Vstm3 is a Member of the CD28 Family and an Important Modulator of T Cell Function (original) (raw)
. Author manuscript; available in PMC: 2013 Aug 6.
Published in final edited form as: Eur J Immunol. 2011 Mar 18;41(4):902–915. doi: 10.1002/eji.201041136
Summary
Members of the CD28 family play important roles in regulating T cell functions and share a common gene structure profile. We have identified VSTM3 as a protein whose gene structure matches that of the other CD28 family members. This protein (also known as TIGIT and WUCAM) has been previously shown to affect immune responses and is expressed on NK cells, activated and memory T cells, and regulatory T cells. The nectin-family proteins CD155 and CD112 serve as counter-structures for VSTM3 and CD155 and CD112 also bind to the activating receptor CD226 on T cells and NK cells. Hence, this group of interacting proteins forms a network of molecules similar to the well-characterized CD28-CTLA4-CD80-CD86 network. In the same way that soluble CTLA4 can be used to block T cell responses, we show that soluble Vstm3 attenuates T cell responses in vitro and in vivo. Moreover, animals deficient in Vstm3 are more sensitive to autoimmune challenges indicating that this new member of the CD28 family is an important regulator of T cell responses.
Introduction
Members of the B7 and CD28 family regulate T cell responses[1-3]. B7 family proteins expressed on antigen presenting cells (APC) engage CD28 family proteins expressed on T cells, triggering costimulatory or inhibitory signals. However, at least one member of the CD28 family, BTLA, binds to the TNFR family member HVEM[4] indicating that triggering of CD28 family proteins is not restricted to B7 family members.
Positive signaling members of the CD28 family include CD28 and ICOS[5]. Negative signaling proteins from this group include CTLA4, PD1, and BTLA and these molecules inhibit T cell activation by diverse and somewhat ill-defined means. BTLA functions by recruiting protein tyrosine phosphatases such as Shp1[6-8], which counteract the effects of protein tyrosine kinases (PTK) activated by antigen receptor engagement. The inhibitory function of CTLA4 and PD1 is clear, but the precise mechanism remains enigmatic. This network of positive and negative signaling molecules is thought to tightly regulate activation of T cells thereby ensuring activation only under appropriate circumstances.
We have identified a novel member of the CD28 family using a bioinformatic algorithm that utilizes gene structure as a basis for family assignment. This protein, given the HUGO designation Vstm3, has been previously characterized by other groups as a transmembrane protein with a single extra-cellular IgV domain[9-11]. Analysis of the cytoplasmic domain revealed two immunoreceptor tyrosine-based inhibitory motifs (ITIM) suggesting it may be an inhibitory protein and one previous report suggested this[10]. Yu and co-workers suggested this molecule played an immunomodulatory role, but its effects were mediated by engagement of CD155 on dendritic cells (DC) inducing production of IL-10[11]. Other evidence suggested that this molecule may play a role in the adhesion of T follicular helper cells (Tfh) to follicular DC[9]. We present experimental evidence suggesting Vstm3 delivers a negative signal directly to T cells. Moreover, our data reveals that it is part of a network of proteins that seem to contribute globally to T cell responses in a manner analogous to the well-characterized CD28-CTLA4-CD80-CD86 network and that this network is an attractive target for therapeutic intervention in human disease.
Results
Identification of Vstm3 as a Member of the CD28 Family and Characterization of its Expression Pattern
We initiated a search for additional members of the CD28 family using bioinformatic analysis. Among the immunoreceptor families, the known members of the CD28 family have the most sequence diversity and hence sequence similarity is a poor predictor of family membership. However, all known CD28 family members have a similar gene structure based on predicted “motifs” (IgV and IgC domains, transmembrane and cytoplasmic domains, signaling motifs), exon counts and sizes, intron phases, and intron locations amongst domains[12]. We previously established that gene structure base on these criteria is conserved as gene families evolve[13] emphasizing analysis of gene structure as an important means of family classification. Examination of genes not previously assigned to immunoreceptor families identified a gene with the HUGO designation VSTM3 that fits this CD28 family profile ([12]; Supplemental Figure S1a). Two potential ITIM motifs in the cytoplasmic domain suggested VSTM3 could be an inhibitory member of this family. Similar analysis of the mouse genome identified a clear ortholog of human VSTM3 (Supplemental Figure S1b) and importantly one of the most highly conserved regions between the two includes the putative ITIM motifs. Collectively, our analysis indicates that VSTM3 represents a novel member of the CD28 family and conservation over the putative ITIM motif suggests this sequence is functionally relevant.
VSTM3 has been identified by other groups as a protein expressed on NK cells, activated and memory T cells, and regulatory T cells (Tregs) and has been alternatively designated VSTM3[10] , TIGIT[11] , and WUCAM[9] . We generated monoclonal antibodies (Mabs) against both mouse and human proteins and confirmed their specificity on transfected cell lines. Furthermore, soluble forms of Vstm3 could completely block staining of a transfected cells when included in excess (data not shown). We confirmed expression patterns reported by others[9-11] with specific Vstm3 staining primarily on activated and memory T cells, NK cells and Foxp3+ Tregs from mice and humans (Supplemental Figures 2 and 3).
Identification of the Counter-structure for Vstm3
To better understand a role for Vstm3 in T and NK cell activation we searched for cells that bound to soluble Vstm3 protein. Two Vstm3 fusion proteins were generated for use as FACS-binding reagents in these experiments. The first was a dimeric Fc-fusion protein and the other was a tetramer., Both were directly conjugated to fluorochromes for FACS-based binding experiments and used to screen a variety of cell types under different stimulation conditions to identify those that specifically bound Vstm3. We noted specific binding of both mouse Vstm3 proteins to mouse DC derived from bone marrow cultures grown in Flt3L and activated with CD40L and IFNγ (Figures 1a and 1b). Binding was competed by excess unlabeled Vstm3 protein, but not by other fusion proteins (Figure 1a, 1b, data not shown). Since tetramer binding was better than dimeric Fc-fusion protein (Figures 1a and 1b) the tetramer was used to identify the activated DC binding activity.
Figure 1.

CD155 expression is sufficient to convey Vstm3 binding on activated DC and transfected cells. (A and B) DC were generated from mouse BM cells by culturing total BM at 106 cells/mL in 100 ng/mL Flt3-L for 7 days. Resultant DC were activated for 24 h with 10 ng/mL IFN-γ and 1 μg/mL CD40-L and then stained with (A) 2 μg/mL Alexa-647 conjugated Vstm3-Fc or (B) Alexa-647 labeled Vstm3-tetramer alone (solid lines) or in the presence of a tenfold excess of unlabeled Vstm3-Fc or Vstm3 tetramer respectively (dotted lines). Control Fc-fusion protein or an irrelevant tetrameric protein (shaded histograms) served as controls. (C) P815 cells transfected with mouse CD155 were stained with 2 μg/mL of an Alexa-647 conjugated Vstm3 tetramer (solid line) alone or in the presence of a tenfold excess unlabeled Vstm3 protein (dotted line). Staining with an irrelevant tetramer (shaded histogram) served as a negative control. (D) HEK-293 cells transfected with mouse CD155 were stained with 2 μg/mL Alexa-647 conjugated Vstm3-tetramer (solid line) or 2 μg/mL Alexa-647 labeled soluble CD226 (dashed line). Untransfected HEK-293 cells stained with either Vstm3-tetramer or Vstm-Fc served as negative controls. All results are representatives of at least two experiments.
An expression library was prepared from this activated DC population and screened by fluorescence microscopy[14] with the Vstm3 tetramer. Consistent with results obtained by other groups[9-11], a cDNA was identified that encoded the mouse homolog of the human polio virus receptor (PVR) CD155[15]. This cDNA was identified from multiple pools of library plasmids and expression of CD155 was sufficient to convey Vstm3 binding activity to a number of cell lines that did not bind the protein in their untransfected state (Figure 1c and 1d). Also, binding was blocked by excess unlabeled Vstm3 protein (Figure 1c), but not by other proteins (data not shown). These experiments confirm that CD155 serves as a counter-structure for Vstm3.
CD155 and CD112 Bind to Vstm3 and CD226
CD155 is a member of the Nectin family and has been characterized in a number of contexts. The human protein serves as a receptor for polio virus[16] and has been suggested to play roles in adhesion and transendothelial migration[17-19] . In the immune system, it is best studied as a counter-structure for the activating T and NK cell receptor CD226 or DNAM-1[20-23] . Moreover, despite its expression on activated DC, CD155 is more widely expressed outside the hematopoietic system, and it has been suggested to play an important role in T cell costimulation when T cells are activated by non-professional APC[24;25] . Consistent with literature reports, we confirmed that soluble mouse CD226-Fc bound HEK-293 cells transfected with mouse CD155 (Figure 1d) and that the human proteins showed the same binding characteristics (Figure 2a; data not shown). Moreover, soluble VSTM3 and CD226 were able to compete with each other for binding to CD155 indicating that these two proteins bind overlapping if not identical regions of CD155 (Figure 2a).
Figure 2.
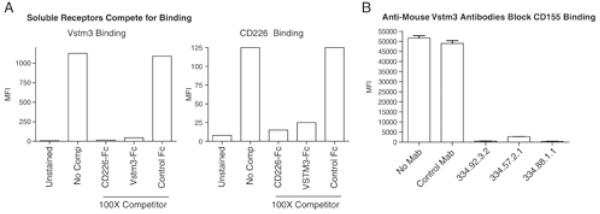
Vstm3 and CD226 cross-compete for binding to CD155 and anti-mouse Vstm3 mAbs inhibit Vstm3 binding to CD155. (A) P815 cells expressing human CD155 were left unstained or stained with 1 μg/mL Alexa-647 labeled human VSTM3 (left graph) or CD226 (right graph) in absence (no comp) or presence of a 100-fold excess of CD226-Fc, VSTM3-Fc or an irrelevant (control) Fc protein. MFI was determined and the results are representatives of at least two experiments. (B) P815-CD155 cells were cultured in medium alone (No mAb) or medium plus Alexa-647 conjugated Vstm3-tetramer (2 μg/mL) and either an irrelevant isotype control (Control mAb), or three independent anti-mouse Vstm3 mAb clones (each at 10 μg/mL). Samples were run in triplicate and bars show mean±SEM. Results are representatives of at least two experiments.
CD226 has also been reported to interact with another member of the Nectin family of proteins, CD112 or Nectin-2[20-23] . We and others (data not shown; [10;11] ) have confirmed that Vstm3 also interacts with CD112. This establishes CD226 and Vstm3 as proteins that interact with the Nectin family members CD155 and CD112. Moreover, since human and mouse forms of both proteins bound to CD155 and CD112 transfected cells, the functional relationships between these molecules has been conserved between species.
Functional Characterization of Soluble Vstm3 Proteins and Anti-Vstm3 Mabs
Expression of CD226 and Vstm3 ([9-11] ; Supplemental Figures 2 and 3; data not shown) overlaps with both molecules expressed on T cells and NK cells. Moreover, CD226 has been identified as a positive signaling molecule in these cells [22;23;26-28],, while the ITIM motifs in Vstm3 suggest an inhibitory function. The observation that positive and negative signaling receptors found on T cells bound to the same two counter-structures on APC is reminiscent of the relationships between CD28, CTLA4, CD80 and CD86 (Figure 3). Soluble forms of CTLA4 antagonize T cell activation by binding to CD80 and CD86 thereby interfering with engagement of CD28[29]. In contrast, blocking anti-CTLA4 Mabs prevent the inhibitory function of CTLA4 leading to enhanced T cell activation[30] . Each of these proteins is currently being used clinically, soluble CTLA4 to antagonize T cell function in autoimmune disease and anti-CTLA4 to enhance T cell function for cancer therapy. If the comparison of the CD226-Vstm3 and CD28-CTLA4 axis proposed in Figure 3 is valid and Vstm3 is in fact an inhibitory molecule opposing CD226-mediated costimulatory signals, we hypothesized that a soluble form of Vstm3 could be used to antagonize T cell function by interfering with costimulatory signals through CD226. Several lines of evidence support the value of antagonizing CD226-mediated signals. First, a polymorphism in the human CD226 gene has recently been associated with increased risk of developing a number of autoimmune diseases including multiple sclerosis (M.S.), type I diabetes, and Graves disease [31-34] . Secondly, a blocking anti-CD226 Mab could prevent development of experimental autoimmune encephalomyelitis (EAE), a mouse model of human M.S. [27]. Finally, CD226 has been established as an important costimulatory molecule for T cells particularly when they are stimulated by non-professional APC[24;25], a situation that pertains in autoimmune disease where T cells are attacking and damaging non-hematopoietic cells in the tissues. Hence, there is rationale for development of a therapeutic that targets CD226-mediated costimulation in human autoimmune disease. Moreover, if Vstm3 is delivering inhibitory signals as hypothesized, a blocking anti-Vstm3 Mab could prevent this putative inhibitory signal and thereby enhance T cell activation, the same way a blocking anti-CTLA4 Mab enhances T cell activation.
Figure 3.
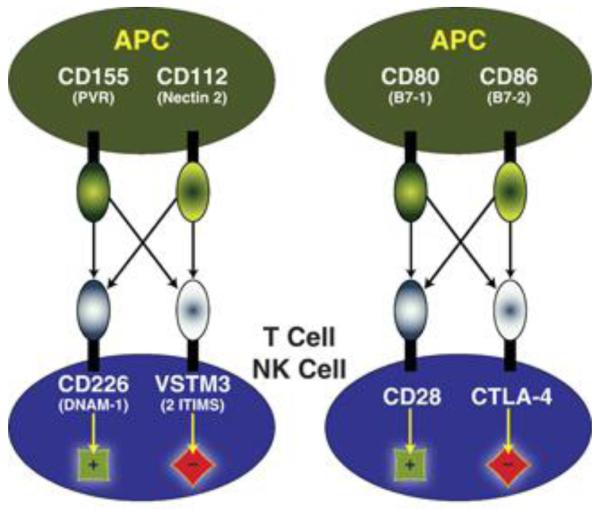
Schematic comparing the CD226-Vstm3 network of costimulation/inhibition between APC and T/NK cells with the CD28-CTLA-4 network. Vstm3 is a putative inhibitory receptor like CTLA-4 whereas CD226 is an activating receptor like CD28. Both sets of molecules are engaged by two sets of counter-structures, Vstm3 and CD226 by CD155 (PVR) and CD112 (nectin-2) and CTLA-4 and CD28 by CD80 (B7.1) and CD86 (B7.2). Activation is indicated by +; inhibition by −; arrows indicate protein–protein interactions.
As shown in Figure 2a, soluble Vstm3 prevented binding of Vstm3 or CD226 to CD155 expressing cells. To further examine interplay of these molecules, we tested whether our anti-Vstm3 Mabs could interfere with CD155 binding. P815 cells transfected with mouse or human CD155 expression vectors were incubated with each anti-Vstm3 Mab plus fluorochrome labeled Vstm3 and binding was assessed by FACS. None of the five Mabs against human VSTM3 generated were capable of blocking the binding of soluble human VSTM3 to cells expressing human CD155, although inclusion of excess human VSTM3 protein did prevent staining (data not shown). However, all of the anti-mouse Vstm3 Mabs generated prevented binding of mouse Vstm3 to P815 expressing mouse CD155 (Figure 2b; data not shown). We obtained the same results incubating fluorochrome labeled soluble CD155 with P815 cells expressing Vstm3 – all mouse Vstm3 Mabs prevented binding, but none of the human Mabs did (data not shown).
We next assessed effects of soluble Vstm3 or blocking Vstm3 antibodies on T cell responses in vitro to dissect out the roles of CD226 and Vstm3 in T cell activation. Professional APC express a large number of costimulatory and inhibitory proteins and T cells typically integrate signals from all molecules engaged during an immune response. Since we were interested in examining contributions of specific molecules, we developed an artificial APC-T cell response assay that allowed examination of the effects of single costimulatory or inhibitory molecules. BHK were transfected with mouse MHC class II molecules (specifically, the α and β chains of the I-Ad molecule) plus CD80, B7H1 or CD155 to generate artificial APC with limited costimulatory-inhibitory potential. BHK were loaded with an ovalbumin peptide and assessed for their ability to stimulate an antigen specific response in purified CD4+ T cells from DO11.10 TCR transgenic mice. Cells lacking mouse I-Ad did not appreciably stimulate mouse T cells in this assay, whereas cells transfected with I-Ad alone induced a modest level of peptide-dependent (there was no proliferation in the absence of peptide; data not shown) T cell activation as judged by CFSE dilution (Figure 4a). The level of response was diminished using BHK transfected with B7H1 due to the inhibitory effects of B7H1 engagement of PD1 and enhanced using BHK that expressed CD80, due to the costimulatory effect of CD28 engagement (Figure 4a). BHK transfected with CD155 plus I-Ad consistently elicited an enhanced T cell response relative to I-Ad alone (Figure 4a and 4b) suggesting the net effect of CD155-T cell interaction is costimulatory. To confirm this costimulation was attributable to CD155 expression, we used soluble Vstm3 to block the interaction of CD155 with T cells and this blocked the costimulatory properties of the CD155-transfected BHK (Figure 4b). However, as discussed above, CD155 engages both Vstm3 and CD226 and soluble Vstm3 inhibits binding of both molecules to CD155. To examine the nature of their respective signals, a blocking anti-Vstm3 Mab was included in the cultures. This Mab only interferes with CD155–Vstm3 interaction (Figure 2b) so this experiment should reveal the contribution of Vstm3 engagement. Inclusion of the blocking Mab led to a consistent increase in T cell responses in repeated experiments (Figure 4b) suggesting that the net signaling effect of Vstm3 engagement was inhibitory despite the overall costimulatory effect observed with cells expressing CD155. These results are analogous to results with CD80 transfected BHK where CD80 engages both the activating CD28 and the inhibitory CTLA4 proteins and yet the net effect is a costimulation of T cell responses (Figure 4a). Similarly, soluble CTLA4 blocks T cell activation whereas a blocking anti-CTLA4 antibody augments responses.
Figure 4.

CD155 delivers a costimulatory signal to T cells, but Vstm3 is an inhibitory receptor. (A) BHK cells stably transfected with I-Ad and the indicated co-receptors, and untransfected BHK cells, were loaded with 1 μg/mL DO11.10-specific OVA peptide and assessed for their ability to stimulate proliferation of DO11.10 TCR TG T cells. (B) Peptide-loaded BHK expressing I-Ad alone or together with CD155 were tested for their effects on DO11.10 T-cell proliferation in the presence or absence of 5 μg/mL soluble Vstm3-tetramer or blocking anti-Vstm3 antibody. (A and B) Graphs show the mean percentage of proliferating T cells±SEM (triplicate cultures), are derived from separate experiments and are representatives of at least five experiments. The dashed line indicates the mean values for BHK cells expressing only I-Ad; * indicates values significantly different from this value (p<0.05, Student’s _t_-test) while ** indicates values differing both from this value and from the value for cells expressing I-Ad plus CD155 (p<0.05, Student’s _t_-test).
Treatment of Mice with Soluble Vstm3 Inhibits Development of Autoimmune Diseases
CTLA4-Ig suppresses T cell mediated disease in a number of mouse models of human autoimmune disease including CIA[35] and EAE[36;37] . Because soluble Vstm3 blocked T cell activation in vitro (Figure 4), we hypothesized that a soluble form of Vstm3 would inhibit disease development in the same models. We first tested soluble Vstm3 in a CIA model and found that administration of either the mouse Vstm3 tetramer or the Fc-fusion protein significantly inhibited the severity of disease (Figure 5a), with the tetramer showing better efficacy than the dimeric Fc-fusion protein. This is consistent with in vitro experiments showing inhibition of T cell responses by soluble Vstm3 (Figure 4). In contrast, administration of a blocking anti-Vstm3 antibody resulted in more rapid disease onset, consistent with blocking the function of an inhibitory molecule (Figure 5b).
Figure 5.

Soluble Vstm3 inhibits disease in a CIA model whereas a blocking anti-Vstm3 mAb accelerates disease development. (A and B) Mice were treated starting one day before the second collagen injection with PBS, Vstm3-Fc, Vstm3-Tet or anti-Vstm3 mAb and the average paw score (±SEM) was measured on the initial day of treatment and daily thereafter; Graphs show average paw score versus time (in days) after the second collagen injection *p<0.05 by two-way ANOVA. Results shown are representatives of at least three experiments. Serum (C) IL-6 or (D) IL-10 levels were determined by Luminex assay on serum samples harvested on day 12 after CIA induction. Each dot represents the mean of triplicate determinations from a single mouse and the horizontal lines are the mean values from all animals in a treatment group; _p_-values are calculated relative to the PBS-treated mice (one-way ANOVA). UI, untreated, non-CIA mice.
To better understand the mechanism by which soluble Vstm3 was attenuating disease, levels of serum cytokines were assessed at the end of each experiment and the numbers and phenotypes of T cells in the draining lymph nodes (DLN) and spleens of individual animals were monitored. The inhibition of disease development correlated with a reduction in the serum levels of the pro-inflammatory cytokine IL-6 (Figure 5c) in treated mice relative to untreated. While both the tetrameric and dimeric proteins produced these effects, the tetramer was more effective. However, inhibition of disease was also associated with a reduction in the serum levels of the anti-inflammatory cytokine IL-10 (Figure 5d). This was somewhat surprising because IL-10 is protective in this challenge model and a previous report suggested that soluble Vstm3 induced IL-10 production in a mouse delayed type hypersensitivity (DTH) challenge model [11] .
To further investigate the effects of soluble Vstm3 in CIA, intracellular cytokine staining was performed after re-stimulation of cells from the DLN of challenged mice. T cells producing the pro-inflammatory cytokines IL-17A and TNFα are involved in pathology in this model and the percentage of CD4+ T cells producing these cytokines was elevated in untreated mice compared to unimmunized control animals (Figures 6a and 6b). However, animals treated with soluble Vstm3 had significantly fewer CD4+ T cells expressing these cytokines in DLN (Figures 6a and 6b). Moreover, mice treated with soluble Vstm3 had significantly fewer cells with a memory phenotype in DLN (Figure 6c) and fewer lymphocytes infiltrating the paws (Figure 6d). Surprisingly, Vstm3 treated animals also showed reductions in the percentage of cells producing IL-10 and IFNγ (Figure 6e and 6f), both of which are protective in CIA. Although the decrease in IL-10+ cells did not attain statistical significance like the percentage of other cytokine producing cells, the result was seen in multiple experiments (Figure 6f, data not shown). Taken together, this data suggests that soluble Vstm3 attenuates disease in CIA through a general dampening of CD4+ T cell responses most likely by interfering with CD226-mediated costimulation.
Figure 6.

Soluble Vstm3 attenuates disease in CIA by inhibiting T-cell activation. On day 12 after CIA induction, cells from the DLN were isolated, acutely restimulated in the presence of Golgi-blockers and surface and the percentage of cells staining for (A) IL-17A, (B) TNF-α, (C) CD4+ cells with a memory (CD44high) phenotype, (E) IFN-γ+ cells, and (F) IL-10+ cells, as well as (D) the number of lymphocytes harvested from the paws of animals, was determined by flow cytometry. Each dot is the mean of triplicate cultures of cells from one animal and the horizontal lines are the mean values from all animals in a treatment group; _p_-values are results from Student’s _t_-test comparing PBS-treated mice to those treated with Vstm3-Tet and only those values showing significant differences from controls have _p_-values associated with them. Results shown are representatives of at least three experiments.
We also tested soluble Vstm3 and a blocking anti-Vstm3 Mab for their effects in EAE induced with MOG peptide. Pathology in this disease model is primarily a consequence of CD4+ T cell responses to components of the myelin sheath. Mice were immunized with MOG peptide and then treated every other day as indicated with vehicle control (PBS) or the Vstm3-tetramer and disease symptoms were assessed daily for 15 days. As in the CIA model, soluble Vstm3 significantly attenuated disease development (Supplemental Figure 4). Moreover, treatment with blocking anti-Vstm3 Mab resulted in an exacerbation of disease (data not shown) consistent with what was seen in CIA studies. Collectively, these data indicate that treatment of mice undergoing autoimmune challenge with soluble Vstm3 can ameliorate disease progression while a blocking anti-Vstm3 Mab exacerbates disease.
Vstm3-Deficient Mice Are More Susceptible to Autoimmune Challenge
To further understand the role of Vstm3 in regulation of immune responses, we generated Vstm3-deficient mice and transgenic mice that over-expressed Vstm3 in T and B cells. As expected, splenocytes from Vstm3-deficient mice failed to stain with anti-Vstm3 antibody (Figure 7a). In the transgenic mice, expression of full-length Vstm3 was regulated by the Lck proximal promoter and the μ-heavy chain enhancer element and over-expression could be detected on splenocytes including all T cells and most B cells (Figure 7a, data not shown). Preliminary analysis of both Vstm3-deficient and transgenic mice showed no developmental defects in hematopoietic cell populations and analysis of T cell responses in vitro showed no evidence of functional defects in terms of proliferation or upregulation of activation antigens in response to T cell stimulation (data not shown). To assess T cell function in vivo in these mice, they were challenged in a MOG peptide-induced EAE model. Consistent with the proposed inhibitory role of Vstm3 in T cell function, the Vstm3-deficient mice developed more severe EAE relative to wild-type mice while the transgenic mice were protected from disease (Figure 7b). Although we believe the protective effect of transgene expression in this disease model is due to the enforced over-expression on T cells, it’s possible that expression on B cells could also play a role. In any case, these data are consistent with what would be expected when deleting or over-expressing a molecule that serves an inhibitory role in the regulation of T cell responses.
Figure 7.

Vstm3-deficient (KO) mice are more sensitive to autoimmune challenge whereas TG mice are protected. (A) Total splenocytes from Vstm3 KO (left panel, dashed line), TG mice (right panel, dotted line) or control WT mice (solid lines) were stained with anti-mouse Vstm3 mAb and and MFI measured. Plots show Vstm3 expression gated on total splenocytes as indicated in the Supporting Information Fig. 5B. (B) Graph shows average disease score (±SEM) versus time after pertussis toxin administration in the MOG EAE model in WT, Vstm3-KO and Vstm3-Tg mice as described in the materials and methods section. * indicates p<0.05 by two-way ANOVA KO/TG versus the respective WT. Results shown are representatives of at least three experiments. (C) T-cell-depleted BM was transferred (day 0) into lethally irradiated B10.BR mice without (BM only) or with purified T cells from Vstm3 KO (KO) or control mice (WT) in the absence/presence of anti-Vatm3 mAb and cell survival was measured over time; p<0.0002 for WT versus KO and WT versus WT + Anti-vstm3 (log-rank test). Results shown are representatives of two experiments.
To further demonstrate that the enhanced disease noted in the knockout mice was a T cell intrinsic effect, we extended our findings in MOG EAE to a model of graft-versus-host disease (GVHD). Purified T cells from either Vstm3 deficient or wild type mice were transferred into lethally irradiated B10.BR mice in a GVHD model. Mice that received the Vstm3 deficient T cells succumbed to GVHD significantly faster than mice that received wild type cells (Figure 7c). Moreover, administration of a blocking anti-Vstm3 antibody to mice that had received wild type cells accelerated mortality (Figure 7c) suggesting that engagement of Vstm3 by its counter-structures contributed to the extended survival of mice receiving wild type cells. Together, these data are consistent with the MOG EAE studies and suggest that the engagement of Vstm3 provides an inhibitory signal to the immune system whereas blockade or absence of Vstm3 leads to accelerated GVHD.
Agonistic Anti-VSTM3 Antibodies Inhibit T cell Responses
The experiments described above suggest that engagement of Vstm3 triggers an inhibitory signal directly to T cells. However, because CD155 engages both CD226 and Vstm3, we sought to examine consequences of specifically engaging only Vstm3. It is widely appreciated that Mabs with agonistic activity (including Mabs to CD3) more readily induce signaling when they are cross-linked, although it is also clear that not all antibodies against immunoreceptors possess agonistic activity. Moreover the properties of agonistic versus non-agonistic antibodies are currently undefined and this makes prediction of an antibody’s behavior difficult. Therefore, we tested the properties of the anti-Vstm3 Mabs we generated directly in T cell proliferation assays. To do this we coupled anti-CD3 Mabs at sub-optimal levels to beads and co-coupled either anti-Vstm3 Mabs or isotype controls to the same beads. Antibody coated beads were then used to stimulate purified mouse or human T cells. Proliferation was monitored by CFSE dilution and activation was assessed by measuring cytokine secretion or upregulation of the activation antigens CD25 and CD69. Beads coated with anti-CD3 Mabs and an irrelevant antibody or isotype control induced a reasonable level of activation and proliferation (Figure 8). Examining responses of mouse T cells, none of the five anti-Vstm3 Mabs generated affected responses relative to isotype control coated beads even though all of these Mabs blocked Vstm3 binding to CD155 (Figure 2; data not shown). Moreover, three out of the five anti-human VSTM3 Mabs also had no effect on human T cell responses when co-coupled to beads with anti-CD3 Mabs (data not shown). However, two of five anti-human VSTM3 Mabs significantly inhibited T cell responses as judged by CFSE dilution, upregulation of CD25 and CD69, and secretion of the cytokines IL-2 and IFNγ (Figure 8; data not shown). These Mabs do not block binding of Vstm3 to CD155, so they could not be functioning in this way, and they did not affect T cell responses when presented in soluble form to T cells, but only showed the inhibitory effect when cross-linked. The simplest explanation for this inhibitory effect is that these Mabs have unique agonistic properties that the other Mabs generated lack. This suggests that engaging VSTM3 can trigger an inhibitory signal in T cells but not all Mabs that bind to VSTM3 exhibit this effect. This inhibition was apparent when analyzing responses of both CD4+ and CD8+ T cells (Figure 8, data not shown) and seemed to represent a general inhibition of activation since induction of the early activation markers CD25 (Figure 8) and CD69 (data not shown) was inhibited as was the release of the cytokines IL-2 and IFNγ (data not shown). Inhibition is most evident at lower concentrations of anti-CD3 Mab whereas increasing anti-CD3 concentrations overcame the anti-VSTM3 effects to some extent. Collectively, these data and data in the preceding section support the idea that VSTM3 functions as an inhibitory receptor in the responses of T cells in both mice and humans.
Figure 8.

Agonistic activity of an anti-VSTM3 mAb inhibits T-cell responses. mAb were coupled to beads (anti-CD3 at the indicated percentage of bead capacity and anti-VSTM3 mAb or an isotype-matched control antibody each at 80%) and the beads incubated at a bead: cell ration of 1:1 with purified T cells. Proliferation was judged by CFSE dilution on T cells (gated as indicated in the Supporting Information Fig. 5C) for anti-CD3 plus isotype control (left hand panels) or anti-CD3 plus anti-VSTM3 (center panels). Upregulation of CD25 was also assessed by FACS (right panels) using the beads coated with anti-CD3 plus the isotype control (dark line) or anti-CD3 plus anti-VSTM3 (grey line). Profiles shown are gated on CD4+ T cells (Supporting Information Fig. 5B), but similar results were obtained examining the same parameters on CD8+ cells in the same cultures. Results are representatives of at least three experiments.
Discussion
Vstm3 was recently described by other groups and alternatively designated Vstm3, TIGIT, or WUCAM[9-11] . These three groups differ regarding the proposed function of Vstm3. Stanietsky et al suggest it is an inhibitory receptor and that the ITIMs are important in function[10] , whereas Boles and co-workers[9] suggest it facilitates adhesion of follicular helper T cells (Tfh) to follicular DC. Yu et al[11] suggest it has an inhibitory function, but that the inhibitory effect is due to Vstm3 signaling to DC to induce IL-10 production, which then inhibits immune cell responses. We identified Vstm3 as a member of the CD28 family based on intron-exon patterning and the predicted domain structure of the encoded protein. Two predicted ITIM motifs in the cytoplasmic domain suggested it could serve a direct inhibitory role in T cell function. The conservation of amino acids around and comprising the putative ITIMs further supports their role in Vstm3 activity. Thus far, we have been unable to generate evidence that the ITIMs are required or that they recruit phosphatases. Stanietsky et al[10] demonstrated that the ITIM was important, but similarly could not see evidence of SHP recruitment. Regardless of the specific mechanism, our data suggests that Vstm3 does serve a T cell intrinsic inhibitory function. Among the members of the CD28 family associated with inhibitory function, only BTLA has been clearly shown to recruit SHP1/2 to the phosphorylated ITIMs[8] . PD1 inhibitory signals involve SHP1/2 but the ITIM motif appears dispensable [6]. The inhibitory nature of CTLA4 is clear, but the means by which it exerts these effects is incompletely understood. Similarly, although the mechanism whereby Vstm3 exerts its effects is unclear, several lines of evidence indicate it functions as an inhibitory receptor in T cells. First, blocking Mabs against mouse Vstm3 enhance T cell activation in vitro (Figure 4) and in vivo (Figures 5b and 7c). Secondly, Vstm3 deficient mice develop exacerbated disease in a MOG EAE model. Thirdly, transfer of Vstm3 deficient T cells results in accelerated GVHD compared to transfer of wild type cells (Figure 7). Finally, a subset of (but not all) anti-human VSTM3 Mabs inhibit T cell responses when co-crosslinked with anti-CD3 Mabs on beads, but not when presented in soluble form. Such behavior is typically attributed to Mabs exhibiting agonistic activity and hence engagement of VSTM3 by these Mabs appears to trigger its inhibitory activity. As discussed above, literature reports make clear that not all antibodies against inhibitory receptors can trigger the inhibitory activity of their receptors and the properties that allow this activity have not been defined. However, it is likely that the anti-human VSTM3 Mabs that do apparently trigger inhibitory function recognize a unique epitope or set of epitopes that allow induction of an appropriate conformational change that mimics the effects of CD155 or CD112 engagement. Additional experimentation will be required to address these properties.
The predominance of Vstm3 expression on activated T cells led us to functionally clone a counter-structure from an activated DC expression library. The cDNA we identified encoded the same Nectin family protein (CD155) previously identified by other groups[9-11]. CD155 also interacts with the activating receptor CD226 and CD226 binds another member of the nectin family, CD112. Vstm3 also bound to CD112 and we have demonstrated that Vstm3 and CD226 cross-compete for binding to CD155 and CD112. The activating properties of CD226, the apparent inhibitory nature of Vstm3, plus the identification of two counter-structures for each was reminiscent of the network of interactions noted for the activating receptor CD28, its inhibitory counterpart CTLA4 and their two ligands, CD80 and CD86. Therefore, we postulated that Vstm3, CD226, CD155, and CD112 formed a network of proteins that have the potential to regulate T cell responses in much the same way as CD28, CTLA4, CD80, and CD86 do (Figure 3). The analogy of Vstm3-CD226-CD155-CD112 with the CTLA4-CD28-CD80-CD86 co-stimulatory/inhibitory network led us to hypothesize that soluble Vstm3 may inhibit T cell responses in the same way that soluble CTLA4 does. Previous work confirmed that soluble Vstm3 could inhibit immune responses (specifically DTH), but the mechanism was proposed to involve Vstm3 engagement of CD155 on DC inducing IL-10 production by the DC[11]. We also demonstrated that soluble Vstm3 blocked T cell responses in vitro (Figure 4) and in vivo (Figure 5, Supplemental Figure 4), but our data suggests that these effects are through prevention of CD226 costimulation. CD226 has recently become an attractive target for therapeutic intervention in autoimmune disease because of its linkage to increased risk of developing multiple autoimmune syndromes[31-34] . Moreover, CD226-mediated costimulation has been shown to be important under conditions where T cells are being activated by non-professional APC[24;25] , which can be the case for T cells infiltrating tissues in auto-inflammatory conditions, and therefore soluble Vstm3 has the potential to be an effective therapeutic agent in the treatment of autoimmune diseases.
Collectively, available information suggests that the Vstm3 network of proteins forms a regulatory axis functionally similar to the CD28-CTLA4-CD80-CD86 axis. However, it is also worth noting that CD96 has also been reported to bind to CD155[38;39] . CD96 includes two putative ITIM in its cytoplasmic domain as well and yet is reported to deliver a positive signal when engaged[38]. This observation emphasized the need to confirm the inhibitory nature of Vstm3. The potential contribution of CD96 in this axis of receptors remains to be elucidated.
Although we do not yet know the extent of Vstm3′s function(s) in vivo, it is likely to play a role unique from other members of the CD28 family. This suggestion is based on the observation that even though the expression pattern of PD1 and BTLA overlap, the ability of each of these molecules to regulate immune responses varies because of the differential expression of their counter-structures[40] . In this respect, it is of interest that CD155 has been reported to be expressed at high levels on a variety of tumor cells[41-43]. While it has been reported that this facilitates rejection by immune-mediated mechanisms through CD226-dependent signals, this does not explain the high propensity of tumor cells to express CD155. Transformed cells tend to acquire ways to subvert the immune system, and it is possible that the acquisition of high level expression of CD155 can actually have an inhibitory effect on T or NK cell activation through Vstm3 engagement. Moreover, CD155 is much more widely expressed outside of the immune system than CD80/CD86 indicating it could also have functions apart from its expression on activated DC. In particular, it could have more significant effects on regulating inflammation and consequent damage to tissues where non-professional APC play a significant role.
Our data indicate that Vstm3 is an inhibitory component of a network of proteins that plays a significant role in regulating the activation and function of T cells. Current understanding indicates that there could be therapeutic benefit in interfering with this axis, but the specific point of intervention would depend upon the indication being targeted. For example, oncology may benefit most by specific intervention in Vstm3 signaling while leaving CD226 signaling intact. On the other hand, autoimmune or alloimmune diseases may benefit from antagonizing CD226 activation or by specifically triggering the inhibitory properties of Vstm3. In particular, there may be benefit in specifically targeting T cell responses in tissues containing activated, pathogenic T cells as opposed to global T cell inhibition induced by therapeutic agents such as CTLA4-Ig. Such targeted intervention may lead to tissue-specific inhibition of immune reactions while leaving global immune responses relatively unaltered.
Materials and Methods
Expression and purification of Vstm3 fusion proteins
An in-frame fusion of the extra-cellular domain of mouse Vstm3 (amino acids 1-138) and an effector negative version of the mouse IgG2a Fc region (aa 216 through the C-terminal lysine) was constructed in a mammalian expression vector. This plasmid was introduced into CHO cells and secreted Fc-fusion protein was purified by affinity chromatography on Protein A Sepharose FF (GE Healthcare). A similar approach was used to generate the human Fc-fusion protein.
Tetrameric protein was generated by fusing the extracellular domain of Vstm3 in frame with amino acids 343 to 375 from the human Vasodilator-stimulated phosphoprotein protein (VASP) and a C-terminal histidine tag to facilitate purification. The peptide from the VASP protein mediates tetramerization. This construct was transfected into CHO cells and secreted tetramer was purified using NiNTA Agarose (Novagen / EMD Biosciences) and size exclusion chromatography on Superdex 200 (GE Healthcare). A similar approach was used to generate the human tetrameric protein.
Anti-Vstm3 Monoclonal Antibodies
Sprague-Dawley rats (Charles River Laboratories) were immunized with recombinant mouse Vstm3 protein and B cells from the rat showing the greatest anti-Vstm3 titer were fused to the P3-X63-Ag8.653 mouse myeloma cell line (ATCC CRL1580). Master wells were screened for antibodies that bound to Vstm3-Fc protein by ELISA and were positive by FACS on BHK cells expressing Vstm3. A total of five different monoclonal antibodies (Mabs) were generated that were specific for mouse Vstm3.
Antibodies to human VSTM3 were generated by immunizing BALB/C mice with soluble human VSTM3 protein and fusions were performed and screened as described above using human VSTM3-Fc protein. A total of five different Mabs were generated that were specific for human VSTM3.
Generation and Screening of an Expression Library from Activated Mouse Bone Marrow DC
Bone marrow cells from the femurs of 6 week old BALB/C mice were cultured 7 days in 100 ng/ml Flt3L (R&D Systems). Cells were then stimulated with 1μg/ml CD40L (R&D Systems) and 10ng/ml IFNγ (R&D Systems). Analysis of Vstm3 binding activity was done using Alexa-647 conjugated Vstm3-Fc fusion protein or Vstm3-tetramer. For generation of an expression library, cells were collected at 4h and 8h post stimulation and pooled for RNA isolation. PolyA+ RNA was purified using RNeasy (Qiagen) and Dynabead mRNA (Dynal) kits according to manufacturer’s recommendations. A cDNA expression library was constructed using mammalian expression vectors as previously described ([44]).
The expression library was split into pools of 200 clones, pools were transiently transfected into COS7 cells and screened as described [14;44] with biotinylated Vstm3-tetramer at 2 μg/ml. Pools showing positive binding were broken down sequentially until single cDNA clones could be identified that conveyed Vstm3-specific binding activity.
Assessment of T cell responses using engineered APC
Baby Hamster Kidney cells (BHK) were transfected with a plasmid encoding the α and β chains of the murine MHC class II allele I-Ad. Stably transfected cells were drug selected and expression was confirmed by FACS. These cells were then transfected with a plasmid encoding mouse CD155, B7H1, or CD80 and stable lines expressing both I-Ad and each coreceptor were generated. B7H1 and CD80 expression was verified by antibody staining and CD155 expression was confirmed by FACS analysis using recombinant Vstm3 protein.
For T cell assays BHK transfectants were seeded in 96-well flat-bottom plates (25,000 cells/well) and allowed to adhere for four hours. Cells were loaded with DO11.10 specific OVA peptide. Purified CD4+ T cells from DO11.10 TCR transgenic mice were CFSE-labeled (Invitrogen) according to manufacturer’s instructions. T cells (105/well) were added to peptide loaded BHK transfectants and incubated three days at 37°C. For some experiments Vstm3-tetramer or a blocking anti-Vstm3 Mab were included at 1 μg/ml. After three days, cells were harvested, stained for CD4, CD25, and CD69, and analyzed by FACS. Proliferation was assessed by CFSE dilution and expression of CD25 and CD69 was quantified.
Coupling Antibodies to Beads
Antibodies were coupled to Dynabeads (M-450 Tosyl-activated; Dynal Biotech) following manufacturer’s protocol. Anti-CD3 Mabs (B-D Biosciences) were used at the indicated percentage of bead capacity and were allowed to bind at room temperature for 30 min. A second Mab (e.g. anti-VSTM3) or control IgG was then added at 80% of bead capacity and beads were incubated overnight at 37°C. Beads were washed and used at a 1:1 ratio of beads to CFSE labeled purified T cells. After 3 days in culture, cells were harvested and stained for surface markers as indicated and proliferation was assessed by CFSE dilution as above.
Genetically Manipulated Animals
Vstm3 deficient mice were generated by Ozgene using ES cells derived from C57BL/6 mice. In brief, most of exons 1-3 were deleted from the genomic sequence and a β-galactosidase coding sequence was inserted in exon 1 in-frame with the original ATG start codon. A neomycin resistance gene with expression driven by the PGK promoter followed the β-gal sequence.
Vstm3 transgenic mice were generated by injection of a DNA fragment including the mouse Vstm3 cDNA in an expression vector, which includes the Lck proximal promoter, the Igμ heavy chain enhancer, and the human growth hormone gene (hGH) [45] . Transgene positive animals were identified by the presence of hGH sequences and five independent lines were generated. Transgenic mice used in the experiments described had been back-crossed at least eight generations onto the C57BL/6 background.
All mice were maintained in a specific pathogen-free AAALAC accredited facility. All animal procedures were conducted in accordance with approved ethical guidelines under the auspices of the ZymoGenetics Institutional Animal Care and Use Committee.
CIA Induction
CIA was induced as described previously [46]. Briefly, male DBA/1J mice (Jackson Laboratories) were injected in the tail intradermally with 100 μl of chick II collagen (1 mg/ml) in complete Freund’s adjuvant, followed 21 days later by a boost using the same amount of collagen in incomplete Freund’s. Disease severity was assessed daily as described [46]. Mice were treated as indicated with 150 μg of soluble Vstm3 protein or blocking anti-mouse Vstm3 antibody or an equal volume of vehicle (PBS) via i.p. injection every other day, beginning the day prior to the second collagen injection.
MOG Peptide Induced EAE
Female C57BL/6 mice were immunized s.c. with MOG35-55 peptide emulsified in complete Freund’s adjuvant and administered 200 ng pertussis toxin i.v. two days later. Disease symptoms were assessed daily for 15 days as described [47]. For some experiments, mice were treated every other day with vehicle control (PBS) or the Vstm3 tetramer.
GVHD Induction
B10.BR (H-2k) recipients were purchased from Jackson Laboratories and received total body irradiation at a lethal dose (11Gy) by single exposure to a 137Cs source 24 hrs before cell infusion. T cells were purified from splenocytes that had been shipped overnight on ice. After red cell lysis T cells were negatively selected using antibodies against CD19, CD11b, NK1.1 (eBioscience), and TCRγδ (BD) and LD columns (Miltenyi Biotech). T cell purity was >90%. 106 or 2×106 T cells were injected together with T depleted bone marrow (BM) cells (107) from 6 mice i.v. on day 0. BMT recipients were monitored daily for survival, weighed twice weekly for 30 days and once weekly thereafter. Where indicated, 150 μg irrelevant rat IgG (Rockland) or 100μg anti-Vstm3 Mab 334.57.2.1 in 200μl PBS, were injected i.p. every other day. Survival was analyzed by life-table methods and actuarial survival data are shown. Comparison between groups was done by log-rank test. P values < 0.05 were considered significant.
Supplementary Material
Supplement
Acknowledgements
The authors wish to thank Harald Haugen for production of transgenic mice, Suzanne Nicholson and Wendy Curtis for help in maintaining and breeding transgenic and knockout animals, and Margo Rogers for help in preparing the manuscript.
Footnotes
All authors except BRB and CB were full-time employees of ZymoGenetics, Inc. during the course of the work detailed in this manuscript and hence declare a competing financial interest.
References
- 1.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu.Rev.Immunol. 2005;23:515–548. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 2.Keir ME, Sharpe AH. The B7/CD28 costimulatory family in autoimmunity. Immunol.Rev. 2005;204:128–143. doi: 10.1111/j.0105-2896.2005.00242.x. [DOI] [PubMed] [Google Scholar]
- 3.Sharpe AH, Freeman GJ. The B7-CD28 superfamily. Nat.Rev.Immunol. 2002;2:116–126. doi: 10.1038/nri727. [DOI] [PubMed] [Google Scholar]
- 4.Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, Scheu S, Pfeffer K, Ware CF, Murphy TL, Murphy KM. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat.Immunol. 2005;6:90–98. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 5.Parry RV, Riley JL, Ward SG. Signalling to suit function: tailoring phosphoinositide 3-kinase during T-cell activation. Trends Immunol. 2007;28:161–168. doi: 10.1016/j.it.2007.02.004. [DOI] [PubMed] [Google Scholar]
- 6.Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J.Immunol. 2004;173:945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- 7.Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to phosphotyrosine. Proc.Natl.Acad.Sci.U.S.A. 2001;98:13866–13871. doi: 10.1073/pnas.231486598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, Murphy TL, Russell JH, Allison JP, Murphy KM. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat.Immunol. 2003;4:670–679. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 9.Boles KS, Vermi W, Facchetti F, Fuchs A, Wilson TJ, Diacovo TG, Cella M, Colonna M. A novel molecular interaction for the adhesion of follicular CD4 T cells to follicular DC. Eur.J.Immunol. 2009;39:695–703. doi: 10.1002/eji.200839116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stanietsky N, Simic H, Arapovic J, Toporik A, Levy O, Novik A, Levine Z, Beiman M, Dassa L, Achdout H, Stern-Ginossar N, Tsukerman P, Jonjic S, Mandelboim O. The interaction of TIGIT with PVR and PVRL2 inhibits human NK cell cytotoxicity. Proc.Natl.Acad.Sci.U.S.A. 2009;106:17858–17863. doi: 10.1073/pnas.0903474106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yu X, Harden K, Gonzalez LC, Francesco M, Chiang E, Irving B, Tom I, Ivelja S, Refino CJ, Clark H, Eaton D, Grogan JL. The surface protein TIGIT suppresses T cell activation by promoting the generation of mature immunoregulatory dendritic cells. Nat.Immunol. 2009;10:48–57. doi: 10.1038/ni.1674. [DOI] [PubMed] [Google Scholar]
- 12.Brandt CS, Baratin M, Yi EC, Kennedy J, Gao Z, Fox B, Haldeman B, Ostrander CD, Kaifu T, Chabannon C, Moretta A, West R, Xu W, Vivier E, Levin SD. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J.Exp.Med. 2009;206:1495–1503. doi: 10.1084/jem.20090681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conklin D, Haldeman B, Gao Z. Gene finding for the helical cytokines. Bioinformatics. 2005;21:1776–1781. doi: 10.1093/bioinformatics/bti283. [DOI] [PubMed] [Google Scholar]
- 14.Gross JA, Johnston J, Mudri S, Enselman R, Dillon SR, Madden K, Xu W, Parrish-Novak J, Foster D, Lofton-Day C, Moore M, Littau A, Grossman A, Haugen H, Foley K, Blumberg H, Harrison K, Kindsvogel W, Clegg CH. TACI and BCMA are receptors for a TNF homologue implicated in B-cell autoimmune disease. Nature. 2000;404:995–999. doi: 10.1038/35010115. [DOI] [PubMed] [Google Scholar]
- 15.Ravens I, Seth S, Forster R, Bernhardt G. Characterization and identification of Tage4 as the murine orthologue of human poliovirus receptor/CD155. Biochem.Biophys.Res.Commun. 2003;312:1364–1371. doi: 10.1016/j.bbrc.2003.11.067. [DOI] [PubMed] [Google Scholar]
- 16.Mendelsohn CL, Wimmer E, Racaniello VR. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56:855–865. doi: 10.1016/0092-8674(89)90690-9. [DOI] [PubMed] [Google Scholar]
- 17.Reymond N, Imbert AM, Devilard E, Fabre S, Chabannon C, Xerri L, Farnarier C, Cantoni C, Bottino C, Moretta A, Dubreuil P, Lopez M. DNAM-1 and PVR regulate monocyte migration through endothelial junctions. J.Exp.Med. 2004;199:1331–1341. doi: 10.1084/jem.20032206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sato T, Irie K, Ooshio T, Ikeda W, Takai Y. Involvement of heterophilic trans-interaction of Necl-5/Tage4/PVR/CD155 with nectin-3 in formation of nectin- and cadherin-based adherens junctions. Genes Cells. 2004;9:791–799. doi: 10.1111/j.1365-2443.2004.00763.x. [DOI] [PubMed] [Google Scholar]
- 19.Sloan KE, Eustace BK, Stewart JK, Zehetmeier C, Torella C, Simeone M, Roy JE, Unger C, Louis DN, Ilag LL, Jay DG. CD155/PVR plays a key role in cell motility during tumor cell invasion and migration. BMC.Cancer. 2004;4:73. doi: 10.1186/1471-2407-4-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bottino C, Castriconi R, Pende D, Rivera P, Nanni M, Carnemolla B, Cantoni C, Grassi J, Marcenaro S, Reymond N, Vitale M, Moretta L, Lopez M, Moretta A. Identification of PVR (CD155) and Nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J.Exp.Med. 2003;198:557–567. doi: 10.1084/jem.20030788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pende D, Bottino C, Castriconi R, Cantoni C, Marcenaro S, Rivera P, Spaggiari GM, Dondero A, Carnemolla B, Reymond N, Mingari MC, Lopez M, Moretta L, Moretta A. PVR (CD155) and Nectin-2 (CD112) as ligands of the human DNAM-1 (CD226) activating receptor: involvement in tumor cell lysis. Mol.Immunol. 2005;42:463–469. doi: 10.1016/j.molimm.2004.07.028. [DOI] [PubMed] [Google Scholar]
- 22.Tahara-Hanaoka S, Shibuya K, Onoda Y, Zhang H, Yamazaki S, Miyamoto A, Honda S, Lanier LL, Shibuya A. Functional characterization of DNAM-1 (CD226) interaction with its ligands PVR (CD155) and nectin-2 (PRR-2/CD112) Int.Immunol. 2004;16:533–538. doi: 10.1093/intimm/dxh059. [DOI] [PubMed] [Google Scholar]
- 23.Tahara-Hanaoka S, Miyamoto A, Hara A, Honda S, Shibuya K, Shibuya A. Identification and characterization of murine DNAM-1 (CD226) and its poliovirus receptor family ligands. Biochem.Biophys.Res.Commun. 2005;329:996–1000. doi: 10.1016/j.bbrc.2005.02.067. [DOI] [PubMed] [Google Scholar]
- 24.Gilfillan S, Chan CJ, Cella M, Haynes NM, Rapaport AS, Boles KS, Andrews DM, Smyth MJ, Colonna M. DNAM-1 promotes activation of cytotoxic lymphocytes by nonprofessional antigen-presenting cells and tumors. J.Exp.Med. 2008;205:2965–2973. doi: 10.1084/jem.20081752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iguchi-Manaka A, Kai H, Yamashita Y, Shibata K, Tahara-Hanaoka S, Honda S, Yasui T, Kikutani H, Shibuya K, Shibuya A. Accelerated tumor growth in mice deficient in DNAM-1 receptor. J.Exp.Med. 2008;205:2959–2964. doi: 10.1084/jem.20081611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Carlsten M, Bjorkstrom NK, Norell H, Bryceson Y, van HT, Baumann BC, Hanson M, Schedvins K, Kiessling R, Ljunggren HG, Malmberg KJ. DNAX accessory molecule-1 mediated recognition of freshly isolated ovarian carcinoma by resting natural killer cells. Cancer Res. 2007;67:1317–1325. doi: 10.1158/0008-5472.CAN-06-2264. [DOI] [PubMed] [Google Scholar]
- 27.Dardalhon V, Schubart AS, Reddy J, Meyers JH, Monney L, Sabatos CA, Ahuja R, Nguyen K, Freeman GJ, Greenfield EA, Sobel RA, Kuchroo VK. CD226 is specifically expressed on the surface of Th1 cells and regulates their expansion and effector functions. J.Immunol. 2005;175:1558–1565. doi: 10.4049/jimmunol.175.3.1558. [DOI] [PubMed] [Google Scholar]
- 28.Tahara-Hanaoka S, Shibuya K, Kai H, Miyamoto A, Morikawa Y, Ohkochi N, Honda S, Shibuya A. Tumor rejection by the poliovirus receptor family ligands of the DNAM-1 (CD226) receptor. Blood. 2006;107:1491–1496. doi: 10.1182/blood-2005-04-1684. [DOI] [PubMed] [Google Scholar]
- 29.Abbas AK, Lohr J, Knoechel B, Nagabhushanam V. T cell tolerance and autoimmunity. Autoimmun.Rev. 2004;3:471–475. doi: 10.1016/j.autrev.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 30.Peggs KS, Quezada SA, Korman AJ, Allison JP. Principles and use of anti-CTLA4 antibody in human cancer immunotherapy. Curr.Opin.Immunol. 2006;18:206–213. doi: 10.1016/j.coi.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 31.The expanding genetic overlap between multiple sclerosis and type I diabetes. Genes Immun. 2009;10:11–14. doi: 10.1038/gene.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hafler JP, Maier LM, Cooper JD, Plagnol V, Hinks A, Simmonds MJ, Stevens HE, Walker NM, Healy B, Howson JM, Maisuria M, Duley S, Coleman G, Gough SC, Worthington J, Kuchroo VK, Wicker LS, Todd JA. CD226 Gly307Ser association with multiple autoimmune diseases. Genes Immun. 2009;10:5–10. doi: 10.1038/gene.2008.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maier LM, Hafler DA. Autoimmunity risk alleles in costimulation pathways. Immunol.Rev. 2009;229:322–336. doi: 10.1111/j.1600-065X.2009.00777.x. [DOI] [PubMed] [Google Scholar]
- 34.Maiti AK, Kim-Howard X, Viswanathan P, Guillen L, Qian X, Rojas-Villarraga A, Sun C, Canas C, Tobon GJ, Matsuda K, Shen N, Chernavsky AC, Anaya JM, Nath SK. Non-synonymous variant (Gly307Ser) in CD226 is associated with susceptibility to multiple autoimmune diseases. Rheumatology.(Oxford) 2010;49:1239–1244. doi: 10.1093/rheumatology/kep470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Webb LM, Walmsley MJ, Feldmann M. Prevention and amelioration of collagen-induced arthritis by blockade of the CD28 co-stimulatory pathway: requirement for both B7-1 and B7-2. Eur.J.Immunol. 1996;26:2320–2328. doi: 10.1002/eji.1830261008. [DOI] [PubMed] [Google Scholar]
- 36.Arima T, Rehman A, Hickey WF, Flye MW. Inhibition by CTLA4Ig of experimental allergic encephalomyelitis. J.Immunol. 1996;156:4916–4924. [PubMed] [Google Scholar]
- 37.Cross AH, San M, Keeling RM, Karr RW. CTLA-4-Fc treatment of ongoing EAE improves recovery, but has no effect upon relapse rate. Implications for the mechanisms involved in disease perpetuation. J.Neuroimmunol. 1999;96:144–147. doi: 10.1016/s0165-5728(99)00015-6. [DOI] [PubMed] [Google Scholar]
- 38.Fuchs A, Cella M, Giurisato E, Shaw AS, Colonna M. Cutting edge: CD96 (tactile) promotes NK cell-target cell adhesion by interacting with the poliovirus receptor (CD155) J.Immunol. 2004;172:3994–3998. doi: 10.4049/jimmunol.172.7.3994. [DOI] [PubMed] [Google Scholar]
- 39.Seth S, Maier MK, Qiu Q, Ravens I, Kremmer E, Forster R, Bernhardt G. The murine pan T cell marker CD96 is an adhesion receptor for CD155 and nectin-1. Biochem.Biophys.Res.Commun. 2007;364:959–965. doi: 10.1016/j.bbrc.2007.10.102. [DOI] [PubMed] [Google Scholar]
- 40.Tao R, Wang L, Han R, Wang T, Ye Q, Honjo T, Murphy TL, Murphy KM, Hancock WW. Differential effects of B and T lymphocyte attenuator and programmed death-1 on acceptance of partially versus fully MHC-mismatched cardiac allografts. J.Immunol. 2005;175:5774–5782. doi: 10.4049/jimmunol.175.9.5774. [DOI] [PubMed] [Google Scholar]
- 41.Masson D, Jarry A, Baury B, Blanchardie P, Laboisse C, Lustenberger P, Denis MG. Overexpression of the CD155 gene in human colorectal carcinoma. Gut. 2001;49:236–240. doi: 10.1136/gut.49.2.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merrill MK, Bernhardt G, Sampson JH, Wikstrand CJ, Bigner DD, Gromeier M. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuro.Oncol. 2004;6:208–217. doi: 10.1215/S1152851703000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pende D, Spaggiari GM, Marcenaro S, Martini S, Rivera P, Capobianco A, Falco M, Lanino E, Pierri I, Zambello R, Bacigalupo A, Mingari MC, Moretta A, Moretta L. Analysis of the receptor-ligand interactions in the natural killer-mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: evidence for the involvement of the Poliovirus receptor (CD155) and Nectin-2 (CD112) Blood. 2005;105:2066–2073. doi: 10.1182/blood-2004-09-3548. [DOI] [PubMed] [Google Scholar]
- 44.Jelinek LJ, Lok S, Rosenberg GB, Smith RA, Grant FJ, Biggs S, Bensch PA, Kuijper JL, Sheppard PO, Sprecher CA. Expression cloning and signaling properties of the rat glucagon receptor. Science. 1993;259:1614–1616. doi: 10.1126/science.8384375. [DOI] [PubMed] [Google Scholar]
- 45.Iritani BM, berola-Ila J, Forbush KA, Perimutter RM. Distinct signals mediate maturation and allelic exclusion in lymphocyte progenitors. Immunity. 1999;10:713–722. doi: 10.1016/s1074-7613(00)80070-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ellsworth JL, Hamacher N, Harder B, Bannink K, Bukowski TR, Byrnes-Blake K, Underwood S, Oliver C, Waggie KS, Noriega C, Hebb L, Rixon MW, Lewis KE. Recombinant soluble human FcgammaR1A (CD64A) reduces inflammation in murine collagen-induced arthritis. J.Immunol. 2009;182:7272–7279. doi: 10.4049/jimmunol.0803497. [DOI] [PubMed] [Google Scholar]
- 47.Svensson L, bdul-Majid KB, Bauer J, Lassmann H, Harris RA, Holmdahl R. A comparative analysis of B cell-mediated myelin oligodendrocyte glycoprotein-experimental autoimmune encephalomyelitis pathogenesis in B cell-deficient mice reveals an effect on demyelination. Eur.J.Immunol. 2002;32:1939–1946. doi: 10.1002/1521-4141(200207)32:7<1939::AID-IMMU1939>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplement