: a review. Crit. Rev. Food Sci. Nutr. 48**:**442-457. [DOI] [PubMed] [Google Scholar]
- 33.Omsland, A., D. C. Cockrell, D. Howe, E. R. Fischer, K. Virtaneva, D. E. Sturdevant, S. F. Porcella, and R. A. Heinzen. 2009. Host cell-free growth of the Q fever bacterium Coxiella burnetii. Proc. Natl. Acad. Sci. U. S. A. 106**:**4430-4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Parker, N. R., J. H. Barralet, and A. M. Bell. 2006. Q fever. Lancet 367**:**679-688. [DOI] [PubMed] [Google Scholar]
- 35.Raoult, D., T. Marrie, and J. Mege. 2005. Natural history and pathophysiology of Q fever. Lancet Infect. Dis. 5**:**219-226. [DOI] [PubMed] [Google Scholar]
- 36.Reilly, T. J., G. S. Baron, F. E. Nano, and M. S. Kuhlenschmidt. 1996. Characterization and sequencing of a respiratory burst-inhibiting acid phosphatase from Francisella tularensis. J. Biol. Chem. 271**:**10973-10983. [DOI] [PubMed] [Google Scholar]
- 37.Robinson, J. M. 2009. Phagocytic leukocytes and reactive oxygen species. Histochem. Cell Biol. 131**:**465-469. [DOI] [PubMed] [Google Scholar]
- 38.Segal, A. W. W. 1999. Components and organization of the NADPH oxidase of phagocytic cells, p. 441-483. In S. Gordon (ed.), Phagocytosis: the host. JAI Press, Greenwich, CT.
- 39.Segal, A. W. 2005. How neutrophils kill microbes. Annu. Rev. Immunol. 23**:**197-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Seshadri, R., I. T. Paulsen, J. A. Eisen, T. D. Read, K. E. Nelson, W. C. Nelson, N. L. Ward, H. Tettelin, T. M. Davidsen, M. J. Beanan, R. T. Deboy, S. C. Daugherty, L. M. Brinkac, R. Madupu, R. J. Dodson, H. M. Khouri, K. H. Lee, H. A. Carty, D. Scanlan, R. A. Heinzen, H. A. Thompson, J. E. Samuel, C. M. Fraser, and J. F. Heidelberg. 2003. Complete genome sequence of the Q-fever pathogen Coxiella burnetii. Proc. Natl. Acad. Sci. U. S. A. 100**:**5455-5460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sheppard, F. R., M. R. Kelher, E. E. Moore, N. J. D. McLaughlin, A. Banerjee, and C. C. Silliman. 2005. Structural organization of the neutrophil NADPH oxidase: phosphorylation and translocation during priming and activation. J. Leukoc. Biol. 78**:**1025-1042. [DOI] [PubMed] [Google Scholar]
- 42.Siemsen, D. W., L. N. Kirpotina, M. A. Jutila, and M. T. Quinn. 2009. Inhibition of the human neutrophil NADPH oxidase by Coxiella burnetii. Microbes Infect. 11**:**671-679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tissot Dupont, H., D. Raoult, P. Brouqui, F. Janbon, D. Peyramond, P. J. Weiller, C. Chicheportiche, M. Nezri, and R. Poirier. 1992. Epidemiologic features and clinical presentation of acute Q fever in hospitalized patients: 323 French cases. Am. J. Med. 93**:**427-434. [DOI] [PubMed] [Google Scholar]
- 44.Wang, T., S. Malawista, U. Pal, M. Grey, J. Meek, M. Akkoyunlu, V. Thomas, and E. Fikrig. 2002. Superoxide anion production during Anaplasma phagocytophila infection. J. Infect. Dis. 186**:**274-280. [DOI] [PubMed] [Google Scholar]
- 45.Wang, X., F. Meng, and H. Zhou. 2004. Unfolding and inactivation during thermal denaturation of an enzyme that exhibits phytase and acid phosphatase activities. Int. J. Biochem. Cell Biol. 36**:**447-459. [DOI] [PubMed] [Google Scholar]
- 46.Zamboni, D. S., and M. Rabinovitch. 2003. Nitric oxide partially controls Coxiella burnetii phase II infection in mouse primary macrophages. Infect. Immun. 71**:**1225-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang, Y. X., Y. Zhu, and H. M. Zhou. 2000. Conformational changes and inactivation of calf intestinal alkaline phosphatase in trifluoroethanol solutions. Int. J. Biochem. Cell Biol. 32**:**887-894. [DOI] [PubMed] [Google Scholar]